

Abstract
Herein, we describe the X-ray structure-based design and optimization of biaryl mannoside FimH inhibitors. Diverse modifications to the biaryl ring to improve drug-like physical and pharmacokinetic properties of mannosides were assessed for FimH binding affinity based on their effects on hemagglutination and biofilm formation along with direct FimH binding assays. Substitution on the mannoside phenyl ring ortho to the glycosidic bond results in large potency enhancements of several-fold higher than corresponding unsubstituted matched pairs and can be rationalized from increased hydrophobic interactions with the FimH hydrophobic ridge (Ile13) or “tyrosine gate” (Tyr137 and Tyr48) also lined by Ile52. The lead mannosides have increased metabolic stability and oral bioavailability as determined from in vitro PAMPA predictive model of cellular permeability and in vivo pharmacokinetic studies in mice, thereby representing advanced preclinical candidates with promising potential as novel therapeutics for the clinical treatment and prevention of recurring urinary tract infections.
Introduction
FimH is a mannose-specific bacterial lectin located at the tip of type 1 pili, an adhesive fiber produced by uropathogenic E. coli (UPEC).1 FimH is known to bind to mannosylated human uroplakins that coat the luminal surface of the bladder2 and has also been shown to be involved in invasion of human bladder cells3 and mast cells4, triggering apoptosis and exfoliation5 and inducing elevated levels of cAMP6. Furthermore, FimH recognizes N-linked oligosaccharides on beta1 and alpha3 integrins, which are expressed throughout the urothelium.7 Murine uroplakin is highly homologous to human and FimH has been shown to facilitate bacterial colonization and invasion of the bladder epithelium in murine models.8 Internalized UPEC are exocytosed in a TLR-4 dependent process;9 however, bacteria can escape into the host cell cytoplasm, where they are able to subvert expulsion and innate defenses by aggregating into biofilm-like intracellular bacterial communities (IBCs) in a FimH dependent process.8b,8c,10 Subsequently, UPEC disperse from the IBC, escape into the bladder lumen, and re-initiate the process by binding and invading naive epithelial cells where they are able to establish quiescent intracellular reservoirs that can persist in a dormant state, tolerant to antibiotic therapy and subsequently serve as seeds for recurrent infection.11 In humans, the severity of UTI was increased and the immunological response was greater in children with infections caused by type 1 piliated UPEC strains and type 1 pilus expression has been shown to be essential for UTI in mouse models.12 In addition, a recent study concluded that type 1 pili play an important role in human cystitis13 and it has been reported that type 1 pili fulfill “Molecular Koch’s postulates of microbial pathogenesis14. In agreement with these findings and in support of a role for FimH in humans, it has been shown that the fimH gene is under positive selection in human clinical isolates of UPEC.8a,15 Aspects of the UPEC pathogenic cascade extensively characterized in a murine model of infection8b,8e,10 have been documented in samples from human clinical studies such as filamentation and IBC formation16. Targeted inhibitors of FimH adhesion which block both E. coli invasion and biofilm formation thus hold promising therapeutic potential as new antibacterials for the treatment of UTI and the prevention of recurrence.17,18
The discovery of simple D-mannose derivatives as inhibitors of bacterial adherence was first reported almost three decades ago19 but early mannosides showed only weak inhibition of bacterial adhesion. Consequently, the vast majority of research in this area has been focused on multivalent mannosides20, which have been pursued in an effort to improve binding avidity to type 1 pili, which can be expressed present in large numbers on a single bacterium (up to hundreds). While substantial progress has been made with this approach, these high molecular weight structures are not suitable for in vivo evaluation or clinical development as oral drugs. The recent X-ray crystal structures of D-mannose21, butyl mannoside22, and mannotriose23 bound to FimH have enabled the rational structure-based design of tighter binding alkyl-22, phenyl-24 and biphenyl-25,26 mannoside FimH inhibitors. The urgency for developing new orally bioavailable FimH inhibitors26 as a targeted strategy for the treatment of UTI alternative to broad spectrum antibiotics is reinforced by the rate of recurrence seen in these type of infections as well as increasing clinical resistance of UPEC to first line antibiotic treatments.27
Results and Discussion
In an earlier study we reported the discovery of biphenyl mannosides 1–3 (Figure 1a) which make strong hydrophobic interactions to residues forming the outer ‘gate’ of the FimH binding pocket. X-ray crystallographic data of compound 1 bound to FimH revealed both a key π-π interaction of Tyr48 with the second phenyl ring of 1 and a tight H-bond between Arg98 and the ester carbonyl.25 In this communication we describe the lead optimization of biphenyl mannoside 3 following the detailed strategy outlined in Figure 1b. Part of the plan was to improve FimH binding affinity through increased interactions with FimH using the structure of the FiimH-1 complex to guide our compound design. Directed by these structure-activity relationships (SAR), the final goal of optimization was to improve the metabolic stability, bioavailability, bladder tissue exposure and in general the drug-like properties of the mannosides through a multifaceted approach including heterocyclic replacements of both phenyl rings, ortho-substitution off of each ring, and amide substitutions with higher pKa moieties.
Figure 1.

a) Biphenyl mannoside FimH inhibitors with meta-substituted H- bond acceptor; b) Lead optimization strategy of lead biphenyl mannoside 3 for increased FimH binding affinity, good drug-like physical properties and improved pharmacokinetics.
ortho-Substituted Phenyl Mannosides
In addition to our work on mannoside FimH inhibitors Ernst26 has subsequently described biphenyl mannosides as FimH antagonists and demonstrated that substitution with chlorine on the ortho-position of the phenyl ring directly attached to D-mannose resulted in a substantial increase in FimH inhibition. This key piece of SAR for phenyl mannosides was actually first observed in 1987 by Firon et al.19 but has gone largely unnoticed until recently. Shown in Figure 2, Firon reported that p-nitro-o-chlorophenyl mannoside had a 10-fold increase in potency relative to p-nitrophenyl mannoside using a yeast hemagglutination assay and later in 2006 Sperling et. al. reported a 4-fold increase in activity from ortho-chlorine substitution on a different phenyl mannoside24. We also previously reported an 8-fold and 5-fold increase in activity, respectively, from ortho chlorine and nitrile substitution on phenyl mannoside but found larger groups (e.g. phenyl) decreased potency.25 Intrigued by these results from us and others, we were interested in elucidating the molecular basis for this increased potency derived from ortho substitution on the biphenyl ring.
Figure 2.

Potency enhancement from ortho-chloro substitution of phenyl mannosides.
Structure-Guided Design of Tight-Binding ortho-Substituted Biaryl Mannosides
The structure of 1 bound to FimH (Figure 1b) suggested that the ortho-position of the ring attached to mannose is aimed at Tyr137 residue at the ridge of FimH binding pocket and improved hydrophobic contact and binding affinity to FimH could be achieved by substitution with small groups but would be sterically encumbered by larger groups such as an aryl ring.25 Thus, we performed a matched pair analysis of monoester 1 compared to ortho-substituted analogs bearing halogen and small alkyl groups shown in Figure 3. The compounds were evaluated for their potency in the hemagglutination inhibition (HAI) assay28 and we discovered that all biphenyl ring substitutions yielded more potent inhibitors (Table 1). Ortho-Cl mannoside 4b had a HAI Titer EC90 of 30 nM which is more than 30-fold better than matched pair 1 while the Me analog 4c was 8-fold more active (HAI Titer EC90 = 120 nM). Substitution with CF3 gave the most potent analog 4d with an HAI Titer EC90 of 30 nM whereas the OMe (4e) and F (4a) analogs showed smaller improvements in activity following the trend CF3 > Cl = Me > OMe > F. This data suggests that increased hydrophobic contact with the tyrosine gate and Ile52, or with Ile13 at the opposite ridge of the mannose binding pocket could explain this enhanced potency since better activity correlates well with increased hydrophobicity as evidenced by the fact that fluoro analog 4a shows no improvement in activity relative to unsubstituted matched pair 1 and the trifluoromethyl analog 4c which has the largest hydrophobic surface area shows the highest activity. However, it is possible that the orientation of both phenyl rings are altered slightly26 and are restricted to a conformation more conducive to improved FimH binding with Tyr137, Tyr48, Ile52, Ile13 and/or Arg98 residues.
Figure 3.

Substituted biphenyl mannosides
Table 1.
Potency enhancement and PAMPA data from ortho-substitution of biphenyl mannosides.
| Compound | HAI Titer EC>90 (μM) | Biofilm Prevention IC50 (μM) | MW (g/mol) | PSA | CLogD7.4 | PAMPA LogPe (cm2/sec) |
|---|---|---|---|---|---|---|
| 1 (ester) | 1.00 | 0.94 | 390.4 | 126 | 1.17 | −5.42 |
| 4a (F) | 0.75 | 408.4 | 126 | 1.00 | ||
| 4b (Cl) | 0.03 | 0.26 | 424.8 | 126 | 1.64 | −4.29 |
| 4c (Me) | 0.12 | 0.33 | 404.4 | 126 | 1.82 | |
| 4d (CF3) | 0.03 | 0.17 | 458.4 | 126 | 1.62 | −3.91 |
| 4e (OMe) | 0.19 | 0.89 | 420.4 | 135 | 0.47 | |
| 6 (m-CO2Me) | 1.0 | 448.4 | 152 | 0.93 | −4.08 | |
| 2 (amide) | 0.50 | 1.35 | 389.4 | 128 | 0.28 | |
| 5a (Cl) | 0.12 | 0.52 | 423.8 | 128 | 0.75 | |
| 5b (Me) | 0.06 | 0.16 | 403.4 | 128 | 0.93 | −3.89 |
| 5c (CF3) | 0.03 | 0.13 | 457.4 | 128 | 0.73 | |
| 3 (di-amide) | 0.37 | 0.74 | 446.4 | 158 | 0.71 | −4.51 |
| 7 (CF3) | 0.01 | 0.043 | 514.5 | 158 | 1.15 | −6.27 |
| 8 (Me) | 0.02 | 0.073 | 460.5 | 158 | 1.35 | −8.46 |
The outcome of this preliminary study directed us to pursue analogs which were more metabolically stable and soluble than esters such as amides 5a–c (Figure 3 and Table 1). A similar trend was observed (CF3 > Me > Cl) with CF3 amide 5c having the best activity but in this case the Me analog 5b showed better activity than Cl analog 5a. We also explored analogs with substitution on the meta position, exemplified by ester 6, which retains potency relative to 1 but did not lead to any enhancement. We next developed ortho-CF3 7 and Me 8 diamide matched pairs to original lead compound 3 which were exponentially more potent than any previously reported mannoside FimH inhibitors with an HAI Titer EC90 of 8 nM and 16 nM, respectively. This unprecedented level of cellular activity corresponds to a 200,000-fold improvement over α-D-mannose and a 15,000-fold improvement over an early reported inhibitor butyl-α-D-mannoside (HAI Titer EC90 = 125 μM) and 50-fold better than previous lead compound 3.
The biofilm inhibition assay17b,17c,29 was utilized to test these mannosides’ ability to prevent bacteria from forming IBCs, a critical pathogenic process in the development of UTIs. As shown in Table 1, the Biofilm Prevention IC50s correlate quite well with the potencies determined by the HAI assay. Introduction of an ortho-substituent (e.g. methyl) to the biphenyl mannoside improved the biofilm activity by 8-fold from mannoside 2 (IC50 = 1.35 μM) to mannoside 5b (IC50 = 0.16 μM). This data confirmed the mannoside’s functional effect and activity on UPEC derived from FimH inhibition with a secondary assay. Biofilm IC50s were utilized in conjunction with pharmacokinetic parameters as a key measure of the predicted lowest effective mannoside concentration in the urine required for efficacy in vivo to be discussed vide infra..
Synthesis of ortho-substituted Biaryl Mannosides
Mannosides were synthesized by traditional Lewis acid mediated glycosylation of mannose penta-acetate by reaction with 2-substituted 4-bromophenols using BF3 etherate (Scheme 1).25,30 Suzuki cross-coupling with commercially available 3-substituted phenyl boronic acid derivatives gave protected ortho-substituted 4′-biphenyl mannosides in excellent yields and subsequent deprotection with NaOMe gave mannosides 4–5. 6 was prepared following the procedure previously described.25 Synthesis of di-amides 7–8 followed a similar procedure but required the synthesis of 3,5-di-(N-methyl aminocarbonyl)phenyl boronic acid pinacol ester 9. Shown in Scheme 1, amidation of dimethyl 5-bromoisophthalate by reaction of methylamine in ethanol proceeded in quantitative yield to give N,N-dimethyl 5-bromoisophthalamide. Installation of the boronate ester was accomplished by Pd-mediated coupling with bis (pinacolato) diboron to give 9.31 Suzuki coupling and deprotection as before yielded compounds 7–8.
