

Abstract
Improvement in protein thermostability was often found to be associated with increase in its proteolytic resistance as revealed by comparative studies of homologous proteins from extremophiles or mutational studies. Structural elements of protein responsible for this association are not firmly established although loops are implicated indirectly due to their structural role in protein stability. To get a better insight, a detailed study of protein wide mutants and their influence on stability and proteolytic resistance would be helpful. To generate such a data set, a model protein, Bacillus subtilis lipase was subjected to loop scanning site-saturation mutagenesis on 86 positions spanning all loops including termini. Upon screening of ∼16,000 clones, 17 single mutants with improved thermostability were identified with increment in apparent melting temperature (Tmapp) by 1–6°C resulting in an increase in free energy of unfolding (ΔGunf) by 0.04–1.16 kcal/mol. Proteolytic resistance of all single mutants upon incubation with nonspecific protease, Subtilisin A, was determined. Upon comparison, post-proteolysis residual activities as well as kinetics of proteolysis of mutants showed excellent correlation with ΔGunf, (r > 0.9), suggesting that proteolysis was strongly correlated with the global stability of this protein. This significant correlation in this set, with least possible sequence changes (single aa substitution), while covering >60% of protein surface strongly argues for the covariance of these two variables. Compared to studies from extremophiles, with large sequence heterogeneity, the observed correlation in such a narrow sequence space (ΔΔGunf = 1.57 kcal−1) justifies the robustness of this relation.
Keywords: loop scanning, site-saturation mutagenesis, loops, protein stability, thermostability, proteolytic resistance, lipase
Introduction
Proteolytic resistance of a protein and its dependence on protein stability is a subject of investigation since long.1–3 Most of the investigations into protein stability-proteolytic resistance revealed that proteolytic resistance is dependent on stability of the protein and found to increase with increase in stability2, 4 suggesting that partially or completely unfolded structures of proteins are the major proteolytic susceptible conformations. It is well established that the native state of a protein is a mixture of isoenergetic conformational states which remain in equilibrium with the unfolded state.1, 2 Proteolysis depends on the stability of the protein, which is a free energy difference between native and unfolded state of the protein (ΔGunf) and also on the nature of the unfolded form of the protein. Unfolding, a prerequisite for proteolysis may be a subglobal or a global event in a protein. Since unfolding pathways are protein specific, it is expected that susceptibility to proteases would be protein specific. While information on energy difference could be obtained by kinetic analysis, information on the structure of the partially cleaved product is difficult. Since partially cleaved products do not accumulate during proteolysis, careful kinetic and thermodynamic analysis of proteolysis was gainfully used to identify partially unfolded regions during proteolysis.5 Current understanding of relation between stability and proteolysis is supported by comparative studies on homologous proteins from extreme phyla, which not only differ widely from each other in terms of physical properties but are also well separated in terms of sequence space.2, 6 Mutational approaches to study this relation are based on limited set of mutants, both in terms of numbers as well as positions.7–9 Clearly, a systematic probing into the phenomenon of protease susceptibility/resistance and its relation to protein stability using protein wide mutations, with least possible sequence differences that alter the physical properties of native and unfolded states to minimum, will be advantageous.
Proteolytic susceptibility of the protein has been exploited in structural characterization using limited proteolysis.3, 10 Moreover, under certain conditions, its relationship with protein stability has been utilized to determine physical parameters such as free energy of unfolding and binding constants of ligands using pulse proteolysis.11–13 Also, the linkage of proteolytic resistance with protein stability has been extensively utilized to evolve stable variants of a numbers of proteins and peptides using protein stability increase by directed evolution (Proside).14–18 In addition, proteolytic resistance has also been exploited for protein purification.19
The susceptibility of a peptide bond to cleavage by a protease is determined by: (a) the flexibility of the protein chain region in which it is located, (b) the extent to which the bond is exposed, and (c) the nature of the local interactions made by the side chains of its flanking residues.20 Owing to these requirements, the unfolded state of the protein is the most preferred target for proteolysis, mainly due to its associated high entropy.
Among all secondary structural elements, loops and termini in the native structure have dynamics associated that are significantly larger than the rest of the protein.21–23 Loops and unordered regions of the protein display conformational dynamics, ranging from angstrom to nanometer in length and femtosecond to seconds in timescale.24 Under denaturing conditions, loops are most prone to “unfolding” owing to limited noncovalent interactions within as well as with other secondary structural elements forming core of the protein molecule, thus causing local unfolding which can be readily attacked by the proteases.20, 24, 25 Also, growing number of studies on protein structure-stability relationship have shown the emerging role of loops and surface residues in protein stability.26–30 Hence, in the present study, all loops and termini positions (86 of 181) of a model protein, Bacillus subtilis lipase, spanning ∼60% of the native structure surface area were subjected to site-saturation mutagenesis. After screening for improved thermostability a set of 17 single mutants were identified and characterized for thermostability and proteolytic resistance against nonspecific protease Subtilisin A. Excellent correlation was observed between protein stability and proteolytic resistance. Moreover, the study also highlights the importance of loops in imparting stability to the protein.
Results
Generation of thermostable mutants
Thermostable mutants, each having only single substitution, were generated by performing site-saturation mutagenesis at 86 positions spanning all loops (10) as well as N- and C- termini of the lipase as shown in Figure 1(A). Bacillus subtilis lipase structure shows a compact domain that consists of six β-strands in a parallel β-sheet, surrounded by α-helices. The fold of the lipase resembles that of the core of other α/β hydrolase fold enzymes.31 The target positions on all loops and termini were selected using highest resolution wild-type lipase structure available as template (1.3 Å; PDB ID: 1ISP).32 These include position 4 in N-terminus; positions 10–19, between β3 and αA; positions 30–34, between αA and β4; positions 40–47, between β4 and αB; positions 67–71, between αB and β5; position 78 between β5 and αC; positions 90–96, between αC and β6; positions 104–123, between β6 and β7; positions 132–138, between β7 and αE; positions 142–146, between αE and β8; positions 152–162, between β8 and αF and positions 174–181 of C-terminus (Supporting Information Fig. 1). The schematic representation of in vitro evolution of lipase for thermostability is depicted in Figure 1(B). Wild-type lipase gene was chosen as parent and subjected to saturation mutagenesis at all 86 positions. For each position mutant population was generated by overlap-extension PCR using mutagenic primers encoding target position by degenerate codon NNK (Supporting Information Table I). The mutagenic library so obtained was used for periplasmic expression of protein in E. coli. The criteria for selecting a mutation depended on its contribution to thermostability and retention of activity at room temperature comparable to that of parent. Only mutants showing higher residual activity upon exposure to higher temperature, without affecting their catalytic activities at room temperatures were selected.
Figure 1.
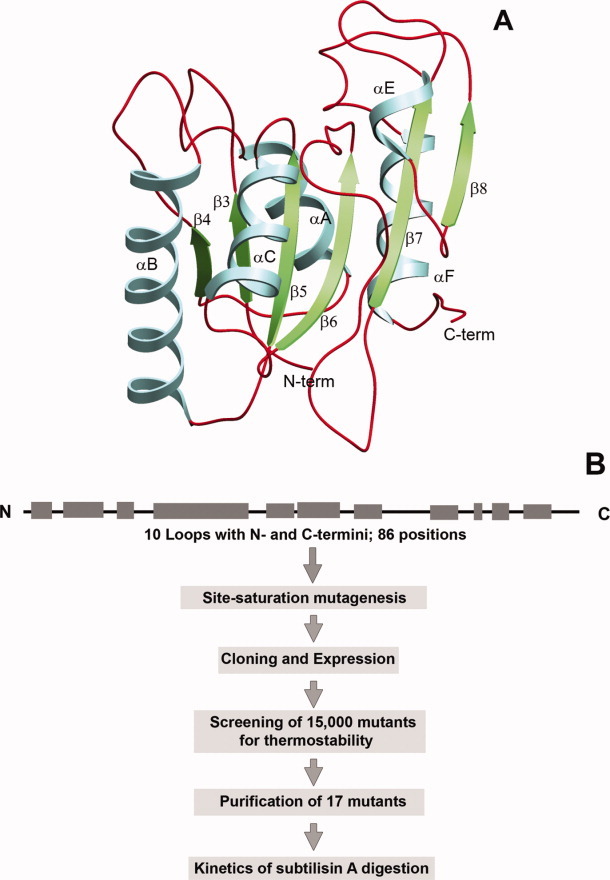
A: The ribbon diagram of B. subtilis lipase with the loop regions highlighted in dark red. B: Schematic representation of thermostable mutant generation and characterization. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Screening was performed using semi-high-throughput assay format in 96-well plates, upon thermal challenge at elevated temperature. Approximately 15,500 clones, 180 clones for each position, were screened for thermostability after exposure to 53°C for 20 min, a temperature at which wild-type lipase shows less than 20% residual activity. After multiwell-plate based screening, clones which displayed significantly higher residual activities compared to parent (>50%) were further tested in tube assays. Thermal inactivation profile of mutants was generated by monitoring residual activities in crude culture supernatant upon incubation over a range of temperature. T50, the temperature at which 50% of activity was retained, was used as a selection criterion. Mutants displaying more than 1°C increment in T50 were subjected to sequencing to determine the mutation occurred (Supporting Information Table II). On the basis of the thermostability properties of mutants over the wild-type, 17 mutants were selected for further study (Table I).
