

Abstract
Aurachin RE (1) is a strong antibiotic that was recently found to possess MenA (1,4-dihydroxy-2-naphthoate prenyltransferase) and bacterial electron transport inhibitory activities. Aurachin RE is the only molecule in a series of aurachin natural products that has the chiral center in the alkyl side chain at C9′-position. To identify selective MenA inhibitors against Mycobacterium tuberculosis, a series of chiral molecules were designed based on the structures of previously identified MenA inhibitors and 1. The synthesized molecules were evaluated in in vitro assays including MenA enzyme and bacterial growth inhibitory assays. We could identify novel MenA inhibitors that showed significant increase in potency of killing non-replicating M. tuberculosis in the low oxygen recovery assay (LORA) without inhibiting other Gram-positive bacterial growth even at high concentrations. The MenA inhibitors reported here are useful new pharmacophores for the development of selective antimycobacterial agents with strong activity against non-replicating M. tuberculosis.
Keywords: New antimycobacterial agent, Menaquinone biosynthesis inhibitor, MDR Mycobacterium tuberculosis, Non-replicating Mycobacterium tuberculosis, TB drugs, quinolone alkaloid, aurachin RE
INTRODUCTION
Mycobacterium tuberculosis (Mtb) causes tuberculosis (TB) and is responsible for nearly two million deaths annually.1,2 Moreover, the emergence of multidrug-resistant (MDR) strains of Mtb seriously threatens TB control and prevention efforts. One-third of the 42 million people living with HIV/AIDS worldwide are co-infected with Mtb.3 Recent studies have shown that infection with Mtb enhances replication of HIV and may accelerate the progression of HIV infection to AIDS. There are significant problems associated with treatment of AIDS and Mtb co-infected patients.4 Rifampicin and isoniazid (key components of the local directly observed treatment strategy) induce the cytochrome P450 3A4 enzyme which shows significant interactions with anti-HIV drugs such as protease inhibitors. In addition, rifampicin strongly interacts with non-nucleoside reverse transcriptase and protease inhibitors for HIV infections. Therefore, clinicians avoid starting Highly Active Antiretroviral Therapy (HAART), which consists of three or more highly potent reverse transcriptase inhibitors and protease inhibitors, until the TB infection has been cleared.5,6 M. tuberculosis is recognized to lie in a non-replicating state (dormancy), particularly in the caseous pulmonary nodules where the lesions have little access to oxygen, and can survive for many years in the host by entering a dormant state. About 10% of patients with latent Mtb are reactivated, causing the risk of fatal diseases.7,8,9,10 Thus, in addition to the necessity of drugs for the treatment of MDR-Mtb, the development of drugs that kill Mtb in any state is very important. However, no current TB drugs are effective in killing the dormant form of Mtb in vivo. Therefore, ideal novel antituberculosis compounds should show: 1) compatibility and lack of cross-resistance with other anti-Mtb agents, because combination therapy remains mandatory to combat Mtb, 2) antimicrobial spectrum focused against Mtb, 3) strong growth inhibitory activity against non-replicating Mtb, and 4) no interactions with HAART.
Function of ubiquinone (coenzyme Q10) as a component of the mitochondrial respiratory chain in human is well established (“the chemiosmotic theory”, Mitchell, 1978).11,12,13 In prokaryotes, especially in Gram-positive bacteria, menaquinone transfers two electrons in a process of either aerobic or anaerobic respiration (Figure 1). On the other hand, a majority of Gram-negative organisms utilize ubiquinone under aerobic conditions and menaquinone under anaerobic conditions in their electron transport systems.14,15,16 Therefore, inhibitors of menaquinone biosynthesis or specific inhibitors of enzymes associated with electron transport systems have great potential for the development of novel and selective drugs against MDR Gram-positive bacteria.17 Recently, Dr. Schnappinger successfully generated menA knockdown mutant M. tuberculosis possessing TetON (tetracycline-inducible expression system). It was unequivocally demonstrated that MenA is essential for growth of Mtb via mouse infection experiments with the menA knockdown Mtb mutant.18 The electron transport system couples with ATP synthase to produce ATP through oxidative phosphorylation. Bacterial ATP synthase, F1F0-ATPase, is a viable target for treatment of MDR Mtb infections. A diarylquinolone, a Phase II clinical drug, is an inhibitor of Mtb ATP synthase that exhibited a remarkable activity against Mtb.19 However, only few studies have investigated the electron transport system for development of new antibacterial drugs.17 Weinstein and co-workers reported the inhibitors of type II NADH:menaquinone oxidoreductase that effectively killed Mtb in vitro and they concluded that type II NADH dehydrogenase could be a unique and interesting antimicrobial target.20 We have reported that inhibition of MenA (1,4-dihydroxy-2-naphthoate prenyltransferase), which catalyzes a formal decarboxylative prenylation of 1,4-dihydroxy-2-naphthoate (DHNA) to form demethylmenaquinone (DMMK) in menaquinone biosynthesis (Figure 2), showed significant growth inhibitory activities against drug resistant Gram-positive bacteria including M. tuberculosis.21,22 In menaquinone biosynthesis, MenD (2-succinyl-5-enoylpyruvyl-6-hydroxy-3-cyclohexane-1-carboxylic acid synthase), MenE (an acyl-CoA synthase), and MenB (1,4-dihydroxynaphthoyl-CoA synthase) have recently been studied for the development of novel drug lead for Gram-positive pathogens including M. tuberculosis.23,24,25,26
Figure 1.

The electron flow system of M. tuberculosis. Menaquinone (MK) mediated energetic pathway (blue).
Figure 2.

Biosynthesis of menaquinone from chorismate (classic pathway).
In an attempt at finding a new pharmacophore for MenA inhibitors, we recently identified a new quinolone natural product, aurachin RE (1, Figure 3),27 which exhibited MenA enzyme inhibitory activity as well as a wide antimicrobial spectrum against Gram-positive bacteria. As there is an overall structural resemblance between aurachin RE and our 1st generation MenA inhibitors (e.g. 10 in Figure 4) or menaquinone, aurachin RE’s antibacterial activity could be attributed to a synergistic effect of respiratory chain and menaquinone biosynthesis inhibitory activities. The identification of aurachin RE’s MenA enzyme inhibitory activity encouraged us to redesign and synthesize chiral MenA inhibitor molecules, in which primary or secondary alcohol was introduced in the side chain of the 1st generation MenA inhibitor molecules.21,22 To date, we have synthesized over 400 molecules with >95% purity either in solution or on polymer-support, and these molecules were evaluated in an enzymatic assay in vitro (IC50) against MenA and in bacterial growth inhibitory assays (MIC). Figure 3 illustrates our assay scheme to identify selective MenA inhibitors against M. tuberculosis. In these molecules 26 compounds were identified to exhibit the in vitro biological activities which met the assay criteria summarized in Figure 3. Based on obtained SAR from a 400-membered library, it became evident that the topology of the N atom in the inhibitor molecules plays an important role in selectivity of the MenA enzymatic and bactericidal activities (Mtb vs. S. aureus). As summarized in Figure 4, selective mycobactericidal molecules (2–6) possess the secondary or tertiary amine in the near center of the molecules (highlighted moieties in 1–6 in Figure 4), whereas the topology of the N atom of the molecules possessing antibacterial activities against both Mtb and S. aureus (7–10) locates the right half of the molecules (highlighted moieties in 7–10 in Figure 4). We have identified selective antimycobacterial MenA inhibitors in their racemic forms. In order to obtain insight into the effect of chirality of new MenA inhibitors (2–6), we commenced syntheses of the optically active forms of the identified inhibitors. Herein we report the synthesis and in vitro biological activity evaluation of optically active molecules of 2–6 and their derivatives. The results disclosed in this article identify novel chiral antimycobacterial MenA inhibitors with significant activity in killing non-replicating Mtb.
Figure 3.

Assay to identify selective MenA inhibitors against M. tuberculosis.
Figure 4.

Rational of selective antimycobacterial MenA inhibitors.
RESULTS AND DISCUSSION
Selective M. tuberculosis MenA Inhibitors-Assay Strategy
Antimicrobial spectrum focused against Mtb (selective antimycobacterial agent) is preferable for TB chemotherapy.28 We realized that the peptide sequences of the Mtb menA and S. aureus menA gene products are only 32% identity and 50% similarity in the BLAST experiment.29 Indeed, we have identified several molecules that exhibit selective MenA enzyme and bacterial growth inhibitory activities against Mtb; more than a 10-fold higher inhibitory activity against Mtb than S. aureus. In order to develop MenA inhibitors which are selective against Mtb, molecules generated in this program were first evaluated in MenA enzyme inhibitory assays. Only molecules exhibited Mtb MenA activity over S. aureus MenA (IC50 < 20 μM, >60 μM against Mtb MenA and S. aureus MenA, respectively) were evaluated in bacterial growth inhibitory assays (MICs) using Mtb, S. aureus, and E. coli (Figure 3). The molecules exhibited good activity only against Mtb (MICs for Mtb, S. aureus, and E. coli are <12.5, >60, and >125μg/mL, respectively) were evaluated in E. coli growth inhibitory assays under anaerobic conditions followed by menaquinone supplementation assays (E. coli utilize only menaquinone in their electron transport system under anaerobic conditions).30,31 This assay confirmed that the molecules killed bacteria by targeting MenA enzyme or electron transport systems (vide infra). Oxygen consumption assay using Mtb identifies electron transport system inhibitors;32,33 in this assay, selective MenA inhibitors should not show activity at concentrations below the MIC against Mtb. Selective antimycobacterial MenA inhibitors confirmed via E. coli growth inhibitory (under anaerobic conditions) and oxygen consumption assays were evaluated for their activity against non-replicating Mtb via the low oxygen recovery assays (LORA).34
Chemistry
As mentioned above, we have identified several antimycobacterial MenA inhibitors in their racemic forms. In order to obtain insight into the effect of chirality of 2–6 (Figure 4) and functional groups around the chiral center on biological activity, we synthesized both enantiomers of 2–6 and their analogs in which the secondary alcohols were functionalized with acyl, carbamate, and benzyl groups. Benzophenone functional group shows a moderate electrophilicity and thus may require chemical modification to improve its physico-chemical property in early stage of drug development. In this study we have introduced benzophenone O-methyl oxime derivatives to obtain preliminary SAR and their cytotoxicity data.
Syntheses of optically active molecules of 2–6 and their analogs are summarized in Scheme 1~5. The Cl-substituted benzophenone derivatives, 21 and 23, were synthesized according to the previously reported procedures.21 O-Methyl oxime derivatives 22 and 24 were prepared by the treatment of 21 or 23 with MeONH2 HCl in pyridine at 105 °C. The phenolic alcohol of 21 was subjected to the Mitsunobu reaction with Boc-protected piperidin-4-ylmethanol 25 to provide the piperidinyl ethers 26 in over 90% yield, after deprotection of the Boc group.35,36 Optically active (2R)- or (2S)-alkyloxiranes, 28 and 29, were synthesized via Jacobsen’s kinetic resolutions of the racemic epoxides.37 Opening of the epoxides (S)-29 with the piperidine derivatives 26 was achieved by using a stoichiometric amount of AlMe3 at room temperature to yield the amino-alcohols (S)-11. The generated optically active alcohol (S)-11 was functionalized with Ac2O, BnBr, and TMSNCO to afford the corresponding acetate (S)-38, benzyl ether (S)-40, and carbamate (S)-12 (class A molecules, Scheme 1). In order to synthesize optically active piperidinyl-benzyl alcohol derivatives 3 (class B molecules), (S)- and (R)-benzaldehyde derivatives 45 were synthesized in 4 steps from 44 via CBS reductions (Scheme 2).38 The starting material 44 was readily synthesized via a Grignard reaction of 42 with the Weinreb amide 43.39 Reductive aminations of 26 with (S)-45 was achieved with NaBH(OAc)3 in the absence of acid to provide the desired tertiary amine (S)-46 without contamination of the diphenylmethanol by-product. Deacetylation of (S)-46 afforded the secondary alcohol (S)-3 whose alcohol was functionalized with TMSNCO to afford the corresponding carbamate (S)-14. Optically active amino-alcohols (S)- and (R)-4 (class C molecules) were synthesized via the resolution of rac-epoxide 49 followed by Zn(ClO4)2-catalyzed selective opening of epoxide with the primary amine (Scheme 3).40 The other optically active secondary alcohols (class D and E molecules) were successfully synthesized via the same synthetic procedures developed for class A and B molecules (Scheme 4 and 5). Syntheses of the molecules having (R)-configuration were also achieved by using the antipodes of the building blocks utilized for the synthesis for the (S)-configuration molecules. Similarly, syntheses of optically active benzophenone O-methyl oxime derivatives 13, 15, 18, 19, 59, 63, and 65 were successfully achieved. Thus, we have synthesized both (S)- and (R)- forms of 2–7 and their derivatives in short number of steps. Optical purity and purity of each molecule were established via HPLC analyses of Mosher esters of secondary alcohols or alcohols using a chiral column.41
Scheme 1.

Syntheses of optically active MenA inhibitors-class Aa.
a Reagents and conditions: (a) Ac2O/Py. (1:1), room temperature; (b) BnBr, NaH, DMF/THF (4:1). 0 °C; (c) TMSNCO, DMAP, CH2Cl2, room temperature.
Scheme 5.

Syntheses of optically active MenA inhibitors-class Ea..
a Reagents and conditions: (c) TMSNCO, DMAP, CH2Cl2, room temperature; (d) 1N NaOH, CH3CN, 0 °C.
Scheme 2.

Syntheses of optically active MenA inhibitors-class Ba.
a Reagents and conditions: (c) TMSNCO, DMAP, CH2Cl2, room temperature; (d) 1N NaOH, CH3CN, 0 °C.
Scheme 3.

Syntheses of optically active MenA inhibitors-Class C
Scheme 4.

Syntheses of optically active MenA inhibitors-class Da.
a Reagents and conditions: (a) Ac2O/Py. (1:1), room temperature; (b) BnBr, NaH, DMF/THF (4:1). 0 °C; (c) TMSNCO, DMAP, CH2Cl2, room temperature
MenA enzyme inhibitory assays
MenA enzymatic assays of generated molecules were originally performed with [3H]rated farnesyl diphosphate and MenA containing membrane fraction.42 This assay requires a significant effort to separate demethylmenaquinone (DMMK) from the reaction mixtures and includes the significant cost for [3H]farnesyl diphosphate. In order to simplify the procedure and to reduce the cost of MenA enzyme inhibitory assays, we have recently developed a MenA assay using HPLC. In new MenA assay, the MenA product, demethylmenaquinone can readily be quantitated via UV absorbance (DMMK; λ2 325 nm).43
We performed a preliminary screening of the activity of compounds synthesized in Scheme 1~5 at the single concentrations of 100 μM. In these MenA enzyme inhibitory assays, the optically active molecules were assayed against M. tuberculosis and S. aureus MenA. IC50 values of the molecules which exhibited activity against Mtb MenA and inactivity against S. aureus MenA were calculated. Of 86 optically active molecules synthesized in Scheme 1~5, 26 molecules exhibited Mtb MenA inhibitory activity and were inactive against S. aureus MenA at 100 μM concentrations. Dose-response plots (DMMK formation vs. concentrations of inhibitor) were obtained for 26 molecules to determine IC50 values. Significant effect of chirality in the MenA enzyme inhibitory activity was observed in the molecules in classes A and D. On the contrary, an obvious effect of chirality in MenA enzyme inhibitory activity of the molecules in classes B, C, and E was not observed; the racemic forms and each enantiomer did not show noticeable difference in MenA enzyme inhibitory activity. It is noteworthy that the carbamate analog (R)-12 group showed 6-fold better MenA enzyme inhibitory activity compared to the alcohol from (R)-11. Similar improvement of enzymatic inhibition by modification of the alcohol with the carbamate group is observed in the molecules in class D (Table 1).
