

Background: Ribose-1,5-bisphosphate isomerase (R15Pi) converts ribose 1,5-bisphosphate into ribulose 1,5-bisphosphate in a novel AMP metabolic pathway.
Results: Crystal structures of reaction-ready and -completed states are determined.
Conclusion: R15Pi undergoes an open-closed conformational change upon substrate binding, and the reaction proceeds via a cis-phosphoenolate intermediate.
Significance: The mechanism of ribose isomerization revealed in this study could be applied on other 1-phosphorylated ribose isomerases.
Keywords: Archaea; Crystal Structure; Enzyme Mechanisms; Protein Conformation; Rubisco; AMP Metabolism; Carbon Fixation; cis-Phosphoenolate Intermediate; Ribose-1,5-bisphosphate Isomerase
Abstract
Ribose-1,5-bisphosphate isomerase (R15Pi) is a novel enzyme recently identified as a member of an AMP metabolic pathway in archaea. The enzyme converts d-ribose 1,5-bisphosphate into ribulose 1,5-bisphosphate, providing the substrate for archaeal ribulose-1,5-bisphosphate carboxylase/oxygenases. We here report the crystal structures of R15Pi from Thermococcus kodakarensis KOD1 (Tk-R15Pi) with and without its substrate or product. Tk-R15Pi is a hexameric enzyme formed by the trimerization of dimer units. Biochemical analyses show that Tk-R15Pi only accepts the α-anomer of d-ribose 1,5-bisphosphate and that Cys133 and Asp202 residues are essential for ribulose 1,5-bisphosphate production. Comparison of the determined structures reveals that the unliganded and product-binding structures are in an open form, whereas the substrate-binding structure adopts a closed form, indicating domain movement upon substrate binding. The conformational change to the closed form optimizes active site configuration and also isolates the active site from the solvent, which may allow deprotonation of Cys133 and protonation of Asp202 to occur. The structural features of the substrate-binding form and biochemical evidence lead us to propose that the isomerase reaction proceeds via a cis-phosphoenolate intermediate.
Introduction
Ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco)2 (EC 4.1.1.39) has long been known as the key enzyme of the Calvin-Benson-Bassham cycle in eukaryotes and bacteria, catalyzing the conversion of ribulose 1,5-bisphosphate (RuBP), CO2, and H2O to two molecules of 3-phosphoglycerate (1, 2). The substrate RuBP is provided by phosphoribulokinase (EC 2.7.1.19), which catalyzes the phosphorylation of ribulose 5-phosphate (3). Rubiscos are also present in the archaea (4) but function in a pathway involved in AMP metabolism along with two novel enzymes, AMP phosphorylase and ribose-1,5-bisphosphate isomerase (R15Pi) (5). AMP phosphorylase catalyzes the conversion of AMP and phosphate to adenine and ribose 1,5-bisphosphate (R15P) (supplemental Fig. 1). R15Pi then converts the R15P to RuBP through an intriguing isomerization reaction, providing the substrate for archaeal Rubisco (Fig. 1). Although the unique activities of these two enzymes have been demonstrated, the biochemical and structural features of AMP phosphorylase and R15Pi are yet to be clarified.
FIGURE 1.
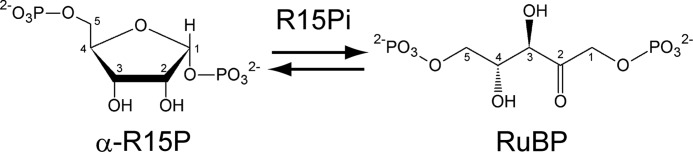
Reaction scheme of the aldose-ketose interconversion catalyzed by R15Pi. The carbon ID numbers for each molecule are shown.
R15Pi from Thermococcus kodakarensis KOD1 (Tk-R15Pi) was originally annotated as a homolog of the eukaryotic translation initiation factor 2B (IF-2B) belonging to the PF01008 family in the PFAM database. The PF01008 family contains eukaryotic IF-2B α, β, and δ subunits and archaeal IF-2B 1 and 2 subunits (6). IF-2B is responsible for the GDP-GTP exchange reaction of the translation initiation factor 2 (7). 5-Methylthioribose-1-phosphate (MTR-1P) isomerase (M1Pi) (EC 5.3.1.23) (8) is also included in the PF01008 family and shares high amino acid sequence similarity with Tk-R15Pi (9). The reactions catalyzed by R15Pi and M1Pi resemble one another (supplemental Fig. 2), both catalyzing the isomerization of pentose molecules with phosphorylated 1-hydroxy groups. The enzymatic isomerization of sugars with C1 hydroxy groups have been well investigated, such as the reactions by triose-phosphate isomerase (EC 5.3.1.1) (10), phosphoglucose isomerase (EC 5.3.1.9) (11), ribose-5-phosphate isomerase (EC 5.3.1.6) (12), and xylose isomerase (EC 5.3.1.5) (13) (supplemental Fig. 3). All of the substrates for these enzymes have open-chain forms, and the ring opening is the first and the essential step of these isomerizations (supplemental Fig. 3) (14–16). However, R15P and MTR-1P cannot adopt the conventional open-chain forms due to the presence of the 1-phosphate, suggesting a different reaction mechanism from the known sugar isomerases.
The crystal structures of M1Pi from Bacillus subtilis and M1Pi homolog from Archaeoglobus fulgidus have been determined (17). The former structure bound with the reaction product displays a closed form, whereas the latter structure of an apoenzyme adopts an open form, suggesting domain movement upon substrate binding in these proteins. Two candidate mechanisms have been proposed for the enzyme from B. subtilis (9, 17). One mechanism considers cis-phosphoenolate as an intermediate (supplemental Fig. 4, top), and the other involves hydride transfer (supplemental Fig. 4, bottom). Both mechanisms initiate by a common donation of a proton to the ring oxygen. In the mechanism via cis-phosphoenolate, proton abstraction from C2 by a deprotonated cysteine occurs, resulting in the intermediate. The proton is then redonated to the C1 carbon, generating the product 5-methylthioribulose 1-phosphate (MTRu-1P). The latter mechanism suggests that the cysteine thiol group may stabilize the C1 carbocation prior to the hydride shift without utilizing a metal ion. Thus far, however, the reaction mechanisms of the enzymes isomerizing sugars with phosphorylated 1-hydroxy groups, including M1Pi and R15Pi, have not yet been conclusively established, and a protein structure bound to substrate would contribute in understanding the mechanism.
Here, we have determined the crystal structures of the substrate-binding form of Tk-R15Pi in addition to structures of the protein in the presence or absence of the reaction product. A comparison of the substrate-binding structure and the other two structures shows a drastic induced-fit rigid-body domain motion. The domain movement isolates the reaction center from the solvent. This structural information allows us to elucidate the reaction mechanism of Tk-R15Pi, which most likely applies for other isomerases that act on sugars with 1-phosphates.
