

Background: Competitors of LEDGF binding to HIV-1 integrase could prevent targeted integration to chromatin.
Results: LEDGF competitors like tBPQAs were also found to inhibit integrase enzyme activity by preventing proper integrase-viral DNA assembly.
Conclusion: tBPQAs are allosteric inhibitors of integrase with a dual mode of action.
Significance: Interference with two distinct steps of integration through the same binding site represents a new antiviral paradigm.
Keywords: Antiviral Agents, Crystal Structure, Fluorescence Resonance Energy Transfer (FRET), HIV-1, Integrase, Allosteric Inhibitor, Antiviral Mechanism
Abstract
tert-Butoxy-(4-phenyl-quinolin-3-yl)-acetic acids (tBPQA) are a new class of HIV-1 integrase (IN) inhibitors that are structurally distinct from IN strand transfer inhibitors but analogous to LEDGINs. LEDGINs are a class of potent antiviral compounds that interacts with the lens epithelium-derived growth factor (LEDGF) binding pocket on IN and were identified through competition binding against LEDGF. LEDGF tethers IN to the host chromatin and enables targeted integration of viral DNA. The prevailing understanding of the antiviral mechanism of LEDGINs is that they inhibit LEDGF binding to IN, which prevents targeted integration of HIV-1. We showed that in addition to the properties already known for LEDGINs, the binding of tBPQAs to the IN dimer interface inhibits IN enzymatic activity in a LEDGF-independent manner. Using the analysis of two long terminal repeat junctions in HIV-infected cells, we showed that the inhibition by tBPQAs occurs at or prior to the viral DNA 3′-processing step. Biochemical studies revealed that this inhibition operates by compound-induced conformational changes in the IN dimer that prevent proper assembly of IN onto viral DNA. For the first time, tBPQAs were demonstrated to be allosteric inhibitors of HIV-1 IN displaying a dual mode of action: inhibition of IN-viral DNA assembly and inhibition of IN-LEDGF interaction.
Introduction
Efforts toward the design and discovery of IN2 inhibitors have been focused principally on targeting the catalytic site of IN with the early development of in vitro enzymatic assays (1–3). Over a decade of these drug design efforts has led to the discovery of the strand transfer-specific class of small molecule IN inhibitors, with three compounds demonstrating clinical success (4, 5). One IN strand transfer inhibitor (INSTI), raltegravir (MK-0518), was approved by the FDA in 2007 for the treatment of HIV-1 infection (6–8), and a second INSTI, elvitegravir (GS-9137), which is in late-stage clinical development, is dosed once daily with a new pharmacokinetic enhancer, cobicistat (9–11). More recently, dolutegravir (S/GSK1349572), a third potent unboosted HIV INSTI with a resistance profile markedly different from that of raltegravir and elvitegravir, has entered phase III clinical testing (12, 13).
Despite the achievements in the design of the INSTI class, resistance development is still one of the limitations of this antiretroviral class. The emergence of raltegravir and elvitegravir resistance has been observed clinically, and the IN mutations that have emerged confer cross-resistance to both drugs (14–17). Although the introduction into the clinic of a second-generation INSTI represented by dolutegravir is expected in the near future (13), all INSTIs function with essentially the same mode of action, through binding to the catalytic site of IN. Therefore, the design and discovery of other classes of IN inhibitors with a mechanism of action distinct from that of INSTIs still represent a highly attractive antiretroviral strategy. Such allosteric inhibitors of IN potentially can be used in combination with active site-based INSTIs and other clinically approved antiviral drugs to provide effective suppression of viral replication with minimal resistance development.