Table 1.
Antibacterial activities against M. tuberculosis and S. aureus, and MenA enzyme inhibitory activities of the selected molecules.
| Compounds | Structure
|
MIC (g/mL)a |
MenA Inhibition
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| X | R1 | R2 | S. aureus | M. tuberculosis (MABA)b | M. tuberculosis (LORA)c | S. aureus (at 100 μM) | M. tuberculosis(at 100 μM) | IC50 (μM) | |
| 2 |
Class A X, R1, R2 = O, CH3, H |
>60 | 12.5 | 5.2 | − | + | 15.0 | ||
| (S)-2 | X, R1, R2 = O, CH3, H | >60 | 12.5 | 5.59 | − | + | 7.5 | ||
| (R)-2 | X, R1, R2 = O, CH3, H | >60 | 6.2 | 4.91 | − | + | 17.0 | ||
| (S)-11 | X, R1, R2 = O, C3H7, H | >60 | 12.5 | 4.91 | − | + | 16.0 | ||
| (R)-11 | X, R1, R2 = O, C3H7, H | >60 | 6.2 | 5.59 | − | + | 9.0 | ||
| (S)-12 | X, R1, R2 = O, C3H7, CONH2 | >60 | 12.5 | 2.94 | − | + | 9.0 | ||
| (R)-12 | X, R1, R2 = O, C3H7, CONH2 | >60 | 2.31 | 0.85 | − | + | 1.5 | ||
| (R)-13 | X, R1, R2 = NOMe, C3H7, CONH2 | >60 | 2.31 | 0.85 | − | + | 1.2 | ||
| 3 |
Class B X, R2 = O, H |
>60 | 12.5 | 1.88 | − | + | 15.0 | ||
| (S)-3 | X, R2 = O, H | >60 | 12.5 | 1.88 | − | + | 25.0 | ||
| (S)-3 | X, R2 = O, H | >60 | 12.5 | 1.46 | − | + | 20.0 | ||
| (S)-14 | X, R2 = O, CONH2 | >60 | 12.5 | 4.93 | − | + | 20.0 | ||
| (R)-14 | X, R2 = O, CONH2 | >60 | 12.5 | 4.93 | − | + | 15.0 | ||
| (R)-15 | X, R2 = NOMe, OH | >60 | 6.25 | 1.43 | − | + | 15.0 | ||
| 4 |
Class C |
>60 | 12.5 | 3.00 | − | + | 15.0 | ||
| (S)-4 | >60 | 12.5 | 3.00 | − | + | 17.0 | |||
| (R)-4 | >60 | 12.5 | 3.00 | − | + | 14.0 | |||
| rac-5 |
Class D X, R1, R2 = O, CH3, H |
>60 | 6.25 | 2.82 | − | + | 15.0 | ||
| rac-16 | X, R1, R2 = O, CH3, CONH2 | >60 | 3.25 | 2.83 | − | + | 7.5 | ||
| (S)-5 | X, R1, R2 = O, CH3, H | >60 | 1.50 | 1.43 | − | + | 4.5 | ||
| (R)-5 | X, R1, R2 = O, CH3, H | >60 | 6.25 | 2.85 | − | + | 7.5 | ||
| (S)-16 | X, R1, R2 = O, C3H7, H | >60 | 1.50 | 1.43 | − | + | 3.5 | ||
| (R)-16 | X, R1, R2 = O, C3H7, H | >60 | 6.25 | 5.20 | − | + | 9.5 | ||
| (S)-17 | X, R1, R2 = O, C3H7, CONH2 | >60 | 1.50 | 1.45 | − | + | 1.5 | ||
| (S)-18 | X, R1, R2 = NOMe, C3H7, CONH2 | >60 | 1.50 | 1.40 | − | + | 1.5 | ||
| rac-6 |
Class E X, R2 = O, H |
>60 | 12.5 | 1.88 | − | + | 20.0 | ||
| (S)-6 | X, R2 = O, H | >60 | 12.5 | 1.88 | − | + | 15.0 | ||
| (R)-6 | X, R2 = O, H | >60 | 12.5 | 1.46 | − | + | 15.0 | ||
| (S)-19 | X, R2 = O, CONH2 | >60 | 12.5 | 2.93 | − | + | 20.0 | ||
| (R)-19 | X, R2 = O, CONH2 | >60 | 12.5 | 4.93 | − | + | 20.0 | ||
| (S)-20 | X, R2 = NOMe, OH | >60 | 12.5 | 5.54 | − | + | 20.0 | ||
| (R)-20 | X, R2 = NOMe, OH | >60 | 12.5 | 5.54 | − | + | 15.0 | ||
| RFPd | - | 0.2 | 1.47 | ||||||
| INHe | - | 0.1 | >128 | ||||||
| EMBf | - | 0.78 | >128 | ||||||
The agar plate dilution method was used.;
MABA: microplate alamar blue assay.;
LORA: Low-oxygen recovery assay.;
RFP: rifampicin.;
INH: isoniazid.;
EMB: ethanbutol.
Bacterial Growth Inhibitory Assays
All molecules that showed activity against MenA in a preliminary assay at 100 μM concentrations were evaluated for their mycobacterial growth inhibitory activity via the microplate alamar blue assay (MABA) and low oxygen recovery assay (LORA).34,44,45 Briefly, the MABA assay is a colorimetric assay that uses the color change of rezasurin to evaluate M. tuberculosis (Mtb) growth inhibitory activity under aerobic conditions. On the other hand, the LORA assay evaluates potency against non-replicating Mtb cells under low oxygen conditions using a luminescent stain. Significantly, in all cases the MIC values obtained from the LORA assays are lower than those from the MABA assays. To the best of our knowledge, it is the first observations that the molecules killed non-replicating Mtb at concentration below the MIC against Mtb grown under aerobic conditions; in all cases the values of MICLORA/MICMABA were <1 (Table 1). In the class A molecules (R)-2 was two-fold more potent than rac-2 and (S)-2 in the MABA assays. Similar to an observed trend in MenA enzyme inhibitory assays, the carbamate (R)-12 could improve over 5-fold increase in mycobactericidal activity compared to rac-2. (R)-12 exhibited a significant activity in the LORA assay; the MIC value of (R)-12 is 1.7-fold more effective in killing non-replicating bacteria than rifampicin (MICLORA 1.47 μg/mL). It is worth mentioning that (R)-12 was the most active in killing non-replicating Mtb in vitro among antimycobacterial drugs (approved by FDA) and preclinical drugs tested in our laboratory. Regardless of the stereochemistry of the chiral centers, the molecules in class B, C, and E did not show noticeable difference in antimycobacterial activity in the both MABA and LORA assays. The molecules in class D are regioisomers at the benzophenone moiety of the class A molecules. Similar to the molecules in class A, pronounced effect of the stereochemistry of secondary alcohol on antimycobacterial activity was observed in the molecules in class D. Contrary to the chirality effect observed in the molecules in class A, the molecules possessing S-configuration (in class D) exhibited better antimycobacterial activity than the corresponding R-configuration molecules. The effect of carbamate group on enzyme and bacterial growth inhibitory activities was not observed in the molecules in class D; the MABA MIC value of the carbamate (S)-16 was equal to the corresponding alcohol (S)-17. Nonetheless, the MABA MIC for the rac-alcohol 5 could be improved two-fold by the formation of carbamate; the MABA MICs of rac-5 and rac-16 were 6.25 and 3.25μg/mL, respectively. Antimycobacterial activity was improved by increasing the hydrophobicity of the side chain (C8 vs. C6) in the molecules of class A, whereas noticeable bactericidal effect by increasing hydrophobicity of the side chain was not observed in the molecules of class D. All molecules summarized in Table 1 did not exhibit bactericidal activity against S. aureus at 60 μg/mL concentrations; lack of anti-staphylococcal activity of these molecules was well-correlated with their S. aureus MenA enzyme inhibitory activity (IC50 >60 μM against S. aureus MenA).46
E. coli Growth Inhibitory Assays under Anaerobic Conditions
M. tuberculosis or S. aureus treated with the MenA inhibitors could not be rescued completely even at higher concentrations of exogenous menaquinone (VK2). In contrast, growth inhibition of E. coli by the MenA inhibitor could be rescued by supplementation of VK2 (50 μM) under “anaerobic conditions” (vide supra).47 E. coli growth inhibition rescued by addition of VK2 may be attributed to the degree of permeability of VK2 through cell envelop. Although lack of activity of MenA inhibitors against Gram-negative bacteria grown under aerobic conditions have been demonstrated, all MenA inhibitors identified in this program showed growth inhibition of E. coli at 5~10 μg/mL concentrations under anaerobic conditions, and the inhibitory effect of MenA inhibitor was rescued by supplementation of VK2. Therefore, these convenient cell-based assays using E. coli under anaerobic conditions can be utilized to confirm that the inhibitor molecules kill Gram-positive bacteria including Mtb by targeting MenA biosynthesis or bacterial electron transport systems.
Oxygen Consumption Assays
The basic concept underlying bacterial oxygen consumption assay is that changes in the rate of oxygen uptake result in a change in the oxygen concentrations. The oxygen consumption by bacterial concentrations greater than 108 cfu/mL bacteria is proportional to the concentration of bacteria. Effect of the inhibitor molecule on electron transport by the quantitation of decolorization of methylene blue, which is a well-known redox dye, has been unambiguously demonstrated.31 Oxygen-consumption assays of (R)-12 and (S)-17 showed decolorization of methylene blue at concentrations (12.5μg/mL) above the MIC of each molecule (MICs 2.31 and 1.50 μg/mL, respectively) against Mtb. Thus, we have concluded that (R)-12 and (R)-17 are very weak (or are not) electron transport system inhibitors, and thus, antimycobacterial activity of these molecules are attributed by targeting menaquinone biosynthesis.
Cytotoxicity of MenA Inhibitors
In order to obtain insight into potential toxicity of identified inhibitor molecules, all antimycobacterial MenA inhibitors were evaluated in in vitro cytotoxicity assays using Vero monkey kidney cells and HepG2 human hepatoblastoma cells.48 Most MenA inhibitors possessing the benzophenone group showed IC50 of 1–6.5μg/mL against two mammalian cell lines; selectivity indexes (IC50 in mammalian cells/MIC against Mtb) of a series of benzophenone MenA inhibitors identified in this program were less than 2.49 On the other hand, O-methyl oxime derivatives in class A showed approximately 10-fold less cytotoxic than the corresponding benzophenone derivatives in vitro cytotoxicity assays (Figure 5); (R)-13 showed an encouraging in vitro activity/toxicity ratio (a SI value >10). It is believed that electrophilicity of the benzopheneone moiety of (R)-12 can be diminished by O-methyl oxime formation of the benzopheneone. Thus, the benzophenone O-methyl oxime may not be a good electron acceptor that interferes with redox systems of mammalian cells.
Figure 5.
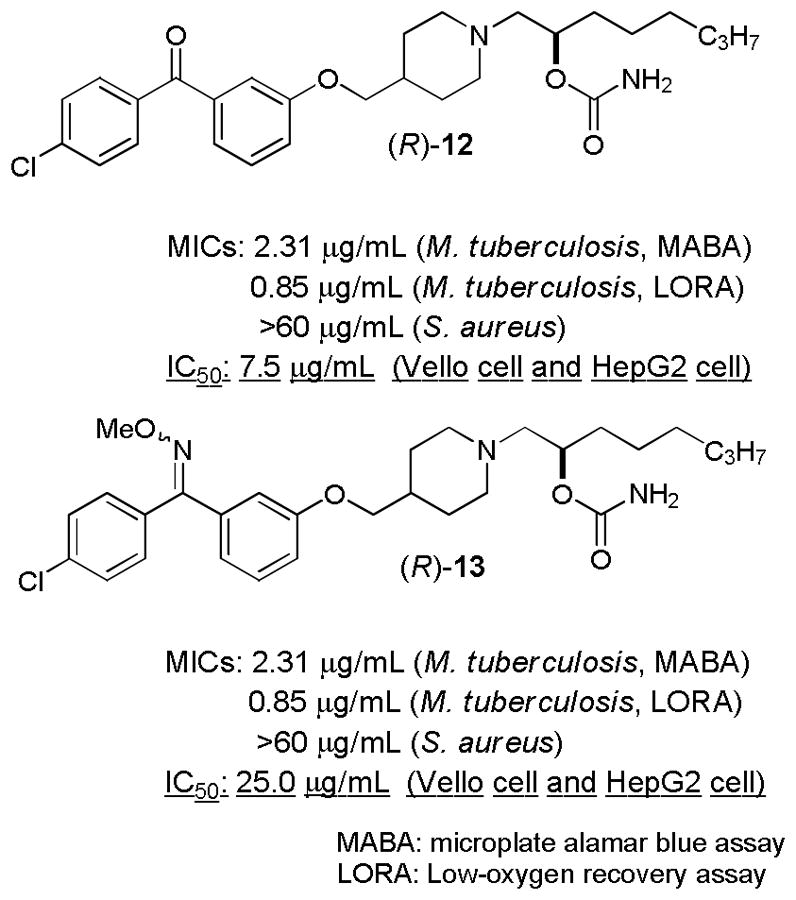
In vitro assay data for (R)-12 and (R)-13.
Antibacterial Activities of (R)-12 and (R)-13 against Drug-Resistant M. tuberculosis and other Mycobacterium Species
Two MenA inhibitors molecules (R)-12 and (R)-13 showed MIC values of 2.31 μg/mL against Mtb (H37Rv, a common laboratory strain). We resynthesized (R)-12 and (R)-13 and determined MICs against several other Mycobacterium species and drug-resistant Mtb. As summarized in Table 2, (R)-12 and (R)-13 showed mycobactericidal activity against rifampicin- and isoniazid-resistant strains at slightly lower concentrations (entries 2 and 3). (R)-12 and (R)-13 also killed other Mycobacterium species such as M. bovis, M. intracellulare, and M. smegmatis at relatively low concentrations (3.0~6.5 μg/mL). Thus, we concluded that new MenA inhibitors identified in this program kills Mycobacterium species selectively and are especially effective in killing Mtb at low concentrations. MenA inhibitors (R)-12 and (R)-13 effectively inhibited growth of drug-resistant Mycobacterium species, indicating that MenA is a valid drug target to develop new drugs for MDR-Mycobacterium tuberculosis.
Table 2.
MICs of (R)-12, (R)-13, and representative antimycobacterial agents (clinically used) for Mycobacterium species including drug-resistant strains.
| Entry | Species and strain | MIC (μg/mL)a | |||
|---|---|---|---|---|---|
| (R)-12 | (R)-13 | RFP | INH | ||
| 1 | M. tuberculosis H37Rv | 2.31 | 2.31 | 0.07 | 0.24 |
| 2 | M. tuberculosis H37Rv INHrb | 2.91 | 1.52 | 0.03 | >8 |
| 3 | M. tuberculosis H37Rv RFPrc | 2.91 | 1.51 | >4 | 0.05 |
| 4 | M. bovis BCG | 3.00 | 2.95 | - | - |
| 6 | M. intracellulare ATCC15984 | 6.50 | 6.50 | - | - |
| 7 | M. smegmatis | 6.50 | 6.50 | - | - |
The agar plate dilution method was used.;
INH-resistant M. tuberculosis.; RFP-resistant M. tuberculosis.; RFP: rifampicin.; INH: isoniazid.