EXPERIMENTAL PROCEDURES
Construction of Expression Plasmids
The expression plasmids for the His6-tagged wild-type and mutant R15P isomerase recombinant proteins were constructed as follows. The DNA fragment including the wild-type Tk-R15Pi gene (Tk-e2b2) was excised from pET-e2b2 (5) with NdeI and BamHI and inserted into a pCold I vector (Takara Bio, Shiga, Japan) digested with the same restriction enzymes. Using the resulting plasmid pCold-His-e2b2 as a template, expression plasmids for four R15Pi mutant proteins, C133S (pCold-His-e2b2-c133s), C133A (pCold-His-e2b2-c133a), D202N (pCold-His-e2b2-d202n), and R227E (pCold-His-e2b2-r227e), were constructed with a QuikChange II XL site-directed mutagenesis kit (Stratagene, La Jolla, CA). Site-directed mutagenesis for the preparation of plasmids for C133S, C133A, D202N, and R227E mutant proteins was performed using the primer sets C133S-F/C133S-R (5′-GCGACGTCATAATGACTCACTCCCACAGCAAGGCTGCCATAAG-3′/5′-CTTATGGCAGCCTTGCTGTGGGAGTGAGTCATTATGACGTCGC-3′), C133A-F/C133A-R (5′-GCGACGTCATAATGACTCACGCCCACAGCAAGGCTGCCATAAG-3′/5′-CTTATGGCAGCCTTGCTGTGGGCGTGAGTCATTATGACGTCGC-3′), D202N-F/D202N-R (5′-GACAAGGTCGTTATGGGGGCTAACTCGATAACCGTAAACGGCGC-3′/5′-GCGCCGTTTACGGTTATCGAGTTAGCCCCCATAACGACCTTGTC-3′), and R227E-F/R227E-R (5′-GCCTTGACCGCGAAGGAACACGAGGTTTGGACTATGATCGCTGC-3′/5′-GCAGCGATCATAGTCCAAACCTCGTGTTCCTTCGCGGTCAAGGC-3′) respectively (underlines indicate the points for mutation). The expression plasmid for the His6-tagged AMP phosphorylase recombinant protein was constructed as follows. A DNA fragment containing the gene encoding AMP phosphorylase (Tk-deoA) was amplified from T. kodakarensis genomic DNA using the primers NdeI-deoA-4f/EcoRI-b-deoA-1509r (5′-CGCATATGAAAGCCAAGATCCGC-3′/5′-CGGAATTCCCGATGTTGCCGATTCGCTG-3′). After digestion with NdeI and EcoRI, the fragment was inserted into pET-21a(+) (Novagen, Madison, WI) digested with the same restriction enzymes, resulting in the plasmid pET-deoA-His.
Gene Expression and Purification of Recombinant Proteins
BL21 CodonPlus(DE3)-RIL Escherichia coli competent cells (Stratagene) were transformed by the recombinant plasmids. After the E. coli cells were grown at 37 °C in LB medium containing 50 μg/ml ampicillin until their optical density at 660 nm reached 0.4–0.8, gene expression was induced by the addition of 0.1 mm isopropyl-1-thio-β-d-thiogalactopyranoside. For cells expressing the R15Pi protein, the culture was further incubated for 40 h at 15 °C. For AMP phosphorylase expression, cells treated with isopropyl-1-thio-β-d-thiogalactopyranoside were cultured for 4 h at 37 °C. Cells were then harvested by centrifugation (5,000 × g, 15 min), washed with 1% NaCl in 50 mm Tris-HCl, pH 8.0, and collected by centrifugation (5,000 × g, 15 min).
For R15Pi proteins, harvested cells were resuspended with NiC buffer (20 mm sodium phosphate, 500 mm NaCl, and 40 mm imidazole, pH 7.4) and disrupted by sonication. The crude extract was heat-treated at 85 °C for 30 min, cooled on ice, and then centrifuged at 20,000 × g for 30 min in order to remove thermolabile proteins derived from the host cells. The supernatant was applied to an Ni2+ column, His GraviTrap (GE Healthcare Japan, Tokyo, Japan), equilibrated with NiC buffer. The column was washed with NiC buffer, and then Tk-R15Pi was eluted with NiC buffer containing 500 mm imidazole. Buffer exchange to HIC buffer (100 mm Bicine-NaOH, 10 mm MgCl2, and 1.2 m ammonium sulfate, pH 8.3) was carried out using a PD-10 column (GE Healthcare). The protein solution containing Tk-R15Pi was loaded onto a Resource ISO hydrophobic interaction column (GE Healthcare) equilibrated with HIC buffer and then eluted with a linear gradient of ammonium sulfate concentration from 1.2 to 0 m.
For enzymatic analysis, the concentration of ammonium sulfate was reduced by ultrafiltration with 100 mm Bicine-NaOH, 10 mm MgCl2, and 100 mm NaCl, pH 8.3, using an Amicon Ultra device (MWCO 10,000) (Millipore, Bedford, MA).
Before crystallization, the fractions of the Resource ISO elution including Tk-R15Pi were merged and concentrated to 5–9 mg/ml. The buffer composition was simultaneously exchanged to 100 mm Bicine-NaOH, pH 8.3, 200 mm MgCl2, 2 mm DTT. The protein concentration of Tk-R15Pi was determined by UV absorbance at 280 nm with an extinction coefficient of 57,800 m−1 cm−1.
Selenomethionine (SeMet)-substituted Tk-R15Pi was expressed in E. coli B834(DE3) strain cells (Takara Bio) cultured in minimal medium supplemented with l-selenomethionine. SeMet Tk-R15Pi was purified with the same protocol as for the unlabeled protein and concentrated to 4 mg/ml for crystallization.
In order to obtain recombinant AMP phosphorylase protein, cells were resuspended with the NiC buffer and sonicated. After heat treatment at 90 °C for 60 min and centrifugation (20,000 × g, 30 min) to remove thermolabile proteins derived from the host cells, the supernatant was applied to His GraviTrap, washed with the NiC buffer, and eluted with the NiC buffer containing 500 mm imidazole. The eluate was applied to a Resource Q anion exchange column (GE Healthcare), and proteins were eluted with a linear gradient of NaCl (0–1.0 m) in 20 mm sodium phosphate buffer (pH 7.4). The fractions containing the recombinant AMP phosphorylase protein were concentrated using an Amicon Ultra device (MWCO 30,000), and further purified by gel filtration using a Superdex 200 HR 10/30 column (GE Healthcare) with a mobile phase of 100 mm NaCl in 20 mm sodium phosphate buffer (pH 7.4).
For the preparation of Rubisco, the recombinant protein was expressed as previously reported (18). Cells were resuspended in 100 mm Bicine-NaOH and 10 mm MgCl2, pH 8.3, and sonicated. Soluble protein solution was heat-treated for 30 min at 85 °C and centrifuged (20,000 × g, 30 min) to remove thermolabile proteins derived from the host cells. The supernatant was applied to a Resource Q anion exchange column, and proteins were eluted with a linear gradient of NaCl (0–1.0 m) in 100 mm Bicine-NaOH and 10 mm MgCl2, pH 8.3. Ammonium sulfate was added to the fractions containing the recombinant Rubisco protein at a final concentration of 1.2 m. The protein solution was applied to a hydrophobic interaction column Resource PHE (GE Healthcare), and proteins were eluted with a linear gradient of ammonium sulfate (1.2–0 m) in 100 mm Bicine-NaOH and 10 mm MgCl2, pH 8.3. The fractions containing the recombinant Rubisco were concentrated using an Amicon Ultra device (MWCO 30,000) and further purified by gel filtration using a Superdex 200 HR 10/30 column with a mobile phase of 100 mm Bicine-NaOH, 10 mm MgCl2, and 100 mm NaCl, pH 8.3.
The sizes of the molecules in solution were estimated using a size exclusion column (Superdex 200 HR 10/30) equilibrated with 100 mm Bicine-NaOH, 10 mm MgCl2, and 100 mm NaCl, pH 8.3. The retention volumes of the samples were compared with those of marker proteins. The applied marker proteins were thyroglobulin (669 kDa), ferritin (440 kDa), catalase (232 kDa), aldolase (158 kDa), albumin (67 kDa), ovalbumin (46 kDa), and chymotrypsinogen A (25 kDa). All marker proteins were purchased from GE Healthcare.
Enzymatic Synthesis of R15P, Substrate for R15Pi, with AMP Phosphorylase
The AMP phosphorylase reaction was performed in a 500-μl mixture containing 15 μg of purified AMP phosphorylase, 100 mm Tris-HCl (pH 7.5), 30 mm sodium phosphate (pH 7.5), and 30 mm AMP. The assay mixture without AMP was preincubated at 85 °C for 3 min, and the reaction was initiated with the addition of AMP. After 10 min, the reaction was terminated by rapid cooling on ice for 5 min, and purified AMP phosphorylase was removed by ultrafiltration with an Amicon Ultra device (MWCO 30,000). The R15P mixture produced with AMP phosphorylase was concentrated by vacuum dry centrifugation twice and used as the substrate for the R15Pi reaction (see below).