Table I.
Stability Parameters of Single Thermostable Mutants (STM) of Lipase
| Thermostability | Equilibrium unfolding | Protease resistance | ||||||
|---|---|---|---|---|---|---|---|---|
| Mutanta | T50b(°C) | Half-lifec (min) | Tmappd |
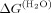 e (kcal.mol−1) e (kcal.mol−1) |
ΔG/m (M) | Residualf activity (%) | Proteolyticg rate (min−1) | Half-lifeg (min) |
| Wild-type | 52.8 | 3.1 | 56.3 | 10.39 | 4.54 | 55.2 | 0.0124 | 55.7 |
| 112A | 55.0 (2.2) | 22.8 | 58.7 (2.4) | 10.91 | 4.77 | 68.3 | — | — |
| A15S | 54.8 (2.0) | 19.5 | 58.7 (2.4) | 10.57 | 4.62 | 60.0 | 0.0190 | 36.4 |
| F17T | 55.4 (2.6) | 26.3 | 59.0 (2.7) | 10.91 | 4.77 | 75.0 | 0.0061 | 114.0 |
| R33P | 53.8 (1.0) | 5.9 | 57.3 (1.0) | 10.43 | 4.56 | 59.9 | — | — |
| T47S | 55.0 (2.2) | 25.3 | 58.8 (2.5) | 11.35 | 4.96 | 86.6 | — | — |
| T109V | 55.4 (2.6) | 36.0 | 59.6 (3.3) | 10.73 | 4.69 | 71.4 | — | — |
| G111S | 53.5 (0.7) | 6.2 | 57.4 (1.1) | 10.50 | 4.59 | 57.2 | 0.0136 | 50.8 |
| L114P | 54.2 (1.4) | 8.7 | 58.2 (1.9) | 10.62 | 4.64 | 66.0 | — | — |
| A132D | 54.5 (1.7) | 6.0 | 57.5 (1.2) | 9.98 | 4.36 | 45.3 | 0.0441 | 15.7 |
| M134R | 55.8 (3.0) | 43.4 | 59.8 (3.5) | 10.64 | 4.65 | 68.5 | 0.0028 | 244.0 |
| M134E | 57.8 (5.0) | 196.0 | 60.3 (4.0) | 11.03 | 4.82 | 85.6 | 0.0055 | 125.5 |
| M137P | >65 (>12) | 2617.0 | 62.4 (6.1) | 11.55 | 5.05 | 92.5 | 0.0073 | 955.8 |
| M134D | 57.8 (5.0) | 212.0 | 60.5 (4.2) | 10.55 | 4.61 | 70.6 | — | — |
| G155S | 55.0 (2.2) | 22.7 | 58.7 (2.4) | 11.44 | 5.00 | 88.4 | — | — |
| I157M | 55.3 (2.5) | 22.4 | 58.6 (2.3) | 10.94 | 4.78 | 83.8 | — | — |
| Y161N | 55.6 (2.8) | 34.0 | 59.3 (3.0) | 10.30 | 4.50 | 62.2 | — | — |
| N174T | 53.8 (1.0) | 9.4 | 57.7 (1.4) | 10.75 | 4.70 | 73.9 | 0.0076 | 91.0 |
Mutant's name is same as that of the corresponding mutation.
Temperature at which enzyme looses 50% of its initial activity upon incubation for 20 min. Difference in T50 value of mutant and wild-type is given in parenthesis.
Half-life of thermal inactivation at 55°C.
Apparent melting temperature as calculated by circular dichroism. Difference in Tmapp value of mutant and valid-type is given in parenthesis.
Free energy of unfolding in the absence of urea was calculated by taking average of m values (2.288 kcal mol−1 M−1) of all mutants and wild-type protein.
Residual activity after incubating lipase with subtilisin A (50:1; molar ratio) for 4 h at 37°C.
Rate of proteolysis and half-life of proteolytic degradation of lipase with subtilisin A at equimolar ratio upon incubation at 40°C.
Of 86 positions, activity was severely compromised upon mutagenesis at 12 locations. These are positions 10, 11, 14, and 19 in loop connecting β3 with αA; position 30 on loop connecting αA with β4; 78 on loop connecting β5 with αC; 92 on loop connecting αC with β6; 104 and 106 on loop connecting β6 and β7; 136 on loop connecting β7 with αE and 141 and 145 on loop connecting αE with β8. Out of these 12 positions 11, 14, 30, 92, 104, and 145 were occupied by glycine in wild-type protein. Substitution of glycine by any other residue might have compromised the required conformational freedom, either affecting the folding of the protein to its native state or perturbing the local dynamics of the protein, thus severely affecting the activity of the mutant enzyme. This indicated the importance of glycine at these locations in maintaining the native tertiary structure of the protein.
Of the 86 positions, substitutions at 16 locations were found to increase protein thermostability during initial rounds of screening. These are positions 10, 12, 15, 17, 33, 47, 109, 111, 114, 132, 134, 137, 155, 157, 161, and 174 (Supporting Information Table II). On six positions out of these, more than one kind of substitution improved thermostability. F17 on the first loop in the wild-type protein was found to be the most liberal by accommodating five different substitutions. On rest of the positions, though redundant, only one substitution was found to be thermostabilizing (Supporting Information Table II). After reconfirming the results of the tube assays, 17 single mutants on 15 locations, conferring maximal increase in thermostability, were selected for further study, as listed in Table I.
The most thermostabilizing mutation identified was M137P, which increases the midpoint of inactivation transition (T50), as calculated using crude culture supernatant, by 6.8 degrees compared to the wild-type. The second best mutation was at the same position (M137D), which increased the T50 by 3.6 degrees. Two moderately thermostabilizing mutations identified were M134E and F17T, which increase T50 by 2.4 and 2.2 degrees, respectively. The other single thermostabilizing mutations on rest of the locations show improvements in T50 by 1.1–1.8 degrees (Supporting Information Table II).
Characterization of single thermostabilizing mutants
All 17 single thermostabilizing mutant genes were over-expressed in E. coli and proteins were purified using standard procedures.30, 33, 34 Far-UV CD spectra of all mutants were identical to that of the wild-type protein suggesting that the presence of mutation did not alter the secondary structure of the mutants. The tertiary structure of mutants, as probed by intrinsic tryptophan fluorescence, was also found to be highly similar to that of the wild-type protein, suggesting proper folding with little perturbation of the native structure of mutants (Supporting Information Fig. 2). Specific activities of the mutants were found to be 70–110% that of the wild-type enzyme except for two mutants (I12A and Y161N), that had specific activities half that of wild-type enzyme. On the other hand, only one mutation M134E was found to positively contribute to the activity, having 1.5-fold higher specific activity than that of wild-type. (Supporting Information Fig. 3).
Thermal inactivation of mutants
To determine resistance of lipase mutants to thermal inactivation, purified enzymes were heated in the temperatures range of 48–65°C for 20 min followed by cooling to room temperature and residual activity measurements at room temperature. The thermal inactivation profiles of selected mutants are shown in Figure 2(A) (also see Table I and Supporting Information Fig. 4). Except mutant M137P, the inactivation transitions of all the mutants are similar, with subtle differences in cooperativity. Wild-type enzyme starts losing activity at 49°C, undergoes cooperative transition which gets over by 57°C, with the midpoint of thermal inactivation (T50) of 52.8°C. The most thermostabilizing mutant was M137P which shows gradual loss in activity beyond 58°C, a transition which goes well beyond 65°C. The T50 value of M137P is more than 65°C which is more than 12 degrees higher as compared to wild-type enzyme. Mutant M137P was followed by M137D and M134E, both having T50 values 5 degrees higher than wild-type lipase. Another mutant, M134R, was also found to be thermostabilizing, with T50 value 3 degree higher than that of the wild-type enzyme (Table I). All other 13 mutants were found to be moderately thermostable with temperature of half-inactivation (T50) 1°C–2.8°C higher as compared to wild-type lipase [Table I; Fig. 2(A) and Supporting Information Fig. 4].
Figure 2.
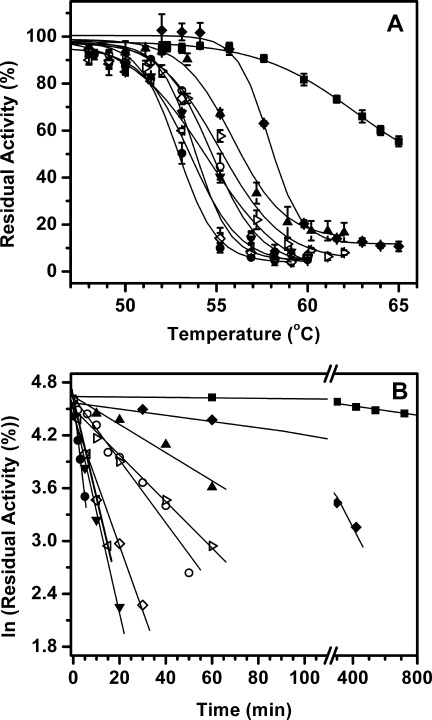
A: Thermal inactivation profiles and (B) thermal inactivation kinetics of wild-type lipase and selected single thermostable mutants upon incubation at 55°C. Inactivation profiles of other mutants are not shown for the sake of clarity. Symbols used: wild-type (•), A15S (–––), F17T (χ), G111S (◃), A132D (▾), M134R (▴), M134E (♦), M137P (▪), and N174T (⋄).