CONCLUSIONS
Through asymmetric synthesis of a series of optically active molecules followed by screening these molecules by the assay methods described here, we have identified new MenA (1,4-dihydroxy-2-naphthoate prenyltransferase) biosynthesis inhibitors that showed very weak (or no) inhibitory activities of bacterial electron transport systems. A series of MenA inhibitors identified in this program exhibited antimicrobial spectrum focused against Mtb. Selective activity against Mtb is ideal in TB drug discovery due to the fact that TB chemotherapy requires long regimen, so that broad-spectrum anti-TB agents may cause resistant to other bacteria during TB chemotherapy. The carbamates (R)-12 and (R)-13 in class A (Scheme 1) showed significant growth inhibitory activities against non-replicating Mtb (MICLORA, 0.85 μg/mL) with the MICLORA/MICMABA/value of 0.37 (MICLORA/MICMABA = 7.35 for rifampin). Effectiveness in non-replicating Mtb was also confirmed via assays using a modified Wayne model.50,51,52 Among antimycobacterial agents tested in our laboratories, the inhibitor (R)-12 and (R)-13 were the most active in killing non-replicating Mtb in vitro. MenA inhibitors identified in this program strongly suggested that menaquinone biosynthesis is important in maintaining mycobacterial viability under conditions of restricted oxygen.53 MenA inhibitors seem to be able to block the electron flow, consequently inhibiting the bacterial growth. It is conceptually very interesting that MenA inhibitors can be developed as indirect ATP synthesis inhibitors.17 The assay data for the identified MenA inhibitors indicate that menaquinone biosynthesis is a unique and new antimycobacterial target. In addition, in tuberculosis, the key to shortening the long regimen lies in targeting the non-replicating persistence subpopulation (vide supra). Thus, the discovery of molecules that kill non-replicating Mtb at lower concentrations than MIC against Mtb under aerobic conditions is expected to be of significance in terms of discovering new lead molecules that can be developed into new drugs to kill Mtb in any state. Moreover, over the last several years, we are unable to isolate resistant bacteria to the MenA inhibitors in the mutation frequency studies. Therefore, we concluded that unlike the other known bacterial molecular targets menA shows extremely low mutation frequency.
In conclusion, robustness of our in vitro assay approaches to identify novel and selective MenA inhibitors against M. tuberculosis (summarized in Figure 3) has been demonstrated by the identification of a lead molecule (R)-13 (MICLORA/MICMABA value of 0.37, SI >10). Further studies are underway to thoroughly characterize the activity of (R)-13 against multidrug resistant strains of Mtb, and to investigate activities of (R)-13 and its related molecules against the other menaquinone biosynthesis enzymes.54 The MenA inhibitors described here can be synthesized in short steps with high yield, and structural modifications to improve in vitro efficacy will be achieved by modifying the hydrophobic side chain and benzophenone O-methyl oxime moieties of (R)-13. Thorough in vitro biological evaluation of (R)-13 and its analogs and in vivo evaluation of promising menaquinone biosynthesis inhibitors will be reported elsewhere.54
EXPERIMENTAL SECTION
Chemistry
General Information
All glassware were oven dried, assembled hot and cooled under a stream of nitrogen before use. Reactions with air sensitive materials were carried out by standard syringe techniques. Commercially available reagents were used as received without further purification. Thin layer chromatography was performed using 0.25 mm silica gel 60 (F254, Merck) plates visualizing at 254 nm, or developed with ceric ammonium molybdate or anisaldehyde solutions by heating with a hot-air gun. Specified products were purified by flash column chromatography using silica gel 60 (230–400 mesh, Merck). IR absorptions on NaCl plates were run on a Perkin Elmer FT-IR 1600. 1H-NMR spectral data were obtained using Varian 300, 400, and 500 MHz instruments. The residual solvent signal was utilized as an internal reference. 13C NMR spectral data were obtained using a Varian 100, 125 MHz spectrometer. Chemical shifts were reported in parts per million (ppm) downfield from TMS, using the middle resonance of CDCl3 (77.0 ppm) as an internal standard. For all NMR spectra, δ values are given in ppm and J values in Hz. Mass spectra were obtained at University of Tennessee Central Instrument Facility. HPLC analyses were performed with a Shimadzu LC-20AD HPLC system. All compounds were purified by PTLC or reverse HPLC to be ≥95% purity whose purities were established by HPLC.
(4-Chlorophenyl)(3-methoxyphenyl)methanone
4-Chlorobenzoyl chloride (5.00 g, 28.58 mmol) was dissolved in CH2Cl2 (240 mL) and cooled to 0 °C. N,O-Dimethylhydroxyl amine (3.07 g, 31.43 mmol) and triethylamine (6.36 g, 62.85 mmol) were added into the reaction mixture. After 4h at r.t., the reaction mixture was quenched with water and the organic phase was washed with 1N HCl. The combined organic phase was dried over Na2SO4 and evaporated in vacuo to afford crude 4-chloro-N-methoxy-N-methylbenzamide (6.41 g). This was used further purification. To a stirred solution of 1-iodo-3-methoxybenzene (21.48 g, 91.80 mmol) in THF (10 mL) was added isopropyl magnesium chloride (2M, 1.50 mmol) at −20 °C. After 1h at the same temperature, the reaction mixture was cooled to −78 °C and 4-chloro-N-methoxy-N-methylbenzamide (100 mg, 0.50 mmol) in THF (1 mL). The reaction mixture was warmed to 0 °C over 30 min. After 3h at 0 °C, the reaction mixture was quenched with aq. sat. NH4Cl. The water phase was extracted with EtOAc and the combined organic phase was dried over Na2SO4 and evaporated in vacuo. Purification by silica gel chromatography (4:1, Hexanes:EtOAc) afforded (4-Chlorophenyl)(3-methoxyphenyl)methanone (97.0 mg, 79%) as a white powder; 1H NMR (500 MHz, CDCl3): δ 3.88 (s, 3H), 7.16 (dd, J = 1.5, 8 Hz, 1H), 7.31–7.35 (m, 2H), 7.40 (t, J = 8 Hz, 1H), 7.47 (d, J = 8.5 Hz, 2H), 7.78 (d, J = 8.5 Hz, 2H); 13C NMR (125 MHz, CDCl3):δ 55.6, 114.4, 119.2, 122.8, 128.8, 129.5, 131.6, 136.1, 138.7, 139.1, 159.8, 195.4; LRMS (ESI) m/z: 247.0 (M+H)+.
(4-Chlorophenyl)(3-hydroxyphenyl)methanone (21)
To a stirred solution of (4-chlorophenyl)(3-methoxyphenyl)methanone (5.30 g, 21.5 mmol) in AcOH (15 mL) was added HBr (48% in water, 200 mL). The reaction mixture was gently refluxed at 125 °C for 36h. The reaction mixture was cooled to r.t. and all volatiles were distilled off. Purification by silica gel chromatography (4:1, Hexanes:EtOAc) afforded 21 (3.84 g, 78%) as a white solid and the unreacted starting material (15%) was recovered. 1H NMR (500 MHz, CDCl3): δ 5.84 (s, 1H), 7.02–7.04 (m, 1H), 7.19–7.21 (m, 1H), 7.25–7.29 (m, 2H), 7.37–7.39 (m, 2H), 7.67–7.69 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 116.6, 120.4, 122.9, 128.9, 129.9, 131.8, 135.8, 138.7, 139.4, 156.2, 195.9; LRMS (ESI) m/z: 233.0 (M+H)+.
tert-Butyl 4-((3-(4-chlorobenzoyl)phenoxy)methyl)piperidine-1-carboxylate
To a stirred solution of (4-chlorophenyl)(3-hydroxyphenyl)methanone (21, 2.00 g, 8.71 mmol), tert-butyl 4-(hydroxymethyl)piperidine-1-carboxylate (25, 3.75 g, 17.4 mmol), and TPP (3.42 g, 13.05 mmol) in THF (40 mL) was added DIAD (4.79 g, 13.05 mmol). After 3h, all volatiles were evaporated in vacuo. Purification by silica gel chromatography (4:1, hexanes:EtOAc) provided tert-butyl 4-((3-(4-chlorobenzoyl)phenoxy)methyl)piperidine-1-carboxylate (3.61 g, 97%) as a colorless oil. 1H NMR (500 MHz, CDCl3): δ 1.64-1.25 (m, 2H), 1.39 (s, 9H), 1.75 (d, J = 13.5 Hz, 2H), 1.87–1.90 (m, 1H), 2.67 (bs, 2H), 3.77 (d, J = 6.5 Hz, 2H), 4.08 (bs, 2H), 7.04 (dd, J = 2, 8 Hz, 1H), 7.20–7.22 (m, 2H), 7.29 (t, J = 8 Hz, 1H), 7.37 (d, J = 8.5 Hz, 2H), 7.66–7.68 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 28.6, 29.0, 36.4, 72.7, 79.6, 115.0, 119.6, 122.8, 128.8, 129.5, 131.6, 136.0, 138.7, 139.1, 155.0, 159.2, 195.4; LRMS (ESI) m/z: 430.1 (M+H)+.
(4-Chlorophenyl)(3-(piperidin-4-ylmethoxy)phenyl)methanone (26)
tert-Butyl 4-((3-(4-chlorobenzoyl)phenoxy)methyl)piperidine-1-carboxylate (3.61 g, 8.50 mmol) was dissolved in 50% trifluoroacetic acid (TFA) in CH2Cl2 (35 mL) and stirred for 2h at r.t. All volatiles were evaporated in vacuo. The reaction mixture was dissolved in CH2Cl2 and washed with 1N NaOH (twice). The combined organic phase was dried over Na2SO4 and evaporated in vacuo. Purification by silica gel chromatography (4:1, CHCl3:MeOH) afforded 26 (2.66 g, 95%) as a color less oil. 1H NMR (500 MHz, CDCl3): δ 1.24–1.32 (m, 2H), 1.79 (d, J = 13 Hz, 2H), 1.86–2.10 (m, 1H), 2.79-2.64 (m, 2H), 3.11 (d, J = 12.5 Hz, 2H), 3.78 (d, J = 6.5 Hz, 2H), 7.05 (dd, J = 2, 8 Hz, 1H), 7.20–7.23 (m, 2H), 7.30 (t, J = 8 Hz, 1H), 7.39 (d, J = 9 Hz, 2H), 7.69 (d, J = 8.5 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 29.2, 29.9, 31.1, 36.3, 46.2, 73.2, 115.1, 119.7, 122.8, 128.8, 129.5, 131.6, 136.1, 138.7, 139.1, 159.4, 195.5; LRMS (ESI) m/z: 330.1 (M+H)+.
(S)-(4-Chlorophenyl)(3-((1-(2-hydroxyoctyl)piperidin-4-yl)methoxy)phenyl)methanone ((S)-11)
To a stirred solution of 26 (100.0 mg, 0.31 mmol) in CH2Cl2 was added Al(CH3)3 (0.2 M in CH2Cl2, 0.43 mmol) at 0 °C. After 15 min., (S)-1,2-epoxyoctane (29, 60.0 mg, 0.43 mmol) was added. After 4h at r.t., the reaction mixture was quenched with aq. NaHCO3 and extracted with CH2Cl2. The combined organic phase was dried over Na2SO4 and evaporated in vacuo. Purification by silica gel chromatography (3:2, CHCl3:MeOH) afforded (S)-2 (103.0 mg, 73%) as a colorless oil. [α]20D = +13.9 (c 1.0 in CHCl3); 1H NMR (300 MHz, CDCl3): δ 0.90 (s, 3H), 1.31–1.49 (m, 11H), 1.84–1.99 (m, 4H), 2.28–2.39 (m, 3H), 2.87 (d, J = 10.2 Hz, 1H), 3.10 (d, J = 10.2 Hz, 1H), 3.69 (s, 1H), 3087 (d, J = 3.9 Hz, 2H), 7.14 (d, J = 7.2 Hz, 1H), 7.29–7.49 (m, 5H), 7.77 (dd, J = 2.1, 8.4 Hz, 2H); 13C NMR (75 MHz, CDCl3): δ 13.5, 22.0, 25.0, 28.5, 28.8, 28.9, 31.2, 34.5, 35.3, 51.3, 54.7, 64.0, 65.8, 72.3, 114.4, 118.8, 122.0, 128.1, 128.8, 130.8, 135.4, 138.1, 138.3, 158.7, 194.6; HRMS (ESI+): m/z Calcd. for C27H36ClNO3 (M+H)+: 458.2405; found: 458.2403. The purity of (S)-11 was determined to be >99% by reverse HPLC analysis (Phenominex kinetex 2.6 μ C18 100A, 100 × 4.60 mm; CH3CN/0.05%TFA in H2O = 25/1). The optical purity of (S)-11 was determined to be >99% by HPLC analyses (Daicel Chiralcel OD-H (0.46 × 25 cm; Hexanes/tBuOH = 20/1 with flow rate=1.0 mL/min. and a UV detector at 245 nm; (S)-enantiomer: tR=21.0 min and (R)-enantiomer: tR = 26.7 min).
(R)-(4-chlorophenyl)(3-((1-(2-hydroxyoctyl)piperidin-4-yl)methoxy)phenyl)methanone ((R)-11)
Colorless oil. [α]20D = −12.8 (c 0.5 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 0.81 (t, J = 7 Hz, 3H), 1.18–1.42 (m, 12H), 1.76–1.78 (m, 3H), 1.90 (t, J = 11 Hz, 1H), 2.17–2.28 (m, 3H), 2.77 (d, J = 11 Hz, 1H), 3.00 (d, J = 11.5 Hz, 1H), 3.58–3.62 (m, 1H), 3.78 (d, J = 6 Hz, 2H), 7.05 (dd, J = 2, 8 Hz, 1H), 7.19–7.23 (m, 2H), 7.30 (t, J = 8 Hz, 1H), 7.39 (d, J = 8.5 Hz, 2H), 7.68 (d, J = 8.5 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ14.3, 14.3, 22.8, 22.9, 25.9, 29.3, 29.6, 29.7, 32.1, 32.1, 34.5, 35.3, 36.1, 52.0, 55.5, 64.7, 66.5, 73.0, 110.2, 115.1, 119.7, 122.8, 128.9, 129.6, 131.7, 136.2, 138.8, 139.1, 159.4, 195.5. HRMS (ESI+): m/z Calcd. for C27H36ClNO3 (M+H)+: 458.2404; found: 458.2403. The purity of (R)-11 was determined to be >99% by reverse HPLC analysis (Phenominex kinetex 2.6 μ C18 100A, 100 × 4.60 mm; CH3CN/0.05%TFA in H2O = 25/1). The optical purity of (R)-11 was determined to be >99% by HPLC analyses (Daicel Chiralcel OD-H (0.46 cm × 25 cm; Hexanes/tBuOH = 20/1 with flow rate=1.0 mL/min. and a UV detector at 245 nm; (S)-enantiomer: tR=21.0 min and (R)-enantiomer: tR = 26.7 min).