Enzymatic Activity Measurements of Tk-R15Pi Variants
R15Pi activity was measured by coupling to the carboxylase activity of Rubisco. The reaction mixture (100 μl) was composed of 0.3 μg of purified R15Pi, 5 μg of purified Rubisco, 100 mm NaHCO3, and 30 μl of enzymatically prepared R15P mixture (see above) in the Bicine buffer (100 mm Bicine-NaOH supplemented with 10 mm MgCl2). After preincubation at 85 °C for 3 min in the absence of the NaHCO3 and R15P, the reaction was initiated by adding these substrates. The reaction was carried out at 85 °C for 5 min and terminated by rapid cooling on ice for 5 min, and then the enzymes were removed by ultrafiltration with an Amicon Ultra device (MWCO 30,000). We confirmed that only negligible levels of product were generated during the procedures after the reaction, with sufficient reaction termination by ice-cooling and ultrafiltration (data not shown). When chemically synthesized α-R15P or β-R15P was used as a substrate instead of the enzymatically prepared R15P mixture, the reaction was initiated by adding 3 mm AMP and 10 mm α-R15P or β-R15P and terminated by the same procedure described above. After the appropriate dilution, the amount of 3-phosphoglycerate synthesized by the reaction was determined by a second coupling reaction as described elsewhere (18). The reaction mixture (100 μl) was composed of 5 mm ATP, 0.2 mm NADH, 20 μl of diluted 3-phosphoglycerate mixture, and 20 μl of a coupling enzyme solution in the Bicine buffer. The coupling enzyme solution contained 563 units ml−1 3-phosphoglycerate phosphokinase, 125 units ml−1 glyceraldehyde-3-phosphate dehydrogenase, 260 units ml−1 triose-phosphate isomerase, 22.5 units ml−1 glycerophosphate dehydrogenase, 5 mm reduced glutathione, 0.1 mm EDTA, and 20% glycerol in 50 mm Bicine-NaOH (pH 8.0). The assay mixture without coupling enzyme solution was preincubated at 25 °C for 3 min, and the reaction was initiated with the addition of coupling enzyme solution. The difference in absorbance at 340 nm due to the conversion of NADH into NAD+ by the coupling reaction was measured.
HPLC Analysis
To evaluate the equilibrium constant of ([RuBP]/[α-R15P]), RuBP and chemically synthesized α-R15P were individually used as the substrates for R15Pi. The reaction mixture (100 μl) was composed of 5 μg of purified R15Pi, 3 mm AMP supplemented with 10 mm α-R15P, 10 mm RuBP, or 30 mm RuBP in the Bicine buffer. After preincubation at 85 °C for 3 min in the absence of RuBP or α-R15P, the reaction was initiated by adding these substrates. The reaction was carried out at 85 °C for various periods of time and terminated by rapid cooling on ice for 5 min, and then R15Pi was removed by ultrafiltration with an Amicon Ultra device (MWCO 30,000). After the addition of 30 μl of 600 mm sodium phosphate buffer (pH 4.4) into 30 μl of the mixture, the sample was applied to HPLC analysis using an amino column, Asahipak NH2P-50 4E (Shodex, Tokyo, Japan), with 300 mm sodium phosphate buffer (pH 4.4) as a mobile phase. The column temperature was set at 40 °C, and compounds were detected with a refractive index detector.
Crystallization
Crystals of unliganded Tk-R15Pi were obtained by the sitting drop vapor diffusion method at 20 °C using equal volumes of protein solution (4–9 mg/ml) and precipitant solution containing 100 mm MES-NaOH, pH 6.4, 16–20% (v/v) PEG 1000, and 200 mm MgCl2. Long prismatic crystals were grown within 1 week, with typical dimensions of 300 × 50 × 50 μm3.
Crystals of the complex with ligands were obtained by means of co-crystallization. RuBP was purchased from Sigma-Aldrich. Chemical synthesis of pentosebisphosphates, α-R15P and β-R15P, was performed by Tokyo Chemical Industry Co., Ltd. The ligands were independently mixed with wild-type (WT) Tk-R15Pi solution at final concentrations of 50 mm. Crystallization of the protein-ligand mixed solution was performed under the same conditions as for the unliganded crystal (apo) as described above. For the crystallization of mutants (C133S and D202N), the final concentrations of the mutant protein and the ligands were 6 mg/ml and 10 mm, respectively. They were also crystallized using the precipitant solutions for WT (unliganded) crystals.
Structure Determination
The structure of Tk-R15Pi was solved at 2.5 Å resolution by the single-wavelength anomalous dispersion method using a SeMet-labeled crystal. X-ray diffraction data of native and SeMet Tk-R15Pi crystals were collected using synchrotron radiation at beamlines BL41XU of the SPring-8 (Hyogo, Japan) and BL-17A of the Photon Factory (Tsukuba, Japan), respectively (Table 1). All data were processed and scaled with the program HKL-2000 (19). 62 of a possible 72 selenium sites including first methionine residues in a Tk-R15Pi hexamer were successfully located by SOLVE (20) at 3.4 Å resolution. Subsequent density modification and model building were carried out using RESOLVE (21, 22) at 2.7 Å resolution, resulting in automatic construction of 77% of the Tk-R15Pi residues. Iterated cycles of manual model building and computational refinement toward the structural factor amplitude of the native crystal were performed using Coot (23) and CNS (24, 25), respectively, until the crystallographic R-factor was equal to 20.4% (Rfree = 26.6%) at 2.5 Å resolution.
TABLE 1.
Data collection and refinement statistics
| SeMet | Native (unliganded) | WT·RuBP complex | C133S·α-R15P complex | |
|---|---|---|---|---|
| Data collection | ||||
| Space group | P212121 | P212121 | P212121 | P41212 |
| Cell dimensions (a, b, c) (Å) | 117.4, 130.9, 132.8 | 117.3, 130.8, 132.8 | 117.3, 130.6, 132.7 | 146.5, 146.5, 99.6 |
| X-ray source | PF BL-17A | SPring-8 BL41XU | PF BL-5A | PF AR NW12A |
| Wavelength (Å) | 0.97896 | 1.0000 | 1.0000 | 1.0000 |
| Resolution (Å)a | 50-2.70 (2.80-2.70) | 50-2.50 (2.59-2.50) | 50-2.60 (2.69-2.60) | 50-2.85 (2.95-2.85) |
| Total reflections | 424,789 | 510,338 | 454,465 | 376,565 |
| Unique reflections | 56,920 | 71,476 | 63,161 | 26,012 |
| Completeness (%)a | 100.0 (100.0) | 100.0 (99.8) | 99.9 (100.0) | 100.0 (100.0) |
| 〈I〉/〈σ(I)〉a | 17.3 (6.2) | 26.3 (4.7) | 23.8 (4.6) | 20.1 (5.6) |
| Rsyma,b | 0.093 (0.330) | 0.071 (0.396) | 0.076 (0.377) | 0.129 (0.462) |
| Phasing | SADc | |||
| Resolution (Å) | 50-2.70 | |||
| Figure of merit after SOLVE | 0.46 (∼3.4 Å) | |||
| Figure of merit after RESOLVE | 0.67 | |||
| Refinement | ||||
| Resolution (Å) | 43.66-2.50 (2.66-2.50) | 43.64-2.60 (2.76-2.60) | 45.97-2.85 (3.03-2.85) | |
| Rwork/Rfreed | 0.204/0.266 | 0.201/0.255 | 0.201/0.262 | |
| No. of atoms | ||||
| Protein, Water, Mg, Cl, PEG | 15183, 308, 2, 0, 42 | 15368, 268, 2, 0, 42 | 7635, 153, 2, 3, 83 | |
| RuBP or α-R15P | 108 | 54 | ||
| Average B-factors | ||||
| Protein, Water, Mg, Cl, PEG (Å2) | 45.8, 33.3, 54.1, −, 56.3 | 43.3, 31.2, 48.6, −, 56.7 | 28.4, 20.7, 35.9, 39.5, 47.7 | |
| RuBP or α-R15P (Å2) | 38.0 | 24.1 | ||
| r.m.s. deviation bond length (Å) | 0.006 | 0.007 | 0.009 | |
| r.m.s. deviation bond angles (degrees) | 1.20 | 1.20 | 1.30 | |
| Ramachandran analysisd | ||||
| Favored, allowed, outliers (%) | 95.5, 4.3, 0.2 | 96.1, 3.9, 0.0 | 95.1, 4.8, 0.1 | |
| Protein Data Bank entry | 3A11 | 3A9C | 3VM6 | |
a Values in parentheses are for the highest resolution shell.