Scheme 1a.

Synthesis of ortho-substituted biphenyl mannosides
a. Reagents and conditions: (a) BF3-Et2O, CH2Cl2, reflux; (b) 3-substituted phenyl boronic acid derivatives, Cat. Pd(PPh3)4, Cs2CO3, dioxane/water(5/1), 80 °C; (c) Cat. MeONa, MeOH, rt; (d) MeNH2/EtOH, rt; (e) bis(pinacolato)diboron, Cat. Pd(dppf)Cl2, KOAc, DMSO, 80 °C.
Optimization of Mannosides for Increased Tissue Penetration and Half-Life
To help improve tissue penetration and identify mannosides with prolonged exposure in the bladder, we explored amide derivatives containing functional groups with higher pKa (Table 2). Higher pKa compounds containing basic moieties such as amines tend to have increased tissue penetration. Therefore, mannosides 10a–f were prepared via standard 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU)-mediated coupling reaction of 4′-(α-D-mannopyranosyloxy)biphenyl-3-carboxylic acid25 with various amines. Unexpectedly, aminoethyl amide 10b showed a disappointing 6-fold drop in activity relative to methyl amide 2. However, hydroxyethyl amide 10a was equipotent to 2. We also found more hydrophobic tertiary amide piperazine analogs 10c and 10d had largely decreased potency (HAI EC90 = 4 μM). Interestingly, pyridyl amides 10e and 10f showed slightly improved activity most pronounced with 4-pyridyl derivative 10e. While it is unclear to the reason for decreased potency of higher pKa substituents it is plausible that this effect could originate from charge-charge repulsion with Arg98 side chain at the edge of FimH binding pocket.
Table 2.
Exploration of amide substitution with increased pKa
 | ||
|---|---|---|
| Compound | R | HAI Titer EC>90 (μM) |
| 10a | 0.50 | |
| 10b | 3.0 | |
| 10c | 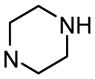 |
4.0 |
| 10d | 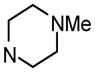 |
4.0 |
| 10e | 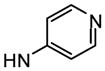 |
0.25 |
| 10f | 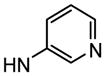 |
0.37 |
Exploration of Heterocyclic Mannosides for Improved Pharmacokinetics
We next pursued heterocyclic replacements of the terminal biphenyl ring as an alternate approach to improve the drug-like properties of lead mannosides and potentially decrease the metabolic clearance. In our target compounds we retained the key H-bond donor to Arg98 of FimH so we could directly compare the effects of heterocyclic ring replacement. In order to synthesize a library of heterocycles in a divergent fashion, we developed a new Suzuki synthesis using a 4-mannopyranosyloxyphenyl boronate intermediate 1132 in place of 4-bromophenyl α-D-mannoside. Only a limited number of heteroaryl boronates are commercially available and this new methodology allows for the use of more readily obtainable heteroaryl bromides as coupling partners. All heteroaryl bromides used in Table 3 were commercially available with reasonable prices except bromothiophene derivatives 16 and 17 (Scheme 2). 16 was prepared via Curtius reaction by first treatment of 5-bromothiophene-3-carboxylic acid with diphenylphosphoryl azide (DPPA) to form isocyanate intermediate, then quenching with ammonia. 17 was synthesized according to previous method.33 As shown in Scheme 2, Starting from 4-bromophenyl-α-D-mannoside, bis(pinacolato)diboron and Pd(dppf)Cl2 in DMSO, intermediate 11 was prepared in good yield. Suzuki cross coupling of 11 with various aryl bromides, followed by acetyl deprotection gave target compounds. Compounds synthesized, shown in Table 3, include pyridyl esters 12a and 12b which showed excellent activity in the HAI titer with improvement over phenyl ester 1. Furthermore, ring replacement with a thiophene urea carboxylate led to dramatic advancements in FimH activity as exemplified by compound 13a which has an HAI EC90 = 16 nM. In order to ascertain whether the carboxylate ester group or urea were responsible for this large increase in potency, we synthesized ester 13b and urea 13c to discover that the enhancement results from a combined effect of both functional since 13b or 13c have much decreased activity. It is unknown why there is a synergistic effect from the compound with both urea and carboxylate but from previous work on thiophene carboxamide ureas33b we have shown that an intramolecular H-bond exists between the internal NH of the urea and the carbonyl of the ester. This conformational restriction might enhance binding entropically to FimH likely from the urea carbonyl. We also pursued several fused rings such as isoquinoline derivatives 14 and 15 to examine the effects of isosteric replacement for the aryl carbonyl H-bond acceptor25 where the heterocyclic ring nitrogen is designed to accept a H-bond from the FimH Arg98 side chain. The promising HAI assay results for 14a–b to 15a–b clearly provides much evidence that the orientation of the C=N in 15a or C=O in 15b is likely the same as the conformation of C=O of mannoside 1 in crystal structure of FimH- mannoside 1 (Figure 1b)25 bringing the potency up by as much as 10-fold over mannoside 1.
Table 3.
Heterocyclic modifications to biphenyl ring for improving drug-like properties
| Compound | R | HAI Titer EC>90 (μM) |
|---|---|---|
| 12a | 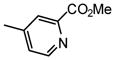 |
0.50 |
| 12b | 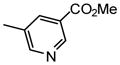 |
0.19 |
| 13a | 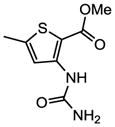 |
0.02 |
| 13b | 1.0 | |
| 13c | 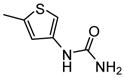 |
1.0 |
| 13d | 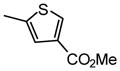 |
2.0 |
| 14a | 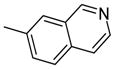 |
0.75 |
| 14b | 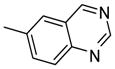 |
0.38 |
| 15a | 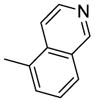 |
0.25 |
| 15b | 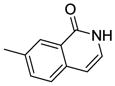 |
0.10 |
Scheme 2a.

Synthesis of heterocyclic mannosides
a. Reagents and conditions: (a) bis(pinacolato)diboron, Cat. Pd(dppf)Cl2, KOAc, DMSO, 80 °C; (b) heteroaryl bromide derivatives, Cat. Pd(PPh3)4, Cs2CO3, dioxane/water(5/1), 80 °C; (c) Cat. MeONa, MeOH, rt; (d) DPPA, iPr2NEt, dioxane, 85 °C; (e) NH3/dioxane (0.5 M), rt.
Direct FimH Binding Studies and Pharmacophore Model for Mannosides
In order to better understand how the excellent potency in cell-based HAI and biofilm assays is correlated with FimH binding by biaryl mannosides and to more precisely select the best lead compounds for in vivo pharmacokinetic (PK) studies, we have developed a biolayer interferometry method to directly measure the binding affinities (Kd) of FimH inhibitors. As shown in Table 4, earlier mannosides 19 ~ 3325 showing moderate potency in the HAI assay had Kd values as low as 1~10 nM. Strikingly, for compounds 1 and 2, Kd values were in the picomolar range. For mannosides 5, 7, and 8, however, Kd could not be calculated because the off-rates were too low to measure. In order to overcome this obstacle, we utilized differential scanning fluorimetry (DSF) to rank the high-affinity mannosides. DSF measures the melting temperature change of protein when binding to small molecules.34 Melting temperature shifts are proportional to the free energy of binding, and melting temperatures increase even as ligand concentration exceeds the Kd.35 As illustrated in Table 4 and Figure 4, the melting temperature of FimH without mannoside was about 60 °C and rose to between 68 °C and 74 °C when binding to mannosides 19 ~ 33 of moderate potency. With tight binding mannosides 5b, 7, 8 the melting temperature of FimH ranged from 74 °C to 76 °C, suggesting that improved mannosides likely bind FimH with low picomolar affinity. Figure 4 illustrates that DSF ranks mannosides in a similar fashion to the HAI assay except for 5c and 25, demonstrating that this is a general and reliable method to qualitatively rank FimH-mannoside binding when Kds span many orders of magnitude. Thus, these direct FimH binding studies solidified that the high potencies stemming from mannosides 5b, 7, 8 derives directly from extremely tight binding to the FimH lectin and not other non-specific effects from the cell assays.
Table 4.
Results of Octet assay and DSF assay
| Compound | Structure of
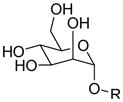 R |
HAI Titer EC>90 (μM) | Octet Kd (nM) | DSF Melting Temp. (°C) |
|---|---|---|---|---|
| 7 | 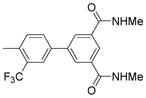 |
0.008 | N.D.* | 76.15 |
| 8 | 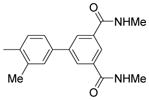 |
0.016 | N.D.* | 75.76 |
| 5c | 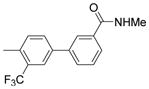 |
0.032 | N.D.* | 72.29 |
| 5b | 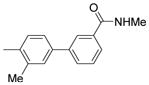 |
0.060 | N.D.* | 74.46 |
| 1825 | 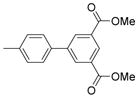 |
0.150 | N.D.* | 72.53 |
| 10f | 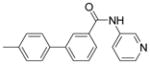 |
0.375 | N.D.* | 72.53 |
| 3 | 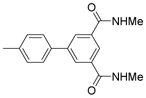 |
0.375 | 0.14 | 74.39 |
| 12a | 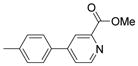 |
0.500 | N.D.* | 73.68 |
| 2 | 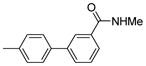 |
0.500 | 0.01 | 73.38 |
| 1925 |  |
1.000 | 0.49 | 72.05 |
| 1 | 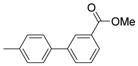 |
1.000 | 0.08 | 73.39 |
| 2025 | 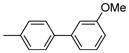 |
1.500 | 1.25 | 71.77 |
| 2125 | 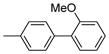 |
2.000 | N.D.* | 70.72 |
| 2225 | 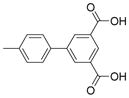 |
2.000 | 14.5 | 72.39 |
| 2325 | 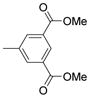 |
2.000 | 3.45 | 73.25 |
| 2425 | 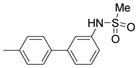 |
2.000 | 2.00 | 70.90 |
| 2525 | 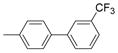 |
2.000 | 1.56 | 68.32 |
| 2625 | 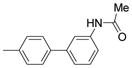 |
2.000 | 1.14 | 72.30 |
| 2725 |  |
2.000 | 1.00 | 71.69 |
| 2825 |  |
3.000 | 2.61 | 71.16 |
| 2925 | 4.000 | 3.99 | 68.69 | |
| 3025 |  |
4.000 | 2.21 | 70.93 |
| 3125 |  |
6.000 | 2.72 | 70.75 |
| 3225 | 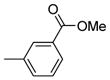 |
6.000 | 2.72 | 70.75 |
| 3325 | 8.000 | 3.00 | 71.17 | |
| Phenyl-α-D- mannoside | 30.000 | N.D.* | 68.93 | |
| 3425 | 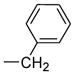 |
60.000 | 43.66 | 68.11 |
N.D. = not determined
Figure 4.

Curves of HAI, Octet and DSF assay results
Although thus far we have not yet been succesful in aquiring an X-ray structure of one of these new ortho-substituted mannosides bound to FimH, we have generated an electrostatic surface with the most potent mannoside 7 docked to FimH shown in Figure 5. The large boost in binding affinity to FimH can be attributed to the fact that in our model the ortho trifluorometyl group orients directly at Ile13 resulting in a very strong hydrophobic interaction with FimH. The added hydrophobicity encompassed by the fluorine atoms in mannoside 7 results in further augmented binding to FimH relative to ortho methyl mannoside 8. X-ray crystallographic studies are aggressively being pursued to confirm this proposed hypothetical model and to elucidate potential other reasons for the improved binding affinity with regards to either compound or FimH conformation.
Figure 5.