(S)-1-(4-((3-(4-chlorobenzoyl)phenoxy)methyl)piperidin-1-yl)octan-2-yl carbamate ((S)-12)
To a stirred solution of (S)-11 (10 mg, 0.02 mmol) and DMAP (5.0 mg, 0.04 mmol) in CH2Cl2 was added trimethylsilylisocyanate (TMSNCO) (7.0 mg, 0.06 mmol). After 4h at r.t., the reaction mixture was quenched with aq. NaHCO3, and extracted with CH2Cl2. The combined organic phase was dried over Na2SO4 and evaporated in vacuo. Purification by silica gel chromatography (9:1, CHCl3:MeOH) afforded (S)-12 (7.0 mg, 63%) as a colorless oil. [α]20D = +9.4° (c 0.9 in CHCl3); 1H NMR (300 MHz, CDCl3): δ 0.89 (d, J = 7.2 Hz, 3H), 1.31–1.58 (m, 12H), 1.90 (d, J = 10.8 Hz, 3H), 2.09 (m, 1H), 2.37–2.44 (m, 3H), 2.96 (m, 1H), 3.15 (m, 1H) 3.76–3.89 (m, 3H), 7.13–7.16 (m, 1H), 7.28–7.50 (m, 5H), 7.78(dd, J = 1.8, 8.4 Hz, 2H); 13C NMR (75 MHz, CDCl3): δ 13.5, 22.0, 25.0, 28.1, 28.4, 28.9, 29.1, 31.2, 34.5, 35.1, 51.5, 54.7, 64.1, 65.7, 72.1, 114.5, 118.8, 122.1, 128.1, 128.8, 130.8, 135.4, 138.1, 138.4, 158.6, 194.6; MS (ESI+): m/z Calculated for C28H37ClN2O4 (M+H)+: 501.2400; found: 501.2309. The purity of (S)-12 was determined to be >99% by reverse HPLC analysis (Phenominex kinetex 2.6 μ C18 100A, 100 × 4.60 mm; CH3CN/0.05%TFA in H2O = 25/1). The optical purity of (S)-12 was determined to be >99% by HPLC analyses (Daicel Chiralcel OD-H (0.46 cm × 25 cm; Hexanes/tBuOH = 20/1 with flow rate=1.0 mL/min. and a UV detector at 245 nm; (S)-enantiomer: tR=18.0 min and (R)-enantiomer: tR = 20.0 min).
(R)-1-(4-((3-(4-chlorobenzoyl)phenoxy)methyl)piperidin-1-yl)octan-2-yl carbamate ((R)-12)
A colorless oil. [α]20D = +−8.6 (c 0.8 in CHCl3); 1H NMR (300 MHz, CDCl3): δ 0.88–0.97 (m, 3H), 1.28–1.49 (m, 13H), 1.93 (d, J = 10.8 Hz, 3H), 2.21 (s, 2H), 2.49 (s, 2H), 3.10 (s, 1H), 3.27 (s, 1H), 3.90 (d, J = 5.4 Hz, 2H), 7.13–7.15 (m, 1H), 7.25–7.50 (m, 5H), 7.76–7.79 (m, 2H); 13C NMR (75 MHz, CDCl3): δ 13.5, 22.0, 23.3, 24.9, 27.7, 28.8, 29.1, 31.2, 34.5, 34.9, 51.7, 54.7, 64.1, 65.6, 71.9, 114.5, 118.8, 122.1, 128.1, 128.8, 130.8, 138.2, 138.4, 158.5, 194.7; HRMS (ESI+): m/z Calculated for C28H37ClN2O4 (M+H)+: 501.2400; found: 501.2309. The purity of (R)-12 was determined to be >99% by reverse HPLC analysis (Phenominex kinetex 2.6 μ C18 100A, 100 × 4.60 mm; CH3CN/0.05%TFA in H2O = 25/1). The optical purity of (R)-12 was determined to be >99% by HPLC analyses (Daicel Chiralcel OD-H (0.46 cm × 25 cm; Hexanes/tBuOH = 20/1 with flow rate=1.0 mL/min. and a UV detector at 245 nm; (S)-enantiomer: tR=18.0 min and (R)-enantiomer: tR = 20.0 min).
1-(3-(((tert-Butyldimethylsilyl)oxy)methyl)phenyl)octan-1-one (44)
To a stirred solution of tert-butyl-(3-iodo-benzyloxy)-dimethylsilane (42, 0.70 g, 2.0 mmol) in THF (3 mL) was added isopropylmagnesium chloride (2M in THF, 3.0 mmol) at 0 °C. After 2.5h, the reaction mixture was cooled to −78 °C and N-methoxy-N-methyloctanamide (43, 0.12 g, 0.66 mmol) in THF (1 mL) was added. After 3h at 0 °C, the reaction mixture was quenched with aq. NH4Cl. The water phase was extracted with CH2Cl2. The combined organic phase was dried over Na2SO4 and evaporated in vacuo. Purification by silica gel chromatography (9.8:0.2, Hexanes:EtOAc) afforded 44 (0.21 g, 91%) as a colorless oil. 1H NMR (500 MHz, CDCl3): δ 0.11 (s, 6H), 0.88 (t, J = 7.5 Hz, 3H), 0.96 (s, 9H), 1.29–1.39 (m, 8H), 1.73 (d, J = 7 Hz, 2H), 2.95 (t, J = 7 Hz, 2H), 4.79 (s, 2H), 7.42 (t, J = 7 Hz, 1H), 7.52 (d, J = 7.5 Hz, 1H), 7.84 (d, J = 8 Hz, 1H), 7.91 (s, 1H); 13C NMR (125 MHz, CDCl3): δ −5.3, 14.1, 18.4, 22.6, 24.5, 25.9, 29.1, 29.4, 31.7, 38.7, 64.5, 125.6, 126.7, 128.5, 130.4, 137.1, 142.0, 200.7; LRMS (ESI) m/z: 336.2 (M+H)+.
(R)-1-(3-(((tert-butyldimethylsilyl)oxy)methyl)phenyl)octan-1-ol
To a stirred solution of 44 (0.2 g, 0.57 mmol) in THF (2 mL) at −78 °C was added (S)-2-methyl-CBS (0.12 g, 0.43 mmol) and BH3-Me2S (43.0 mg, 0.57 mmol). The reaction mixture was kept at −15 °C for an additional 1.5h and quenched with MeOH followed by water. The water phase was extracted with EtOAc. The combined organic phase was dried over Na2SO4 and evaporated in vacuo. Purification by silica gel chromatography (9:1, Hexanes:EtOAc) afforded (R)-1-(3-(((tert-butyldimethylsilyl)oxy)methyl)phenyl)octan-1-ol (180.0 mg, 89%) as a colorless oil. [α]20D = +35.1(c 1 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 0.10 (s, 6H), 0.87 (t, J = 6.5 Hz, 3H), 0.94 (s, 9H), 1.25–1.31 (m, 9H), 1.37–1.42 (m, 1H), 1.67–1.83 (m, 3H), 4.66 (t, J = 6.5 Hz, 1H), 4.74 (s, 3H), 7.21–7.26 (m, 2H), 7.29–7.32 (m, 2H); 13C NMR (125 MHz, CDCl3): δ −5.2, 14.1, 18.4, 22.6, 25.8, 26.0, 29.2, 29.5, 31.8, 39.1, 65.0, 74.8, 123.6, 124.5, 125.3, 128.3, 141.7, 144.9; LRMS (ESI) m/z: 338.2 (M+H)+.
(S)-1-(3-(((tert-Butyldimethylsilyl)oxy)methyl)phenyl)octan-1-ol
Colorless oil. [α]20D = −34.8 (c 1 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 0.10 (s, 6H), 0.87 (t, J = 6.5 Hz, 3H), 0.94 (s, 9H), 1.25–1.31 (m, 9H), 1.37–1.42 (m, 1H), 1.67–1.83 (m, 3H), 4.66 (t, J = 6.5 Hz, 1H), 4.74 (s, 3H), 7.21–7.26 (m, 2H), 7.29–7.32 (m, 2H); 13C NMR (125 MHz, CDCl3): δ −5.2, 14.1, 18.4, 22.6, 25.8, 26.0, 29.2, 29.5, 31.8, 39.1, 65.0, 74.8, 123.6, 124.5, 125.3, 128.3, 141.7, 144.9; LRMS (ESI) m/z: 338.2 (M+H)+.
(R)-1-(3-(Hydroxymethyl)phenyl)octyl acetate
(R)-1-(3-(((tert-butyldimethylsilyl)oxy)methyl)phenyl)octan-1-ol (70 mg, 0.2 mmol) was dissolved in pyridine (1 mL) and acetic anhydride (1 mL). After 5h at r.t., all volatiles were evaporated in vaccuo. Purification by silica gel chromatography (9.5:0.5, Hexanes:EtOAc) afforded (R)-1-(3-(((tert-butyldimethylsilyl)oxy)methyl)phenyl)octyl acetate (78.4 mg, 100%) as a colorless oil. To a stirred solution of (R)-1-(3-(((tert-butyldimethylsilyl)oxy)methyl)phenyl)octyl acetate (25 mg, 0.06 mmol) in THF (0.5 mL) was added TBAF (1M in THF, 0.12 mmol). After 4h at r.t., the reaction was quenched with water. The water phase was extracted with EtOAc, and the combined extract was washed with brine, dried over Na2SO4, and concentrated in vacuo. Purification by silica gel chromatography (7:3, Hexane:EtOAc) afforded (R)-1-(3-(hydroxymethyl)phenyl)octyl acetate (14.0 mg, 79%) as a colorless oil. [α]20D = +38.0(c 0.7 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 0.87 (t, J = 7 Hz, 3H), 1.20–1.34 (m, 10H), 1.73–1.79 (m, 1H), 1.86–1.93 (m, 1H), 2.06 (s, 3H), 4.70 (s, 2H), 5.71 (t, J = 7 Hz, 1H), 7.24–7.29 (m, 2H), 7.32–7.35 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 14.1, 21.3, 22.6, 25.6, 29.1, 29.3, 31.8, 36.3, 65.3, 76.2, 125.1, 125.8, 126.4, 128.7, 141.1, 141.3, 170.5; LRMS (ESI) m/z: 279.2 (M+H)+.
(S)-1-(3-(Hydroxymethyl)phenyl)octyl acetate
Colorless oil. [α]20D = −35.0 (c 0.8 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 0.87 (t, J = 7 Hz, 3H), 1.20–1.34 (m, 10H), 1.73–1.79 (m, 1H), 1.86–1.93 (m, 1H), 2.06 (s, 3H), 4.70 (s, 2H), 5.71 (t, J = 7 Hz, 1H), 7.24–7.29 (m, 2H), 7.32–7.35 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 14.1, 21.3, 22.6, 25.6, 29.1, 29.3, 31.8, 36.3, 65.3, 76.2, 125.1, 125.8, 126.4, 128.7, 141.1, 141.3, 170.5; LRMS (ESI) m/z: 279.2 (M+H)+.
(R)-1-(3-((4-((3-(4-chlorobenzoyl)phenoxy)methyl)piperidin-1-yl)methyl)phenyl)octyl acetate ((R)-46)
To a stirred solution of DMSO (113.0 mg, 1.44 mmol) in CH2Cl2 (2 mL) at −78 °C was added oxalyl chloride (91.0 mg, 0.72 mmol). After 30 min., (R)-1-(3-(hydroxymethyl)phenyl)octyl acetate (100.0 mg, 0.36 mmol) in CH2Cl2 (0.5 mL) was added. After 30 min. at −78 °C, Et3N (220.0 mg, 2.16 mmol) was added and the reaction mixture was warmed to r.t. over 1h. The reaction mixture was quenched with water and the water phase was extracted with CH2Cl2. The combined extract was washed with brine, dried over Na2S2O4, and evaporated in vacuo. Purification by silica gel chromatography afforded the corresponding aldehyde (R)-1-(3-formylphenyl)octyl acetate ((R)-45, 95.0 mg) as a colorless oil. To a stirred solution of (R)-45 (95.0 mg, 0.34 mmol) and (4-chlorophenyl)(3-(piperidin-4-ylmethoxy)phenyl)methanone (26, 98.7 mg, 0.30 mmol) in CH2Cl2 was added NaBH(OAc)3 (150.0 mg, 0.72 mmol). After 8h at r.t., the reaction was quenched with aq. sat. NaHCO3 and the water phase was extracted with CH2Cl2. The combined extract was washed with brine, dried over Na2S2O4, and evaporated in vacuo. Purification by silica gel chromatography (3:7, Hexanes:EtOAc) afforded (R)-46. (170.0 mg, 96%) as a colorless oil. [α]20D = −17.9 (c 0.8 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 0.86 (t, J = 7 Hz, 3H), 1.23–1.29 (m, 10H), 1.43–1.45 (m, 2H), 1.73–1.91 (m, 5H), 1.99–2.04 (m, 2H), 2.07 (s, 3H), 2.93 (d, J = 10 Hz, 2H), 3.53 (d, J = 1.5 Hz, 2H), 3.85 (d, J = 6 Hz, 2H), 5.72 (t, J = 7 Hz, 1H), 7.12 (dd, J = 1.5, 7.5 Hz, 1H), 7.21–7.30 (m, 6H), 7.36 (t, J = 8 Hz, 1H), 7.45 (d, J = 8.5 Hz, 2H), 7.75 (d, J = 8.5 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 14.1, 21.4, 22.6, 25.6, 29.0, 31.8, 35.9, 36.4, 53.3, 53.3, 63.2, 72.9, 76.2, 114.9, 119.5, 122.5, 125.1, 127.3, 128.3, 128.6, 128.6, 129.3, 131.4, 135.9, 138.5, 138.9, 140.8, 159.2, 170.4, 195.3; HRMS (ESI+): m/z Calcd. for C36H44ClNO4 (M+H)+: 590.3010; found: 590.3015.
(R)-(4-chlorophenyl)(3-((1-(3-(1-hydroxyoctyl)benzyl)piperidin-4-yl)methoxy)phenyl) methanone ((R)-3)
To a stirred solution of (R)-46 (20.0 mg, 0.03 mmol) in acetonitrile (1 mL) was added 1N NaOH (1 mL). After 2h at r.t., the reaction mixture was quenched with water and extracted with CHCl3. The combined organic phase was washed with brine, Na2S2O4, and evaporated in vacuo. Purification by silica gel chromatography (1:3, Hexanes:EtOAc) afforded (R)-3 (10.0 mg, 54%) as a colorless oil. [α]20D = +25.8 (c 0.8 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 0.86 (t, J = 7 Hz, 3H), 1.21–1.28 (m, 10H), 1.40–1.47 (m, 3H), 1.68–1.82 (m, 5H), 2.02 (t, J = 12 Hz, 2H), 2.94 (d, J = 11 Hz, 2H), 3.53 (s, 2H), 3.85 (d, J = 6 Hz, 2H), 4.66 (t, J = 7 Hz, 1H), 7.12 (dd, J = 2.5, 8 Hz, 1H), 7.22–7.31 (m, 6H), 7.36 (t, J = 8 Hz, 1H), 7.46 (d, J = 8.5 Hz, 2H), 7.75 (d, J = 8.5 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 14.1, 22.7, 25.9, 29.0, 29.2, 29.5, 31.8, 35.9, 39.2, 45.6, 53.3, 53.3, 63.3, 72.9, 74.7, 114.9, 119.5, 122.6, 124.6, 126.8, 128.3, 128.4, 128.6, 129.3, 131.4, 135.9, 138.5, 138.9, 145.0, 159.2, 195.3. HRMS (ESI+): m/z Calcd. for C34H42ClNO3 (M+H)+: 548.2906; found: 548.2909. The purity of (R)-3 was determined to be >95% by reverse HPLC analysis (Phenominex kinetex 2.6 μ C18 100A, 100 × 4.60 mm; CH3CN/0.05%TFA in H2O = 25/1). The optical purity of (R)-3 was determined to be 95% by HPLC analyses (Daicel Chiralcel OD-H (0.46 cm × 25 cm; Hexanes/tBuOH = 20/1 with flow rate=1.0 mL/min. and a UV detector at 245 nm; (S)-enantiomer: tR=15.0 min and (R)-enantiomer: tR = 16.0 min).