There are multiple approaches to targeting the IN enzyme allosterically for drug development (18). The N-terminal domain (NTD) of IN (residues 1–50) contains a zinc finger motif, HHCC, that mediates viral DNA binding and is essential for viral replication (19–23). A natural product, hyrtiosal, has been shown to inhibit IN through direct binding to the NTD of IN (24). Such a targeting of the NTD of IN could disrupt Zn2+ coordination by the zinc finger motif and have a detrimental effect on viral DNA binding, IN multimerization, and reverse transcription and/or integration. The C-terminal domain (CTD) of IN (residues 220–288) is much less conserved than the NTD, possesses nonspecific DNA binding activity, and could have a role in binding genomic DNA during integration (25, 26). A molecule such as pyridoxal 5′-phosphate, the binding site of which is mapped to the CTD, could be used as a lead for the design of more potent inhibitors disrupting DNA binding to the CTD (27). Another allosteric IN inhibitor approach consists of disrupting IN multimerization using either peptides derived from the interfacial region between two IN monomers (28–30) or small molecules binding to the IN dimer interface (31–34). In this latter category, an acetyl bis-caffeoyl compound binding at the dimer interface has been shown to stabilize an IN multimeric complex by inhibiting the IN subunit exchange required for IN-DNA complex formation (33). This inhibition of IN subunit exchange has also been observed with LEDGF and LEDGF-derived peptides (35, 36).
The host protein, LEDGF/p75, was identified as a cofactor of IN critical for HIV-1 replication by tethering IN to the host chromatin to facilitate integration (37–46). Efforts at disrupting the IN-LEDGF/p75 interaction as a therapeutic target have resulted in a new class of allosteric inhibitors binding to the LEDGF/p75 binding pocket at the IN dimer interface (47–50). Of these allosteric inhibitors of IN-LEDGF/p75 interaction, the 2-(quinolin-3-yl)acetic acid series, termed LEDGINs, has shown the best antiviral potency with an EC50 of ∼1 μm for the lead molecule and high specificity for IN-LEDGF/p75 interaction (50). In addition, this molecule retains its antiviral potency against various raltegravir-resistant viruses, showing that they are a class of inhibitors distinct from the INSTIs (50). Another series of LEDGIN structural analogs with potent in vitro antiretroviral activity, tert-butoxy-(4-phenyl-quinolin-3-yl)-acetic acids (tBPQA), was designed originally by Boehringer Ingelheim Pharmaceuticals Inc. (51–53) and licensed subsequently by Gilead Sciences, Inc. for further development.
In this report, biochemical and biological assays were used to systematically elucidate the mechanism of action of the tBPQAs. It was found that tBPQAs interact with IN at the LEDGF binding pocket located at the IN dimer interface, and we show that this interaction can have two consequences: (i) competition with LEDGF, which would disrupt chromatin tethering of IN; and (ii) induction of conformational change in the IN dimer, which inhibits proper assembly of donor DNA-IN complex. Both actions can contribute to the potent antiviral effect of the tBPQAs.
EXPERIMENTAL PROCEDURES
Peptides
All of the peptides used in this report were custom synthesized and purified to >90% using the services of Anaspec Inc. (San Jose, CA).
Proteins
Expression vectors and methods of expression and purification of His6-IN and IN-FLAG proteins have been described previously (54).
Oligonucleotides
All of the deoxyoligonucleotides were HPLC-purified and purchased from Trilink Biotechnologies (San Diego). For the strand transfer assay, the 3′-processed top donor DNA strand is ST1 (5′-Cy5(C6-NH)ATGTGGAAAATCTCTAGCA-3′); the non-processed top donor DNA strand is ST7 (5′-Cy5(C6-NH)ATGTGGAAAATCTCTAGCAGT-3′); the bottom donor DNA strand is ST2 (5′-Cy5(C6-NH)ACTGCTAGAGATTTTCCACAT-3′); the top target DNA strand is ST5 (5′-ACAGGCCTAGCAAAACGCGTCG-(biotin BB)-3′); and the bottom target DNA strand is ST6 (5′-CGACGCGTTTTGCTAGGCCTGT-(biotin BB)-3′). For the 3′ processing assay, the non-processed top donor DNA strand is ST10 (5′-ATGTGGAAAATCTCTAGCAGT(C6-NH)-Cy5–3′), and the bottom donor DNA strand is ST11 (5′-ACTGCTAGAGATTTTCCACAT-(biotin BB)-3′).
Antiviral, Cytotoxicity, 2-LTR Circle Accumulation, and Alu-PCR Assays
Antiviral and cytotoxicity assays in MT-2, MT-4, and human peripheral blood mononuclear cells have been described elsewhere (55). 2-LTR circle accumulation and Alu-PCR assays were also performed as described previously (55).