Proposed model of mannoside 7 bound to FimH calculated with APBS (Adaptive Poisson-Boltzmann Solver) software using PDB code: 3MCY25.
Stability and Elimination Kinetics of Mannosides
We recently reported that mannoside 3 shows efficacy in vivo36 in the treatment and prevention of established UTIs in mice when dosed orally but the compound displayed some metabolic instability from hydrolysis of the glycosidic bond (Figure 6a) and very rapid elimination kinetics to the urine partly due to its low CLogD value (Table 1). While renal clearance is an attractive feature for UTI therapy, we still wanted to develop improved mannosides encompassing lower clearance rates as well as increased oral bioavailability and bladder tissue permeability. In order to have a general idea of the elimination rate of mannoside 3, PK studies of its urinary clearance were conducted in mice (Figure 6b). These experiments demonstrated that the lower oral dosing of 20 mg/kg was unlikely to maintain effective concentrations (as determined by Biofilm IC50) due to rapid clearance. Maintenance of mannoside 3 levels above the minimal effective level of 0.74 μM (based on the Biofilm Prevention IC50 in Table 1) during in an eight-hour period required a larger, 100 mg/kg dose. During the PK study, we detected a small amount of a phenolic biphenyl metabolite (R) (Figure 6a) in the urine indicating some glycoside bond hydrolysis takes place upon oral dosing. Urine levels of the R group were unchanged from the 100 and 200 mg/kg doses, suggesting that metabolism by glycoside bond hydrolysis is saturated between the 20 and 100 mg/kg doses (Figure 6b).
Figure 6.

a) Metabolic lability of the mannoside 3 glycosidic bond from hydrolysis to mannose and biphenol; b) Elimination kinetics and clearance of mannoside 3 and biphenol (R) hydrolysis product in mouse urine.
In vitro Parallel Artificial Membrane Permeability Assay (PAMPA)
The low LogD of polyhydroxylated sugar-based mannosides and other carbohydrate-derived compounds37 can limit their ability to cross cell membranes in the absence of active transport mechanisms and so increasing their hydrophobicity is one strategy to help improve cell permeability and oral bioavailability of this class of molecules. The ortho-substituted mannosides described above were designed for increased hydrophobic contact with FimH but also in part to increase the LogD and were predicted to improve the solubility, oral bioavailability and bladder tissue penetration relative to starting mannosides 1–3. It was anticipated that the newly designed inhibitors would also have increased metabolic stability via protection of the glycosidic bond from acidic hydrolysis in the gut and enzymatic hydrolysis by α-mannosidases in blood and tissues. In order to test these hypotheses experimentally, we used the PAMPA. PAMPA is commonly used as an in vitro model of passive, transcellular permeability to predict oral bioavailability for drug candidates.38 We tested the most potent biphenyl mannosides in this model for prioritizing compounds to evaluate further in animal PK and efficacy studies. Shown in Table 1, Compound 5b with LogPe of −3.89 cm2/sec proved to have the highest oral absorption, while the most potent mannosides (determined by HAI assay) 7 and 8 with LogPe of −6.27 and −8.46 cm2/sec exhibited significantly lower oral bioavailability.
In vivo Pharmacokinetic Studies of ortho-Biphenyl Mannoside Inhibitors
Based on these results, oral PK studies were performed in mice to assess any improvements in the PK of these ortho-substituted mannosides. Compounds were dosed at 50 mg/kg and plasma and urine samples were taken at 30 min and 1, 2, 3, 4, 6 hours after dosing. As demonstrated in Figure 7, a generally 10-fold higher mannoside concentration was observed in urine (Figure 7b) compared to plasma (Figure 7a), indicating a high clearance rate for these mannosides, which in this case aids in clearing uropathogens on the bladder surface. We found that compounds 8 and 5b consistently maintain a high level of concentration in both urine and plasma, which is well above the predicted minimum effective concentration within a 6-hour period. While 100 mg/kg dosing of mannoside 3 was required to achieve effective mannoside concentrations, only 50 mg/kg dosing of mannoside 5b was required, permitting a much larger therapeutic window for treatment. Taken together with PAMPA results, high oral bioavailability and in vivo efficacy in recently reported animal studies36 support mannoside 5b as the most promising therapeutic candidate for UTI treatment/prevention.
Figure 7.

a). Plasma pharmacokinetics of optimized ortho-substituted mannosides 5a–c, 7, 8 and mannoside 3. b) Elimination clearance kinetics of optimized ortho-substituted mannosides 5a–c, 7, 8 and mannoside 3 in urine.
Conclusions
Using a combination of traditional ligand-based and X-ray structure-guided approaches with SAR driven by cell-based hemagglutination and biofilm assays in combination with direct FimH binding assays, we have identified a diverse array of biaryl mannoside FimH inhibitors that exhibit binding affinities into the picomolar range. While we found the most potent mannoside 7 with respect to FimH binding affinity and activity in cell assays contains an ortho-trifluoromethyl group off the phenyl ring adjacent to the mannose ring group, the most promising inhibitor from in vivo studies is the ortho-methyl analog 8 showing prolonged compound exposure in plasma and urine PK studies. We rationalize the large FimH binding affinity improvement. We have also discovered a variety of heterocyclic biaryl mannosides that either retain or improve FimH binding activity. The novel inhibitors of UPEC type 1 mediated bacterial adhesion reported herein show unprecedented activity in hemagglutination and biofilm in vitro assays in addition to desirable PK properties in vivo. Further optimization of lead mannosides is currently being focused on the identification of mannose modifications with reduced sugar-like character.39 Biaryl mannosides have high potential as innovative therapeutics for the clinical treatment and prevention of UTIs. This report describes the most potent mannoside FimH inhibitors to date which display good oral bioavailability and drug-like properties in vivo. The unique mechanism of action of targeting the pilus tip adhesin, FimH, circumvents the conventional requirement for drug penetration of the outer membrane and the potential for development of resistance by porin mutations, efflux or degradative enzymes, all mechanisms that promote resistance to antibiotics. Current efforts are directed at the selection of one or more clinical candidate drugs through rigorous preclinical evaluation in several models of recurrent UTI with antibiotic resistant forms of UPEC. These preclinical models will facilitate further optimization of current lead compounds to clinical candidate drugs which promise to provide a new and more selective therapy to treat and prevent chronic and recurrent urinary tract infections for which there is currently no effective treatment. Furthermore, since mannoside FimH inhibitors function outside the cell and are not cytotoxic to bacteria in contrast to all commonly prescribed antibiotics susceptible to resistance this innovative therapeutic strategy could dramatically reduce resistant forms of uropathogenic E. coli.
Experimental Section
General synthesis, purification, and analytical chemistry procedures
Starting materials, reagents, and solvents were purchased from commercial vendors unless otherwise noted. 1H NMR spectra were measured on a Varian 300 MHz NMR instrument. The chemical shifts were reported as δ ppm relative to TMS using residual solvent peak as the reference unless otherwise noted. The following abbreviations were used to express the multiplicities: s = singlet; d = doublet; t = triplet; q = quartet; m = multiplet; br = broad. High-performed liquid chromatography (HPLC) was carried out on GILSON GX-281 using Waters C18 5μM, 4.6*50mm and Waters Prep C18 5μM, 19*150mm reverse phase columns, eluted with a gradient system of 5:95 to 95:5 acetonitrile:water with a buffer consisting of 0.05% TFA. Mass spectra (MS) were performed on HPLC/MSD using electrospray ionization (ESI) for detection. All reactions were monitored by thin layer chromatography (TLC) carried out on Merck silica gel plates (0.25 mm thick, 60F254), visualized by using UV (254 nm) or dyes such as KMnO4, p-Anisaldehyde and CAM. Silica gel chromatography was carried out on a Teledyne ISCO CombiFlash purification system using pre-packed silica gel columns (12g~330g sizes). All compounds used for biological assays are greater than 95% purity based on NMR and HPLC by absorbance at 220 nm and 254 nm wavelengths.
Procedures for the preparation of biphenyl mannoside derivatives through Suzuki coupling reaction with bromophenyl mannoside derivatives as intermediates: 4′-(α-D-mannopyranosyloxy)-N,3′-dimethylbiphenyl-3-carboxamide (5b)
4-Bromo-2-methylphenyl 2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside: Under nitrogen atmosphere and at room temperature, boron trifluoride diethyl etherate (3.41 g, 24 mmol) was added dropwise into the solution of α-D-mannose pentaacetate (3.12 g, 8 mmol) and 4-bromo-2-methylphenol (2.99 g, 16 mmol) in 100 ml of anhydrous CH2Cl2. After a few mins the mixture was heated to reflux and kept stirring for 45 hrs. The reaction was then quenched with water and extracted with CH2Cl2. The CH2Cl2 layer was collected dried with Na2SO4, concentrated. The resulting residue was purified by silica gel chromatography with hexane/ethyl acetate combinations as eluent, giving 4-bromo-2-methylphenyl 2,3,4,6-tetra-O-acetyl-αD-mannopyranoside (3.22 g) in 77% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 7.18 – 7.38 (m, 2H), 6.97 (d, J = 8.79 Hz, 1H), 5.50 – 5.59 (m, 1H), 5.43 – 5.50 (m, 2H), 5.32 – 5.42 (m, 1H), 4.28 (dd, J = 5.63, 12.50 Hz, 1H), 3.99 – 4.15 (m, 2H), 2.27 (s, 3H), 2.20 (s, 3H), 2.02 – 2.11 (three singlets, 9H). MS (ESI): found: [M + Na]+, 539.0.
4′-(α-D-Mannopyranosyloxy)-N,3′-dimethylbiphenyl-3-carboxamide (5b)
Under nitrogen atmosphere, the mixture of 4-bromo-2-methylphenyl 2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside (0.517 g, 1 mmol), 3-(N-methylaminocarbonyl)phenylboronic acid pinacol ester (0.392g, 1.5 mmol), cesium carbonate (0.977 g, 3 mmol) and tetrakis(triphenylphosphine)palladium (0.116 g, 0.1 mmol) in dioxane/water (15 mL/3 mL) was heated at 80 °C with stirring for 1 h under a nitrogen atmosphere. After cooling to RT, the mixture was filtered through silica gel column to remove the metal catalyst and salts with hexane/ethyl acetate combinations as eluent. The filtrate was concentrated, and then dried in vacuo. The residue was diluted with 15 mL of methanol containing a catalytic amount of sodium methoxide (0.02 M) and the mixture was stirred at RT overnight. H+ exchange resin (DOWEX 50WX4-100) was added to neutralize the mixture. The resin was filtered off and the filtrate was concentrated. The resulting residue was purified by silica gel chromatography with CH2Cl2/MeOH combinations as eluent, giving the title compound (0.260 g) in 64% yield for two steps. 1H NMR (300 MHz, METHANOL-d4) δ ppm 7.94 (t, J = 1.65 Hz, 1H), 7.57 – 7.72 (m, 2H), 7.33 – 7.50 (m, 3H), 7.23 (d, J = 8.52 Hz, 1H), 5.48 (d, J = 1.92 Hz, 1H), 4.00 (dd, J = 1.79, 3.43 Hz, 1H), 3.83 – 3.94 (m, 1H), 3.60 – 3.76 (m, 3H), 3.46 – 3.58 (m, 1H), 2.87 (s, 3H), 2.24 (s, 3H). MS (ESI): found: [M + H]+, 404.2.
Methyl 3′-fluoro-4′-(α-D-mannopyranosyloxy)biphenyl-3-carboxylate (4a)
4-Bromo-2-fluorophenyl 2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside: it was prepared using the same procedure as for 4-bromo-2-methylphenyl 2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside in the synthesis of 5b. Yield: 25%. 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 7.30 (dd, J = 2.34, 10.03 Hz, 1H), 7.21 (td, J = 1.79, 8.79 Hz, 1H), 7.08 (t, J = 8.52 Hz, 1H), 5.48 – 5.58 (m, 2H), 5.46 (d, J = 1.65 Hz, 1H), 5.31 – 5.41 (m, 1H), 4.23 – 4.31 (m, 1H), 4.13 – 4.22 (m, 1H), 4.05 – 4.13 (m, 1H), 2.20 (s, 3H), 2.02 – 2.08 (three singlets, 9H). MS (ESI): found: [M + Na]+, 543.0.