(S)-(4-chlorophenyl)(3-((1-(3-(1-hydroxyoctyl)benzyl)piperidin-4-yl)methoxy)phenyl) methanone ((S)-3)
Colorless oil, [α]20D = −25.6 (c 0.5 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 1H NMR (500 MHz, CDCl3): δ 0.86 (t, J = 7 Hz, 3H), 1.21–1.28 (m, 10H), 1.40–1.47 (m, 3H), 1.68–1.82 (m, 5H), 2.02 (t, J = 12 Hz, 2H), 2.94 (d, J = 11 Hz, 2H), 3.53 (s, 2H), 3.85 (d, J = 6 Hz, 2H), 4.66 (t, J = 7 Hz, 1H), 7.12 (dd, J = 2.5, 8 Hz, 1H), 7.22–7.31 (m, 6H), 7.36 (t, J = 8 Hz, 1H), 7.46 (d, J = 8.5 Hz, 2H), 7.75 (d, J = 8.5 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 14.1, 22.7, 25.9, 29.0, 29.2, 29.5, 31.8, 35.9, 39.2, 45.6, 53.3, 53.3, 63.3, 72.9, 74.7, 114.9, 119.5, 122.6, 124.6, 126.8, 128.3, 128.4, 128.6, 129.3, 131.4, 135.9, 138.5, 138.9, 145.0, 159.2, 195.3. HRMS (ESI+): m/z Calcd. for C34H42ClNO3 (M+H)+: 548.2907; found: 548.3001. The purity of (S)-3 was determined to be >95% by reverse HPLC analysis (Phenominex kinetex 2.6 μ C18 100A, 100 × 4.60 mm; CH3CN/0.05%TFA in H2O = 25/1). The optical purity of (S)-3 was determined to be 95% by HPLC analyses (Daicel Chiralcel OD-H (0.46 cm × 25 cm; Hexanes/tBuOH = 20/1 with flow rate=1.0 mL/min. and a UV detector at 245 nm; (S)-enantiomer: tR=15.0 min and (R)-enantiomer: tR = 16.0 min).
(R)-1-(3-((4-((3-(4-chlorobenzoyl)phenoxy)methyl)piperidin-1-yl)methyl)phenyl)octyl carbamate ((R)-14)
To a stirred solution of (R)-3 (20.0 mg, 0.04 mmol) and DMAP (100.0 mg, 0.08 mmol) in CH2Cl2 (0.5 mL) was added TMSNCO (100.0 mg, 0.09 mmol). After 4h at r.t., all volatiles were evaporated in vauo. Purification by silica gel chromatography (9:1, CHCl3:MeOH) afforded (R)-14. (17.3 mg, 80%) as a colorless oil. [α]20D = +21.1 (c 0.5 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 0.85 (t, J = 7 Hz, 3H), 1.26 (m, 9H), 1.35–1.46 (m, 3H), 1.59–1.74 (m, 2H), 1.82 (d, J = 10.5 Hz, 3H), 2.02 (m, 2H), 2.94 (d, J = 8 Hz, 2H), 3.54 (s, 2H), 3.85 (d, J = 6 Hz, 2H), 4.60 (q, J = 5 Hz, 1H), 7.11–7.12 (m, 1H), 7.18–7.19 (m, 2H), 7.24–7.29 (m, 4H), 7.36 (t, J = 8.5 Hz, 1H), 7.46 (d, J = 8 Hz, 2H), 7.75 (d, J = 9 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 14.1, 22.7, 26.0, 29.0, 29.3, 29.5, 31.9, 35.9, 40.7, 53.2, 72.9, 75.1, 114.9, 119.4, 122.5, 126.8, 128.0, 128.6, 129.3, 131.4, 135.9, 138.5, 138.9, 159.2, 195.3; HRMS (ESI+): m/z Calcd. for C35H43ClN2O4 (M+H)+: 590.2901; found: 590.2898. The purity of (R)-14 was determined to be >95% by reverse HPLC analysis (Phenominex kinetex 2.6 μ C18 100A, 100 × 4.60 mm; CH3CN/0.05%TFA in H2O = 25/1). The optical purity of (R)-14 was determined to be 95% by HPLC analyses (Daicel Chiralcel OD-H (0.46 cm × 25 cm; Hexanes/tBuOH = 20/1 with flow rate=1.0 mL/min. and a UV detector at 245 nm; (S)-enantiomer: tR=14.0 min and (R)-enantiomer: tR = 15.0 min).
(S)-1-(3-((4-((3-(4-chlorobenzoyl)phenoxy)methyl)piperidin-1-yl)methyl)phenyl)octyl carbamate (S)-14
Colorless oil. [α]20D = −21.9 (c 0.5 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 0.85 (t, J = 7 Hz, 3H), 1.26 (m, 9H), 1.35–1.46 (m, 3H), 1.59–1.74 (m, 2H), 1.82 (d, J = 10.5 Hz, 3H), 2.02 (m, 2H), 2.94 (d, J = 8 Hz, 2H), 3.54 (s, 2H), 3.85 (d, J = 6 Hz, 2H), 4.60 (q, J = 5 Hz, 1H), 7.11–7.12 (m, 1H), 7.18–7.19 (m, 2H), 7.24–7.29 (m, 4H), 7.36 (t, J = 8.5 Hz, 1H), 7.46 (d, J = 8 Hz, 2H), 7.75 (d, J = 9 Hz, 2H). 13C NMR (125 MHz, CDCl3): δ 14.1, 22.7, 26.0, 29.0, 29.3, 29.5, 31.9, 35.9, 40.7, 53.2, 72.9, 75.1, 114.9, 119.4, 122.5, 126.8, 128.0, 128.6, 129.3, 131.4, 135.9, 138.5, 138.9, 159.2, 195.3; HRMS (ESI+): m/z Calcd. for C35H43ClN2O4(M+H)+: 590.2901; found: 590.2899. The purity of (S)-14 was determined to be >95% by reverse HPLC analysis (Phenominex kinetex 2.6 μ C18 100A, 100 × 4.60 mm; CH3CN/0.05%TFA in H2O = 25/1). The optical purity of (S)-14 was determined to be 95% by HPLC analyses (Daicel Chiralcel OD-H (0.46 cm × 25 cm; Hexanes/tBuOH = 20/1 with flow rate=1.0 mL/min. and a UV detector at 245 nm; (S)-enantiomer: tR=14.0 min and (R)-enantiomer: tR = 15.0 min).
(4-Chlorophenyl)(3-((3-vinylbenzyl)oxy)phenyl)methanone
To a stirred solution of (4-chlorophenyl)(3-hydroxyphenyl)methanone (21, 26.0 mg, 1.13 mmol), (3-vinylphenyl)methanol (48, 230.0 mg, 1.7 mmol), TPP (0.45 g, 1.7 mmol) in THF (10 mL) was added DIAD (0.62 g, 1.7 mmol). After 4h at r.t., all volatiles were evaporated in vacuo to afford the crude product. Purification by silica gel chromatography (4:1, hexanes:EtOAc) provided (4-chlorophenyl)(3-((3-vinylbenzyl)oxy)phenyl)methanone (334.3 mg, 85%) as a yellow oil. 1H NMR (500 MHz, CDCl3): δ 5.12 (s, 3H), 5.28 (d, J = 10.5 Hz, 1H), 5.77 (d, J = 17.5 Hz, 1H), 6.73 (q, J = 11 Hz, 1H), 7.22 (dd, J = 2.5, 8.5 Hz, 1H), 7.31–7.46 (m, 8H), 7.71–7.73 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 69.9, 70.1, 70.4, 114.3, 114.4, 114.5, 114.5, 114.5, 114.6, 115.3, 115.6, 119.8, 120.0, 122.7, 123.0, 125.2, 125.5, 125.8, 126.1, 126.7, 127.0, 128.5, 128.8, 129.0, 129.4, 129.6, 131.3, 131.5, 135.8, 136.5, 136.5, 136.8, 138.1, 138.5, 138.9, 158.7, 195.1; LRMS (ESI) m/z: 349.1 (M+H)+.
(4-Chlorophenyl)(3-((3-(oxiran-2-yl)benzyl)oxy)phenyl)methanone (rac-49)
To a stirred solution of (4-chlorophenyl)(3-((3-vinylbenzyl)oxy)phenyl)methanone (250.0 mg, 0.70 mmol) in acetonitrile (60 mL) was added Na2EDTA (0.09%, 8 mL), oxone (86.0 mg, 0.28 mmol), NaHCO3 (0.056 g, 0.67 mmol), and trifluoroacetone (250.0 mg, 2.24 mmol) at 0 °C. After 1h, an additional oxone (86.0 mg, 0.28 mmol) and NaHCO3 (56.0 mg, 0.67 mmol) were added every 1h; this process was repeated 8 times during which the reaction temperature was kept at 0 °C. After completion of the reaction all volatiles were evaporated in vacuo. The residue was dissolved in water and the water phase was extracted with CH2Cl2. The combined organic phase was dried over Na2SO4 and evaporated in vacuo. Purification by silica gel chromatography (9:1, Hexane:EtOAc) afford rac-49 (148.0 mg, 74%) as a colorless oil. rac-49 was resolute via Jocobsen’s kinetic resolution with (S,S)-salene Co(III) or (R,R)-salene Co(III) to afford (R)-49 and (S)-49, respectively. (R)-49. [α]20D = +11.3° (c 0.5 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 2.79 (dd, J = 2.5, 5.5 Hz, 1H), 3.15 (t, J = 4 Hz, 1H), 3.88 (t, J = 3 Hz, 1H), 5.10 (s, 2H), 7.21 (dd, J = 4.5, 8 Hz, 1H), 7.26–7.27 (m, 1H), 7.33–7.41 (m, 6H), 7.44 (d, J = 8.5 Hz, 2H), 7.73 (d, J = 8.5 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 51.5, 52.4, 70.2, 115.6, 120.0, 123.1, 124.7, 125.5, 127.5, 128.8, 129.1, 129.7, 131.6, 136.0, 137.1, 138.5, 138.8, 139.1, 158.8, 195.3. LRMS (ESI) m/z: 365.1 (M+H)+. (S)-49. Colorless oil. [α]20D = −9.3° (c 0.5 in CHCl3).
(R)-(4-Chlorophenyl)(3-((3-(2-hydroxy-1-((4-methoxyphenethyl)amino)ethyl)benzyl)oxy) phenyl)methanone ((R)-4)
A mixture of (R)-49 (700.0 mg, 0.2 mmol), p-methoxyphenethyl amine (30.0 mg, 0.20 mmol) and Zn(ClO4)2.6H2O (10.0 mg) is heated at 90 °C for 1h. Purification by silica gel chromatography (7:3, Hexanes:EtOAc) afforded (R)-4 (27.0 mg, 27%) as a colorless oil and the corresponding regiodisomer (15%). (R)-4. [α]20D = +20.3(c 0.5 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 2.68–2.76 (m, 3H), 2.84–2.95 (m, 3H), 3.78 (s, 3H), 4.72 (dd, J = 3.5, 9.5 Hz, 1H), 5.09 (s, 2H), 6.83 (d, J = 8 Hz, 2H), 7.10 (d, J = 8.5 Hz, 2H), 7.20 (dd, J = 1.5, 8 Hz, 1H), 7.32–7.44 (m, 9H), 7.72 (d, J = 8.5 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 30.9, 35.2, 50.7, 55.3, 56.8, 70.2, 71.2, 114.0, 115.5, 119.8, 122.9, 124.9, 125.7, 126.7, 128.6, 128.7, 129.5, 129.6, 131.4, 135.8, 136.6, 138.5, 138.9, 143.0, 158.2, 158.7, 195.2; HRMS (ESI+): m/z Calcd. for C31H30ClNO4 (M+H)+: 516.1905; found: 516.2101. The purity of (R)-4 was determined to be >95% by reverse HPLC analysis (Phenominex kinetex 2.6 μ C18 100A, 100 × 4.60 mm; CH3CN/0.05%TFA in H2O = 25/1). The optical purity of (R)-4 was determined to be >99% by HPLC analyses (Daicel Chiralcel OD-H (0.46 cm × 25 cm; Hexanes/tBuOH = 20/1 with flow rate=1.0 mL/min. and a UV detector at 245 nm; (S)-enantiomer: tR=13.5 min and (R)-enantiomer: tR = 15.0 min).
(S)-(4-Chlorophenyl)(3-((3-(2-hydroxy-1-((4-methoxyphenethyl)amino)ethyl)benzyl)oxy) phenyl)methanone ((S)-4)
A colorless oil. [α]20D = −19.5 (c 0.5 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 2.68–2.76 (m, 3H), 2.84–2.95 (m, 3H), 3.78 (s, 3H), 4.72 (dd, J = 3.5, 9.5 Hz, 1H), 5.09 (s, 2H), 6.83 (d, J = 8 Hz, 2H), 7.10 (d, J = 8.5 Hz, 2H), 7.20 (dd, J = 1.5, 8 Hz, 1H), 7.32–7.44 (m, 9H), 7.72 (d, J = 8.5 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 30.9, 35.2, 50.7, 55.3, 56.8, 70.2, 71.2, 114.0, 115.5, 119.8, 122.9, 124.9, 125.7, 126.7, 128.6, 128.7, 129.5, 129.6, 131.4, 135.8, 136.6, 138.5, 138.9, 143.0, 158.2, 158.7, 195.2; HRMS (ESI+): m/z Calcd. for C31H30ClNO4 (M+H)+: 516.1895; found: 516.19051. The purity of (S)-4 was determined to be >95% by reverse HPLC analysis (Phenominex kinetex 2.6 μ C18 100A, 100 × 4.60 mm; CH3CN/0.05%TFA in H2O = 25/1). The optical purity of (S)-4 was determined to be >99% by HPLC analyses (Daicel Chiralcel OD-H (0.46 cm × 25 cm; Hexanes/tBuOH = 20/1 with flow rate=1.0 mL/min. and a UV detector at 245 nm; (S)-enantiomer: tR=13.5 min and (R)-enantiomer: tR = 15.0 min).
tert-Butyl 4-((3-chloro-4-(4-chlorobenzoyl)phenoxy)methyl)piperidine-1-carboxylate
Colorless oil. 1H NMR (500 MHz, CDCl3): δ 1.29 (m, 2H), 1.48 (s, 9H), 1.83 (d, 12.5 Hz, 2H), 1.98 (s, 1H), 2.76 (bs, 2H), 3.86 (d, J = 6.5 Hz, 2H), 4.18 (bs, 2H), 6.87 (dd, J = 1, 8.5 Hz, 1H), 6.97 (d, J = 1.5 Hz, 1H), 7.34 (d, J = 8.5 Hz, 1H), 7.42 (d, J = 8.5 Hz, 2H), 7.72 (d, J = 8.5 Hz); 13C NMR (125 MHz, CDCl3): δ 28.6, 28.9, 36.2, 72.9, 79.5, 113.2, 116.2, 128.9, 130.2, 131.4, 131.4, 133.3, 135.8, 139.8, 154.9, 161.3, 193.7; LRMS (ESI) m/z: 465.4 (M+H)+.
(2-Chloro-4-(piperidin-4-ylmethoxy)phenyl)(4-chlorophenyl)methanone (50)
Colorless oil. 1H NMR (500 MHz, CDCl3): δ 1.38–1.29 (m, 2H), 1.84 (s, 2H), 2.01–1.95 (m, 1H), 2.70 (m, 2H), 3.18 (d, J = 12 Hz, 2H), 3.87 (d, J = 6.5 Hz, 2H), 6.89 (dd, J = 2, 8.5 Hz, 1H), 6.99 (d, J = 2.5 Hz, 1H), 7.37 (d, J = 8.5 Hz, 1H), 7.45 (d, J = 8.5 Hz, 2H), 7.75 (d, J = 8.5 Hz, 2H). 13C NMR (125 MHz, CDCl3): δ 30.2, 36.5, 46.4, 73.6, 113.3, 116.3, 129.0, 130.2, 131.5, 131.6, 133.5, 136.0, 140.0, 161.6, 193.9. LRMS (ESI) m/z: 364.3 (M+H)+.