b Rsym = ΣhklΣi|Ii(hkl) − 〈I(hkl)〉|/ΣhklΣiIi(hkl), where Ii(hkl) is the ith intensity measurement of reflection hkl, and 〈I(hkl)〉 is the mean intensity obtained from multiple observations of symmetry-related reflections.
c Single-wavelength anomalous dispersion.
d Rwork = Σ‖Fo| − |Fc|/Σ|Fo|. Rfree is the same as Rwork but for the 5% subset of all reflections that were never used in crystallographic refinement.
d Calculated by MolProbity.
X-ray diffraction data for the WT·RuBP complex and the C133S·α-R15P crystal were collected to 2.6 and 2.85 Å resolutions at beamlines BL-5A and AR-NW12A at the Photon Factory, respectively, and processed by the same protocol as for the native crystal described above (Table 1). Model building and crystallographic refinement were performed using Coot and CNS, respectively, until the crystallographic R-factors were dropped to 20.1% (Rfree = 25.5%) at 2.6 Å resolution for the WT·RuBP complex and 20.1% (Rfree = 26.2%) at 2.85 Å resolution for the C133S·α-R15P complex. The figures of structures were prepared with PyMOL (Schrödinger, LLC).
RESULTS AND DISCUSSION
Structure Determination of Tk-R15Pi Hexamer without Ligands
Tk-R15Pi was expressed in E. coli, purified to homogeneity, and crystallized as described under “Experimental Procedures.” The crystal structure of unliganded Tk-R15Pi was solved by means of the single-wavelength anomalous dispersion method using SeMet-substituted Tk-R15Pi and refined at 2.5 Å resolution (Fig. 2 and Table 1). We found six Tk-R15Pi molecules in an asymmetric unit of the crystal, forming a homohexamer (Fig. 2). This oligomerization was comparable with the result of gel filtration analysis (Fig. 3A). The biological unit of Tk-R15Pi is therefore suggested to be a hexamer in aqueous solution.
FIGURE 2.

Crystal structure of Tk-R15Pi. Shown are the monomer (A), dimer (subunits A and B) (B), and hexamer (subunits A–F) (C) structures of the nonliganded enzyme. The non-crystallographic 3-fold axis is shown by a broken line in C. D, electrostatic potential map of the monomer in the Tk-R15Pi·RuBP complex. The map was drawn on the solvent-accessible molecular surface (red, −10 kT (the Boltzmann constant multipied by absolute temperature) e−1; blue, 10 kT e−1) calculated with a probe radius of 1.4 Å using the program APBS (39). The molecular orientation is the same as in A.
FIGURE 3.

Intersubunit interactions. A, size exclusion column analyses for the WT and R227E mutant of Tk-R15Pi. The molecular masses were calculated to be 260 and 140 kDa, respectively. The mass for WT was slightly larger than that of the calculated mass (217 kDa) of the hexamer from the amino acid sequence, probably because the shape of the Tk-R15Pi hexamer was oval. B, additional β-strand (β8(B)) for the Rossmann fold-like conformation and salt bridges in the dimerization surface. C, contacts of Tyr300 around the non-crystallographic 3-fold axis. D, intersubunit salt bridges by Arg227, which are responsible for the hexamerization.
Monomer Structure
The Tk-R15Pi monomer (protomer) consists of two domains (i.e. an N-terminal α-helical domain and a C-terminal αβα-sandwich domain) (Fig. 2A). Five α-helices (α1–α5) form a helix bundle and constitute the N-terminal moiety. The last α-helix of the N-terminal domain, α5, is kinked in the middle by about 45° and bridges the two domains. The C-terminal domain contains a Rossmann fold-like conformation as its core structure. The conformations of six crystallographically independent molecules are closely similar, with the root mean square (r.m.s.) deviations from 0.43 to 1.05 Å for 315 Cα atoms of 322 residues.
As predicted by amino acid sequence alignment, the results of a three-dimensional structure database search with the DALI server (26, 27) showed that the structure of Tk-R15Pi closely resembles those of the homologous proteins in the PF01008 family, with r.m.s. deviation values of 1.7–2.3 Å for 91–104 Cα atoms of the N-terminal domain (Z-score > 11) and r.m.s. deviation values of 1.6–2.1 Å for 152–196 Cα atoms of the C-terminal domain (Z-score > 15). The bend angles of α5 helices of these proteins are ∼20°, which is quite a bit smaller than that in R15Pi (supplemental Fig. 5). In addition to members of the PF01008 family, Pheleum pretense Pollen allergen PHL P 6 (Z-score 7.6) and E. coli GroEL (Z-score 6.6) have helix bundles similar to that in the N-terminal half of Tk-R15Pi. The C-terminal domain structure of Tk-R15Pi closely resembles the domain of several enzymes that accommodate phosphoribose moieties (e.g. ribose-5-phosphate isomerase (Z-score 13.6) and 4-hydroxybutyrate CoA transferase (Z-score 12.1)). These enzymes also possess the Rossmann fold-like conformation.
Quaternary Structure
The two monomers aligned along a non-crystallographic 3-fold axis form a dimer with strong interactions through their C-terminal domains (Fig. 2, B and C). The dimers correspond to the physiologically relevant dimers in other PF01008 family proteins (17, 28, 29). The β8 strand from one subunit is utilized as an additional β-strand to the parallel β-sheet of the Rossmann fold-like conformation in another subunit (Fig. 3B). In addition, several intersubunit ion pairs, especially those formed by Glu158–Arg160 residues in the loop region between β2 and α7, bundle the two subunits (Fig. 3B). Approximately 4,200 Å2 of the molecular surface area per dimer (13% in total surface) is involved in the dimer formation.
The hexameric assembly of Tk-R15Pi forms a Rugby football-like shape (Fig. 2C), and is unique among structurally related PF01008 family proteins. Three Tyr300 residues make van der Waals contacts around the non-crystallographic 3-fold axis, tethering neighboring dimers to each other (Fig. 3C). Arg227 on the α9-β7 loop connects with Glu225 and Glu285 from distinct polypeptide chains to gather three adjacent protomers (Fig. 3D). Substitution of this Arg into Glu (R227E) exhibits a decrease in molecular size of the Tk-R15Pi protein (Fig. 3A) and significantly decreases the enzymatic activity (Table 2). This Arg plays a key role in hexamerization, and the hexameric form may be essential for R15Pi activity at 85 °C. In contrast to the tight interaction between the C-terminal domains, no intersubunit interactions are found between the axial N-terminal domains. The contact surface area for trimerization is 3,100 Å2/dimer (10% in total surface), which is relatively smaller than the area for dimerization (4,200 Å2/dimer).
TABLE 2.