Kinetics of irreversible thermal inactivation was monitored by incubating the enzymes at elevated temperature followed by residual activity measurements at room temperature with respect to the time of incubation. All thermal inactivation traces were found to follow first-order kinetics (Supporting Information Fig. 5). Time course of thermal inactivation of selected mutants are shown in Figure 2(B), while half-life of inactivation at 55°C of all mutants are listed in Table I. Mutant M137P has the longest half-life of more than 2600 min (∼ 43 h/2 days) at 55°C, which is more than 800-fold compared to wild-type enzyme with 3.1 min. To the best of our knowledge, this is the largest improvement in thermostability conferred by a single amino acid substitution. The second best improvement was shown by M137D followed by M134E with half-lives at 55°C of 212 and 196 min, respectively, more than 60-fold as compared to wild-type lipase. The R33P, G111S, L114P, A132D, and N174T mutants showed modest improvement in inactivation half-lives, which ranges between 6 and 10 min. The remaining mutants including I12A, A15S, F17T, T47S, T109V, M134R, G155S, I157M, and Y161N showed moderate improvement in their half-lives which lies between 13 and 43 min at 55°C (Table I).
Thermostability of mutants
The thermostability of lipase variants was tested by monitoring temperature-induced unfolding using circular dichroism35 [Fig. 3(A), Table I, and Supporting Information Fig. 6(A)]. The melting profiles of the mutants largely reflect the thermal inactivation profiles of the mutants. The mutants and the wild type lipase aggregate upon heating. In our experiments for determination of midpoint of thermal transition by CD measurement, we observed that midpoint may be influenced by partial loss of protein during heating ramp. Since this may alter the slope of the transition we are treating the Tm as Tmapp.36 The most thermostable mutant again was M137P, which melts at 62.4°C, ∼6 degrees higher as compared to wild-type protein (Tmapp = 56.3°C). It was followed by M134E and M137D mutants with apparent melting temperatures (Tmapp) of 60.4 and 60.2°C, respectively, more than 4 degrees higher than wild-type lipase. Other mutants displaying moderate improvement in thermostability are I12A, A15S, F17T, T47S, T109V, M134R, G155S, I157M, and Y161N with apparent melting temperatures of 58.7°C, 58.7°C, 59.0°C, 58.8°C, 59.6°C, 59.8°C, 58.7°C, 58.6°C, and 59.3°C, respectively, which are 2–3.5 degrees higher as compared to wild-type protein. Rest of the mutants including R33P, G111S, L114P, A132D, and N174T show only modest improvement in thermostability, with increment in Tmapp by 1°C–1.9°C. For most of the mutants, the improvement in T50 of inactivation matches very well with the improvement in their apparent melting temperatures. For all 17 mutants, the correlation coefficient between T50 of inactivation and Tmapp, as monitored by CD, was found to 0.94, which is highly significant. We also investigated whether the enhanced stability, indicated by the increase in melting temperature, also reflected in the kinetic stability.37 It was observed that the correlation coefficient between the logarithm of kinetics of denaturation (inactivation) and the Tmapp, as determined by CD was 0.98, which signifies the stabilization is both thermodynamic and kinetic in nature. Individually, each single substitution stabilizes wild-type protein, increasing its Tmapp by 1–6°C. To test the cumulative behavior of stabilization of these mutations, 13 best mutations covering all loops were recombined. This recombinant mutant, named 13x, not only retains the activity as wild-type but also melts at 78.9°C, which is ∼23 degrees higher than wild-type protein [Supporting Information Fig. 6(B)].
Figure 3.
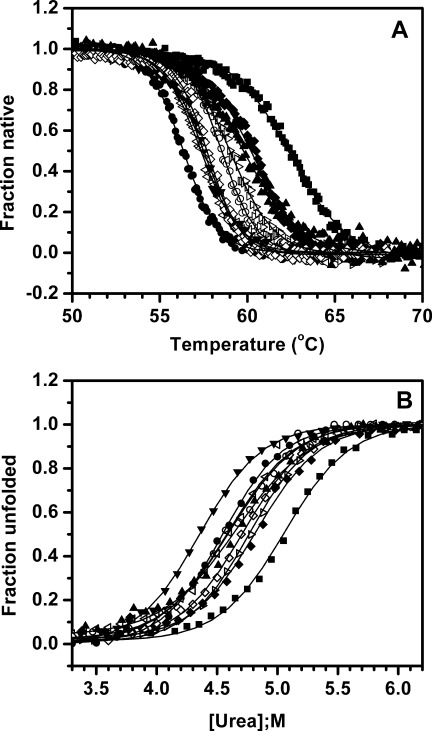
A: Thermal unfolding profiles of wild-type lipase along with selected single thermostable mutants as monitored by circular dichroism. Change in ellipticity at 222 nm (Θ222) with temperature was used to calculate unfolding transitions. B: Equilibrium unfolding profiles of wild-type lipase along with selected single thermostable mutants in the presence of urea as observed by circular dichroism. Fraction unfolded was calculated based on changes in ellipticity at 222 nm (Θ222) with urea concentration. Unfolding transitions of other mutants are not shown for the sake of clarity. Symbols used: wild-type (•), A15S (–), F17T (χ), G111S (◃), A132D (▾), M134R (▴), M134E (♦), M137P (▪), and N174T (⋄).
Equilibrium unfolding of lipase variants in urea
Wild-type lipase and its all single thermostable mutants undergo irreversible thermal unfolding. This irreversibility is mainly attributed to protein aggregation upon thermal unfolding (Supporting Information Fig. 7). Due to associated irreversibility, the apparent melting temperature only grossly reflects protein stability as it is affected by the rate of aggregation, which may be different in case of each mutant. To avoid such ambiguity, equilibrium unfolding of lipase variants was performed in the presence of urea using fluorescence and CD spectroscopy. The shift in tryptophan fluorescence emission (λmax) and ellipticity at 222 nm (Θ222) were monitored with increasing concentration of denaturant [Fig. 3(B) and Supporting Information Fig. 8]. The unfolding transitions in urea monitored either by fluorescence or far-UV CD were indistinguishable. Using the approach described by Pace et al.,38 the data was normalized and free energy of unfolding was determined (Table I). Mutant M137P was most stable with an increase of 0.51 M in midpoint of unfolding transition (Cm) followed by G155S, with significant improvement by 0.46 M compared to wild-type. This corresponds to 1.16 and 1.05 kcal/mol increment in free energy of unfolding (ΔGunf), respectively, as compared to wild-type protein (ΔGunf = 10.39 kcal/mol). All other mutants showed moderate improvement of < 0.96 kcal/mol with free energy of unfolding between 10.3 and 11.35 kcal/mol. It is interesting to note that one mutant, A132D, is actually less stable as compared to wild-type protein, although it shows higher thermostability as measured by thermal inactivation (T50 and half-life of inactivation) and thermal unfolding (Tmapp). The midpoint of unfolding transition (Cm) for A132D was 4.36 M, which is 0.18 M less as compared to wild-type. Correspondingly, its free energy of unfolding is 9.98 kcal/mol, which is 0.41 kcal/mol lower than wild-type protein. The fact that mutant A132D, despite being conformationally less stable than wild-type, got selected during screening based on residual activity measurement can be attributed to its slower aggregation kinetics, thus showing higher recovery of activity upon cooling.
Proteolytic resistance of lipase variants
If proteolysis of a folded protein occurs due to global unfolding, proteolytic susceptibility is directly linked to its thermodynamic stability.22, 39 Such linkage is apparent since both thermostability and protease susceptibility originate from the dynamics associated with the protein.9, 22–24 Moreover, proteolytic susceptibility could be independent of stability when proteolysis is due to subglobal fluctuations.24 To get better insight into the phenomenon, proteolytic resistance of the selected 17 single thermostable mutants (STMs) of lipase was probed by measuring residual activities upon incubation with a nonspecific protease, Subtilisin A. On the basis of the dependence of proteolysis on protease concentration and concentration of Subtilisin A used in the study, it is indicative that for proteolysis, concentration of Subtilisin A was not rate limiting. Therefore we presume that the conformational change is rate limiting and proteolysis is governed by EX1 like mechanism. Upon incubation with protease for 4 h at 37°C, most of the mutants had distinctly different residual activities compared to that of wild-type lipase (Fig. 4; Table I). The most thermostable single mutant, M137P, was found to be most resistant to proteolytic attack, retaining more than 90% of its initial activity when wild-type loses its activity by 50%. Similar rank order of the mutants was observed at various lipases: subtilisin weight ratios and incubation times. The other mutants displaying enhanced proteolytic resistance include F17T, T47S, M134E, G155S, and I157M, with proteolytic residual activities between 75 and 88%. On the other hand, mutant A132D, displaying significant improved thermostability, as based on thermal inactivation (T50) and unfolding (Tmapp) compared to wild-type, but was found conformationally less stable than wild-type, was found to be more prone to proteolytic attack. Mutant A132D shows proteolytic residual activity of 45% which is 10% lesser than wild-type at 55%. All other single thermostable mutants showed moderate post proteolysis residual activities between 57 and 74%.