(R)-(2-chloro-4-((1-(2-hydroxyoctyl)piperidin-4-yl)methoxy)phenyl)(4-chlorophenyl)methanone ((R)-16)
Colorless oil. [α]20D = +28.6 (c 1.0 in CHCl3); 1H NMR (300 MHz, CDCl3): δ 0.90 (bs, 3H), 1.31–1.56 (m, 12H), 1.88 (d, J = 10.5 Hz, 3H), 2.08 (bs, 1H), 2.36–2.44 (m, 3H), 2.94–3.18 (m, 2H), 3.75 (s, 1H), 3.88 (d, J = 4.5 Hz, 2H), 6.87–6.90 (m, 1H), 6.99 (t, J = 2.1 Hz, 1H), 7.34–7.46 (m, 3H), 7.73–7.76 (m, 2H); 13C NMR (75 MHz, CDCl3): δ 8.3, 13.5, 22.0, 25.0, 28.1, 28.4, 28.9, 31.2, 34.5, 35.1, 45.4, 51.4, 54.6, 64.0, 65.7, 72.4, 112.5, 115.7, 128.3, 129.7, 130.7, 130.8, 132.7, 135.2, 139.2, 160.7, 194.0; MS (ESI+): m/z Calcd. for C27H35Cl2NO3 (M+H)+: 492.2063; found: 492.2068. The purity of (R)-16 was determined to be >95% by reverse HPLC analysis (Phenominex kinetex 2.6 μ C18 100A, 100 × 4.60 mm; CH3CN/0.05%TFA in H2O = 25/1). The optical purity of (R)-16 was determined to be >99% by HPLC analyses (Daicel Chiralcel OD-H (0.46 cm × 25 cm; Hexanes/tBuOH = 20/1 with flow rate=1.0 mL/min. and a UV detector at 245 nm; (S)-enantiomer: tR=16.5 min and (R)-enantiomer: tR = 17.5 min).
(R)-1-(4-((3-chloro-4-(4-chlorobenzoyl)phenoxy)methyl)piperidin-1-yl)octan-2-yl carbamate ((R)-17)
Colorless oil. [α]20D = +21.3 (c 1.0 in CHCl3); 1H NMR (300 MHz, CDCl3): δ 0.9 (bs, 3H), 1.31–1.50 (m, 12H), 1.85–2.00 (m, 4H), 2.29–2.40 (m, 3H), 2.88 (d, J = 10.8 Hz, 1H), 3.11 (d, J = 10.8 Hz, 1H), 3.70 (s, 1H), 3.87 (d, J = 5.7 Hz, 2H), 6.87–6.99 (m, 2H), 7.28–7.46 (m, 3H), 7.73–7.76 (m, 2H); 13C NMR (75 MHz, CDCl3): δ13.5, 22.0, 25.0, 28.4, 28.7, 28.9, 31.3, 34.5, 35.2, 51.3, 54.6, 64.0, 65.8, 72.5, 112.6, 115.7, 128.3, 129.7, 130.7, 130.8, 132.7, 135.3, 139.2, 160.8, 193.0; HRMS (ESI+): m/z Calcd. for C28H36Cl2N2O4 (M+H)+: 535.2106; found: 535.2109. The purity of (R)-17 was determined to be >95% by reverse HPLC analysis (Phenominex kinetex 2.6 μ C18 100A, 100 × 4.60 mm; CH3CN/0.05%TFA in H2O = 25/1). The optical purity of (R)-17 was determined to be >99% by HPLC analyses (Daicel Chiralcel OD-H (0.46 cm × 25 cm; Hexanes/tBuOH = 20/1 with flow rate=1.0 mL/min. and a UV detector at 245 nm; (S)-enantiomer: tR=18.0 min and (R)-enantiomer: tR = 19.5 min).
(S)-(2-Chloro-4-((1-(3-(1-hydroxyoctyl)benzyl)piperidin-4-yl)methoxy)phenyl)(4-chlorophenyl) methanone ((S)-6)
Colorless oil. [α]20D = −31.7 (c 0.2 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 1H NMR (500 MHz, CDCl3): δ 0.87 (t, J = 6.5 Hz, 3H), 1.25–1.50 (m, 12H), 1.68–1.86 (m, 5H), 2.05 (t, J = 11.5 Hz, 3H), 2.97 (d, J = 11.5 Hz, 2H), 3.56 (s, 2H), 3.85 (d, J = 6 Hz, 2H), 4.67 (t, J = 7 Hz, 1H), 6.86 (dd, J = 2, 8.5 Hz, 1H), 6.96 (d, J = 2.5 Hz, 1H), 7.24–7.26 (m, 2H), 7.29–7.35 (m, 3H), 7.42 (d, J = 8.5 Hz, 2H), 7.73 (d, J = 9 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 14.1, 22.7, 25.9, 28.8, 29.3, 29.5, 31.8, 35.7, 39.2, 53.2, 53.2, 63.3, 73.1, 76.6, 113.1, 116.1, 124.7, 126.8, 128.4, 128.4, 128.8, 030.0, 131.3, 131.4, 133.2, 135.8, 139.7, 145.0, 161.4, 193.7; HRMS (ESI+): m/z Calcd. for C34H41Cl2NO3 (M+H)+: 582.2504; found: 582.2302. The purity of (S)-6 was determined to be >95% by reverse HPLC analysis (Phenominex kinetex 2.6 μ C18 100A, 100 × 4.60 mm; CH3CN/0.05%TFA in H2O = 25/1). The optical purity of (S)-6 was determined to be 95% by HPLC analyses (Daicel Chiralcel OD-H (0.46 cm × 25 cm; Hexanes/tBuOH = 20/1 with flow rate=1.0 mL/min. and a UV detector at 245 nm; (S)-enantiomer: tR=14.5 min and (R)-enantiomer: tR = 15.5 min).
(R)-(2-Chloro-4-((1-(3-(1-hydroxyoctyl)benzyl)piperidin-4-yl)methoxy)phenyl)(4-chlorophenyl) methanone ((R)-6)
Colorless oil. [α]20D = +32.5 (c 0.2 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 0.87 (t, J = 6.5 Hz, 3H), 1.25–1.50 (m, 12H), 1.68–1.86 (m, 5H), 2.05 (t, J = 11.5 Hz, 3H), 2.97 (d, J = 11.5 Hz, 2H), 3.56 (s, 2H), 3.85 (d, J = 6 Hz, 2H), 4.67 (t, J = 7 Hz, 1H), 6.86 (dd, J = 2, 8.5 Hz, 1H), 6.96 (d, J = 2.5 Hz, 1H), 7.24–7.26 (m, 2H), 7.29–7.35 (m, 3H), 7.42 (d, J = 8.5 Hz, 2H), 7.73 (d, J = 9 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 14.1, 22.7, 25.9, 28.8, 29.3, 29.5, 31.8, 35.7, 39.2, 53.2, 53.2, 63.3, 73.1, 76.6, 113.1, 116.1, 124.7, 126.8, 128.4, 128.4, 128.8, 030.0, 131.3, 131.4, 133.2, 135.8, 139.7, 145.0, 161.4, 193.7; HRMS (ESI+): m/z Calcd. for C34H41Cl2NO3 (M+H)+: 582.2508; found: 582.2509. The purity of (R)-6 was determined to be >95% by reverse HPLC analysis (Phenominex kinetex 2.6 μ C18 100A, 100 × 4.60 mm; CH3CN/0.05%TFA in H2O = 25/1). The optical purity of (R)-6 was determined to be 95% by HPLC analyses (Daicel Chiralcel OD-H (0.46 cm × 25 cm; Hexanes/tBuOH = 20/1 with flow rate=1.0 mL/min. and a UV detector at 245 nm; (S)-enantiomer: tR=14.5 min and (R)-enantiomer: tR = 15.5 min).
(S)-1-(3-((4-((3-chloro-4-(4-chlorobenzoyl)phenoxy)methyl)piperidin-1-yl)methyl)phenyl)octyl carbamate ((S)-19)
Colorless oil. [α]20D = −26.6 (c 0.2 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 1H NMR (500 MHz, CDCl3): δ 0.87 (t, J = 6.5 Hz, 3H), 1.29–1.42 (m, 13H), 1.68–1.88 (m, 6H), 2.17 (bs, 2H), 3.08–3.11 (m, 1H), 3.67 (bs, 1H), 3.87 (d, J = 6 Hz, 2H), 4.68 (q, J = 1.5 Hz, 1H), 6.85 (dd, J = 2.5, 9 Hz, 1H), 6.96 (d, J = 2.5 Hz, 1H), 7.28–7.43 (m, 7H), 7.71–7.74 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 14.1, 16.6, 20.0, 22.6, 24.5, 25.9, 29.5, 31.8, 39.3, 113.0, 116.2, 128.9, 131.3, 131.4, 133.2, 135.6, 139.8, 159.7, 192.4, 193.7; HRMS (ESI+): m/z Calcd. for C35H42Cl2N2O4 (M+H)+: 625.2508; found: 625.2510. The purity of (S)-19 was determined to be >95% by reverse HPLC analysis (Phenominex kinetex 2.6 μ C18 100A, 100 × 4.60 mm; CH3CN/0.05%TFA in H2O = 25/1). The optical purity of (S)-19 was determined to be 95% by HPLC analyses (Daicel Chiralcel OD-H (0.46 cm × 25 cm; Hexanes/tBuOH = 20/1 with flow rate=1.0 mL/min. and a UV detector at 245 nm; (S)-enantiomer: tR=12.5 min and (R)-enantiomer: tR = 14.0 min).
(R)-1-(3-((4-((3-chloro-4-(4-chlorobenzoyl)phenoxy)methyl)piperidin-1-yl)methyl)phenyl)octyl carbamate ((R)-19)
Colorless oil. [α]20D +26.3 (c 0.2 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 0.87 (t, J = 6.5 Hz, 3H), 1.29–1.42 (m, 13H), 1.68–1.88 (m, 6H), 2.17 (bs, 2H), 3.08–3.11 (m, 1H), 3.67 (bs, 1H), 3.87 (d, J = 6 Hz, 2H), 4.68 (q, J = 1.5 Hz, 1H), 6.85 (dd, J = 2.5, 9 Hz, 1H), 6.96 (d, J = 2.5 Hz, 1H), 7.28–7.43 (m, 7H), 7.71–7.74 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 14.1, 16.6, 20.0, 22.6, 24.5, 25.9, 29.5, 31.8, 39.3, 113.0, 116.2, 128.9, 131.3, 131.4, 133.2, 135.6, 139.8, 159.7, 192.4, 193.7; MS (ESI+): m/z Calcd. for C35H42Cl2N2O4 (M+H)+: 625.2508; found: 625.2506. The purity of (R)-19 was determined to be >95% by reverse HPLC analysis (Phenominex kinetex 2.6 μ C18 100A, 100 × 4.60 mm; CH3CN/0.05%TFA in H2O = 25/1). The optical purity of (R)-19 was determined to be 95% by HPLC analyses (Daicel Chiralcel OD-H (0.46 cm × 25 cm; Hexanes/tBuOH = 20/1 with flow rate=1.0 mL/min. and a UV detector at 245 nm; (S)-enantiomer: tR=12.5 min and (R)-enantiomer: tR = 14.0 min).
(4-Chlorophenyl)(3-hydroxyphenyl)methanone O-methyl oxime (22)
To a stirred solution of 21 (800.0 mg, 3.5 mmol) in pyridine (30 mL) was added O-methylhydroxylamine hydrochloride (880 mg, 10.5 mmol). The reaction mixture was heated to 105 °C for 16h. All volatiles were evaporated in vacuo. Purification by silica gel chromatography (7:3, Hexanes:EtOAc) afforded 22 (780.0 mg, 86%) as a colorless oil. 1H NMR (500 MHz, CDCl3): δ 3.96 (s, 3H), 6.77–6.86 (m, 2H), 6.92–6.96 (m, 1H), 7.24–7.28 (m, 3H), 7.37–7.42 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 62.5, 62.5, 114.5, 116.1, 116.3, 116.8, 120.5, 121.3, 128.5, 128.5, 129.1, 129.6, 129.6, 130.8, 131.4, 134.2, 134.6, 135.0, 135.5, 137.3, 155.6, 155.6, 155.8, 155.8; LRMS (ESI) m/z: 262.026(M+H)+.
(2-Chloro-4-hydroxyphenyl)(4-chlorophenyl)methanone O-methyl oxime (24)
To a stirred solution of 23 (500.0 mg, 1.87 mmol) in pyridine (15 mL) was added O-methylhydroxylamine hydrochloride (470.0 mg, 5.60 mmol). The reaction mixture was heated to 105 °C for 16h. All volatiles were evaporated in vacuo. Purification by silica gel chromatography (7:3, Hexanes:EtOAc) afforded 24 (520.0 mg, 93%) as a colorless oil. 1H NMR (500 MHz, CDCl3): δ 3.99 (s, 3H), 6.80 (dd, J = 2.5, 8.5 Hz, 1H), 6.95 (d, J = 2 Hz, 1H), 7.03 (d, J = 8Hz, 1H), 7.29 (d, J = 8.5 Hz, 2H), 7.42 (d, J = 8.5 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 62.7, 114.4, 116.8, 124.8, 128.2, 128.7, 130.7, 133.2, 133.6, 135.5, 153.4, 156.7; LRMS (ESI) m/z: 296.02(M+H)+.
tert-Butyl4-((3-((4-chlorophenyl)(methoxyimino)methyl)phenoxy)methyl)piperidine-1-carboxylate
To a stirred solution of 22 (0.93 g, 3.59 mmol), tert-butyl 4-(hydroxymethyl)piperidine-1-carboxylate (25, 1.55 g, 7.18 mmol), and TPP (1.41 g, 5.38 mmol) in THF (8 mL) was added DIAD (1.98 g, 5.38 mmol). After 4h at r.t., all volatiles were evaporated in vauo. Purification by silica gel chromatography (9:1, Hexanes:EtOAc) provided tert-butyl 4-((3-((4-chlorophenyl)(methoxyimino)methyl)phenoxy)methyl)piperidine-1-carboxylate (1.28 g, 78%; a mixture of isomers) as a yellow color liquid. 1H NMR (500 MHz, CDCl3): δ 1.22–1.31 (m, 2H), 1.46 (s, 9H), 1.81 (d, J = 13 Hz, 2H), 1.93–1.95 (m, 1H), 2.74 (bs, 2H), 3.79 (dd, J = 2.5, 6.5 Hz, 2H), 3.98 (s, 3H), 4.15 (bs, 2H), 6.82–7.04 (m, 3H), 7.20–7.44 (m, 4H); 13C NMR (125 MHz, CDCl3): δ 28.5, 28.9, 36.3, 62.5, 62.6, 72.3, 72.3, 79.4, 113.6, 115.0, 115.1, 120.6, 121.4, 128.4, 128.5, 129.0, 129.3, 129.4, 130.7, 131.5, 134.1, 134.7, 134.9, 135.4, 137.4, 154.9, 155.5, 158.8, 159.0; LRMS (ESI) m/z: 459.20(M+H)+.