Enzymatic activity and assembly of Tk-R15Pi and its mutants
The molecular assembly of these proteins was examined by size exclusion chromatography (Fig. 3 and supplemental Fig. 10).
| Tk-R15Pi | Specific activitya |
|---|---|
| μmol min−1mg−1 | |
| WT | 29.3 ± 2.1 |
| C133S | <0.1 |
| C133A | <0.1 |
| D202N | <0.1 |
| R227E | 0.5 ± 0.2 |
a R15P enzymatically synthesized by AMP phosphorylase was used as the substrate.
The PF01008 family proteins have six conserved sequence motifs (6, 17, 28). Fig. 4 shows an amino acid sequence alignment of the PF01008 family proteins whose crystal structures are available in the Protein Data Bank. All motifs are conserved in Tk-R15Pi, and the key residues responsible for the dimerization (Glu158 and Arg160) are located in motif 4. In contrast, the essential residues involved in hexamerization (blue ellipses and circles in Fig. 4) and the kink region in the α5 helix (green bar in Fig. 4) identified in Tk-R15Pi are not well conserved in the other PF01008 family proteins.
FIGURE 4.

Amino acid sequence alignment of the structure-determined PF01008 family proteins. Sequences were aligned by ClustalW (40), and this figure was produced by ESPript (41) with manual modifications. The names of species and UniProt accession numbers are as follows: Tk-R15Pi, R15Pi from T. kodakarensis KOD1, Q5JFM9; Bs-M1Pi, M1Pi from B. subtilis, O31662; Tm-M1Pi, M1Pi from Thermotoga maritima, Q9X013; Sc-M1Pi, M1Pi from Saccharomyces cerevisiae, Q06489; Af-M1PiRP, M1Pi-related protein from A. fulgidus, O29877; Ph-aIF2Balpha, archaeal IF-2B α-subunit from Pyrococcus horikoshii OT3, O58185; Hs-eIF2Balpha, eukaryotic IF-2B α-subunit from Homo sapiens, Q14232. Identical and similar amino acid residues are highlighted by white characters in red closed boxes and red characters in blue open boxes, respectively. The residues highlighted in blue are the insertion regions related to the substrate binding of M1Pi. The catalytic residues of R15Pi and M1Pi (e.g. Cys133 and Asp202 of Tk-R15Pi) are indicated by red stars and are also highlighted in yellow. Red and green arrowheads represent important residues constructing the active site surrounding Cys133 and Asp202, respectively. These residues are considered to contribute in the deprotonation or protonation of Cys133 or Asp202, respectively. Blue closed circles, residues related to the hexamerization by hydrophilic interactions, including salt bridges. Blue closed ellipses, residues related to the hexamerization by hydrophobic interaction. The bend region of the α5 helix is indicated by the word kink with a green bar. The six sequence motifs described in this work are boxed, and their major roles are indicated. The secondary structures of Tk-R15Pi are indicated above the sequence alignment. Residue names and numbers of Tk-R15Pi described in this work are also shown above the sequence alignment.
Active Site and Ligand Specificity
In order to elucidate the molecular mechanism of Tk-R15Pi, we co-crystallized wild-type Tk-R15Pi with one of the anomers of its substrate, α-R15P. The crystallized Tk-R15Pi did not contain an α-R15P molecule but was found to hold the product RuBP (WT·RuBP complex at 2.6 Å resolution; Table 1, Fig. 5A, and supplemental Fig. 6). The α-R15P molecule was considered to be converted into RuBP during the crystallization process. The equilibrium constant of [RuBP]/[α-R15P] was estimated to be 16.3 ± 0.2 from their concentrations after 14–18 min of the R15Pi reaction (Table 3 and supplemental Fig. 7), indicating that RuBP generation is preferred from a thermodynamic point of view.
FIGURE 5.

Close-up views of the active sites of WT·RuBP (A), C133S·α-R15P (B), and WT (unliganded) (C). Green meshes indicate the Fo − Fc omit maps for RuBP or α-R15P contoured at 4.0σ. Schematic representations of the active site in WT·RuBP and C133S·α-R15P are shown in the right panels. Green broken lines represent the hydrogen bonds between Asp202 and α-R15P (B, right).
TABLE 3.
Enzymatic activity of Tk-R15Pi
| Substrate | Specific activitya | Keq = [RuBP]/[R15P]b |
|---|---|---|
| μmol min−1mg−1 | ||
| α-R15P | 28.6 ± 1.1 | 16.3 ± 0.2 |
| β-R15P | <0.1 |
a Chemically synthesized R15Ps were used as initial substrates.
b Keq was calculated by integration of the peak area from the HPLC experiments (supplemental Fig. 7C).
The RuBP-binding site is positively charged and located between the N- and C-terminal lobes (Fig. 2D and supplemental Fig. 6C, middle). The positive charge is suitable to hold the negative charges on the two phosphates of R15P and RuBP. The location of the active site is consistent with those of other enzymes, such as M1Pi from Bacillus subtilis (Bs-M1Pi) (17) and ribose-5-phosphate isomerase from Thermus thermophilus (15) (supplemental Fig. 8). Cys133 and Asp202 are superposed well on the corresponding conserved residues in M1Pi (supplemental Fig. 9). These two residues are thought to be important for the M1Pi reaction, although no biochemical assays have been performed (17). The residue conservation implies their importance also for the R15Pi reaction.
The roles of the two residues were examined by enzymatic assays. The amino acid substitutions C133S, C133A, and D202N resulted in proteins with undetectable levels of activity for RuBP production (<0.1 μmol min−1 mg−1) compared with that of the WT enzyme (29 μmol min−1 mg−1) (Table 2). Size exclusion column analyses showed that the elution volumes of these mutant Tk-R15Pi proteins were almost the same as that of the wild type (supplemental Fig. 10). These analyses indicate that these mutants form a hexamer like the wild-type enzyme and that both Cys133 and Asp202 are essential residues for catalysis.
Although co-crystallization with the β-type anomer of R15P (β-R15P) was also attempted, there was no electron density representing any compounds, such as β-R15P or RuBP, at the ligand-binding site. This is consistent with the results of an enzymatic activity measurement using β-R15P as a substrate, in which production of RuBP could not be detected (Table 3). These results indicate that Tk-R15Pi distinguishes the chiral isomers and that only α-R15P is utilized as a substrate to generate RuBP.
In order to further understand the active site environment, we prepared crystals of the inactive C133S mutant with α-R15P. In this case, α-R15P was not isomerized, and the C133S·α-R15P complex structure was refined at 2.85 Å resolution (Table 1, Fig. 5B, and supplemental Fig. 6). The overall structure of C133S·α-R15P was significantly different from those of WT (unliganded) and WT·RuBP (see below). We also tried to elucidate the crystal structure of the inert D202N mutant in complex with α-R15P. However, no electron density corresponding to a binding ligand was observed at the active site of the crystal structure (data not shown). Asp202 might play an important role in the substrate binding of Tk-R15Pi.
Open-Closed Conformational Change upon Ligand Binding
Comparison of the three determined structures revealed that Tk-R15Pi can accommodate two types of conformations (Fig. 6 and supplemental Fig. 6). The structures of WT (unliganded) (hereafter referred to as WT) and WT·RuBP are classified here as “open” conformations, whereas that of C133S·α-R15P is considered a “closed” conformation. No significant difference was found between the WT and WT·RuBP structures (r.m.s. deviation 0.56 Å for 314 Cα atoms) except for the side-chain orientations of the residues (Lys136 and Arg254) involved in RuBP-binding (Fig. 5, A and C). The ligand-binding site is exposed to the solvent in this open conformation. In contrast, the closed C133S·α-R15P conformation is quite different from the other two open conformations (r.m.s. deviation 4.12 Å for 314 Cα atoms in comparison with the WT). This conformational change is induced by the alteration of the bend angle of the α5-helix. The bend angle is ∼45° in the open conformation versus ∼25° in the closed conformation. The N-terminal domain moves around 8 Å toward the C-terminal domain, covering the active site cleft (Fig. 6). The domain motion sequesters the substrate-binding site from the aqueous environment (Figs. 5B and 6). Similar domain motion has been predicted based on the small structural difference between two distinct enzymes, Bs-M1Pi and the M1Pi-related protein from A. fulgidus (17). However, the structures obtained here provide the first direct evidence of conformational change in these proteins.