Methyl 3′-fluoro-4′-(α-D-mannopyranosyloxy)biphenyl-3-carboxylate (4a)
4a was prepared using the same procedure as for 5b with 4-bromo-2-fluorophenyl 2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside and 3-methoxycarbonylphenyl boronic acid as the reactants. Yield: 66%. 1H NMR (300 MHz, METHANOL-d4) δ ppm 8.21 (t, J = 1.65 Hz, 1H), 7.99 (td, J = 1.44, 7.83 Hz, 1H), 7.84 (ddd, J = 1.10, 1.92, 7.69 Hz, 1H), 7.35 – 7.62 (m, 4H), 5.55 (d, J = 1.92 Hz, 1H), 4.10 (dd, J = 1.79, 3.43 Hz, 1H), 3.94 (s, 3H), 3.86 – 4.00 (m, 1H), 3.59 – 3.86 (m, 4H). MS (ESI): found [M + Na]+, 431.1.
Methyl 3′-chloro-4′-(α-D-mannopyranosyloxy)biphenyl-3-carboxylate (4b)
4-Bromo-2-chlorophenyl 2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside: it was prepared using the same procedure as for 4-bromo-2-methylphenyl 2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside in the synthesis of 5b. Yield: 46%. 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 7.55 (d, J = 2.47 Hz, 1H), 7.33 (dd, J = 2.47, 8.79 Hz, 1H), 7.06 (d, J = 8.79 Hz, 1H), 5.58 (dd, J = 3.02, 10.16 Hz, 1H), 5.52 (s, 1H), 5.49 – 5.54 (m, 1H), 5.33 – 5.42 (m, 1H), 4.22 – 4.32 (m, 1H), 4.04 – 4.17 (m, 2H), 2.21 (s, 3H), 2.07 (s, 3H), 2.05 (s, 3H), 2.04 (s, 3H). MS (ESI): found [M + Na]+, 561.0.
Methyl 3′-chloro-4′-(α-D-mannopyranosyloxy)biphenyl-3-carboxylate (4b)
4b was prepared using the same procedure as for 5b with 4-bromo-2-chlorophenyl 2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside and 3-methoxycarbonylphenyl boronic acid as the reactants. It was further purified by HPLC (C18, 15*150 mm column; eluent: acetonitrile/water (0.1% TFA)). Yield: 43%. 1H NMR (300 MHz, METHANOL-d4) δ ppm 3.59 – 3.71 (m, 1 H) 3.71 – 3.85 (m, 3 H) 3.94 (s, 3 H) 4.01 (dd, J=9.34, 3.30 Hz, 1 H) 4.12 (dd, J=3.30, 1.92 Hz, 1 H) 5.61 (d, J=1.65 Hz, 1 H) 7.40 – 7.49 (m, 1 H) 7.49 – 7.62 (m, 2 H) 7.68 (d, J=2.20 Hz, 1 H) 7.82 (ddd, J=7.76, 1.85, 1.10 Hz, 1 H) 7.98 (dt, J=7.83, 1.30 Hz, 1 H) 8.14 – 8.25 (m, 1 H). MS (ESI): found [M + H]+, 425.1.
Methyl 4′-(α-D-mannopyranosyloxy)-3′-methylbiphenyl-3-carboxylate (4c)
4c was prepared using the same procedure as for 5b with 4-bromo-2-methylphenyl 2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside and 3-methoxycarbonylphenyl boronic acid as the reactants. Yield: 54%. 1H NMR (300 MHz, METHANOL-d4) δ ppm 8.20 (t, J = 1.51 Hz, 1H), 7.94 (td, J = 1.41, 7.90 Hz, 1H), 7.77 – 7.87 (m, 1H), 7.52 (t, J = 7.55 Hz, 1H), 7.39 – 7.48 (m, 2H), 7.27 – 7.38 (m, 1H), 5.56 (d, J = 1.65 Hz, 1H), 4.08 (dd, J = 1.92, 3.30 Hz, 1H), 3.94 – 4.01 (m, 1H), 3.90 – 3.94 (m, 3H), 3.68 – 3.83 (m, 3H), 3.55 – 3.65 (m, 1H), 2.31 (s, 3H). MS (ESI): found [M + Na]+, 427.1.
Methyl 4′-(α-D-mannopyranosyloxy)-3′-(trifluoromethyl)biphenyl-3-carboxylate (4d)
4-Bromo-2-(trifluoromethyl)phenyl 2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside: it was prepared using the same procedure as for 4-bromo-2-methylphenyl 2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside in the synthesis of 5b. Yield: 54%. 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 7.74 (d, J = 2.20 Hz, 1H), 7.61 (dd, J = 2.33, 8.93 Hz, 1H), 7.15 (d, J = 8.79 Hz, 1H), 5.61 (d, J = 1.92 Hz, 1H), 5.48 – 5.56 (m, 1H), 5.33 – 5.48 (m, 2H), 4.21 – 4.34 (m, 1H), 3.97 – 4.14 (m, 2H), 2.21 (s, 3H), 1.99 – 2.12 (three singlets, 9H). MS (ESI): found [M + Na]+, 593.0.
Methyl 4′-(α-D-mannopyranosyloxy)-3′-(trifluoromethyl)biphenyl-3-carboxylate (4d)
4d was prepared using the same procedure as for 5b with 4-bromo-2-(trifluoromethyl)phenyl 2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside and 3-methoxycarbonylphenyl boronic acid as the reactants. Yield: 53%. 1H NMR (300 MHz, METHANOL-d4) δ ppm 8.23 (t, J = 1.51 Hz, 1H), 8.01 (td, J = 1.37, 7.69 Hz, 1H), 7.80 – 7.93 (m, 3H), 7.52 – 7.67 (m, 2H), 5.66 (d, J = 1.65 Hz, 1H), 4.07 (dd, J = 1.79, 3.43 Hz, 1H), 3.95 (s, 3H), 3.90 – 4.00 (m, 1H), 3.66 – 3.87 (m, 3H), 3.52 – 3.66 (m, 1H). MS (ESI): found [M + Na]+, 481.0.
Methyl 4′-(α-D-mannopyranosyloxy)-3′-methoxybiphenyl-3-carboxylate (4e)
4e was prepared using the same procedure as for 5b. In the first step of glycosidation reaction 4-bromo-2-methoxyphenol was used as glycosidation acceptor and in the second step of Suzuki coupling reaction 3-methoxycarbonylphenyl boronic acid was used instead. All intermediates were directly taken to the next step reaction without further purification. 4e was further purified by HPLC (C18, 15*150 mm column; eluent: acetonitrile/water (0.1% TFA)). Yield: 3%. 1H NMR (300 MHz, METHANOL-d4) δ ppm 8.21 (t, J = 1.51 Hz, 1H), 7.96 (td, J = 1.44, 7.83 Hz, 1H), 7.84 (ddd, J = 1.24, 1.92, 7.83 Hz, 1H), 7.49 – 7.61 (m, 1H), 7.30 (d, J = 8.24 Hz, 1H), 7.24 (d, J = 1.92 Hz, 1H), 7.13 – 7.21 (m, 1H), 5.47 (d, J = 1.92 Hz, 1H), 4.11 (dd, J = 1.79, 3.43 Hz, 1H), 3.94 (s, 3H), 3.92 (s, 3H), 3.86 – 4.03 (m, 1H), 3.67 – 3.86 (m, 4H). MS (ESI): found [M + Na]+, 443.1.
3′-Chloro-4′-(α-D-mannopyranosyloxy)-N-methylbiphenyl-3-carboxamide (5a)
5a was prepared using the same procedure as for 5b. Yield: 59%. 1H NMR (300 MHz, METHANOL-d4) δ ppm 8.03 (t, J = 1.51 Hz, 1H), 7.69 – 7.83 (m, 3H), 7.32 – 7.67 (m, 3H), 5.60 (d, J = 1.65 Hz, 1H), 4.12 (dd, J = 1.79, 3.43 Hz, 1H), 4.01 (dd, J = 3.57, 9.34 Hz, 1H), 3.69 – 3.84 (m, 3H), 3.61 – 3.69 (m, 1H), 2.95 (s, 3H). MS (ESI): found [M + Na]+, 424.1.
4′-(α-D-Mannopyranosyloxy)-N-methyl-3′-(trifluoromethyl)biphenyl-3-carboxamide (5c)
5c was prepared using the same procedure as for 5b. Yield: 65%. 1H NMR (300 MHz, METHANOL-d4) δ ppm 7.98 (t, J = 1.65 Hz, 1H), 7.77 – 7.87 (m, 2H), 7.65 – 7.77 (m, 2H), 7.42 – 7.60 (m, 2H), 5.58 (d, J = 1.65 Hz, 1H), 3.99 (dd, J = 1.79, 3.43 Hz, 1H), 3.81 – 3.91 (m, 1H), 3.59 – 3.80 (m, 3H), 3.45 – 3.58 (m, 1H), 2.87 (s, 3H). MS (ESI): found [M + H]+, 458.1.
Dimethyl 4-(α-D-mannopyranosyloxy)biphenyl-2,3′-dicarboxylate (6)
Methyl 5-hydroxy-2-(3-methoxycarbonylphenyl)benzoate: The reactants of methyl 2-bromo-5-hydroxybenzoate (0.231 g, 1 mmol), 3-methoxycarbonylphenyl boronic acid (0.214 g, 1.2 mmol), palladium acetate (0.022g, 0.1 mmol), potassium carbonate (0.346 g, 2.5 mmol) and tetrabutylammonium bromide (0.322 g, 1 mmol) in 1.2 ml of water was heated with stirring at 70 °C for 1 h and 20 mins in a sealed vial by microwave. Then the mixture was partitioned between AcOEt and 1 N HCl aqueous solution. The organic layer was collected, dried with Na2SO4, then concentrated. The resulting residue was purified by silica gel chromatography with AcOEt/Hex combinations as eluent, giving the title compound (0.240 g) in 84% yield. 1H NMR (300 MHz, DMSO-d6) δ ppm 10.02 (s, 1H), 7.90 (td, J = 2.03, 6.66 Hz, 1H), 7.72 – 7.81 (m, 1H), 7.45 – 7.60 (m, 2H), 7.28 (d, J = 8.52 Hz, 1H), 7.16 (d, J = 2.47 Hz, 1H), 7.03 (dd, J = 2.61, 8.38 Hz, 1H), 3.86 (s, 3H), 3.56 (s, 3H). MS (ESI): found [M + Na]+, 309.2.
Dimethyl 4-(α-D-mannopyranosyloxy)biphenyl-2,3′-dicarboxylate (6)
6 was prepared via glycosidation between α-D-mannose pentaacetate and methyl 5-hydroxy-2-(3-methoxycarbonylphenyl)benzoate following the procedure previously described.25 Yield: 73%. 1H NMR (300 MHz, METHANOL-d4) δ ppm 3.56 – 3.68 (m, 4 H) 3.68 – 3.84 (m, 3 H) 3.86 – 3.98 (m, 4 H) 4.06 (dd, J=3.30, 1.92 Hz, 1 H) 5.60 (d, J=1.92 Hz, 1 H) 7.29 – 7.43 (m, 2 H) 7.45 – 7.53 (m, 2 H) 7.56 (d, J=2.47 Hz, 1 H) 7.84 – 7.92 (m, 1 H) 7.93 – 8.04 (m, 1 H). MS (ESI): found [M + H]+, 449.0.
4′-(α-D-Mannopyranosyloxy)-N,N′-dimethyl-3′-(trifluoromethyl)biphenyl-3,5-dicarboxamide (7)
N1,N3-Dimethyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzene-1,3-dicarboxamide (9): dimethyl 5-bromobenzene-1,3-dicarboxylate (10.6 g, 36.8 mmol) was dissolved in a 33 wt% solution of methylamine in EtOH (30 mL) and stirred for 6h at RT. The precipitate that formed during the reaction was filtered to give 5.3 g (53%) of the intermediate 5-bromo-N1,N3-dimethyl-benzene-1,3-dicarboxamide as a white solid. Concentration of the remaining filtrate yielded an additional 4.6 g (46%) of product. 5-bromo-N1,N3-dimethyl-benzene-1,3-dicarboxamide (5.3 g, 19.5 mmol), Pd(dppf)Cl2 (0.87 g, 1.2 mmol), bis(pinacolato)diboron (6.1 g, 24 mmol), and potassium acetate (7.8 g, 80 mmol) were dissolved in DMSO (100 mL). The solution was stirred under vacuum and then repressurized with nitrogen. This process was repeated 3 times and then the resultant mixture was stirred at 80 °C for 5h under a nitrogen atmosphere. After removal of the solvent under high vacuum, the crude material was purified by silica gel chromatography to give 9 as a light tan solid (2.2 g, 35%). 1H NMR (300 MHz, DMSO-d6) δ ppm 8.63 (m, 2H), 8.41 (t, J = 1.51 Hz, 1H), 8.23 (d, J = 1.65 Hz, 2H), 2.79 (d, J = 4.40 Hz, 6H), 1.33 (s, 12H). MS (ESI): found: [M + H]+, 319.2.