(4-Chlorophenyl)(3-(piperidin-4-ylmethoxy)phenyl)methanone O-methyl oxime (27)
tert-butyl 4-((3-((4-chlorophenyl)(methoxyimino)methyl)phenoxy)methyl)piperidine-1-carboxylate (0.6 g, 1.3 mmol) was subjected to 50% TFA in CH2Cl2 (7 mL). After 1h at r.t., all volatiles were evaporated in vacuo. The residue was dissolved in CHCl3 and washed with 1N NaOH. The combined CHCl3 was washed with brine, dried over Na2SO4, and evaporated in vacuo. Purification by silica gel chromatography (4:1, CHCl3:MeOH) afforded 27 (0.56 g, 79%) as a yellow oil. 1H NMR (500 MHz, CDCl3): δ 1.74(q, J = 12 Hz, 2H), 2.05 (t, J = 14 Hz, 3H), 2.95 (q, J = 11.5 Hz, 2H), 3.47 (d, J = 12.5 Hz, 2H), 3.83 (d, J = 5.5 Hz, 2H), 3.98 (s, 3H), 6.82–7.04 (m, 3H), 7.21–7.43 (m, 5H); 13C NMR (125 MHz, CDCl3): δ 25.7, 34.4, 43.7, 62.6, 62.6, 71.2, 71.3, 113.4, 115.0, 115.1, 115.7, 121.0, 121.7, 128.5, 128.5, 129.0, 129.5, 130.7, 131.4, 134.2, 134.6, 134.9, 135.4, 137.5, 155.3, 155.3, 158.4, 158.6; LRMS (ESI) m/z: 459.14(M+H)+.
(R)-1-(4-((3-((4-Chlorophenyl)(methoxyimino)methyl)phenoxy)methyl)piperidin-1-yl)octan-2-yl carbamate ((R)-13)
To a stirred solution of 27 (180 mg, 0.50 mmol) in CH2Cl2 was added Al(CH3)3 (0.2 M in CH2Cl2, 1.50 mmol). After 15 min. at 0 °C, (R)-1,2-epoxyoctane ((R)-29, 220.0 mg, 1.75 mmol) in CH2Cl2 (0.5 mL) was added. After 4h at 0 °C, the reaction mixture was quenched with aq. sat. NaHCO3. The water phase was extracted with CH2Cl2 and the combined extract was dried over Na2SO4, and evaporated in vacuo. Purification by silica gel chromatography (4:1, CHCl3:MeOH) afforded (R)-(4-chlorophenyl)(3-((1-(2-hydroxyoctyl)piperidin-4-yl)methoxy)phenyl)methanone O-methyl oxime ((R)-30, 195.5 mg, 80%) as a colorless oil. To a stirred solution of (R)-(4-chlorophenyl)(3-((1-(2-hydroxyoctyl)piperidin-4-yl)methoxy)phenyl)methanone O-methyl oxime ((R)-30, 40.0 mg, 0.08 mmol) and DMAP (33.0 mg, 0.27 mmol) in CH2Cl2 (1 mL) was added TMSNCO (30.0 mg, 0.27 mmol). After 4h at r.t., all volatiles were evaporated in vacuo. Purification by silica gel chromatography (9:1, 1H CHCl3:MeOH) afforded (R)-13 (30.0 mg, 69%) as a colorless oil. [α]20D +105 (c 0.2 in CHCl3); NMR (400 MHz, CDCl3): δ 0.87 (d, J = 6.8 Hz, 3H), 1.26–1.53 (m, 11H), 1.86 (d, J = 11.6 Hz, 2H), 2.06 (t, J = 9.6 Hz, 1H), 2.39 (t, J = 11.6 Hz, 2H), 2.96 (d, J = 8.4 Hz, 1H), 3.16 (d, J = 10 Hz, 1H), 3.74–3.80 (m, 3H), 3.97 (s, 2H), 1.82–7.04 (m, 2H), 7.13–7.44 (m, 6H); 13C NMR (100 MHz, CDCl3): δ 14.1, 22.6, 25.6, 26.5, 28.6, 28.9, 29.2, 29.4, 31.8, 35.0, 35.7, 52.0, 55.3, 60.5, 62.5, 62.6, 64.5, 66.1, 72.3, 113.6, 115.0, 115.1, 115.6, 120.6, 121.3, 127.8, 128.4, 128.5, 129.0, 129.3, 129.4, 130.7, 131.5, 134.1, 134.7, 134.8, 135.3, 137.4, 155.5, 155.5, 158.8, 159.0. MS (ESI+): m/z Calcd. for C29H40ClN3O4 (M+H)+: 530.2706; found: 530.2801. The purity of (R)-13 was determined to be >95% by reverse HPLC analysis (Phenominex kinetex 2.6 μ C18 100A, 100 × 4.60 mm; CH3CN/0.05%TFA in H2O = 25/1). The optical purity of (R)-13 was determined to be 95% by HPLC analyses (Daicel Chiralcel OD-H (0.46 cm × 25 cm; Hexanes/tBuOH = 20/1 with flow rate=1.0 mL/min. and a UV detector at 245 nm; (S)-enantiomer: tR=20.5 min and (R)-enantiomer: tR = 21.5 min).
(R)-(4-chlorophenyl)(3-((1-(3-(1-hydroxyoctyl)benzyl)piperidin-4-yl)methoxy)phenyl)methanone O-methyl oxime ((R)-15)
Colorless oil. [α]20D +30.5 (c 0.2 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 0.86 (t, J = 6.5 Hz, 3H), 1.24–1.43 (m, 12H), 1.67–1.80 (m, 5H), 1.99 (t, J = 11.5 Hz, 2H), 2.91 (d, J = 11.5 Hz, 2H), 3.51 (s, 2H), 3.77 (d, J = 4 Hz, 2H), 3.97 (s, 3H), 4.65 (t, J = 7 Hz, 1H), 6.81–7.03 (m, 3H), 7.19–7.43 (m, 9H); 13C NMR (125 MHz, CDCl3): δ 14.1, 22.7, 25.9, 29.0, 29.3, 29.5, 31.8, 35.9, 39.2, 53.3, 53.4, 62.5, 62.6, 63.4, 72.7, 74.6, 113.7, 115.0, 115.1, 115.6, 120.5, 121.2, 124.6, 126.8, 128.3, 128.4, 128.4, 128.5, 129.0, 129.3, 129.3, 130.7, 131.5, 134.1, 134.7, 134.8, 135.3, 137.4, 138.4, 145.0, 155.5, 155.6, 159.0, 159.1; HRMS (ESI+): m/z Calcd. for C35H45ClN2O3 (M+H)+: 577.3105; found: 577.3106. The purity of (R)-15 was determined to be >95% by reverse HPLC analysis (Phenominex kinetex 2.6 μ C18 100A, 100 × 4.60 mm; CH3CN/0.05%TFA in H2O = 25/1). The optical purity of (R)-15 was determined to be >99% by HPLC analyses (Daicel Chiralcel OD-H (0.46 cm × 25 cm; Hexanes/tBuOH = 20/1 with flow rate=1.0 mL/min. and a UV detector at 245 nm; (S)-enantiomer: tR = 20.5 min and (R)-enantiomer: tR = 21.0 min)..
(R)-1-(4-((3-chloro-4-((4-chlorophenyl)(methoxyimino)methyl)phenoxy)methyl)piperidin-1-yl)octan-2-yl carbamate ((R)-18)
Colorless oil. [α]20D +21.5 (c 0.5 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 0.89 (t, J = 7 Hz, 3H), 1.29–1.51 (m, 13H), 1.78–1.85 (m, 3H), 1.95–2.08 (m, 1H), 2.23–2.36 (m, 3H), 2.92 (bs, 1H), 3.67–3.75 (1H), 3.82 (t, J = 6 Hz, 2H), 3.98 (s, 3H), 6.88 (dd, J = 1, 8 Hz, , 1H), 7.01 (d, J = 2 Hz, 1H), 7.07 (dd, J = 3, 9 Hz, 1H), 7.29 (d, J = 8.5 Hz, 2H), 7.42 (d, J = 8 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 14.1, 14.1, 22.6, 22.7, 25.6, 25.7, 29.0, 29.1, 29.3, 29.4, 29.5, 30.9, 31.8, 31.9, 35.0, 35.8, 36.1, 51.7, 54.3, 54.4, 55.2, 62.7, 64.5, 65.8, 66.3, 70.6, 72.8, 73.1, 113.5, 113.5, 115.4, 124.6, 124.7, 128.1, 128.6, 130.5, 130.5, 133.2, 133.2, 133.8, 133.8, 135.4, 153.1, 153.2, 159.9, 160.0; HRMS (ESI+): m/z Calculated for C29H39Cl2N3O4 (M+H)+: 564.2308; found: 564.2310. The purity of (R)-18 was determined to be >95% by reverse HPLC analysis (Phenominex kinetex 2.6 μ C18 100A, 100 × 4.60 mm; CH3CN/0.05%TFA in H2O = 25/1). The optical purity of (R)-18 was determined to be >99% by HPLC analyses (Daicel Chiralcel OD-H (0.46 cm × 25 cm; Hexanes/tBuOH = 22/1 with flow rate=1.0 mL/min. and a UV detector at 245 nm; (S)-enantiomer: tR=20.5 min and (R)-enantiomer: tR = 21.0 min).
(S)-(2-Chloro-4-((1-(3-(1-hydroxyoctyl)benzyl)piperidin-4-yl)methoxy)phenyl)(4-chlorophenyl) methanone O-methyl oxime ((S)-20)
Colorless oil. [α]20D −30.5 (c 0.5 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 1H NMR (500 MHz, CDCl3): δ 0.87 (t, J = 7 Hz, 3H), 1.25–1.46 (m, 12H), 1.68–1.74 (m, 1H), 1.81 (d, J = 10.5 Hz, 4H), 2.02 (t, J = 9.5 Hz, 2H), 2.94 (d, J = 10.5 Hz, 2H), 3.53 (s, 2H), 3.81 (d, J = 6 Hz, 2H), 3.97 (s, 3H), 4.67 (t, J = 6.5 Hz, 1H), 6.86 (dd, J = 2.5, 8.5 Hz, 1H), 7.00 (d, J = 2 Hz, 1H), 7.06 (d, J = 8.5 Hz, 1H), 7.23–7.32 (m, 6H), 7.41 (d, J = 8 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 14.1, 22.7, 25.9, 29.0, 29.3, 29.5, 31.8, 35.8, 39.2, 45.9, 53.3, 53.3, 62.7, 63.4, 72.9, 74.7, 113.5, 115.4, 124.6, 126.8, 128.1, 128.3, 128.4, 128.6, 130.5, 133.2, 133.8, 135.8, 145.0, 153.2, 160.0; HRMS (ESI+): m/z Calcd. for C35H44Cl2N2O3 (M+H)+: 611.2712; found: 611.2717. The purity of (S)-20 was determined to be >95% by reverse HPLC analysis (Phenominex kinetex 2.6 μ C18 100A, 100 × 4.60 mm; CH3CN/0.05%TFA in H2O = 25/1). The optical purity of (S)-20 was determined to be 95% by HPLC analyses (Daicel Chiralcel OD-H (0.46 cm × 25 cm; Hexanes/tBuOH = 25/1 with flow rate=1.0 mL/min. and a UV detector at 245 nm; (S)-enantiomer: tR=20.0 min and (R)-enantiomer: tR = 21.5 min).
(R)-(2-Chloro-4-((1-(3-(1-hydroxyoctyl)benzyl)piperidin-4-yl)methoxy)phenyl)(4-chlorophenyl) methanone O-methyl oxime ((R)-20)
Colorless oil. [α]20D +29.5 (c 0.5 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 1H NMR (500 MHz, CDCl3): δ 0.87 (t, J = 7 Hz, 3H), 1.25–1.46 (m, 12H), 1.68–1.74 (m, 1H), 1.81 (d, J = 10.5 Hz, 4H), 2.02 (t, J = 9.5 Hz, 2H), 2.94 (d, J = 10.5 Hz, 2H), 3.53 (s, 2H), 3.81 (d, J = 6 Hz, 2H), 3.97 (s, 3H), 4.67 (t, J = 6.5 Hz, 1H), 6.86 (dd, J = 2.5, 8.5 Hz, 1H), 7.00 (d, J = 2 Hz, 1H), 7.06 (d, J = 8.5 Hz, 1H), 7.23–7.32 (m, 6H), 7.41 (d, J = 8 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 14.1, 22.7, 25.9, 29.0, 29.3, 29.5, 31.8, 35.8, 39.2, 45.9, 53.3, 53.3, 62.7, 63.4, 72.9, 74.7, 113.5, 115.4, 124.6, 126.8, 128.1, 128.3, 128.4, 128.6, 130.5, 133.2, 133.8, 135.8, 145.0, 153.2, 160.0; HRMS (ESI+): m/z Calcd. for C35H44Cl2N2O3 (M+H)+: 611.2712; found: 611.2717. The purity of (R)-20 was determined to be >95% by reverse HPLC analysis (Phenominex kinetex 2.6 μ C18 100A, 100 × 4.60 mm; CH3CN/0.05%TFA in H2O = 25/1). The optical purity of (R)-20 was determined to be 95% by HPLC analyses (Daicel Chiralcel OD-H (0.46 cm × 25 cm; Hexanes/tBuOH = 25/1 with flow rate=1.0 mL/min. and a UV detector at 245 nm; (S)-enantiomer: tR=20.0 min and (R)-enantiomer: tR = 21.5 min).
Preparation of MenA containing Membrane Fraction
M. tuberculosis was grown to mid-log phase in Difco Middlebrook 7H9 nutrient broth (enriched with OADC) and then the cells were harvested by centrifugation at 4 °C followed by washing with 0.9% saline solution (thrice) through centrifugation. The washed cell pellets were suspended in homogenization buffer (containing 50 mM MOPS of pH = 8, 0.25 M sucrose, 10 mM MgCl2 and 5 mM 2-Marcaptoethanol) and disrupted by probe sonication on ice (10 cycles of 60s on and 90s off). The resulting suspension was then centrifuged at 15,000 g for 15 min at 4 °C. The pellet was discarded and the supernatant was centrifuged again at 200,000 g at 4 °C for 1h. The resulting supernatant contain the membrane bound Men A. M. smegmatis and S. aureus MenA containing membrane fractions were obtained by the same procedures.41
MenA Enzyme Inhibitory Assay (IC50)
The substrate 1,4-dihydroxy-2-napthanoic acid (DHNA) (500 μM; 20 μL), MgCl2 (5 μM; 20 μL); CHAPS (0.1%; 20 μl), Tris-buffer (pH = 8; 20 μL), membrane fraction containing Men A (100 μl), inhibitors (0–60 μg/mL of DMSO) and fernesylpyrophosphate (200 μM, 40 μL) were mixed together and incubated for 2h at 37 °C. After the incubation, the reaction mixture was quenched with 0.1 M AcOH in MeOH and the reaction mixture was extracted with hexanes (thrice). The organic portions were then concentrated and diluted with MeOH (300 μL). From each set of enzymatic reaction mixture 20 μl was injected into the HPLC (CH3CN:0.05%TFA in H2O = 90:10, UV: 325 nm, flow rate: 1.5 mL/min) and the area of the peak for DMMK was quantified to obtain the IC50 value for different inhibitor molecules.
Minimum Inhibitory Concentration Assays (MABA and LORA)
These were performed according to the published protocol. A preliminary screening was conducted at 12.5 μg/mL against M. tuberculosis H37Rv (ATCC 27294), at 60 μg/mL against S. aureus Seattle 1945 (ATCC 25923), and at 125 mg/mL against E. coli Seattle 1946 (ATCC 25922). Compounds demonstrated at least 90% inhibitions in the preliminary screen were retested at lower concentrations to determine the MIC.
E. coli Growth Inhibitory Assays under Anaerobic Conditions
E. coli was grown with inhibitor molecule (5μg/mL) under anaerobic conditions in the modified E. coli growth media (see Supporting Information), and the growth curve was monitored photometrically by reading the optical density at 600 nm.55,56 E. coli growth rescue studies were performed by supplementing VK2 (50 μM).