Figure 4.
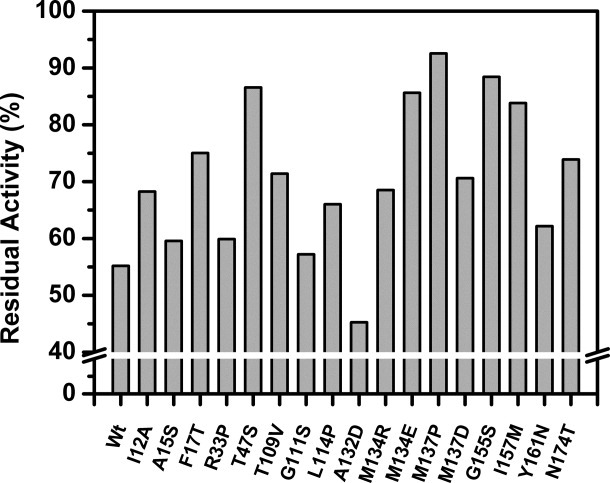
Proteolytic resistance of wild-type lipase and its variants measured as post proteolysis residual activity, after incubation with Subtilisin A.
Kinetics of proteolysis
To get a robust assessment into proteolytic susceptibility, a set of eight mutants with “graded increment in ΔGunf, i.e., alternate mutant when arranged on ΔΔG(Mut-wt) scale, along with wild-type were subjected to incubation with nonspecific protease followed by measurement of the kinetics of proteolysis. Wild-type lipase and eight selected mutants were incubated with Subtilisin A in equimolar ratio at 40°C.12 At regular intervals, an aliquot was removed and reaction was stopped by adding PMSF followed by separation on SDS-PAGE [Fig. 5(A) and Supporting Information Fig. 9]. SDS-PAGE profiles of wild-type along with different mutants clearly show progressive degradation of wild-type protein till 120 min. While mutant A132D degrades rapidly with no visible band corresponding to native protein after 20 min, mutant M137P degrades extremely slowly with no apparent decrease in band intensities corresponding to wild-type protein till 120 min. Other mutants A15S, F17T, G111S, M134R, M134E, and N174T also show progressive decrease in band intensities corresponding to native protein albeit with significantly delayed kinetics as compared to wild-type protein (Supporting Information Fig. 9).
Figure 5.
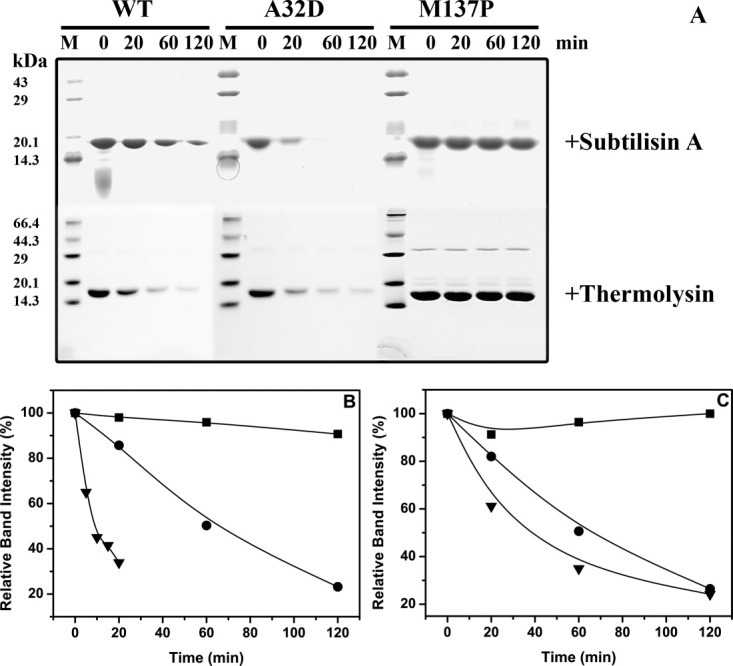
A: SDS-PAGE profiles of proteolytic degradation of wild-type along with least (A132D) and most (M137P) stable variant of the lipase against Subtilisin A (upper panel) and Thermolysin (lower panel). B and C. Kinetics of proteolytic degradation of mutant A132D (▾) and M137P (▪) along with wild-type lipase (•) against Subtilisin A (B) and Thermolysin (C) as quantitated by densitometric scanning of SDS-PAGE shown in (A).
To quantify the degradation kinetics of selected lipase mutants in the presence of Subtilisin A, the residual band intensities calculated by densitometric scanning, corresponding to that of native protein was plotted against time of incubation [Fig. 5(B) and Supporting Information Fig. 10]. The traces of all mutants, including wild-type protein followed first-order kinetics. The half-life of proteolytic degradation of the most resistant mutant, M137P was 956 min (∼16 h), which is ∼17-fold higher as compared to that of wild-type protein of 56 min. The proteolytic half-life of the most susceptible mutant A132D was just 15.7 min; more than threefold shorter than that of wild-type protein. Two mutants, A15S and G111S show comparable half-lives to that of wild-type. Rest of the mutants, F17T, M134R, M134E, and N174T, were found to be significantly more resistant than wild-type protein with proteolytic half-lives between 91 and 244 min (Table I).
To demonstrate that the observed kinetics of proteolysis is not dependent on the protease employed, we have subjected wt, M137P, and A132D to proteolysis by another protease, Thermolysin. Thermolysin is a thermostable metalloprotease with preference to hydrophobic amino acids. We have obtained kinetic susceptibility profiles similar to subtilisin indicating that observed relations are not protease specific [Fig. 5(B)].
Peptide fragment mass analysis of the mutants
To gain mechanistic insight into the proteolytic susceptibility of lipase variants, fragmentation pattern of three proteins i.e., wild-type, A132D, and M137P was analyzed. Digestion with Subtilisin A was done at 1:1 molar ratio for 4 h, followed by separation of the degraded fragments generated by reverse phase HPLC (Fig. 6). The fragments generated by proteolytic degradation of wild-type elute within 20 to 60 min. Mutant A132D was found to be most susceptible to proteolytic degradation as shown by higher intensity of absorption, both at 280 and 216 nm, compared to wild-type. On the other hand, mutant M137P shows very low intensity peaks under the identical digestion conditions. This suggests that A132D was extensively cleaved upon digestion, whereas the extent of cleavage was limited in case of mutant M137P. The elution profiles of wild-type and the two mutant digests were similar, but with considerable variation in the distribution of the peaks indicating randomness in the digestion pattern resulting in differences in the chemical properties of the generated peptides.
Figure 6.
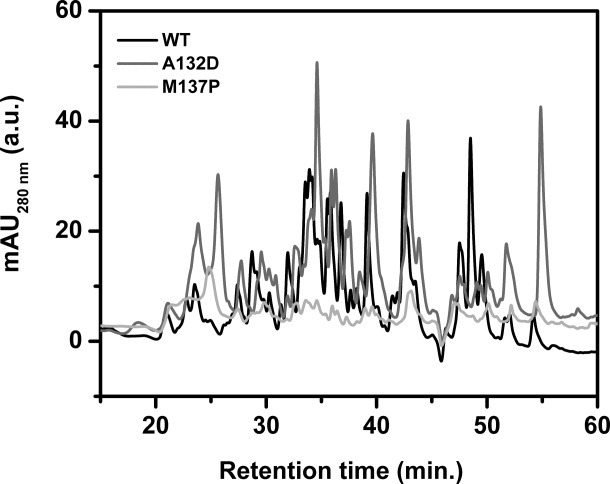
Reverse phase HPLC profiles of wild-type lipase along with mutant A132D and M137P preincubated with Subtilisin A. Chromatogram of wild-type lipase is shown in black while that of mutant A132D and M137P are in gray and light gray, respectively.
To further probe the differences into the peptides generated upon digestion, mass analysis of the peptides eluted from the reverse phase column after Subtilisin A digestion of the three proteins was performed using MALDI-TOF (Supporting Information Table III). The analysis was repeated at various signal to-noise ratios to ascertain the reproducibility of the data. The fragments are referred to the database generated from respective protein sequences. The data base has all the peptides of the length 3–30 amino acids and also masses of side group modifications, if occurred. MALDI analysis shows that, fragmentation of all three proteins covered the entire sequence, suggesting that cleavage occurs over the entire protein length without any protection of a particular region.
Variation of proteolytic resistance with stability
To gain insights into protein thermostability and proteolytic resistance relationship, proteolytic residual activities of all seventeen single thermostable mutants, along with wild-type, were compared with free energy of unfolding, a thermodynamic parameter as determined by equilibrium unfolding in the presence of urea. As shown in Figure 7(A), proteolytic resistance shows excellent correlation with free energy of unfolding of mutants (r = 0.95; P < 0.0001). To rule out the errors associated with end point assay during activity measurement, kinetics of proteolysis of wild-type along with eight selected mutants was performed. As shown in Figure 7(B), rate of proteolysis of selected variants also shows an excellent negative correlation with the free energy of unfolding (r = 0.9; P < 0.0009; regression coefficient = 1.04). Overall, this data suggest that proteolytic resistance of this lipase is very closely linked to its global stability as even little perturbation in one parameter (stability) by a single substitution in protein is clearly reflected in the other (proteolytic resistance).