4′-(α-D-Mannopyranosyloxy)-N,N′-dimethyl-3′-(trifluoromethyl) biphenyl-3, 5-dicarboxamide (7)
Using the procedure outlined in the synthesis of 5b with 4-bromo-2-(trifluoromethyl)phenyl 2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside (0.57 g) and 9 (0.48 g), the title compound (0.340 g) was obtained in 69% yield for the two steps. 1H NMR (300 MHz, METHANOL-d4) δ ppm 8.17 – 8.24 (m, 1H), 8.14 (d, J = 1.65 Hz, 2H), 7.92 (d, J = 1.92 Hz, 1H), 7.87 (dd, J = 2.20, 8.79 Hz, 1H), 7.57 (d, J = 8.79 Hz, 1H), 5.64 (d, J = 1.65 Hz, 1H), 4.04 (dd, J = 1.65, 3.30 Hz, 1H), 3.87 – 3.96 (dd, 1H), 3.64 – 3.83 (m, 3H), 3.48 – 3.63 (m, 1H), 2.93 (s, 6H). MS (ESI): found: [M + H]+, 515.1.
4′-(α-D-Mannopyranosyloxy)-N,N′,3′-trimethylbiphenyl-3,5-dicarboxamide (8)
8 was prepared using the same procedure as for 5b with 9 as the Suzuki coupling partner. Yield: 56%. 1H NMR (300 MHz, METHANOL-d4) δ ppm 8.09 – 8.23 (m, 3H), 7.46 – 7.59 (m, 2H), 7.33 (d, J = 8.52 Hz, 1H), 5.57 (d, J = 1.92 Hz, 1H), 4.08 (dd, J = 1.92, 3.30 Hz, 1H), 3.97 (dd, J = 3.43, 9.48 Hz, 1H), 3.67 – 3.85 (m, 3H), 3.60 (ddd, J = 2.47, 5.01, 7.35 Hz, 1H), 2.96 (s, 6H), 2.32 (s, 3H). MS (ESI): found: [M + H]+, 461.2.
Procedure for the preparation of mannosides via amide coupling reaction: N-(2-hydroxyethyl)-4′-(α-D-mannopyranosyloxy)biphenyl-3-carboxamide (10a)
Under nitrogen atmosphere, at 0 °C anhydrous DMF (5 mL) was added into the RB flask containing 4′-(α-D-mannopyranosyloxy)biphenyl-3-carboxylic acid25 (0.038 g, 0.1 mmol) and HATU (0.046 g, 0.12 mmol). After stirring for 10 min, aminoethanol (0.007 g, 0.12 mmol), then N,N-diisopropylethylamine (0.039 g, 0.3 mmol) were added. The mixture was stirred overnight while being warmed to RT naturally. The solvent was removed and the residue was purified by HPLC (C18, 15*150 mm column; eluent: acetonitrile/water (0.1% TFA)) to give the title compound (0.032 g) in 76% yield. 1H NMR (300 MHz, DEUTERIUM OXIDE) δ ppm 3.53 – 3.63 (m, 2 H) 3.68 – 3.94 (m, 6 H) 4.11 (dd, J=9.20, 3.43 Hz, 1 H) 4.22 (dd, J=3.30, 1.92 Hz, 1 H) 5.66 (d, J=1.65 Hz, 1 H) 7.21 (d, J=8.79 Hz, 2 H) 7.47 – 7.62 (m, 3 H) 7.66 – 7.78 (m, 2 H) 7.87 (t, J=1.65 Hz, 1 H). MS (ESI): found [M + H]+, 420.1.
N-(2-Aminoethyl)-4′-(α-D-mannopyranosyloxy)biphenyl-3-carboxamide (10b)
10b was prepared using the same procedure as for 10a. Yield: 60%. 1H NMR (300 MHz, METHANOL-d4) δ ppm 3.14 – 3.26 (m, 2 H) 3.57 – 3.66 (m, 1 H) 3.66 – 3.83 (m, 5 H) 3.87 – 4.00 (m, 1 H) 4.03 (dd, J=3.30, 1.92 Hz, 1 H) 5.49 – 5.62 (m, 1 H) 7.19 – 7.31 (m, 2 H) 7.49 – 7.59 (m, 1 H) 7.59 – 7.72 (m, 2 H) 7.75 – 7.89 (m, 2 H) 8.03 – 8.21 (m, 1 H). MS (ESI): found [M + H]+, 419.2.
3′-(Piperazin-1-ylcarbonyl)biphenyl-4-yl α-D-mannopyranoside (10c)
10c was prepared using the same procedure as for 10a. Yield: 65%. 1H NMR (300 MHz, METHANOL-d4) δ ppm 3.54 – 3.67 (m, 1 H) 3.67 – 3.85 (m, 4 H) 3.85 – 3.99 (m, 4 H) 4.03 (dd, J=3.30, 1.92 Hz, 1 H) 5.54 (d, J=1.65 Hz, 1 H) 7.19 – 7.29 (m, 2 H) 7.42 (dt, J=7.69, 1.24 Hz, 1 H) 7.50 – 7.64 (m, 3 H) 7.67 – 7.79 (m, 2 H). MS (ESI): found [M + H]+, 445.3.
3′-[(4-Methylpiperazin-1-yl)carbonyl]biphenyl-4-yl α-D-mannopyranoside (10d)
10d was prepared using the same procedure as for 10a. Yield: 87%. 1H NMR (300 MHz, METHANOL-d4) δ ppm 2.96 (s, 3 H) 3.05~3.65 (m, 9 H) 3.68 – 3.85 (m, 3 H) 3.87 – 3.97 (m, 1 H) 4.03 (dd, J=3.30, 1.92 Hz, 1 H) 5.54 (d, J=1.65 Hz, 1 H) 7.17 – 7.30 (m, 2 H) 7.42 (dt, J=7.62, 1.27 Hz, 1 H) 7.50 – 7.66 (m, 3 H) 7.67 – 7.80 (m, 2 H). MS (ESI): found [M + H]+, 459.0.
4′-(α-D-Mannopyranosyloxy)-N-(pyridin-4-yl)biphenyl-3-carboxamide (10e)
10e was prepared using the same procedure as for 10a. Yield: 93%. 1H NMR (300 MHz, METHANOL-d4) δ ppm 3.57 – 3.69 (m, 1 H) 3.69 – 3.84 (m, 3 H) 3.93 (dd, J=9.34, 3.30 Hz, 1 H) 4.04 (dd, J=3.30, 1.92 Hz, 1 H) 5.56 (d, J=1.65 Hz, 1 H) 7.18 – 7.37 (m, 2 H) 7.57 – 7.78 (m, 3 H) 7.82 – 8.06 (m, 2 H) 8.24 (t, J=1.65 Hz, 1 H) 8.36 – 8.51 (m, 2 H) 8.68 (d, J=7.42 Hz, 2 H). MS (ESI): found [M + H]+, 453.1.
4′-(α-D-Mannopyranosyloxy)-N-(pyridin-3-yl)biphenyl-3-carboxamide (10f)
10f was prepared using the same procedure as for 10a. Yield: 75%. 1H NMR (300 MHz, METHANOL-d4) δ ppm 3.55 – 3.68 (m, 1 H) 3.68 – 3.85 (m, 3 H) 3.88 – 3.98 (m, 1 H) 4.04 (dd, J=3.43, 1.79 Hz, 1 H) 5.56 (d, J=1.92 Hz, 1 H) 7.20 – 7.32 (m, 2 H) 7.52 – 7.73 (m, 3 H) 7.80 – 7.91 (m, 1 H) 7.91 – 7.99 (m, 1 H) 8.04 (dd, J=8.65, 5.63 Hz, 1 H) 8.23 (t, J=1.65 Hz, 1 H) 8.59 (d, J=5.49 Hz, 1 H) 8.67 – 8.79 (m, 1 H) 9.55 (s, 1 H). MS (ESI): found [M + H]+, 453.2.
Procedure for the preparation of biphenyl mannoside derivatives through Suzuki coupling reaction with 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl 2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside (11) as intermediates: methyl 5-[4-(α-D-mannopyranosyloxy)phenyl]pyridine-3-carboxylate (12b)
4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl 2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside (11): under nitrogen atmosphere, the mixture of 4-bromophenyl 2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside (2.791 g, 5.55 mmol), bis(pinacolato)diboron (1.690g, 6.66 mmol), potassium acetate (2.177 g, 22.18 mmol) and (1.1′-bis(diphenylphosohino)ferrocene)dichloropalladium(II) (0.244 g, 0.33 mmol) in DMSO (50 ml) was heated at 80 °C with stirring for 2.5 h. The solvent was removed and the resulting residue was purified by silica gel chromatography with hexane/ethyl acetate combinations as eluent to give 11 (2.48 g) in 81% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 1.33 (s, 12 H) 1.98 – 2.12 (m, 9 H) 2.20 (s, 3 H) 3.93 – 4.19 (m, 2 H) 4.21 – 4.36 (m, 1 H) 5.32 – 5.42 (m, 1 H) 5.45 (dd, J=3.57, 1.92 Hz, 1 H) 5.51 – 5.62 (m, 2 H) 7.00 – 7.15 (m, 2 H) 7.67 – 7.84 (m, 2 H). MS (ESI): found: [M + Na]+, 573.2.
Methyl 5-[4-(α-D-mannopyranosyloxy)phenyl]pyridine-3-carboxylate (12b)
Under nitrogen atmosphere, the mixture of 11 (0.132 g, 0.24 mmol), methyl 5-bromonicotinate (0.043g, 0.2 mmol), cesium carbonate (0.196 g, 0.6 mmol) and tetrakis(triphenylphosphine)palladium (0.023 g, 0.02 mmol) in dioxane/water (5 mL/1 mL) was heated at 80 °C with stirring for 1 h. After cooling down, the mixture was filtered through silica gel column to remove the metal catalyst and salts with hexane/ethyl acetate (2/1) containing 2% triethylamine as eluent. The filtrate was concentrated, then dried in vacuo. Into the residue, 6 mL of methanol with catalytic amount of sodium methoxide (0.02 M) was added and the mixture was stirred at room temperature overnight. The solvent was removed. The resulting residue was purified by silica gel chromatography with CH2Cl2/MeOH combinations containing 2% NH3/H2O as eluent, giving rise to 12b (0.031 g) in 40% yield. 1H NMR (300 MHz, CD3OD) δ ppm 3.53 – 3.65 (m, 1 H) 3.67 – 3.83 (m, 3 H) 3.89 – 3.96 (m, 1 H) 3.99 (s, 3 H) 4.04 (dd, J=3.43, 1.79 Hz, 1 H) 5.57 (d, J=1.92 Hz, 1 H) 7.22 – 7.37 (m, 2 H) 7.58 – 7.73 (m, 2 H) 8.54 (t, J=2.06 Hz, 1 H) 8.97 (d, J=2.20 Hz, 1 H) 9.04 (d, J=1.92 Hz, 1 H). MS (ESI): found: [M + H]+, 392.1.
Methyl 4-[4-(α-D-mannopyranosyloxy)phenyl]pyridine-2-carboxylate (12a)
12a was prepared using the same procedure as for 12b and was purified by HPLC (C18, 15*150 mm column; eluent: acetonitrile/water (0.1% TFA)). Yield: 15%. 1H NMR (300 MHz, METHANOL-d4) δ ppm 3.51 – 3.63 (m, 1 H) 3.65 – 3.84 (m, 3 H) 3.88 – 3.95 (m, 1 H) 4.00 – 4.13 (m, 4 H) 5.62 (d, J=1.65 Hz, 1 H) 7.28 – 7.40 (m, 2 H) 7.82 – 7.95 (m, 2 H) 8.13 (dd, J=5.49, 1.92 Hz, 1 H) 8.55 (d, J=1.65 Hz, 1 H) 8.73 (d, J=5.49 Hz, 1 H). MS (ESI): found [M + H]+, 392.2.