Viral Resistance Selections, Clonal Sequencing of Viral DNA, and Construction of Infectious HIV-1 DNA with IN Mutations
Viral resistance selections using GS-A and GS-B, clonal sequencing of the selected resistant viral pools, and reintroduction of selected IN mutations into the infectious wild-type HIV-1 DNA clone, HXB2, were all performed as described previously (55).
Integrase Crystallization, Data Collection, and Structure Refinement
Crystals of the HIV-1 integrase catalytic core domain (CCD) F185K were grown by the hanging drop vapor diffusion method. Equal parts of protein and reservoir solution were mixed with a mother liquor containing 8% PEG 8000, 100 mm sodium cacodylate, pH 6.5, 200 mm (NH4)2SO4, and 5 mm DTT at 20 °C. Crystals were moved to a solution containing 12% PEG 8000, 100 mm sodium cacodylate, pH 6.5, 20% ethylene glycol, 5 mm DTT, and 1 mm GS-A or GS-B for 24 h prior to data collection. Crystals were then cryo-cooled in a stream of liquid nitrogen at 100 K. All data were collected at a temperature of 100 K and processed with HKL2000 (56). All diffraction data were collected on a Rigaku MicroMax 007 rotating copper anode x-ray generator.
Molecular replacement of the integrase CCD was performed with the software package Phenix (57) using the starting model (Protein Data Bank code 1ITG). Rigid body refinement, simulated annealing, energy minimization, and B-factor refinement were likewise carried out with Phenix. Bulk solvent correction and anisotropic B-factor scaling were used during refinement. Model building was performed with the molecular graphics program Coot (58). The final model statistics are listed in Table 4. Coordinates have been deposited in the Protein Data Bank with accession numbers 4E1M and 4E1N for CCD complexed with GS-A and GS-B, respectively.
TABLE 4.
X-ray data collection and refinement statistics
| GS-A | GS-B | |
|---|---|---|
| Data collection | ||
| Unit cell (a/b, c in Å)a | 71.79, 65.33 | 71.71, 64.80 |
| Resolution (Å) | 50.0-1.9 (1.97-1.9) | 50.0-2.0 (2.07-2.00) |
| No. of reflections | 60,916 | 52,818 |
| No. unique | 15,616 | 13,300 |
| I/σ | 12.6 (2.1) | 9.5 (1.9) |
| Rmergeb (%) | 4.2 (49.0) | 5.9 (50.5) |
| Completeness (%) | 99.4 (100.0) | 99.4 (100.0) |
| Refinement statistics | ||
| Resolution (Å) | 30.0-1.9 | 30.0-2.0 |
| No. reflections (F ≥ 0) | 15,556 | 13,250 |
| R-factorc | 23.8 | 24.0 |
| R-freec | 27.3 | 26.5 |
| r.m.s. bond lengths (Å) | 0.007 | 0.008 |
| r.m.s. bond angles (°) | 1.02 | 1.08 |
a All crystals belong to space group P3121.
b Rmerge = [ΣhΣi|Ih − Ihi|/ΣhΣiIhi] where Ih is the mean of Ihi observations of reflection h. Numbers in parentheses represent highest resolution shell.
c R-factor and R-free = Σ‖Fobs| − Fcalc‖/Σ|Fobs| × 100 for 95% of recorded data (R-factor) or 5% of data (R-free).
Competition Binding Assay for IN-LEDGF Interaction
This homogeneous time-resolved FRET (HTRF) assay measures the interaction of His6-IN (N-terminal His6 tag) and LEDGF-FLAG (C-terminal FLAG tag) in the presence of a competitor of LEDGF (54).
IN Dimer Promotion Assay
This homogeneous time-resolved FRET assay measures the interaction of two IN monomers, His6-IN (N-terminal His6 tag) and IN-FLAG (C-terminal FLAG tag), in the presence of a dimer ligand. This assay is used to assess the ability of IN dimer ligands to promote IN dimer formation and inhibit IN dimer subunit exchange (36).