Figure 7.
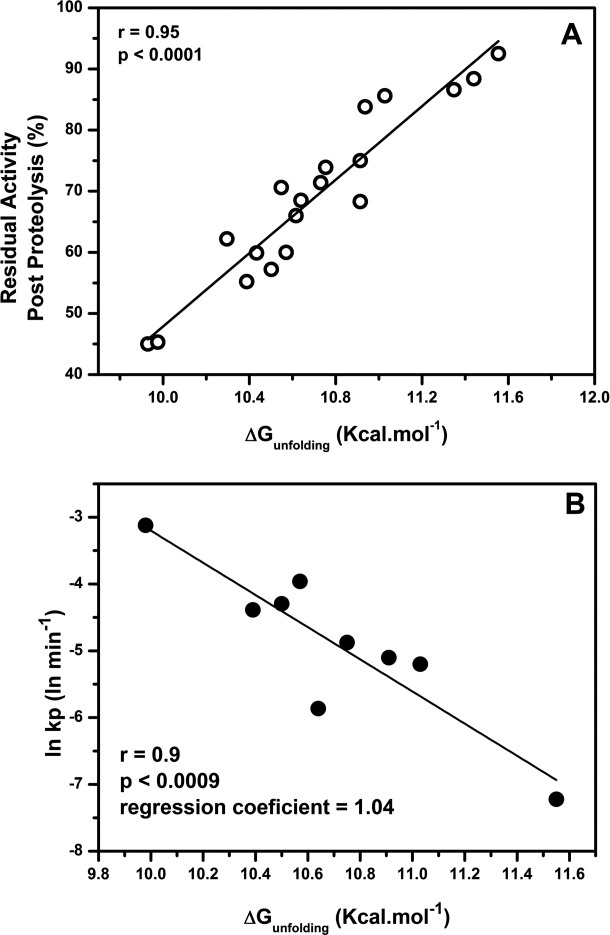
A: Correlation of proteolytic resistance [(residual activity after incubation with Subtilisin A (from Figure 4)] of single thermostable mutants with free energy of unfolding as determined by equilibrium unfolding in urea. B: Correlation of rate of proteolytic degradation of eight graded mutants along with wild type with their free energies of unfolding.
Discussion
The data presented on 17 single mutants, generated from site-saturation mutagenesis on 86 loop positions of the lipase as shown in Figure 8, show that a strong correlation exists between stability of this protein and its proteolytic susceptibility. While such relation is largely being understood to exist in proteins, our data obtained systematically on single substitutions over loops, sampling 60% of the surface area, bring forth the robustness of the correlation. At such a fine analysis, the observed strong correlations strongly argue for a tight relationship, which is independent of loop on which the mutation is located. This data is particularly relevant since a number of reports on stability changes do not translate into changes in proteolytic susceptibility.40, 41
Figure 8.
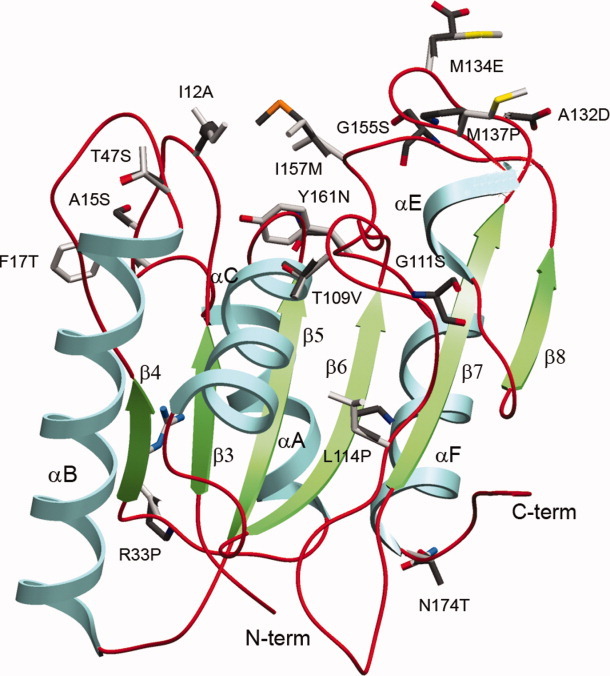
Ribbon diagram of Bacillus subtilis lipase, depicting 15 thermostabilizing positions, along with corresponding preferred substitutions. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The dependence of protease susceptibility on stability was mainly reported from the studies on homologous proteins from extremophiles.2, 6 Comparison of crystal structures of several homologous proteins from extremophiles have shown that the crystallographic B-factors are negatively correlated with the thermophilicity of proteins. Experimentally, B-factor engineering for thermostability was established by decreasing or increasing the B-factor to evolve either thermostable or thermolabile proteins, respectively.42, 43 Recently, proteolysis has been effectively used in screening of mutants for stability to identify the stable proteins in a proteome, in vitro evolution of proteins for thermostability or in screening proteins for enhanced ligand binding.17, 18, 39 However, several reports also indicated such relation between stability and proteolytic susceptibility does not hold always. Examples are presented wherein the mutants of a protein show altered proteolytic susceptibility in the absence of any change in the global stability (ΔG) and vice versa.44 Similarly, enormous differences in proteolytic susceptibilities of yeast and E. coli phosphoglycerate kinase has been reported which posses very similar three-dimensional structure as well as global stability.41 A set of mutants generated in the N-terminal domain of λ repressor showed that thermostable mutants are less susceptible to proteolytic attack.4 Similarly, mutants of ribonuclease barnase from Bacillus amyloliquefaciens evolved for stability displayed great improvement in their proteolytic resistance with little improvement in their thermodynamic stabilities, suggesting factors governing proteolytic resistance might be different from those governing thermodynamic stability.40 In these studies the investigations did not probe the details of structural basis of proteolytic susceptibility. The challenging nature of observed “all-or-none” nature of the protein proteolysis products does not allow identification of temporal profiling of cleaved bonds to determine the partial unfolded structures and locations of the susceptible bonds.
The correlation between stability and proteolytic susceptibility is also a consequence of underlying structural requirements of these two properties. Protein dynamics play a critical role both in proteolytic susceptibility as well as in stability. Proteases readily hydrolyze peptide bonds in a flexible region, a property extensively used to investigate protein folding/unfolding events.20, 25, 45 Several lines of evidence points to the fact that one way of stabilizing a protein is to reduce its dynamics.46, 47 For example hydrogen-deuterium exchange studies in extremophilic proteins indicated that dynamics associated with exchangeable protons are directly related to the stability of the proteins.23 Challenges in isolating partially cleaved products of proteins make it difficult to identify the effects of mutations on proteolytic stability. To overcome this, a detailed study on identifying partially unfolded species under native conditions was reported by using φ analysis on a set of mutations in maltose binding protein.5 Despite variations in stability the peptide profile of the three proteins, i.e., wt, A132D, and M137P, was found to be similar, though the intensity of the peptide reflects the differences in their protease susceptibilities. In addition, we have not observed any bias in the preference of amino acids for improving proteolytic resistance. For example, introduction of proline was considered to be a “simple” strategy to improve proteolytic stability and also thermostability. Proline substitution, based on sequence comparisons, has provided useful thermostability and proteolytic resistance in hirudin48 and in oligo-1,6-glucosidases.49 However, these studies caution that the location of substitution is critical for proline effect. Even in our set of mutants, proline substitution did not always lead to similar improvement in stability or proteolytic resistance. However, for improvement of stability focus on the positions on the loops would be beneficial. In the present study, each single substitution improves Tmapp of wild-type protein by 1–6°C; however, recombination of 13 selected mutations increases it from 56 to 79°C, an increment of 23°C (unpublished data). This huge improvement in stability is equivalent to a highly thermostable variant of the same protein, (6B), which was in vitro evolved, by performing six rounds of random mutagenesis and screening, earlier in the lab.50 Overall this study convincingly suggest that loops generally comprises weak regions of the protein and targeting only loop regions to strengthen these weak locations by residue optimization can substantially increase protein stability.
Proteolysis is responsible for protein turnover in the cells. Presence of unfolded structure is considered to be an early event in protein degradation. Excellent correlation between melting temperature of proteins and their in vivo turnover rates was observed.4, 51 These studies conclude a possibility of protein turnover being thermodynamically controlled. Protein turnover is a complex and regulated property of the cell involving a host of processes. Though lipase is a secreted protein in Bacillus, the observed relation may help in understanding the stability, protein turnover and protease susceptibility link. Overall, this study, which involves sampling of all the loops of a lipase for identification of thermostabilizing mutants and their protease susceptibility, indicates that protease susceptibility is strongly correlated with the global stability of the protein. Since the proteolysis was performed in conditions involving native state of the protein, the variations in the proteolysis kinetics may be due to influence of the mutations on the local fluctuations or global unfolding. It would be interesting to investigate whether these mutations influence the native state or partially unfolded state. In our opinion the strength of the present work is in the quality of the data set, wherein the mutants have least possible sequence variation, i.e., one mutation per variant, along with sampling 60% of protein surface. Observation of strong correlation (r = 0.9), when the ΔΔGunf (between most and least stable variants) is only 1.57 kcal/mol, indicates these two properties are tightly linked.