Methyl 3-(carbamoylamino)-5-[4-(α-D-mannopyranosyloxy)phenyl]thiophene-2-carboxylate (13a)
13a was prepared using the same procedure as for 12b and was purified by HPLC (C18, 15*150 mm column; eluent: acetonitrile/water (0.1% TFA)). Yield: 10%. 1H NMR (300 MHz, METHANOL-d4) δ ppm 3.58 (ddd, J=7.21, 4.88, 2.47 Hz, 1 H) 3.66 – 3.83 (m, 3 H) 3.83 – 3.96 (m, 4 H) 4.02 (dd, J=3.30, 1.92 Hz, 1 H) 5.54 (d, J=1.65 Hz, 1 H) 7.12 – 7.27 (m, 2 H) 7.56 – 7.69 (m, 2 H) 8.12 (s, 1 H). MS (ESI): found [M + H]+, 455.1.
Methyl 5-[4-(α-D-mannopyranosyloxy)phenyl]thiophene-2-carboxylate (13b)
13b was prepared using the same procedure as for 12b and was purified by silica gel chromatography with CH2Cl2/MeOH combinations. Yield: 23%. 1H NMR (300 MHz, METHANOL-d4) δ ppm 3.53 – 3.62 (m, 1 H) 3.68 – 3.81 (m, 3 H) 3.85 – 3.94 (m, 4 H) 4.02 (dd, J=3.60, 2.1 Hz, 1 H) 5.54 (d, J=1.8 Hz, 1 H) 7.19 (m, 2 H) 7.34 (d, J=3.9 Hz, 1 H) 7.64 (m, 2 H) 7.75 (d, J=3.9 Hz, 1 H). MS (ESI): found [M + Na]+, 419.1.
(5-bromo-3-thienyl)urea (16)
Under nitrogen atmosphere N,N-diisopropylethylamine (0.390 g, 3 mmol) was added to the solution of 5-bromothiophene-3-carboxylic acid (0.207g, 1 mmol) and DPPA (0.330 g, 1.2 mmol) in dioxane (5 ml) at RT. After stirring for 30 min, the mixture was heated at 85 for 1.5 h. After the mixture cooled down to RT, 0.5 M of ammonia solution in dioxane (12 ml) was added. 30 min’s later, the solvents was removed and the resulting residue was purified by silica gel chromatography with CH2Cl2/MeOH combinations to give (5-bromo-3-thienyl)urea (0.072g) in 32% yield. 1H NMR (300 MHz, DMSO-d6) δ ppm 8.80 (s, 1H), 7.09 (s, 2H), 5.87 (s, 2H). MS (ESI): found [M + H]+, 223.0.
1-{5-[4-(α-D-Mannopyranosyloxy)phenyl]thiophen-3-yl}urea (13c)
13c was prepared using the same procedure as for 12b and was purified by HPLC (C18, 15*150 mm column; eluent: acetonitrile/water (0.1% TFA)). Yield: 80%. 1H NMR (300 MHz, METHANOL-d4/ACETONITRILE-d3(3/1)) δ 7.50 – 7.62 (m, 2H), 7.11 – 7.21 (m, 3H), 7.08 (d, J = 1.37 Hz, 1H), 5.53 (d, J = 1.65 Hz, 1H), 4.02 (dd, J = 1.92, 3.30 Hz, 1H), 3.82 – 3.95 (m, 1H), 3.66 – 3.81 (m, 3H), 3.51 – 3.64 (m, 1H). MS (ESI): found [M + H]+, 397.1.
Methyl 5-[4-(α-D-mannopyranosyloxy)phenyl]thiophene-3-carboxylate (13d)
13d was prepared using the same procedure as for 12b and was purified by silica gel chromatography with CH2Cl2/MeOH combinations. Yield: 33%. 1H NMR (300 MHz, METHANOL-d4) δ ppm 3.55 – 3.63 (m, 1 H) 3.68 – 3.81 (m, 3 H) 3.84 – 3.94 (m, 4 H) 4.02 (dd, J=3.30, 1.8 Hz, 1 H) 5.53 (d, J=1.8 Hz, 1 H) 7.18 (m, 2 H) 7.59 (m, 2 H) 7.64 (d, J=1.5 Hz, 1 H) 8.11 (d, J=1.5 Hz, 1 H). MS (ESI): found [M + Na]+, 419.1.
4-(Isoquinolin-7-yl)phenyl α-D-mannopyranoside (14a)
14a was prepared using the same procedure as for 12b and was purified by HPLC (C18, 15*150 mm column; eluent: acetonitrile/water (0.1% TFA)). Yield: 73%. 1H NMR (300 MHz, METHANOL-d4) δ 9.73 (s, 1H), 8.59 – 8.71 (m, 1H), 8.47 – 8.58 (m, 2H), 8.42 (d, J = 6.59 Hz, 1H), 8.33 (d, J = 8.79 Hz, 1H), 7.76 – 7.89 (m, 2H), 7.25 – 7.41 (m, 2H), 5.60 (d, J = 1.92 Hz, 1H), 4.06 (dd, J = 1.92, 3.30 Hz, 1H), 3.87 – 4.00 (m, 1H), 3.68 – 3.87 (m, 3H), 3.55 – 3.68 (m, 1H). MS (ESI): found [M + H]+, 384.2.
4-(Quinazolin-6-yl)phenyl α-D-mannopyranoside (14b)
14b was prepared using the same procedure as for 12b. Yield: 28%. 1H NMR (300 MHz, METHANOL-d4) δ 9.51 (s, 1H), 9.15 (s, 1H), 8.21 – 8.35 (m, 2H), 8.02 (d, J = 8.52 Hz, 1H), 7.63 – 7.80 (m, J = 8.79 Hz, 2H), 7.14 – 7.32 (m, J = 8.79 Hz, 2H), 5.50 (d, J = 1.37 Hz, 1H), 3.94 – 4.03 (m, 1H), 3.80 – 3.94 (m, 1H), 3.62 – 3.80 (m, 3H), 3.48 – 3.62 (m, 1H). MS (ESI): found [M + H]+, 385.1.
4-(Isoquinolin-5-yl)phenyl α-D-mannopyranoside (15a)
15a was prepared using the same procedure as for 12b and was purified by HPLC (C18, 15*150 mm column; eluent: acetonitrile/water (0.1% TFA)). Yield: 90%. 1H NMR (300 MHz, METHANOL-d4) δ 9.80 (s, 1H), 8.45 – 8.60 (m, 2H), 8.35 (d, J = 6.59 Hz, 1H), 8.01 – 8.21 (m, 2H), 7.45 – 7.55 (m, 2H), 7.31 – 7.42 (m, 2H), 5.62 (d, J = 1.92 Hz, 1H), 4.07 (dd, J = 1.92, 3.30 Hz, 1H), 3.89 – 3.99 (m, 1H), 3.70 – 3.86 (m, 3H), 3.60 – 3.69 (m, 1H). MS (ESI): found [M + H]+, 384.2.
4-(1-Oxo-1,2-dihydroisoquinolin-7-yl)phenyl α-D-mannopyranoside (15b)
15b was prepared using the same procedure as for 12b and was purified by HPLC (C18, 15*150 mm column; eluent: acetonitrile/water (0.1% TFA)). Yield: 75%. 1H NMR (300 MHz, METHANOL-d4) δ 8.51 (d, J = 2.20 Hz, 1H), 7.93 – 8.05 (m, 1H), 7.62 – 7.77 (m, 3H), 7.21 – 7.31 (m, 2H), 7.18 (d, J = 7.14 Hz, 1H), 6.71 (d, J = 7.14 Hz, 1H), 5.56 (d, J = 1.92 Hz, 1H), 4.04 (dd, J = 1.92, 3.57 Hz, 1H), 3.88 – 4.00 (m, 1H), 3.69 – 3.87 (m, 3H), 3.57 – 3.69 (m, 1H). MS (ESI): found [M + H]+, 400.2.
Affinity Measurement by Bio-Layer Interferometry
Samples or buffer (200 μL per well) were dispensed into 96-well microtiter plates (Greiner Bio-One, Monroe NC) and maintained at 30°C with 1000RPM shaking. Pre-manufactured pins for individual assays were made by biotinylating FimH lectin domain25 at a 1:1 molar ratio with NHS-PEO4-Biotin (Thermo Fisher, Rockford IL), diluting it to 50μg/ml in 20mM HEPES pH 7.5, 150mM NaCl (HBS), immobilizing it on high-binding streptavidin-coated biosensor tips (Super Streptavidin, FortéBio, Inc., Menlo Park, CA) for 10 minutes at 30°C, blocking the pins with 10μg/ml biocytin for 2 minutes, washing in HBS for 1 hour, and rinsing in 15% sucrose in HBS. Pins were then air dried for 30 minutes and stored in their original packaging with a dessicant packet. Assays were performed by re-wetting premade pins with HBS for 15 minutes, then storing them in fresh HBS until use. Individual affinity assays were performed on an Octet Red instrument (FortéBio, Inc.; Menlo Park, CA) and consisted of a short baseline measurement followed by incubation of pins for 10 minutes with 7 twofold dilutions (in HBS) of compound in a concentration range experimentally determined to give well-measured association and dissociation kinetics, then a 30-minute dissociation phase in HBS. Each experimental pin was referenced to a biocytin-blocked pin to control for instrument drift and a second biocytin-blocked pin that was passed through a duplicate experiment in the same 96-well plate to control for nonspecific binding of the compound to the pin. Kinetics data and affinity constants were generated automatically by the global fitting protocol in ForteBio Data Analysis version 6.3. Typical signal for compound binding was approximately 0.2 “nm shift” units, while the noise level of the instrument is around 0.0025nm.
DSF Method
FimH lectin domain (residues 1–158 of UPEC J96 FimH, without an affinity tag)22 was purified as described previously. Five micrograms FimH in 5μl HBS were mixed with HBS to yield a final volume of 50μl containing compound at a final concentration of 100μM and a “5X” final concentration of SYPRO Orange (sold as a “5000X” stock in DMSO and mixed with HBS to a working stock of 50X immediately before use: Life Technologies Inc., Grand Island, NY). Compounds were diluted to 100μM from stocks in DMSO and compared to matched control wells with FimH alone plus 0.2% DMSO. 50μl reactions were placed in 96-well clear-bottom PCR plates and subjected to a melt curve from 20–90°C in 0.5 °C increments of 15 seconds each followed by a fluorescence read of the “HEX” channel in a Bio-Rad CFX96 thermocycler (Bio-Rad, Hercules, CA). Melt curves were fitted to the Boltzmann equation to determine the melting temperature (Tm) (y = A2 + (A1−A2)/(1 + exp((x−x0)/dx)) where x0 is the Tm) using OriginPro 8 (OriginLab, Northampton MA). Melting temperatures are represented as the mean of three replicates plus or minus 1 standard deviation in Figure 4.
PK Studies
Compound levels in mouse urine and plasma were made using an AB Sciex API-4000 QTrap (AB Sciex, Foster City, CA) as previously described.36 Selected reaction monitoring (SRM) mode quantification was performed with using the following MS/MS transitions [precursor mass/charge ratio (m/z)/product m/z]: compound 3, 447/285; compound 5a, 424/262; compound 5b, 404/242; compound 5c, 548/296; compound 7, 515/353; compound 8, 461/299; compound 3 R group, 285/254.
PAMPA Method. Materials
The assay was carried out using Multiscreen PVDF 96-well plates (Millipore, Billerica, MA) using the company’s Transporter Receiver Plate. The lipid (dioleyoylphosphotidylcholine (DOPC):Stearic Acid (80:20, wt%) in dodecane was obtained from Avanti Polar lipids (Alabaster, AL). Hank’s Buffered Salt Solution (HBSS), pH 7.4 was obtained from MediaTech (Manassas, VA).