FIGURE 6.

Dynamic motion upon α-R15P binding. The protomers of WT (open), WT·RuBP (open), and C133S·α-R15P (closed) are colored green, cyan, and magenta, respectively. These structures were superimposed at the C-terminal domain residues.
Among the homologous proteins, the open structure is determined only with Tk-R15Pi. The structures of all of the other homologous proteins are similar to the closed conformation of Tk-R15Pi whether they are bound to ligands or not (i.e. their active sites are not exposed to the solvent) (supplemental Fig. 5). We observed a structural correlation between the ligand-binding site of the “Open” conformation of Tk-R15Pi and the properties of its ligands. α-R15P and RuBP, the ligands of R15Pi, have no hydrophobic part, whereas the methylthio groups in MTR-1P and MTRu-1P, the ligands for M1Pi, are hydrophobic (the chemical structures of the ligands are drawn in supplemental Fig. 2). This difference is reflected in the ligand-binding site moieties of the two enzymes. M1Pi has a hydrophobic patch on the active site to accommodate the methylthio group. The patch is generated by amino acid insertions in motif 3 of the M1Pi proteins (highlighted blue in the box for motif 3 in Fig. 4). The closed conformation in M1Pi may be favorable because it covers the hydrophobic patch from the solvent. In contrast, the ligand-binding site of Tk-R15Pi is hydrophilic and positively charged to interact with the two phosphate groups of the ligands. It can thus be presumed that Tk-R15Pi has a greater preference for the open conformation compared with M1Pi.
The two active site residues, Cys133 and Asp202, were too distant to interact with RuBP in the open conformation; there are 3.9 and 3.6 Å between the γ-sulfur atom of Cys133 and the C1 and C2 atoms of RuBP, respectively, and the δ-oxygen atoms of Asp202 are at least 4.1 Å apart from the O2 and O4 atoms of RuBP (Fig. 5A). In contrast, these values in C133S·α-R15P were 4.6 and 3.3 Å (Ser133 γ-oxygen to α-R15P C1 and C2, respectively) and 3.1 and 2.7 Å (δ1-oxygen of Asp202 to α-R15P O2 and δ2-oxygen to O4, respectively). The substrate-binding structure (C133S·α-R15P) indicates that both δ-oxygen atoms of Asp202 can participate in ribose binding and that Cys133 does not interact with the C1 atom of the ribose but with the C2 atom (Fig. 5B). The conserved Asn212 forms hydrogen bonds with the O2 and O3 oxygen atoms of both α-R15P and RuBP. A water molecule (HOH601) coordinates the O3 of both ligands (α-R15P and RuBP) and three groups in the enzyme (the side chain of Asn212, main-chain carbonyl group of His132, and NH group of Gly200; Fig. 5B). The product-binding structure (WT·RuBP) is similar to the M1Pi·MTRu-1P structure (supplemental Fig. 9), but the substrate-binding structure C133S·α-R15P, which can be considered to better represent the structure of the enzyme-substrate complex, is different from the M1Pi·MTRu-1P structure, especially regarding the Cys and Asp residues.
The large domain motion from open to closed conformations induces two arginine interactions involving Arg20 and Arg63 in the N-terminal domain (Fig. 5, A and B). Arg254 recognizes 1-phosphate of RuBP in the WT·RuBP structure, but it stabilizes the closed conformation by forming a hydrogen bond with the carbonyl oxygen of Lys15 in the N-terminal domain. These closed form-specific interactions are also seen in M1Pi.
The open conformation provides the route for the ligands to access and leave the active site. We assume that the open conformations of WT and WT·RuBP are the prereaction and postreaction (product rebinding for the reverse reaction) forms, respectively. On the other hand, the closed conformation of C133S·α-R15P is a reaction-ready form in which the isomerization reaction is about to be initiated; the bound substrate is almost completely shielded from the solvent, and the catalytic residues directly interact with the substrate.
Roles of Six Conserved Sequence Motifs
The functional roles of the six sequence motifs (Fig. 4) of the PF01008 proteins can be explained based on our structure analyses together with previous reports. We here describe in detail the roles of these motifs in phosphoribose isomerases.
Motif 1 (residues 19–28 of Tk-R15Pi; Fig. 4) contains the α1-α2 loop, which covers the active site in the closed conformation (Figs. 5B and 6). Conserved Arg20 and the main-chain amido NH group of Gly21 participate in the 1-phosphate binding. The α-helix dipole of α2 is appropriate to hold the negatively charged 5-phosphate (30). The four-residue fragment at the N terminus of the α2 helix (23GKIA26) satisfies the features of a structural P-loop (GXXX) that is frequently found in the phosphate-binding site of various protein superfamilies (31). Therefore, motif 1 can serve as the phosphate-binding scaffold.
Motif 2 (residues 61–73) corresponds to the α3-α4 loop. Completely conserved Pro64 and Leu69 residues contribute to the formation of the α3-α4 helix bundle in the N-terminal domain. Further, Arg63 interacts with the 1-phosphate of α-R15P (Fig. 5B). These findings indicate that motif 2 acts to stabilize the N-terminal domain and additionally functions as a binding region for 1-phosphate.
Motif 3 (residues 129–135) includes the catalytic Cys residue (Cys133). In addition, the γ-OH group of Ser135 makes a hydrogen bond with 5-phosphate of α-R15P. Correspondingly, M1Pi proteins have a hydrophobic region next to the catalytic Cys residue, which provides a favorable environment for binding of the hydrophobic methylthio group of the M1Pi ligands. This motif is an essential region for binding the C5 substituent of the ligand and for the catalytic reaction.
The other three motifs, motif 4 (residues 158–172), motif 5 (residues 197–216), and motif 6 (residues 269–296), are located in the C-terminal domain. These motifs show relatively high sequence identity compared with those in the N-terminal domain (motifs 1 and 2). The three motifs (motifs 4–6) are distributed in the Rossmann fold-like motif (α7, β4, β5, β6, β9, and β10) and may play a significant role in maintaining the C-terminal domain structure. In addition, Glu158 and Arg160 in motif 4 is responsible for the dimerization shown in Figs. 2B and 3B. Motif 5 includes the catalytic Asp residue (Asp202) and the conserved Asn212 related to ligand binding. The residues of motif 5 surrounding Asp202 are also highly conserved. These residues most likely contribute to the construction of an environment around Asp202 that promotes the reaction.
Implications for Reaction Mechanism
The enzymatic analysis combined with the structure analysis in this study directly demonstrated that the Cys133 and Asp202 located at the active site are essential residues for the isomerase reaction. Their side chain conformations and electrostatic states are apparently tuned for the reaction by the surrounding residues. The main-chain amide NH group of Cys133 makes a hydrogen bond with the hydroxy group of the Thr159 side chain (Fig. 7A). Thr159 is likely to play a significant role in determining the orientation of the γ-sulfur of Cys133. The thiol group of Cys133 (hydroxy group of C133S) interacts with the main chain amido NH group of the adjacent His134 (Fig. 7A). This environment may be sufficient for lowering the thiol pKa of Cys133 because electrostatic complementarity and hydrogen bonding to the thiol group have been shown to stabilize the thiolate anion (32–34). This would allow the thiol group of Cys133 to be easily deprotonated in the active site. The other catalytic residue, Asp202, is surrounded by Ala22, Val67, Ile211, and Phe279 residues (Fig. 7B). The pKa of a buried Asp residue is considered to be higher than that of one exposed to aqueous solvent (35, 36), suggesting that the carboxylate of Asp202 can be present in its protonated form.