Conclusion
In order to investigate proteolytic resistance of a protein and its dependence on protein stability, a systematic mutagenic study was performed using Bacillus subtilis lipase as the model protein. Since loops, due to their associated high dynamics as compared to other structural elements, are implicated to have a role in protein stability and proteolytic susceptibility, all loops and termini positions were subjected to site-saturation mutagenesis and a set of 17 single mutants was generated with substantial improvement in protein stability. Proteolytic resistance of this mutant set was determined upon incubation with nonspecific protease Subtilisin A and was found to be in excellent correlation with the free energy of unfolding of the mutants. Overall, this study highlighted the role of loops in protein stability and proteolytic resistance. Only targeting of loops leads to the identification of 15 positions, optimization of which leads to substantial improvement in protein stability. In addition, in contrast to the studies based on proteins from extremophiles, the observed correlation between two properties in this mutant set, with least possible sequence change, and in such a narrow sequence space (ΔΔGunf = 1.57 kcal/mol) justifies the robustness of this correlation.
Materials and Methods
Taq DNA polymerase and dNTPs were obtained from Invitrogen, USA. Restriction enzymes were from New England Biolabs (Beverley, MA). Fast link™ DNA Ligation kit was from EPICENTRE Biotechnologies (Madison, WI). QIAquick® gel extraction kit and QIAprep® spin Miniprep kits were from Qiagen (Hilden, Germany). Phenyl Sepharose 6 Fast Flow (High Sub) was from Amersham Biosciences. PMSF, PNPB, Subtilisin A, Thermolysin and Triton-X 100 were from Sigma Chemical Co. Urea was purchased from USB Corporation (Cleveland, OH). IPTG was purchased from Calbiochem (Darmstadt, Germany). Oligonucleotides were purchased from BioServe (Hyderabad, India). All other reagents were of analytical grade or higher.
Construction of vectors used for screening and protein purification
For efficient screening, wild-type lipase gene was cloned in pET22b expression vector (Novagen), under pelB signal sequence for periplasmic expression, essentially as descried earlier.52 The structural gene cloned in pET21b,34 was PCR amplified using primers NcoFOR and T7-terminator, purified by gel extraction followed by digestion with NcoI and HindIII. The digested and gel-purified product was ligated with similarly digested pET22b vector to get pET22-wt, which was used for the screening of thermostable mutants upon transformation of E. coli BL21 (DE3), which directs the expression of protein in periplasm upon induction with IPTG.
For protein over-expression and purification, structural genes of lipase mutants cloned in pET22b, were PCR amplified using primers PET22NDE and T7-terminator, and cloned in pET21b between NdeI and HindIII sites, as described earlier.52
Site-saturation mutagenesis
Site-saturation mutagenesis at selected 86 positions was performed by overlap extension method using a pair of position specific mutagenic primers, along with two vector specific primers, T7-promoter and T7-terminator (Novagen), using pET22-wt as template, as described earlier.52 The target amino acid position was coded by NNK (sense strand) and MNN (antisense strand), where N = A, G, C or T, K = G or T and M = A or C. The PCR amplified products were digested with NcoI and HindIII followed by gel purification, ligated into similarly digested pET22b which were used for transformation of E. coli BL21 (DE3).
Screening for thermostability
The thermostability of mutants was assessed by following a two tier screening protocol as described earlier with some modifications.52 Initial screening was performed using a 96-well plate assay format followed by rigorous assessment of positives in tube assays.
The mutant library obtained after site-saturation mutagenesis was used to transform E. coli BL21 (DE3). Cells were plated on LB (1% tryptone, 0.5% yeast extract and 1% NaCl) agar plates having 100 μg/mL ampicillin and incubated at 37°C for 12 h. Transformants obtained were inoculated in individual wells of 96-well plates (Tarsons, India) containing 200 μL of LB medium having 100 μg/mL ampicillin. Plates were incubated at 37°C with shaking at 200 rpm for 6 h. Grown cultures were used to inoculate fresh medium in identical plates and were incubated at 37°C with shaking at 200 rpm. After 6 h, cultures were induced by adding IPTG in each well to a final concentration of 0.5 mM and incubated further for 12 h. Cells were harvested by spinning the plates at 4000 rpm for 30 min at 4°C in 5810 R Eppendorf centrifuge. Supernatant (100 μL) was mixed 1:1 with 100 mM sodium phosphate buffer (pH 7.2) and splitted into two identical 96-well PCR plates (Axygen Scientific, Union City, CA). One plate was incubated at 53°C for 20 min, cooled at 4°C for 20 min followed by equilibration at 25°C for 15 min in a PCR thermal cycler (GeneAmp 9700, Applied Biosystems, Foster City, CA). The other plate was incubated under identical conditions, except incubation at high temperature. Activity measurements were performed by mixing 80 μL of diluted supernatant with 80 μL of 2 x PNPB- Triton X-100 substrate solution (4 mM PNPB micellized in 40 mM Triton X-100). Increase in absorbance at 405 nm with time was monitored using Spectramax 190 plate reader (Molecular Devices, Sunnyvale, CA) and the data was analyzed by using SoftMaxPro 4.7.1 software provided with the instrument. The ratio of activity of each clone after incubation at higher temperature versus without incubation was taken as residual activity and was used to identify the positives.
Positives were further confirmed by performing tube assays. Ten milliliters of cell culture in LB medium, having 100 μg/mL ampicillin, was inoculated with 1% of overnight grown culture, followed by incubation at 37°C with vigorous shaking (250 rpm). After 6 h, culture was induced with 0.5 mM IPTG and incubated further for 12 h after which cells were separated from culture supernatant by centrifugation at 18,000g for 10 min at 4°C. Culture supernatant was diluted 1:1 with 100 mM sodium phosphate buffer (pH 7.2) and incubated (100 μL) at various temperatures for 20 min followed by cooling at 4°C for 20 min and final equilibration at 25°C for 15 min in a PCR thermal cycler. Activity was measured by adding 80 μL of diluted supernatant to 920 μL of 50 mM sodium phosphate buffer (pH 7.2) containing 2 mM PNPB micellized in 20 mM Triton X-100 as substrate. Increase in absorbance at 405 nm with time was measured using spectrophotometer (U-2000, Hitachi, Japan). Residual activity corresponding to each temperature of incubation was determined by taking the ratio of activities obtained upon incubation with that of without incubation. The temperature at which enzyme loses 50% of its activity after incubation for 20 min (T50) was taken as the thermostability index for the selection of mutants. Plasmids were purified from mutants displaying higher T50 values with respect to parent and genes were sequenced to identify the mutation that had occurred.
DNA sequencing
Plasmid DNA was isolated from E. coli DH5α or BL21 (DE3), using QIAprep® spin Miniprep kit according to the manufacturer's instructions. DNA sequencing was performed using ABI Prism BigDye™ v3.0 reaction kit according to the manufacturer's instructions (Applied Biosystems) as described earlier.52
Protein purification
Lipase mutant proteins, genes of which were cloned in pET21b, were purified upon expression in E. coli BL21 (DE3) as described earlier.34 Purified protein was stored in 2 mM glycine-NaOH buffer (pH 10) at −20°C. Purity was checked by running SDS-PAGE and was found to be more than 95 percent. Protein quantitation was done by the modified Lowry method.53
Activity measurements
Specific activities of all mutants were determined at 25°C. To 1 mL of 50 mM sodium phosphate buffer (pH 7.2) containing 2 mM PNPB, micellized in 20 mM Triton X-100, 1 μg pure enzyme was added. Increase in absorbance at 405 nm with time was recorded on a spectrophotometer (U-2000, Hitachi, Japan).
Spectroscopy
Far-UV CD spectra were recorded in the 250–200 nm range using a JASCO J-815 spectropolarimeter fitted with Jasco Peltier-type temperature controller (CDF-426S/15) as described earlier.52 Similarly, intrinsic tryptophan fluorescence was recorded between 310 and 380 nm using a Hitachi F-4500 fluorimeter by exciting the sample at 295 nm, essentially as described earlier.52
Thermal inactivation and unfolding
Thermal inactivation and unfolding of lipase was performed, as described earlier with some variations.52 Briefly, purified proteins were heated in the range of 48 to 65°C in a thermal cycler for 20 min, cooled at 4°C for 20 min followed by equilibration at 25°C for 15 min. Samples were centrifuged to get rid of any aggregated protein, followed by activity measurement at room temperature. Kinetics of thermal inactivation was monitored by incubating the proteins at 55°C. At various time intervals, an aliquot was removed followed by residual activity measurement at room temperature as described above. Typically, inactivation was followed until >80% of the activity was lost.
Thermal unfolding of lipase mutants was monitored using circular dichroism spectroscopy in a JASCO J-815 spectropolarimeter fitted with Jasco Peltier-type temperature controller (CDF-426S/15). The protein concentration used was 0.05 mg/mL in 50 mM sodium phosphate buffer (pH 7.2) with path length of 1 cm. Temperature dependent unfolding profiles were obtained by heating protein at a constant rate of 1°C per min from 25 to 75°C while measuring change in ellipticity at 222 nm.