Methods
Each of the test compounds was diluted to 2.5 mM in DMSO (Sigma, St Louis, MO) and further diluted prior to testing to 2.5 μM in HBSS, pH 7.4. The assay was performed using a Millipore 96-well Multiscreen-IP PAMPA plate, 5 μl of the lipid suspension was directly added to the PVDF membrane of the filter plate. Immediately following the addition of the lipid to the membrane, 200 μl of HBSS solution containing the test compound was added to the donor (upper) chamber. HBSS (300 μl) is also added to the receiver plate and the filter and receiver plates assembled and incubated overnight at room temperature in a moistened sealed bag to prevent evaporation. The concentration in the receiver plate as well as an equilibrium plate, that represents a theoretical, partition-free sample, was determined by HPLC-tandem mass spectrometry. Analysis of each compound was performed in triplicate.
Analysis
Analysis of both the acceptor and equilibrium samples were performed using an AB 3200 triple quadripole mass spectrometer linked to a Shimadzu DGU-20A HPLC with a Prevail C18 column (3 μm, 2.1×10mm) with a flow rate of 0.35 ml/min. The mobile phase used was A: 0.1% Formic Acid in water, B: 0.1% Formic Acid in methanol. The elation gradient method is described in the table. Data acquisition and peak height determination was performed using Analyst v.1.4.2.
| Time, min | A | B |
| 0.01 | 95 | 5 |
| 0.50 | 95 | 5 |
| 1.00 | 5 | 95 |
| 2.00 | 5 | 95 |
| 2.01 | 95 | 5 |
| 6.01 | Stop | |
Calculations
The log10 of the effective permeability (LogPe) was calculated using the following equation
Where the drug concentration is the peak areas for the analyte and VD and VA is the volume of the donor and acceptor compartment respectively. The area is the surface area of the PVDF membrane (0.11 cm2) and time is the incubation in seconds (64,800 sec). Each value is the mean of triplicates performed on the same day.
Supplementary Material
Acknowledgments
This work was completed in part by funding from the National Institutes of Health (NIH) American Recovery and Reinvestment Act (ARRA) Challenge Grant 1RC1DK086378 and a Washington University OTM Bear Cub Grant.
Abbreviations
- UTI
urinary tract infection
- UPEC
uropathogenic E. coli
- PAMPA
parallel artificial membrane permeability assay
- IBCs
intracellular bacterial communities
- HAI
hemagglutination inhibition
- SAR
structure activity relationship
- PK
pharmacokinetic
- PSA
polar surface area
- DSF
differential scanning fluorimetry
- IC50
half maximal inhibitory concentration
- EC>90
over 90% of maximal effective concentration
- PO
oral
- HATU
2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- DPPA
diphenylphosphoryl azide
- methyl man
methyl α-D-mannoside
- butyl man
butyl α-D-mannoside
- phenyl αD man
phenyl α-D-mannoside
References
- 1.(a) Waksman G, Hultgren SJ. Structural biology of the chaperoneusher pathway of pilus biogenesis. Nat Rev Microbiol. 2009;7:765–774. doi: 10.1038/nrmicro2220. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mulvey MA. Adhesion and entry of uropathogenic Escherichia coli. Cell Microbiol. 2002;4:257–271. doi: 10.1046/j.1462-5822.2002.00193.x. [DOI] [PubMed] [Google Scholar]; (c) Capitani G, Eidam O, Glockshuber R, Grutter MG. Structural and functional insights into the assembly of type 1 pili from Escherichia coli. Microbes Infect. 2006;8:2284–2290. doi: 10.1016/j.micinf.2006.03.013. [DOI] [PubMed] [Google Scholar]; (d) Bouckaert J, Mackenzie J, De Paz JL, Chipwaza B, Choudhury D, Zavialov A, Mannerstedt K, Anderson J, Piérard D, Wyns L, Seeberger PH, Oscarson S, De Greve H, Knight SD. The affinity of the FimH fimbrial adhesin is receptor-driven and quasi-independent of Escherichia coli pathotypes. Mol Microbiol. 2006;61:1556–1568. doi: 10.1111/j.1365-2958.2006.05352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Yu J, Lin JH, Wu XR, Sun TT. Uroplakins Ia and Ib, two major differentiation products of bladder epithelium, belong to a family of four transmembrane domain (4TM) proteins. J Cell Biol. 1994;125:171–182. doi: 10.1083/jcb.125.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sun TT, Zhao H, Provet J, Aebi U, Wu XR. Formation of asymmetric unit membrane during urothelial differentiation. Mol Biol Rep. 1996;23:3–11. doi: 10.1007/BF00357068. [DOI] [PubMed] [Google Scholar]
- 3.Martinez JJ, Mulvey MA, Schilling JD, Pinkner JS, Hultgren SJ. Type 1 pilus-mediated bacterial invasion of bladder epithelial cells. EMBO J. 2000;19:2803–2812. doi: 10.1093/emboj/19.12.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malaviya R, Ross E, MacGregor JI, Ikeda T, Little JR, Jakschik BA, Abraham SN. Mast cell phagocytosis of FimH - expressing Enterobacteria. J Immunol. 1994;152:1907–1914. [PubMed] [Google Scholar]
- 5.Mulvey MA, Lopez-Boado YS, Wilson CL, Roth R, Parks WC, Heuser J, Hultgren SJ. Induction and evasion of host defenses by type 1-piliated uropathogenic Escherichia coli. Science. 1998;282:1494–1497. doi: 10.1126/science.282.5393.1494. [DOI] [PubMed] [Google Scholar]
- 6.Bishop BL, Duncan MJ, Song J, Li G, Zaas D, Abraham SN. Cyclic AMP-regulated exocytosis of Escherichia coli from infected bladder epithelial cells. Nat Med. 2007;13:625–630. doi: 10.1038/nm1572. [DOI] [PubMed] [Google Scholar]
- 7.Eto DS, Jones TA, Sundsbak JL, Mulvey MA. Integrin-mediated host cell invasion by type 1-piliated uropathogenic Escherichia coli. PLoS Pathog. 2007;3:e100. doi: 10.1371/journal.ppat.0030100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Chen SL, Hung CS, Pinkner JS, Walker JN, Cusumano CK, Li Z, Bouckaert J, Gordon JI, Hultgren SJ. Positive selection identifies an in vivo role for FimH during urinary tract infection in addition to mannose binding. Proc Natl Acad Sci USA. 2009;106:22439–22444. doi: 10.1073/pnas.0902179106. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Justice SS, Hung C, Theriot JA, Fletcher DA, Anderson GG, Footer MJ, Hultgren SJ. Differentiation and developmental pathways of uropathogenic Escherichia coli in urinary tract pathogenesis. Proc Natl Acad Sci USA. 2004;101:1333–1338. doi: 10.1073/pnas.0308125100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wright KJ, Seed PC, Hultgren SJ. Development of intracellular bacterial communities of uropathogenic Escherichia coli depends on type 1 pili. Cell Microbiol. 2007;9:2230–2241. doi: 10.1111/j.1462-5822.2007.00952.x. [DOI] [PubMed] [Google Scholar]; (d) Nicholson TF, Watts KM, Hunstad DA. OmpA of uropathogenic Escherichia coli promotes postinvasion pathogenesis of cystitis. Infect Immun. 2009;77:5245–5251. doi: 10.1128/IAI.00670-09. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Justice SS, Lauer SR, Hultgren SJ, Hunstad DA. Maturation of intracellular Escherichia coli communities requires SurA. Infect Immun. 2006;74:4793–4800. doi: 10.1128/IAI.00355-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Anderson GG, Goller CC, Justice SS, Hultgren SJ, Seed PC. Polysaccharide capsule and sialic acid-mediated regulation promote biofilm-like intracellular bacterial communities during cystitis. Infect Immun. 2010;78:963–975. doi: 10.1128/IAI.00925-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song J, Bishop BL, Li G, Grady R, Stapleton A, Abraham SN. TLR4-mediated expulsion of bacteria from infected bladder epithelial cells. Proc Natl Acad Sci USA. 2009;106:14966–14971. doi: 10.1073/pnas.0900527106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson GG, Palermo JJ, Schilling JD, Roth R, Heuser J, Hultgren SJ. Intracellular bacterial biofilm-like pods in urinary tract infections. Science. 2003;301:105–107. doi: 10.1126/science.1084550. [DOI] [PubMed] [Google Scholar]
- 11.(a) Shilling JD, Lorenz RG, Hultgren SJ. Effect of trimethoprim-sulfamethoxazole on recurrent bacteriuria and bacterial persistence in mice infected with uropathogenic Escherichia coli. Infect Immun. 2002;70:7042–7049. doi: 10.1128/IAI.70.12.7042-7049.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mulvey MA, Schilling JD, Hultgren SJ. Establishment of a persistent Escherichia coli reservoir during the acute phase of a bladder infection. Infect Immun. 2001;69:452–459. doi: 10.1128/IAI.69.7.4572-4579.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mysorekar IU, Hultgren SJ. Mechanisms of uropathogenic Escherichia coli persistence and eradication from the urinary tract. Proc Natl Acad Sci USA. 2006;103:14170–14175. doi: 10.1073/pnas.0602136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Connell H, Agace W, Klemm P, Schembri M, Marild S, Svanborg C. Type 1 fimbrial expression enhances. Escherichia coli virulence for the urinary tract. Proc Natl Acad Sci USA. 1996;93:9827–9832. doi: 10.1073/pnas.93.18.9827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Norinder BS, Köves B, Yadav M, Brauner A, Svanborg C. Do Escherichia coli strains causing acute cystitis have a distinct virulence repertoire? Microbial Pathogenesis. 2012;52:10–16. doi: 10.1016/j.micpath.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 14.Snyder JA, Lloyd AL, Lockatell CV, Johnson DE, Mobley HL. Role of phase variation of type 1 fimbriae in a uropathogenic Escherichia coli cystitis isolate during urinary tract infection. Infect Immun. 2006;74:1387–1393. doi: 10.1128/IAI.74.2.1387-1393.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Weissman SJ, Beskhlebnaya V, Chesnokova V, Chattopadhyay S, Stamm WE, Hooton TM, Sokurenko EV. Differential stability and trade-off effects of pathoadaptive mutations in the Escherichia coli FimH adhesin. Infect Immun. 2007;75:3548–3555. doi: 10.1128/IAI.01963-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ronald LS, Yakovenko O, Yazvenko N, Chattopadhyay S, Aprikian P, Thomas WE, Sokurenko EV. Adaptive mutations in the signal peptide of the type 1 fimbrial adhesin of uropathogenic Escherichia coli. Proc Natl Acad Sci USA. 2008;105:10937–10942. doi: 10.1073/pnas.0803158105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosen DA, Hooton TM, Stamm WE, Humphrey PA, Hultgren SJ. Detection of intracellular bacterial communities in human urinary tract infection. PLoS Med. 2007;4:e329. doi: 10.1371/journal.pmed.0040329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Langermann S, Palaszynski S, Barnhart M, Auguste G, Pinkner JS, Burlein J, Barren P, Koenig S, Leath S, Jones CH, Hultgren SJ. Prevention of mucosal escherichia coli infection by FimH-adhesin-based systemic vaccination. Science. 1997;276:607–611. doi: 10.1126/science.276.5312.607. [DOI] [PubMed] [Google Scholar]; (b) Cegelski L, Pinkner JS, Hammer ND, Cusumano CK, Hung CS, Chorell E, Aberg V, Walker JN, Seed PC, Almqvist F, Chapman MR, Hultgren SJ. Small-molecule inhibitors target Escherichia coli amyloid biogenesis and biofilm formation. Nat Chem Biol. 2009;5:913–919. doi: 10.1038/nchembio.242. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Pinkner JS, Remaut H, Buelens F, Miller E, Åberg V, Pemberton N, Hedenström M, Larsson A, Seed P, Waksman G, Hultgren SJ, Almqvist F. Rationally designed small compounds inhibit pilus biogenesis in uropathogenic bacteria. Proc Natl Acad Sci USA. 2006;103:17897–17902. doi: 10.1073/pnas.0606795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.For review of small molecular FimH binding inhibitors: Hartmann M, Lindhorst TK. The bacterial lectin FimH, a target for drug discovery - Carbohydrate inhibitors of type 1 fimbriae-mediated bacterial adhesion. Eur J Org Chem. 2011:3583–3609.