FIGURE 7.

Proposed isomerase reaction model of Tk-R15P. The protein environments around Cys133 (A) and Asp202 (B) are shown. Significant conformational changes from the open (WT, green, left panels) to the closed conformation (C133S·α-R15P, magenta, right panels) allow Tk-R15Pi to enter a reaction-ready state. C, proposed reaction mechanism via a cis-phosphoenolate intermediate. Step 1, proton transfer from the protonated carboxylate of Asp202 to the O4 atom of α-R15P; Step 2, proton abstraction from the C2 atom of α-R15P by the deprotonated thiolate of Cys133 and the cleavage of the C1–O4 bond; Step 3, proton abstraction from the O2 atom of the cis-phosphoenolate intermediate by deprotonated carboxylate of Asp202 and proton transfer from the thiol of Cys133 to the C1 atom of the intermediate; Step 4, production of RuBP by keto-enol tautomerization.
The structure analyses together with the biochemical assays allow us to propose the following catalytic mechanism via a cis-phosphoenolate intermediate triggered by substrate binding (Fig. 7C). Until the substrate α-R15P is supplied to Tk-R15Pi, the active site must be opened as in the WT form. Once α-R15P enters the active site, the induced fit conformational changes shield the substrate α-R15P from the solvent (Fig. 6). Upon α-R15P binding, the side-chain amide NH group of Gln164 moves close to the 133rd residue (Ser133 in the C133S mutant) (Fig. 7A, right). The approach of the side-chain NH group of Gln164 and the guanidinyl group of Arg20 to Cys133 could facilitate the decrease in pKa and deprotonation of Cys133-SH. Also, the shielding of Asp202 by the hydrophobic residues (Fig. 7B, right) could promote Asp202 protonation.
The first stage of our proposed isomerization mechanism is the proton transfer from the protonated carboxylate of Asp202 to the O4 atom of α-R15P (Step 1 in Fig. 7C). Then the C2 hydrogen atom of α-R15P is abstracted by the thiolate of Cys133 (Step 2). The proton abstraction proceeds simultaneously with the cleavage of the O4-C1 bond of α-R15P as a concerted elimination of the pseudo-E2 mechanism, resulting in formation of a C1=C2 double bond (Step 2). This intermediate is the cis-phosphoenolate (Fig. 7B, bottom right). These two steps (Steps 1 and 2) may occur simultaneously like the ring-opening reaction of ribose-5-phosphate isomerase (supplemental Fig. 3A). Finally, the RuBP molecule can be produced by keto-enol tautomerization as follows. Based on the closed α-R15P complex structure, the Asp202 side chain is located close to both the O2 and O4 of the intermediate. Therefore, Asp202 is able to interact with both protons on the O2 and O4 of the ligand without conformational change and can abstract the O2 proton from the intermediate. Thus the appropriate location of Asp202 can be considered vital for catalysis (Step 3). Together with the proton abstraction from O2, the π-electron of the C1=C2 double bond attacks Cys133-SH, resulting in proton addition to the C1 atom of the intermediate (Step 4). If the proton of Cys133-SH is transferred to C2 instead of C1, α-R15P is regenerated (reversed reaction, back to the top right intermediate of Fig. 7C).
We consider that the reaction mechanism of Bs-M1Pi is fundamentally similar to our proposed mechanism of Tk-R15Pi described above. This corresponds to the mechanism proposed for Bs-M1Pi involving a cis-phosphoenolate intermediate. The active site environment sequestered from the solvent in the “Closed” conformation, demonstrated here in Tk-R15Pi, is consistent with the lack of incorporation of the solvent deuterium onto the C1 carbon atom of the Bs-M1Pi reaction product (9). The active sites of the isomerases utilizing the cis-enediolate intermediate, triose-phosphate isomerase, phosphoglucose isomerase, and ribose-5-phosphate isomerase, are expected to be exposed to the solvent even in substrate-binding forms, which provides an opportunity for an exchange between hydrogen and deuterium (37, 38). In contrast, because the catalytic Cys residues of R15Pi and M1Pi are isolated from the solvent, the hydrogen (deuterium) atom between the thiol group and solvent would be difficult to exchange. The other candidate mechanism for the reaction, the hydride shift pathway, is unlikely, because Cys133 (Ser133) is too distant (4.6 Å) from the C1 atom of α-R15P in our crystal structure to stabilize the tentative positive charge at the C1 position (carbocation) (supplemental Fig. 4). The complete loss of activity in the C133S mutant is also inconsistent with the hydride shift pathway. Substitution of this residue is thought to modestly decrease, but not to abolish, the enzyme activity with this mechanism.
In the catalytic reaction, we presume that the closed conformation allows Asp202 and Cys133 to act as the general acid and base catalyst, respectively. Our structure analyses demonstrate that the active site cleft is closed only when the substrate (α-R15P) is present in the active site and that the active site closure is accompanied by formation of multiple interactions with the N- and C-terminal domains of Tk-R15Pi. Therefore, we suggest that the closed conformation is a transient state for R15Pi when the substrate is bound to the active site. In other words, the binding of α-R15P induces domain motion of the enzyme as a reaction trigger, which results in optimal active site formation and initiation of the reaction.
Acknowledgments
We thank the beamline staff at the Photon Factory and SPring-8.
This work was supported by the Targeted Proteins Research Program from the Ministry of Education, Culture, Sports, Science, and Technology, Japan.

This article contains supplemental Figs. 1–10.
The atomic coordinates and structure factors (codes 3A11, 3A9C, and 3VM6) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- Rubisco
- ribulose-1,5-bisphosphate carboxylase/oxygenase
- RuBP
- ribulose 1,5-bisphosphate
- R15Pi
- ribose-1,5-bisphosphate isomerase
- R15P
- d-ribose 1,5-bisphosphate
- Tk-R15Pi
- R15Pi from T. kodakarensis KOD1
- IF-2B
- translation initiation factor 2B
- MTR-1P
- 5-methylthioribose 1-phosphate
- M1Pi
- MTR-1P isomerase
- MTRu-1P
- 5-methylthioribulose 1-phosphate
- r.m.s.
- root mean square
- Bs-M1Pi
- M1Pi from B. subtilis
- Bicine
- N,N-bis(2-hydroxyethyl)glycine
- MWCO
- molecular weight cut-off
- SeMet
- selenomethionine
- WT
- wild-type.