Equilibrium unfolding
Equilibrium unfolding in the presence of urea was monitored using CD and fluorescence spectroscopy. Briefly, purified protein was incubated in varying concentrations of urea for 12 h at room temperature followed by far-UV CD and intrinsic tryptophan fluorescence measurements, as described earlier.52 Unfolding profiles were determined by monitoring the change in ellipticity at 222 nm and shift in tryptophan emission maxima (λmax) with increasing concentration of urea. The concentration of stock urea solution was determined by refractive index measurements. Data was analyzed as described by Pace et al.38, 54
Protease resistance assays
Lipase mutants, at a concentration of 1 mg/mL, were incubated with and without 0.03 mg/mL Subtilisin A (50:1 molar ratio) in 50 mM Tris-HCl buffer pH 8.0 having 2 mM CaCl2 at 37°C in a thermal cycler. After 4 hours of incubation an aliquot was diluted and enzymatic activities were determined as described above. Residual activity was calculated by measuring activities of mutants incubated identically except without protease.
Peptide fingerprints after proteolysis of selected lipase mutants was determined by incubating with Subtilisin A as described above except at 1:1 molar ratio. After 4 h of incubation, the mutants were subjected to reverse phase HPLC to separate the fragments generated using a C-18 column with particle size of 5 μm and pore size of 300 Å (Hi-Pore RP 318, BioRad) on a HPLC system (1200 series, Agilent Technologies). The eluted fractions were vacuum dried and resuspended in 10 μL of 50% acetonitrile in 0.1% trifluoroacetic acid. An aliquot of 0.8 μL from each of the fractions was loaded on a MALDI plate in duplicates, with equal volume of α-cyano-4-hydroxycinnamic acid (5 mg/mL) and MALDI analysis was done on a 4800 TOF/TOF system (Applied Biosystems). Peptide peak list with minimum signal to noise ratio threshold of 50 was generated for each fraction from data explorer program in GPS explorer (Applied Biosystems). Peptide mass lists generated were submitted to MS-nonspecific program with respective protein sequences as templates for generating mass matches. Program was run in MALDI-TOF/TOF mode with average error of 20 ppm. On the basis of amino acid length, matched peptides were grouped into six classes and protein dependent variation in peptide fragment length was studied.
To monitor kinetics of proteolysis, selected lipase mutants at a concentration of 0.5 mg/mL were incubated with Subtilisin A at an equimolar ratio in 50 mM Tris-HCl buffer (pH 8.0), having 2 ms CaCl2 at 40°C in a thermal cycler. At different time intervals aliquots were removed and reaction was stopped by adding PMSF to a final concentration of 5 mM. The samples were subjected to SDS-PAGE and gel was stained with coomassie brilliant blue R-250. Proteolytic degradation kinetics was determined by measuring band intensities corresponding to native protein using densitometric scanning software ImageJ (http://rsbweb.nih.gov/ij/). Band intensities were plotted against time of incubation to monitor degradation kinetics. Rate and half-life of proteolytic degradation was determined by fitting first order exponential decay curve to the above data. To monitor proteolysis by thermolysin we incubated lipase mutants at 0.5 mg/mL concentration with 0.1 mg/mL thermolysin at 40°C in buffer conditions specified by Chang and Park.5 Aliquots were taken at specific time intervals and reaction stopped by 50 mM EDTA. For analysis same methodology was followed as for proteolysis by Subtilisin A specified above.
Acknowledgments
SA is Research Associate in a research project NWP 0044 of Council of Scientific and Industrial Research (CSIR), Govt. of India. Authors VK and KBR acknowledge University Grant Commission, (UGC), Govt. of India, for the Research Fellowships. Assistance provided by Poornima Yedavalli in screening of mutants is deeply acknowledged.
Glossary
Abbreviations:
- CD
circular dichroism
- HPLC
high pressure liquid chromatography
- IPTG
Isopropyl β-d-1-thiogalactopyranoside
- MALDI
matrix assisted laser desorption/ionization
- PAGE
polyacrylamide gel electrophoresis
- PMSF
phenylmethanesulfonyl fluoride
- PNPB
p-nitrophenyl butyrate
- SDS
sodium dodecyl sulfate
- TOF
time of flight
- T50
Temperature of half inactivation
- Tmapp
apparent melting temperature.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Ottesen M. Induction of biological activity by limited proteolysis. Annu Rev Biochem. 1967;36:55–76. doi: 10.1146/annurev.bi.36.070167.000415. [DOI] [PubMed] [Google Scholar]
- 2.Daniel RM, Cowan DA, Morgan HW, Curran MP. A correlation between protein thermostability and resistance to proteolysis. Biochem J. 1982;207:641–644. doi: 10.1042/bj2070641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fontana A, de Laureto PP, Spolaore B, Frare E, Picotti P, Zambonin M. Probing protein structure by limited proteolysis. Acta Biochim Pol. 2004;51:299–321. [PubMed] [Google Scholar]
- 4.Parsell DA, Sauer RT. The structural stability of a protein is an important determinant of its proteolytic susceptibility in Escherichia coli. J Biol Chem. 1989;264:7590–7595. [PubMed] [Google Scholar]
- 5.Chang Y, Park C. Mapping transient partial unfolding by protein engineering and native-state proteolysis. J Mol Biol. 2009;393:543–556. doi: 10.1016/j.jmb.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 6.Khajeh K, Khezre-Barati S, Nemat-Gorgani M. Proteolysis of mesophilic and thermophilic alpha-amylases: a comparative study. Appl Biochem Biotechnol. 2001;94:97–109. doi: 10.1385/abab:94:2:097. [DOI] [PubMed] [Google Scholar]
- 7.Ogasahara K, Tsunasawa S, Soda Y, Yutani K, Sugino Y. Effect of single amino acid substitutions on the protease susceptibility of tryptophan synthase alpha subunit. Eur J Biochem. 1985;150:17–21. doi: 10.1111/j.1432-1033.1985.tb08979.x. [DOI] [PubMed] [Google Scholar]
- 8.Akasako A, Haruki M, Oobatake M, Kanaya S. High resistance of Escherichia coli ribonuclease HI variant with quintuple thermostabilizing mutations to thermal denaturation, acid denaturation, and proteolytic degradation. Biochemistry. 1995;34:8115–8122. doi: 10.1021/bi00025a018. [DOI] [PubMed] [Google Scholar]
- 9.Markert Y, Koditz J, Mansfeld J, Arnold U, Ulbrich-Hofmann R. Increased proteolytic resistance of ribonuclease A by protein engineering. Protein Eng. 2001;14:791–796. doi: 10.1093/protein/14.10.791. [DOI] [PubMed] [Google Scholar]
- 10.Fontana A, Zambonin M, Polverino de LP, De Filippis V, Clementi A, Scaramella E. Probing the conformational state of apomyoglobin by limited proteolysis. J Mol Biol. 1997;266:223–230. doi: 10.1006/jmbi.1996.0787. [DOI] [PubMed] [Google Scholar]
- 11.Park C, Marqusee S. Probing the high energy states in proteins by proteolysis. J Mol Biol. 2004;343:1467–1476. doi: 10.1016/j.jmb.2004.08.085. [DOI] [PubMed] [Google Scholar]
- 12.Park C, Marqusee S. Pulse proteolysis: a simple method for quantitative determination of protein stability and ligand binding. Nat Methods. 2005;2:207–212. doi: 10.1038/nmeth740. [DOI] [PubMed] [Google Scholar]
- 13.Hanes MS, Ratcliff K, Marqusee S, Handel TM. Protein-protein binding affinities by pulse proteolysis: application to TEM-1/BLIP protein complexes. Protein Sci. 2010;19:1996–2000. doi: 10.1002/pro.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sieber V, Pluckthun A, Schmid FX. Selecting proteins with improved stability by a phage-based method. Nat Biotechnol. 1998;16:955–960. doi: 10.1038/nbt1098-955. [DOI] [PubMed] [Google Scholar]
- 15.Martin A, Schmid FX, Sieber V. Proside: a phage-based method for selecting thermostable proteins. Methods Mol Biol. 2003;230:57–70. doi: 10.1385/1-59259-396-8:57. [DOI] [PubMed] [Google Scholar]
- 16.Wunderlich M, Martin A, Schmid FX. Stabilization of the cold shock protein CspB from Bacillus subtilis by evolutionary optimization of Coulombic interactions. J Mol Biol. 2005;347:1063–1076. doi: 10.1016/j.jmb.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 17.Kather I, Jakob RP, Dobbek H, Schmid FX. Increased folding stability of TEM-1 beta-lactamase by in vitro selection. J Mol Biol. 2008;383:238–251. doi: 10.1016/j.jmb.2008.07.082. [DOI] [PubMed] [Google Scholar]
- 18.Eldridge B, Cooley RN, Odegrip R, McGregor DP, Fitzgerald KJ, Ullman CG. An in vitro selection strategy for conferring protease resistance to ligand binding peptides. Protein Eng Des Sel. 2009;22:691–698. doi: 10.1093/protein/gzp052. [DOI] [PubMed] [Google Scholar]