HTRF-based IN Strand Transfer Assay
IN strand transfer activity was measured using an HTRF-based assay adapted from a previous report (59). IN strand transfer activity was tested in the absence and presence of compounds in a 25-μl reaction in a 96-well half-area white flat-bottom plates (catalogue No. 3693, Corning). The FRET signal generating strand transfer product is resistant to treatment with 0.5 m NaCl, which dissociates all DNA from IN and prevents any non-covalently linked donor and target DNAs held together by IN to produce the FRET signal. Dose-response data were analyzed by curve fitting using Equation 1,
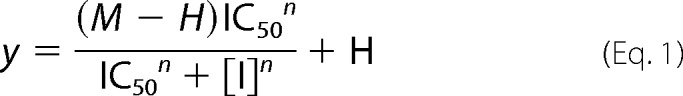 |
where y = a 665/620 ratio, IC50 = 50% inhibitory concentration, [I] = inhibitor concentration, n = Hill coefficient, M = signal in the absence of inhibitor, and H = signal at full inhibition.
The assay details are as follows. Cy5-labeled donor DNA (ST7/ST2) and biotin-labeled target DNA (ST5/ST6) were made by annealing complementary strands at a concentration of 50 μm each in annealing buffer (20 mm Tris, pH 7.4, and 100 mm NaCl). IN strand transfer assay with non-processed donor DNA was performed in the strand transfer buffer (20 mm Tris, pH 7.4, 25 mm NaCl, 7.5 mm MgCl2, 0.05% Brij-35, 10% glycerol, 1 mm DTT, 1% DMSO, and 0.1 mg/ml BSA). Test compounds were first serially diluted 3-fold in DMSO using a 2.5 mm stock (2-fold serial dilution from a 5 mm stock for test peptide) followed by 4-fold dilution in 20 mm Tris, pH 7.4, to obtain the 25× serially diluted compound. Nine microliters of His6-IN (final concentration 250 nm) was preincubated with 1 μl of 25× serially diluted compound for 5 min at room temperature. After preincubation, 15 μl of a mixture containing donor DNA ST7/ST2 (12.5 nm final concentration) and target DNA ST5/ST6 (5 nm final concentration) was added. The strand transfer reaction was incubated at 37 °C for 120 min. The reaction was stopped by the addition of 25 μl of 2× Stop/development buffer (25 mm EDTA, 1 m NaCl, 1 mg/ml BSA, 0.05% Brij-35, 2 nm europium chelate-streptavidin (catalogue No. AD0060, PerkinElmer Life Sciences). Plates were read after a16-h incubation at room temperature on an Envision 2102 multilabel reader (PerkinElmer Life Sciences) using excitation filter UV2 (TRF) 320 nm, emission filter 1 590 nm, and emission filter 2 665 nm with dichroic mirror D400. Raw counts (in cps) at 665 and 620 nm were collected, and the signal was expressed as the ratio of (cps at 665 nm/cps at 620 nm) × 1000.
Time Course of Donor DNA-IN Preassembly
The HTRF-based IN strand transfer assay was used to study the effect of donor DNA-IN preassembly on the inhibitory activity of compounds used at 25 μm. Donor DNA and IN were allowed to preassemble at 37 °C for various lengths of time (0, 5, 10, 15, 30, 45, 60, 90, and 120 min) before compound and target DNA ST5/ST6 addition. Final concentrations of 250, 12.5, and 5 nm were used for His6-IN, 3′-processed donor DNA ST1/ST2, and target DNA ST5/ST5, respectively.
Compounds were first diluted in 100% DMSO to 5 mm followed by an 8-fold dilution in 20 mm Tris, pH 7.4, to obtain an intermediate compound stock of 625 μm. For controls where His6-IN was incubated simultaneously with donor DNA and compound, 900 μl of His6-IN dilution was mixed with 100 μl of intermediate compound stock and 1000 μl of ST1/ST2. For reactions where His6-IN was preassembled with donor DNA in the absence of compound, 900 μl of His6-IN dilution was mixed with 100 μl of 12.5% DMSO in 20 mm Tris, pH 7.4, and 1000 μl of ST1/ST2. After the specified incubation time, 20 μl of the reaction was withdrawn and mixed either with 4 μl of ST5/ST6 target DNA and 1 μl of 12.5% DMSO for the assembly-with-compound controls or with 4 μl of ST5/ST6 target DNA and 1 μl of intermediate compound stock for the preassembly controls. The reaction mixtures were incubated at 37 °C for an additional 30 min. Reactions were stopped by the addition of 25 μl of 2× Stop/development buffer, incubated, and read as described in the previous section.