Oxygen Consumption Assays
In sterile glass vial 1.5 mL of M. tuberculosis or M. Smegmatis (grown to 0.5 OD value) was placed. Inhibitor molecule (80 μL, total concentration ranged from 5–100 μg/mL) and 0.01% methylene blue (100 μL) was added into the culture solution. The culture vials were kept at 37 °C for 22h. The effective concentration of the inhibitor molecule at which the electron transport systems was determined by comparing the color intensity with the control vial (no inhibitor).
Determination of IC50 in Vero and HepG2 cells
Selected molecules were tested for cytotoxicity (IC50) in Vero and HepG2 cells at concentrations 10 times the MIC for M. tuberculosis H37Rv. After 72h of exposure of molecule to these cell lines, viability was assessed on the basis of cellular conversion of MTT into a formazan product. Selectivity index (SI = IC50/MIC) was determines. SI >10 was considered to be significant in this program.
Supplementary Material
Acknowledgments
We are grateful to Drs. Takushi Kaneko, Christopher Cooper, and Khisimuzi Mdluli at Global Alliance for TB Drug Development for useful discussions. MK thanks Dr. Dirk Schnappinger (Weill Carnell Medical College) for invaluable information regarding menA knockdown mutant M. tuberculosis. MK also thanks Dr. Kitagawa for his generous support in providing a sample of natural aurachin RE. This work was supported by the University of Tennessee. We utilized some synthetic intermediates in this study obtained from the program funded by the National Institutes of Health (AI084411-02). NMR data were obtained on instruments supported by the NIH Shared Instrumentation Grant.
ABBREVIATIONS USED
- Mtb
Mycobacterium tuberculosis
- S. aureus
Staphylococcus aureus
- MDR
multi-drug resistant
- VK2
vitamin K2 (menaquinone)
- MenA
1,4-dihydroxy-2-naphthoate prenyltransferase
- Q10
coenzyme Q10 (ubiquinone)
- MIC
minimum inhibitory concentration
- MABA
microplate alamar blue assay
- ROLA
low-oxygen recovery assay
- MK
menaquinone
- DMMK
demethylmenaquinone
- SI
selectivity index
- REF
rifampin
- INH
isoniazid
- EMB
ethanbutol; a diarylquinolone TB drug lead ((1R,2S)-1-(6-bromo-2-methoxyquinolin-3-yl)-4-(dimethylamino)-2-(naphthalen-1-yl)-1-phenylbutan-2-ol)
- Tet
tetracycline
- DIAD
Diisopropyl azodicarboxylate
- TPP
triphenylphosphine
- CBS
Corey-Bakshi-Shibata reduction
- Py
pyridine
- DMAP
dimethylaminopyridine
Footnotes
ASSOCIATED CONTENT
Supporting Information. Some assay data, copies of NMR spectra and HPLC chromatogram of new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
REFRENCES AND NOTES
- 1.Grenet K, Guillemot D, Jarlier V, Moreau B, Dubourdieu S, Ruimy R, Armand-Lefevre L, Brau P, Andremont A. Antibacterial resistance, Wayampis Amerindians, French Guyana. Emerg Infect Dis. 2004;10:1150–1153. doi: 10.3201/eid1006.031015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen J. New TB drug promises shorter, simpler treatment. Science. 2004;306:1872. doi: 10.1126/science.306.5703.1872. [DOI] [PubMed] [Google Scholar]
- 3.Cole ST, Alzari PM. TB-A new target, a new drug. Science. 2005;14:214–215. doi: 10.1126/science.1108379. [DOI] [PubMed] [Google Scholar]
- 4.Stover CK, Warrener P, VanDevater DR, Sherman DR, Arain TM, Langhorne MH, Anderson SW, Towell JA, Yuan Y, McMurray DN, Kreiswirth BN, Barry CE, Baker WR. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature. 2000;405:962–966. doi: 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- 5.Nunn P, Kochi A. A deadly duo-TB and AIDS. World Health. 1993;46:7–8. [Google Scholar]
- 6.Godfrey-Faussett P. District-randomized phased implementation: strengthening the evidence based for contrimoxazole for HIV-positive tuberculosis patients. AIDS. 2003;17:1079–1081. doi: 10.1097/01.aids.0000060354.78202.fe. [DOI] [PubMed] [Google Scholar]
- 7.McKinney JD, Hoener zu Bentrup K, Munoz-Elias EJ, Miczack A, Chen B, Chan WT, Swenson D, Sacchettini C, Jacobs WR, Russell DG. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature. 2000;406:683–685. doi: 10.1038/35021074. [DOI] [PubMed] [Google Scholar]
- 8.Weber I, Fritz C, Ruttkowski S, Kreft A, Bange FC. Anaerobic nitrate reductase (narGHJI) activity of Mycobacterium bovis BCG in vitro and its contribution to virulence in immunodeficient mice. Mol Microbiol. 2000;35:1017–1025. doi: 10.1046/j.1365-2958.2000.01794.x. [DOI] [PubMed] [Google Scholar]
- 9.Rosenkrands I, Slayden RA, Crawford J, Aagaard C, Barry CE, Andersen P. Hypoxic response of Mycobacterium tuberculosis studied by metabolic labeling and proteome analysis of cellular and extracellular proteins. J Bacteriol. 2002;184:3485–3491. doi: 10.1128/JB.184.13.3485-3491.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iona E, Giannoni F, Pardini M, Brunori L, Orefici G, Fattorini L. Metronidazole plus rifampin sterilizes long-term dormant Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2007;51:1537–1540. doi: 10.1128/AAC.01468-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. 1961;191:144–148. doi: 10.1038/191144a0. [DOI] [PubMed] [Google Scholar]
- 12.Mitchell P. Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biol Rev Cambridge Phil Soc. 1966;41:445–502. doi: 10.1111/j.1469-185x.1966.tb01501.x. [DOI] [PubMed] [Google Scholar]
- 13.Mitchell P. Chemiosmotic coupling in energy transduction: A logical development of biochemical knowledge. Bioenergetics. 1972;3:5–24. doi: 10.1007/BF01515993. [DOI] [PubMed] [Google Scholar]
- 14.Bishop DHL, Pandya KP, King HK. Ubiquinone and vitamin K in bacteria. Biochem J. 1962;83:606–614. doi: 10.1042/bj0830606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haddock BA, Colin JW. Bacterial respiration. Bacteriol Rev. 1977;41:47–99. doi: 10.1128/br.41.1.47-99.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yasuhiro A. Bacterial electron transport chains. Ann Rev Biochem. 1988;57:101–312. doi: 10.1146/annurev.bi.57.070188.000533. [DOI] [PubMed] [Google Scholar]
- 17.Kurosu M, Begari E. Vitamin K2 in electron transport system: Are enzymes Involved in vitamin K2 biosynthesis promising drug targets? Molecules. 2010;15:1531–1553. doi: 10.3390/molecules15031531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schnappinger D. unpublished data [Google Scholar]
- 19.Andries K, Verhasselt P, Guillemont J. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005;14:223–727. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 20.Weinstein EAT, Yano LS, Li D, Avarbock A, Avarbock D, Helm AA, McColm K, Duncan J, Lonsdale T, Rubin H. Inhibitors of type II NADH:menaquinone oxidoreductase represent a class of antitubercular drugs. Proc Natl Acad Sci US A. 2005;102:4548–4553. doi: 10.1073/pnas.0500469102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kurosu M, Narayanasamy P, Biswas K, Dhiman R, Crick DC. Discovery of 1, 4-dihydroxy-2-naphthoate prenyltransferase inhibitors: New drug leads for multidrug-resistant Gram-positive pathogens. J Med Chem. 2007;50:3973–3974. doi: 10.1021/jm070638m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurosu M, Crick DC. MenA is a promising drug target for developing novel molecules to combat Mycobacterium tuberculosis. Med Chem. 2009;5:197–207. doi: 10.2174/157340609787582882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu X, Zhang H, Tonge PJ, Tan DS. Mechanism-based inhibitors of MenE, an acyl-CoA synthetase involved in bacterial menaquinone biosynthesis. Bioorg Med Chem Lett. 2008;18:5963–5966. doi: 10.1016/j.bmcl.2008.07.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang H, Tonge PJ. The mechanism of the reactions catalyzed by 1,4-dihydroxynaphthoyl-CoA synthase (MenB) and 2-ketocyclohexanecarboxyl-CoA hydrolase (BadI). Abstract in 234th ACS National Meeting; Boston, MA, United States. 2007, August 19–23. [Google Scholar]
- 25.Li X, Zhang H, Tonge PJ. Inhibition of 1,4-dihydroxynaphthoyl-CoA synthase (MenB), an enzyme drug target bacterial menaquinone biosynthesis pathway. Abstract in 236th ACS National Meeting; Philadelphia, PA, United States. 2008, August 17–21. [Google Scholar]
- 26.Xu H, Graham M, Karelis J, Walker SG, Peter J, Tonge PJ. Mechanistic studies of MenD, 2-succinyl-5-enoylpyruvyl-6-hydroxy-3-cyclohexene-1-carboxylic acid synthase from Staphylococcus aureus. Abstract in 237th ACS National Meeting; Salt Lake City, UT. 2009, March 22–26. [Google Scholar]
- 27.Kitagawa W, Tamura T. A quinoline antibiotic from Rhodococcus ertthropolis JCM 6824. J Antibiot. 2008;61:680–682. doi: 10.1038/ja.2008.96. [DOI] [PubMed] [Google Scholar]
- 28.Barry PJ, O’Connor TM. Novel agents in the management of Mycobacterium tuberculosis disease. Cur Med Chem. 2007;14:2000–2008. doi: 10.2174/092986707781368496. [DOI] [PubMed] [Google Scholar]
- 29.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 30.Unden G, Bongaerts J. Alternative respiratory pathways of Escherichia coli: energetics and transcriptional regulation in response to electron acceptors. Biochim Biophys Acta. 1997;1320:217–234. doi: 10.1016/s0005-2728(97)00034-0. [DOI] [PubMed] [Google Scholar]
- 31.The MenA inhibitors identified in this program inhibit growth of E. coli at 5~10 μg/mL concentrations under anaerobic conditions.
- 32.Boshoff HI, Myers TG, Copp BR, McNeil MR, Wilson MA, Barry CE., III The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: novel insights into drug mechanisms of action. J Biol Chem. 2004;279:40174–40184. doi: 10.1074/jbc.M406796200. [DOI] [PubMed] [Google Scholar]
- 33.Mathew R, Kruthiventi AK, Prasad JV, Kumar SP, Srinu G, Chatterji D. Inhibition of Mycobacterial growth by plumbagin derivatives. Chem Biol Drug Des. 2010;76:34–42. doi: 10.1111/j.1747-0285.2010.00987.x. [DOI] [PubMed] [Google Scholar]
- 34.Cho SH, Warit S, Wan B, Hwang CH, Pauli GF, Franzblau SG. Low-oxygen-recovery assay for high-throughput screening of compounds against nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2007;51:1380–1358. doi: 10.1128/AAC.00055-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mitsunobu O, Yamada Y. Preparation of esters of carboxylic and phosphoric acid via quaternary phosphonium salts. Bull Chem Soc Japan. 1967;40:2380–2382. [Google Scholar]
- 36.Mitsunobu O. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products. Synthesis. 1981:1–28. [Google Scholar]
- 37.Tokunaga M, Larrow JF, Kakiuchi F, Jacobsen EN. Asymmetric catalysis with water: Efficient kinetic resolution of terminal epoxides by means of catalytic hydrolysis. Science. 1997;277:936–938. doi: 10.1126/science.277.5328.936. [DOI] [PubMed] [Google Scholar]
- 38.Corey EJ, Bakshi RK, Shibata S. Highly enantioselective borane reduction of ketones catalyzed by chiral oxazaborolidines. Mechanism and synthetic implications. J Am Chem Soc. 1987;109:5551–5552. [Google Scholar]
- 39.Nahm S, Weinreb SM. N-Methoxy-N-methylamides as effective acylating agents. Tetrahedron Lett. 1981;22:3815–3818. [Google Scholar]
- 40.Pujala SB, Chakraborti AK. Zinc(II) perchlorate hexahydrate catalyzed opening of epoxide ring by amines: applications to synthesis of (RS)/(R)-propranolols and (RS)/(R)/(S)-naftopidils. J Org Chem. 2007;72:3713–3722. doi: 10.1021/jo062674j. [DOI] [PubMed] [Google Scholar]
- 41.Kurosu M, Li K. New chiral derivatizing agents: Convenient determination of absolute configurations of free amino acids by 1H NMR. Org Lett. 2009;11:911–914. doi: 10.1021/ol8028719. references therein. [DOI] [PubMed] [Google Scholar]
- 42.Dhiman RK, Mahapatra S, Slayden RA, Boyne ME, Lenaerts A, Hinshaw JC, Angala SK, Chatterjee D, Biswas K, Narayanasamy P, Kurosu M, Crick DC. Menaquinone synthesis is critical for maintaining Mycobacterial viability during exponential growth and recovery from non-replicating persistence. Mol Microbiol. 2009;72:85–97. doi: 10.1111/j.1365-2958.2009.06625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.UV absorption of DMMK; λ1 265 nm, λ2 325 nm
- 44.Collins LA, Franzblau SG. Distribution of isoprenoid quinone structual types in bacterial and their taxonomic implications. Antimicrob Agents Chemother. 1997;41:1004–1009. doi: 10.1128/aac.41.5.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lenaerts AJ, Gruppo, Marietta KS, Johnson CM, Driscoll DK, Tompkins NM, Rose JD, Reynolds RC, Orme IM. Preclinical testing of the nitroimidazopyran PA-824 for activity against Mycobacterium tuberculosis in a series of in vitro and in vivo models. Antimicrob Agents Chemother. 2005;49:2294–2301. doi: 10.1128/AAC.49.6.2294-2301.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.The molecules summarized in Table 1 did not show bactericidal activities against several other Gram-positive bacteria, Streptococcus salivarius (ATCC 7073), Enterococcus faecalis (ATCC 47077), and Enterococcus durans (ATCC 6056) at 60 μg/mL concentrations.
- 47.Debnath J, Siricilla S, Kurosu M. unpublished data [Google Scholar]
- 48.The IC50 values of the molecules evaluated using Vero and HepG2 cells were identical.
- 49.It is considered that the selectivity index (SI) of >10 is significant in this program.
- 50.Wayne LG, Sramek HA. Antigenic differences between extracts of actively replicating and synchronized resting cells of Mycobacterium tuberculosis. Infect Immun. 1979;24:363–370. doi: 10.1128/iai.24.2.363-370.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wayne LG, Hayes LG. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect Immun. 1996;64:2062–2069. doi: 10.1128/iai.64.6.2062-2069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wayne LG, Sramek HA. Metronidazole is bactericidal to dormant cells of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 1994;38:2054–2058. doi: 10.1128/aac.38.9.2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Honaker RW, Leistikow RL, Bartek IL, Voskuil MI. Unique roles of DosT and DosS in DosR regulon induction and Mycobacterium tuberculosis dormancy. Infect Immun. 2009;77:3258–3263. doi: 10.1128/IAI.01449-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.A series of molecules identified in this program showed strong MenG activity. These data will report elsewhere.
- 55.Futatsugi L, Saito H, Kakegawa T, Kobayashi H. Growth of an Escherichia coli mutant deficient in respiration. FEMS Microbiol Lett. 1997;156:141–145. doi: 10.1111/j.1574-6968.1997.tb12719.x. [DOI] [PubMed] [Google Scholar]
- 56.Bagramyan K, Mnatsakanyan N, Poladyan A, Vassilian A, Trchounian A. The roles of hydrogenases 3 and 4, and the F0F1-ATPase, in H2 production by Escherichia coli at alkaline and acidic pH. FEBS Lett. 2002;516:172–178. doi: 10.1016/s0014-5793(02)02555-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.