- 19.Mukherjee S, Guptasarma P. Direct proteolysis-based purification of an overexpressed hyperthermophile protein from Escherichia coli lysate: a novel exploitation of the link between structural stability and proteolytic resistance. Protein Expr Purif. 2005;40:71–76. doi: 10.1016/j.pep.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 20.Hubbard SJ. The structural aspects of limited proteolysis of native proteins. Biochim Biophys Acta. 1998;1382:191–206. doi: 10.1016/s0167-4838(97)00175-1. [DOI] [PubMed] [Google Scholar]
- 21.Cooper A. Protein fluctuations and the thermodynamic uncertainty principle. Prog Biophys Mol Biol. 1984;44:181–214. doi: 10.1016/0079-6107(84)90008-7. [DOI] [PubMed] [Google Scholar]
- 22.Frauenfelder H, Parak F, Young RD. Conformational substates in proteins. Annu Rev Biophys Biophys Chem. 1988;17:451–479. doi: 10.1146/annurev.bb.17.060188.002315. [DOI] [PubMed] [Google Scholar]
- 23.Zavodszky P, Kardos J, Svingor Petsko GA. Adjustment of conformational flexibility is a key event in the thermal adaptation of proteins. Proc Natl Acad Sci USA. 1998;95:7406–7411. doi: 10.1073/pnas.95.13.7406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamerzell TJ, Middaugh CR. The complex inter-relationships between protein flexibility and stability. J Pharm Sci. 2008;97:3494–3517. doi: 10.1002/jps.21269. [DOI] [PubMed] [Google Scholar]
- 25.Novotny J, Bruccoleri RE. Correlation among sites of limited proteolysis, enzyme accessibility and segmental mobility. FEBS Lett. 1987;211:185–189. doi: 10.1016/0014-5793(87)81433-3. [DOI] [PubMed] [Google Scholar]
- 26.Spiller B, Gershenson A, Arnold FH, Stevens RC. A structural view of evolutionary divergence. Proc Natl Acad Sci USA. 1999;96:12305–12310. doi: 10.1073/pnas.96.22.12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao H, Arnold FH. Directed evolution converts subtilisin E into a functional equivalent of thermitase. Protein Eng. 1999;12:47–53. doi: 10.1093/protein/12.1.47. [DOI] [PubMed] [Google Scholar]
- 28.Numata K, Hayashi-Iwasaki Y, Kawaguchi J, Sakurai M, Moriyama H, Tanaka N, Oshima T. Thermostabilization of a chimeric enzyme by residue substitutions: four amino acid residues in loop regions are responsible for the thermostability of Thermus thermophilus isopropylmalate dehydrogenase. Biochim Biophys Acta. 2001;1545:174–183. doi: 10.1016/s0167-4838(00)00275-2. [DOI] [PubMed] [Google Scholar]
- 29.Max KE, Wunderlich M, Roske Y, Schmid FX, Heinemann U. Optimized variants of the cold shock protein from in vitro selection: structural basis of their high thermostability. J Mol Biol. 2007;369:1087–1097. doi: 10.1016/j.jmb.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 30.Ahmad S, Kamal MZ, Sankaranarayanan R, Rao NM. Thermostable Bacillus subtilis lipases: in vitro evolution and structural insight. J Mol Biol. 2008;381:324–340. doi: 10.1016/j.jmb.2008.05.063. [DOI] [PubMed] [Google Scholar]
- 31.van PG, Eggert T, Jaeger KE, Dijkstra BW. The crystal structure of Bacillus subtilis lipase: a minimal alpha/beta hydrolase fold enzyme. J Mol Biol. 2001;309:215–226. doi: 10.1006/jmbi.2001.4659. [DOI] [PubMed] [Google Scholar]
- 32.Kawasaki K, Kondo H, Suzuki M, Ohgiya S, Tsuda S. Alternate conformations observed in catalytic serine of Bacillus subtilis lipase determined at 1.3 A resolution. Acta Crystallogr D Biol Crystallogr. 2002;58:1168–1174. doi: 10.1107/s090744490200714x. [DOI] [PubMed] [Google Scholar]
- 33.Sambrook J, Russel DW. Molecular cloning: a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 34.Acharya P, Rajakumara E, Sankaranarayanan R, Rao NM. Structural basis of selection and thermostability of laboratory evolved Bacillus subtilis lipase. J Mol Biol. 2004;341:1271–1281. doi: 10.1016/j.jmb.2004.06.059. [DOI] [PubMed] [Google Scholar]
- 35.Kumar S, Tsai CJ, Nussinov R. Factors enhancing protein thermostability. Protein Eng. 2000;13:179–191. doi: 10.1093/protein/13.3.179. [DOI] [PubMed] [Google Scholar]
- 36.Kamal MZ, Ahmad S, Rao NM. Stabilizing effect of polyols is sensitive to inherent stability of protein. Biophys Chem. 2011;156:68–71. doi: 10.1016/j.bpc.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 37.Baker D, Agard DA. Kinetics versus thermodynamics in protein folding. Biochemistry. 1994;33:7505–7509. doi: 10.1021/bi00190a002. [DOI] [PubMed] [Google Scholar]
- 38.Pace CN, Shirley BA, Thompson JA. Mechanism of protein folding. In: Creighton ED, editor. Protein structure: a practical approach. Oxford: Oxford Press; 1989. pp. 311–330. [Google Scholar]
- 39.Park C, Zhou S, Gilmore J, Marqusee S. Energetics-based protein profiling on a proteomic scale: identification of proteins resistant to proteolysis. J Mol Biol. 2007;368:1426–1437. doi: 10.1016/j.jmb.2007.02.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pedersen JS, Otzen DE, Kristensen P. Directed evolution of barnase stability using proteolytic selection. J Mol Biol. 2002;323:115–123. doi: 10.1016/s0022-2836(02)00891-4. [DOI] [PubMed] [Google Scholar]
- 41.Young TA, Skordalakes E, Marqusee S. Comparison of proteolytic susceptibility in phosphoglycerate kinases from yeast and E. coli: modulation of conformational ensembles without altering structure or stability. J Mol Biol. 2007;368:1438–1447. doi: 10.1016/j.jmb.2007.02.077. [DOI] [PubMed] [Google Scholar]
- 42.Reetz MT, Carballeira JD, Vogel A. Iterative saturation mutagenesis on the basis of B factors as a strategy for increasing protein thermostability. Angew Chem Int Ed Engl. 2006;45:7745–7751. doi: 10.1002/anie.200602795. [DOI] [PubMed] [Google Scholar]
- 43.Reetz MT, Soni P, Fernandez L. Knowledge-guided laboratory evolution of protein thermolability. Biotechnol Bioeng. 2009;102:1712–1717. doi: 10.1002/bit.22202. [DOI] [PubMed] [Google Scholar]
- 44.LeMaster DM, Tang J, Paredes DI, Hernandez G. Enhanced thermal stability achieved without increased conformational rigidity at physiological temperatures: spatial propagation of differential flexibility in rubredoxin hybrids. Proteins. 2005;61:608–616. doi: 10.1002/prot.20594. [DOI] [PubMed] [Google Scholar]
- 45.Fontana A, Polverino de LP, De Filippis V, Scaramella E, Zambonin M. Probing the partly folded states of proteins by limited proteolysis. Fold Des. 1997;2:R17–R26. doi: 10.1016/S1359-0278(97)00010-2. [DOI] [PubMed] [Google Scholar]
- 46.Zhou HX. Loops, linkages, rings, catenanes, cages, and crowders: entropy-based strategies for stabilizing proteins. Acc Chem Res. 2004;37:123–130. doi: 10.1021/ar0302282. [DOI] [PubMed] [Google Scholar]
- 47.Vemparala S, Mehrotra S, Balaram H. Role of loop dynamics in thermal stability of mesophilic and thermophilic adenylosuccinate synthetase: a molecular dynamics and normal mode analysis study. Biochim Biophys Acta. 2011;1814:630–637. doi: 10.1016/j.bbapap.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 48.Wirsching F, Keller M, Hildmann C, Riester D, Schwienhorst A. Directed evolution towards protease-resistant hirudin variants. Mol Genet Metab. 2003;80:451–462. doi: 10.1016/j.ymgme.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 49.Watanabe K, Kitamura K, Suzuki Y. Analysis of the critical sites for protein thermostabilization by proline substitution in oligo-1,6-glucosidase from Bacillus coagulans ATCC 7050 and the evolutionary consideration of proline residues. Appl Environ Microbiol. 1996;62:2066–2073. doi: 10.1128/aem.62.6.2066-2073.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kamal MZ, Ahmad S, Molugu TR, Vijayalakshmi A, Deshmukh MV, Sankaranarayanan R, Rao NM. In vitro evolved non-aggregating and thermostable lipase: structural and thermodynamic investigation. J Mol Biol. 2011;413:726–741. doi: 10.1016/j.jmb.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 51.McLendon G, Radany E. Is protein turnover thermodynamically controlled? J Biol Chem. 1978;253:6335–6337. [PubMed] [Google Scholar]
- 52.Ahmad S, Rao NM. Thermally denatured state determines refolding in lipase: mutational analysis. Protein Sci. 2009;18:1183–1196. doi: 10.1002/pro.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Markwell MAK, Hass SM, Tolbert NE, Bieber LL. Protein determination in membrane and lipoprotein samples: manual and automated procedures. Methods Enzymol. 1981;72:296–303. doi: 10.1016/s0076-6879(81)72018-4. [DOI] [PubMed] [Google Scholar]
- 54.Pace CN. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.