REFERENCES
- 1. Watson G. M., Tabita F. R. (1997) Microbial ribulose 1,5-bisphosphate carboxylase/oxygenase: a molecule for phylogenetic and enzymological investigation. FEMS Microbiol. Lett. 146, 13–22 [DOI] [PubMed] [Google Scholar]
- 2. Shively J. M., van Keulen G., Meijer W. G. (1998) Something from almost nothing: Carbon dioxide fixation in chemoautotrophs. Annu. Rev. Microbiol. 52, 191–230 [DOI] [PubMed] [Google Scholar]
- 3. Mueller-Cajar O., Badger M. (2007) New roads lead to Rubisco in archaebacteria. BioEssays 29, 722–724 [DOI] [PubMed] [Google Scholar]
- 4. Ezaki S., Maeda N., Kishimoto T., Atomi H., Imanaka T. (1999) Presence of a structurally novel type ribulose-bisphosphate carboxylase/oxygenase in the hyperthermophilic archaeon, Pyrococcus kodakaraensis KOD1. J. Biol. Chem. 274, 5078–5082 [DOI] [PubMed] [Google Scholar]
- 5. Sato T., Atomi H., Imanaka T. (2007) Archaeal type III RuBisCOs function in a pathway for AMP metabolism. Science 315, 1003–1006 [DOI] [PubMed] [Google Scholar]
- 6. Kyrpides N. C., Woese C. R. (1998) Archaeal translation initiation revisited: The initiation factor 2 and eukaryotic initiation factor 2B α-β-δ subunit families. Proc. Natl. Acad. Sci. U.S.A. 95, 3726–3730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Price N., Proud C. (1994) The guanine nucleotide-exchange factor, eIF-2B. Biochimie 76, 748–760 [DOI] [PubMed] [Google Scholar]
- 8. Ashida H., Saito Y., Kojima C., Kobayashi K., Ogasawara N., Yokota A. (2003) A functional link between RuBisCO-like protein of Bacillus and photosynthetic RuBisCO. Science 302, 286–290 [DOI] [PubMed] [Google Scholar]
- 9. Saito Y., Ashida H., Kojima C., Tamura H., Matsumura H., Kai Y., Yokota A. (2007) Enzymatic characterization of 5-methylthioribose 1-phosphate isomerase from Bacillus subtilis. Biosci. Biotechnol. Biochem. 71, 2021–2028 [DOI] [PubMed] [Google Scholar]
- 10. Rieder S. V., Rose I. A. (1959) The mechanism of the triosephosphate isomerase reaction. J. Biol. Chem. 234, 1007–1010 [PubMed] [Google Scholar]
- 11. Topper Y. J. (1957) On the mechanism of action of phosphoglucose isomerase and phosphomannose isomerase. J. Biol. Chem. 225, 419–425 [PubMed] [Google Scholar]
- 12. Zhang R., Andersson C. E., Savchenko A., Skarina T., Evdokimova E., Beasley S., Arrowsmith C. H., Edwards A. M., Joachimiak A., Mowbray S. L. (2003) Structure of Escherichia coli ribose-5-phosphate isomerase. A ubiquitous enzyme of the pentose phosphate pathway and the Calvin cycle. Structure 11, 31–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Whitlow M., Howard A. J., Finzel B. C., Poulos T. L., Winborne E., Gilliland G. L. (1991) A metal-mediated hydride shift mechanism for xylose isomerase based on the 1.6 Å Streptomyces rubiginosus structures with xylitol and d-xylose. Proteins 9, 153–173 [DOI] [PubMed] [Google Scholar]
- 14. Schray K. J., Benkovic S. J., Benkovic P. A., Rose I. A. (1973) Catalytic reactions of phosphoglucose isomerase with cyclic forms of glucose 6-phosphate and fructose 6-phosphate. J. Biol. Chem. 248, 2219–2224 [PubMed] [Google Scholar]
- 15. Hamada K., Ago H., Sugahara M., Nodake Y., Kuramitsu S., Miyano M. (2003) Oxyanion hole-stabilized stereospecific isomerization in ribose-5-phosphate isomerase (Rpi). J. Biol. Chem. 278, 49183–49190 [DOI] [PubMed] [Google Scholar]
- 16. Collyer C. A., Henrick K., Blow D. M. (1990) Mechanism for aldose-ketose interconversion by d-xylose isomerase involving ring opening followed by a 1,2-hydride shift. J. Mol. Biol. 212, 211–235 [DOI] [PubMed] [Google Scholar]
- 17. Tamura H., Saito Y., Ashida H., Inoue T., Kai Y., Yokota A., Matsumura H. (2008) Crystal structure of 5-methylthioribose 1-phosphate isomerase product complex from Bacillus subtilis: Implications for catalytic mechanism. Protein Sci. 17, 126–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maeda N., Kanai T., Atomi H., Imanaka T. (2002) The unique pentagonal structure of an archaeal Rubisco is essential for its high thermostability. J. Biol. Chem. 277, 31656–31662 [DOI] [PubMed] [Google Scholar]
- 19. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 20. Terwilliger T. C., Berendzen J. (1999) Automated MAD and MIR structure solution. Acta Crystallogr. D 55, 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Terwilliger T. C. (2000) Maximum-likelihood density modification. Acta Crystallogr. D 56, 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Terwilliger T. C. (2003) Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallogr. D 59, 38–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 24. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 25. Brunger A. T. (2007) Version 1.2 of the Crystallography and NMR system. Nat. Protoc. 2, 2728–2733 [DOI] [PubMed] [Google Scholar]
- 26. Holm L., Sander C. (1993) Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233, 123–138 [DOI] [PubMed] [Google Scholar]
- 27. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bumann M., Djafarzadeh S., Oberholzer A. E., Bigler P., Altmann M., Trachsel H., Baumann U. (2004) Crystal structure of yeast Ypr118w, a methylthioribose-1-phosphate isomerase related to regulatory eIF2B subunits. J. Biol. Chem. 279, 37087–37094 [DOI] [PubMed] [Google Scholar]
- 29. Kakuta Y., Tahara M., Maetani S., Yao M., Tanaka I., Kimura M. (2004) Crystal structure of the regulatory subunit of archaeal initiation factor 2B (aIF2B) from hyperthermophilic archaeon Pyrococcus horikoshii OT3: a proposed structure of the regulatory subcomplex of eukaryotic IF2B. Biochem. Biophys. Res. Commun. 319, 725–732 [DOI] [PubMed] [Google Scholar]
- 30. Hol W. G., van Duijnen P. T., Berendsen H. J. (1978) The α-helix dipole and the properties of proteins. Nature 273, 443–446 [DOI] [PubMed] [Google Scholar]
- 31. Kinoshita K., Sadanami K., Kidera A., Go N. (1999) Structural motif of phosphate-binding site common to various protein superfamilies: all-against-all structural comparison of protein-mononucleotide complexes. Protein Eng. 12, 11–14 [DOI] [PubMed] [Google Scholar]
- 32. Kortemme T., Darby N. J., Creighton T. E. (1996) Electrostatic interactions in the active site of the N-terminal thioredoxin-like domain of protein-disulfide isomerase. Biochemistry 35, 14503–14511 [DOI] [PubMed] [Google Scholar]
- 33. Jao S. C., English Ospina S. M., Berdis A. J., Starke D. W., Post C. B., Mieyal J. J. (2006) Computational and mutational analysis of human glutaredoxin (thioltransferase). Probing the molecular basis of the low pKa of cysteine 22 and its role in catalysis. Biochemistry 45, 4785–4796 [DOI] [PubMed] [Google Scholar]
- 34. Naor M. M., Jensen J. H. (2004) Determinants of cysteine pKa values in creatine kinase and α1-antitrypsin. Proteins 57, 799–803 [DOI] [PubMed] [Google Scholar]
- 35. Wilson N. A., Barbar E., Fuchs J. A., Woodward C. (1995) Aspartic acid 26 in reduced Escherichia coli thioredoxin has a pKa > 9. Biochemistry 34, 8931–8939 [DOI] [PubMed] [Google Scholar]
- 36. Mehler E. L., Fuxreiter M., Simon I., Garcia-Moreno E. B. (2002) The role of hydrophobic microenvironments in modulating pKa shifts in proteins. Proteins 48, 283–292 [DOI] [PubMed] [Google Scholar]
- 37. O'Donoghue A. C., Amyes T. L., Richard J. P. (2005) Hydron transfer catalyzed by triosephosphate isomerase. Products of isomerization of (R)-glyceraldehyde 3-phosphate in D2O. Biochemistry 44, 2610–2621 [DOI] [PubMed] [Google Scholar]
- 38. O'Donoghue A. C., Amyes T. L., Richard J. P. (2005) Hydron transfer catalyzed by triosephosphate isomerase. Products of isomerization of dihydroxyacetone phosphate in D2O. Biochemistry 44, 2622–2631 [DOI] [PubMed] [Google Scholar]
- 39. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) ClustalW and ClustalX version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 41. Gouet P., Courcelle E., Stuart D. I., Métoz F. (1999) ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15, 305–308 [DOI] [PubMed] [Google Scholar]