- 19.Firon N, Ashkenazi S, Mirelman D, Ofek I, Sharon N. Aromatic alpha-glycosides of mannose are powerful inhibitors of the adherence of type 1 fimbriated Escherichia coli to yeast and intestinal epithelial cells. Infect Immun. 1987;55:472–476. doi: 10.1128/iai.55.2.472-476.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Almant M, Moreau V, Kovensky J, Bouckaert J, Gouin SG. Clustering of Escherichia coli type-1 fimbrial adhesins by using multimeric heptyl α-D-mannoside probes with a carbohydrate core. Chem Eur J. 2011;17:10029–10038. doi: 10.1002/chem.201100515. [DOI] [PubMed] [Google Scholar]; (b) Schierholt A, Hartmann M, Lindhorst TK. Bi- and trivalent glycopeptide mannopyranosides as inhibitors of type 1 fimbriae-mediated bacterial adhesion: Variation of valency, aglycon and scaffolding. Carbohydrate Res. 2011;346:1519–1526. doi: 10.1016/j.carres.2011.04.023. [DOI] [PubMed] [Google Scholar]; (c) Schierholt A, Hartmann M, Schwekendiek K, Lindhorst TK. Cysteine-based mannoside glycoclusters: Synthetic routes and antiadhesive properties. Eur J Org Chem. 2010:3120–3128. [Google Scholar]; (d) Gouin SG, Wellens A, Bouckaert J, Kovensky J. Synthetic multimeric heptyl mannosides as potent antiadhesives of uropathogenic Escherichia coli. ChemMedChem. 2009;4:749–755. doi: 10.1002/cmdc.200900034. [DOI] [PubMed] [Google Scholar]; (e) Touaibia M, Wellens A, Shiao TC, Wang Q, Sirois S, Bouckaert J, Roy R. Mannosylated G(0) dendrimers with nanomolar affinities to Escherichia coli FimH. ChemMedChem. 2007;2:1190–1201. doi: 10.1002/cmdc.200700063. [DOI] [PubMed] [Google Scholar]; (f) Touaibia M, Shiao TC, Papadopoulos A, Vaucher J, Wang QG, Benhamioud K, Roy R. Tri- and hexavalent mannoside clusters as potential inhibitors of type 1 fimbriated bacteria using pentaerythritol and triazole linkages. Chem Commun. 2007:380–382. doi: 10.1039/b612471b. [DOI] [PubMed] [Google Scholar]; (g) Touaibia M, Roy R. Glycodendrimers as anti-adhesion drugs against type 1 fimbriated E. coli uropathogenic infections. Mini-Rev Med Chem. 2007;7:1270–1283. doi: 10.2174/138955707782795610. [DOI] [PubMed] [Google Scholar]; (h) Nagahori N, Lee RT, Nishimura S, Page D, Roy R, Lee YC. Inhibition of adhesion of type 1 fimbriated Escherichia coli to highly mannosylated ligands. Chem BioChem. 2002;3:836–844. doi: 10.1002/1439-7633(20020902)3:9<836::AID-CBIC836>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]; (i) Furneaux RH, Pakulski Z, Tyler PC. New mannotriosides and trimannosides as potential ligands for mannose-specific binding proteins. Can J Chem. 2002;80:964–972. [Google Scholar]; (j) Lindhorst TK, Kieburg C, Krallmann-Wenzel U. Inhibition of the type 1 fimbriae-mediated adhesion of Escherichia coli to erythrocytes by multiantennary alpha-mannosyl clusters: the effect of multivalency. Glycoconjugate J. 1998;15:605–613. doi: 10.1023/a:1006920027641. [DOI] [PubMed] [Google Scholar]; (k) Kotter S, Krallmann-Wenzel U, Ehlers S, Lindhorst TK. Multivalent ligands for the mannose-specific lectin on type 1 fimbriae of Escherichia coli: syntheses and testing of trivalent alpha-D-mannoside clusters. J Chem Soc, Perkin Trans. 1998;1:2193–2200. [Google Scholar]; (l) Dubber M, Sperling O, Lindhorst TK. Oligomannoside mimetics by glycosylation of ‘octopus glycosides’ and their investigation as inhibitors of type 1 fimbriae-mediated adhesion of Escherichia coli. Org Biomol Chem. 2006;4:3901–3912. doi: 10.1039/b610741a. [DOI] [PubMed] [Google Scholar]; (m) Papadopoulos A, Shiao TC, Roy R. Diazo Transfer and Click Chemistry in the Solid Phase Syntheses of Lysine-Based Glycodendrimers as Antagonists against Escherichia coli FimH. Mol Pharm. 2012 doi: 10.1021/mp200490b. [DOI] [PubMed] [Google Scholar]
- 21.Hung CS, Bouckaert J, Hung D, Pinkner J, Widberg C, DeFusco A, Auguste CG, Strouse R, Langermann S, Waksman G, Hultgren SJ. Structural basis of tropism of Escherichia coli to the bladder during urinary tract infection. Mol Microbiol. 2002;44:903–915. doi: 10.1046/j.1365-2958.2002.02915.x. [DOI] [PubMed] [Google Scholar]
- 22.Bouckaert J, Berglund J, Schembri M, de Genst E, Cools L, Wuhrer M, Hung CS, Pinkner J, Slattegard R, Zavialov A, Choudhury D, Langermann S, Hultgren SJ, Wyns L, Klemm P, Oscarson S, Knight SD, De Greve H. Receptor binding studies disclose a novel class of high-affinity inhibitors of the Escherichia coli FimH adhesion. Mol Microbiol. 2005;55:441–455. doi: 10.1111/j.1365-2958.2004.04415.x. [DOI] [PubMed] [Google Scholar]
- 23.Wellens A, Garofalo C, Nguyen H, Van Gerven N, Slattegård R, Hernalsteens JP, Wyns L, Oscarson S, De Greve H, Hultgren S, Bouckaert J. Intervening with urinary tract infections using antiadhesives based on the crystal structure of the FimH-oligomannose-3 complex. PLoS One. 2008;3:e2040. doi: 10.1371/journal.pone.0002040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sperling O, Fuchs A, Lindhorst TK. Evaluation of the carbohydrate recognition domain of the bacterial adhesin FimH: design, synthesis and binding properties of mannoside ligands. Org Biomol Chem. 2006;4:3913–3922. doi: 10.1039/b610745a. [DOI] [PubMed] [Google Scholar]
- 25.Han Z, Pinkner JS, Ford B, Obermann R, Nolan W, Wildman SA, Hobbs D, Ellenberger T, Cusumano CK, Hultgren SJ, Janetka JW. Structure-based drug design and optimization of mannoside bacterial fimH antagonists. J Med Chem. 2010;53:4779–4792. doi: 10.1021/jm100438s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klein T, Abgottspon D, Wittwer M, Rabbani S, Herold J, Jiang X, Kleeb S, Lüthi C, Scharenberg M, Bezençon J, Gubler E, Pang L, Smiesko M, Cutting B, Schwardt O, Ernst B. FimH antagonists for the oral treatment of urinary tract infections: From design and synthesis to in vitro and in vivo evaluation. J Med Chem. 2010;53:8627–8641. doi: 10.1021/jm101011y. [DOI] [PubMed] [Google Scholar]
- 27.(a) Gupta K, Hooton TM, Stamm WE. Increasing antimicrobial resistance and the management of uncomplicated community-acquired urinary tract infections. Ann Intern Med. 2001;135:41–50. doi: 10.7326/0003-4819-135-1-200107030-00012. [DOI] [PubMed] [Google Scholar]; (b) Blango MG, Mulvey MA. Persistence of uropathogenic Escherichia coli in the face of multiple antibiotics. Antimicrob Agents Chemother. 2010;54:1855–1863. doi: 10.1128/AAC.00014-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hultgren SJ, Schwan WR, Schaeffer AJ, Duncan JL. Regulation of production of type 1 pili among urinary tract isolates of Escherichia coli. Infect Immun. 1986;54:613–620. doi: 10.1128/iai.54.3.613-620.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ren D, Zuo R, Barrios AFG, Bedzyk LA, Eldridge GR, Pasmore ME, Wood TK. Differential gene expression for investigation of Escherichia coli biofilm inhibition by plant extract ursolic acid. Appl and Environ Microbiol. 2005;71:4022–4034. doi: 10.1128/AEM.71.7.4022-4034.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.France RR, Compton RG, Davis BG, Fairbanks AJ, Rees NV, Wadhawan JD. Selective electrochemical glycosylation by reactivity tuning. Org Biomol Chem. 2004;2:2195–2202. doi: 10.1039/b316728c. [DOI] [PubMed] [Google Scholar]
- 31.(a) Ishiyama T, Murata M, Miyaura N. Palladium(0)-catalyzed cross-coupling reaction of alkoxydiboron with haloarenes: A direct procedure for arylboronic esters. J Org Chem. 1995;60:7508–7510. [Google Scholar]; (b) Takagi J, Takahashi K, Ishiyama T, Miyaura N. Palladium-Catalyzed Cross-Coupling Reaction of Bis(pinacolato)diboron with 1-Alkenyl Halides or Triflates: Convenient Synthesis of Unsymmetrical 1,3-Dienes via the Borylation-Coupling Sequence. J Am Chem Soc. 2002;124:8001–8006. doi: 10.1021/ja0202255. [DOI] [PubMed] [Google Scholar]; (c) Thompson ALS, Kabalka GW, Akula MR, Huffman JW. The Conversion of Phenols to the Corresponding Aryl Halides Under Mild Conditions. Synthesis. 2005:547–550. [Google Scholar]
- 32.Schwizer D, Gäthje H, Kelm S, Porro M, Schwardt O, Ernst B. Antagonists of the myelin-associated glycoprotein: A new class of tetrasaccharide mimics. Bioorg Med Chem. 2006;14:4944–4957. doi: 10.1016/j.bmc.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 33.(a) Ashwell S, Gero T, Ioannidis S, Janetka J, Lyne P, Su M, Toader D, Yu D, Yu Y. Preparation of substituted heterocycles, particularly ureidothiophenes, as CHK1 kinase inhibitors for treating neoplasm. WO 2005066163 PCT Int Appl. 2005; (b) Janetka JW, Almeida L, Ashwell S, Brassil PJ, Daly K, Deng C, Gero T, Glynn RE, Horn CL, Ioannidis S, Lyne P, Newcombe NJ, Oza VB, Pass M, Springer SK, Su M, Toader D, Vasbinder MM, Yu D, Yu Y, Zabludoff SD. Discovery of a novel class of 2-ureido thiophene carboxamide checkpoint kinase inhibitors. Bioorg Med Chem Lett. 2008;18:4242–4248. doi: 10.1016/j.bmcl.2008.05.016. [DOI] [PubMed] [Google Scholar]
- 34.Niesen F, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nature Protocols. 2007;2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 35.Kranz JK, Schalk-Hihi C. Protein Thermal Shifts to Identify Low Molecular Weight Fragments. Methods in Enzymology. 2011;493:277–298. doi: 10.1016/B978-0-12-381274-2.00011-X. [DOI] [PubMed] [Google Scholar]
- 36.Cusumano CK, Pinkner JS, Han Z, Greene SE, Ford BA, Crowley JR, Henderson JP, Janetka JW, Hultgren SJ. Treatment and prevention of urinary tract infection with orally active FimH inhibitors. Sci Transl Med. 2011;3:109ra115. doi: 10.1126/scitranslmed.3003021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ernst B, Magnani JL. From carbohydrate leads to glycomimetic drugs. Nature Reviews Drug Discovery. 2009;8:661–677. doi: 10.1038/nrd2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.(a) Kansy M, Senner F, Gubernator K. Physicochemical High Throughput Screening: Parallel Artificial Membrane Permeation Assay in the Description of Passive Absorption Processes. J Med Chem. 1998;41:1007–1010. doi: 10.1021/jm970530e. [DOI] [PubMed] [Google Scholar]; (b) Avdeef A, Bendels S, Di L, Faller B, Kansy M, Sugano K, Yamauchi Y. Parallel artificial membrane permeability assay (PAMPA)-critical factors for better predictions of absorption. J Pharm Sci. 2007;96:2893–2909. doi: 10.1002/jps.21068. [DOI] [PubMed] [Google Scholar]
- 39.Schwardt O, Rabbani S, Hartmann M, Abgottspon D, Wittwer M, Kleeb S, Zalewski A, Smieko M, Cutting B, Ernst B. Design, synthesis and biological evaluation of mannosyl triazoles as FimH antagonists. Bioorg Med Chem. 2011;19:6454–6473. doi: 10.1016/j.bmc.2011.08.057. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.