Two additional controls were prepared: a no inhibition control, where the compound was omitted from both incubation steps, and a full inhibition control, where His6-IN was omitted from the reaction. For the first control, 2000 μl of ST1/ST2 was mixed with 200 μl of 12.5% DMSO and 1800 μl of His6-IN dilution. Time points were sampled for the addition to target DNA, incubated, developed, and read as described above. For the second control, 400 μl of donor DNA ST1/ST2 was mixed with 40 μl of intermediate compound stock and 360 μl of buffer. Only a 45-min time point was sampled for the addition to target DNA, incubated, developed, and read as described above. Each data point was performed in triplicate.
HTRF-based IN 3′-Processing Assay
The HTRF-based 3′-processing assay was performed essentially as described for the IN strand transfer assay using a specially designed non-processed donor DNA in the absence of target DNA. The specially designed non-processed blunt-ended donor DNA (ST10/ST11) was labeled with Cy5 at the dinucleotide-removable 3′-end and with biotin at the other 3′-end. Removal of the dinucleotide by 3′-processing concomitantly removes the Cy5 fluorophore, resulting in a decrease in the FRET signal. The ST10/ST11 donor DNA duplex was used at 12.5 nm final concentration. All other reagents were used at the concentrations described for strand transfer assay with non-processed donor DNA.
2-LTR Junction Sequencing
Half a million MT-2 cells were infected with HIV-1 IIIb virus (catalogue No. 10-124-000, Advanced Biotechnologies Inc., Columbia, MD) at a multiplicity of infection of 10 at 37 °C. After 3 h of viral adsorption, inhibitors at the lowest concentrations determined previously to give the maximum accumulation of 2LTR circles were added. Specifically, raltegravir, elvitegravir, GS-A, and GS-B were used at concentrations of 240 nm, 120 nm, 10 μm, and 1 μm, respectively. Infected cells with no drug added were used as a negative control. Viral DNA was isolated 24 h post-drug addition using a QIAamp DNA mini kit (catalogue No. 51304, Qiagen, Valencia, CA). The 2-LTR junction was PCR-amplified using primers FP9600 (5′-GCTTAAGCCTCAATAAAGCTTGCCT-3′) and RP71 (5′-GCCTTGTGTGTGGTAGATCC-3′) to generate a 191-bp fragment. The gel-purified fragment was cloned into the pCR-XL-TOPO vector using the TOPO XL PCR cloning kit (catalogue No. K4750-20, Invitrogen). Ninety-six colonies were picked for each drug treatment condition, and mini prep DNA from each colony was isolated using DirectPrep 96 MiniPrep kit (catalogue No. 27361, Qiagen). Sequencing was performed using the M13 Forward(−20) primer (5′-GTAAAACGACGGCCAG-3′) located 96 bp upstream from the 2-LTR junction PCR product insert.
FRET Efficiency Measurement
FRET efficiency measurement was performed using the IN dimer promotion assay. The goal of this measurement is to determine whether the addition of a compound that binds to the LEDGF binding pocket at the IN dimer interface can induce the movement of one monomer relative to the other; this translates into a distance change between the europium and XL665 of the anti-FLAG and anti-His6 antibody conjugates binding to the tags of the His6-IN-IN-FLAG heterodimer. By measuring the efficiency of resonance energy transfer from donor (europium) to acceptor (XL665) the distance (r) between donor and acceptor fluorophores can be calculated using Equation 2,
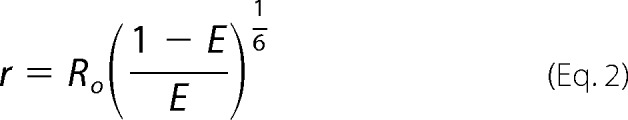 |
where E is the efficiency of resonance energy transfer and Ro is the Förster radius of a donor-acceptor pair (Ro = 90 Å for europium/XL665). The efficiency of resonance energy transfer can be calculated from the measurement of donor fluorescence in the absence and presence of acceptor using Equation 3,
 |
where FD is donor fluorescence in the absence of acceptor, FDA is donor fluorescence in the presence of acceptor, and fa is fractional labeling with the acceptor. FD is measured by adding only the anti-FLAG-europium cryptate conjugate to the reaction, whereas FDA is measured by adding both the anti-FLAG-europium cryptate and anti-His6-XL665 conjugates. As a negative control for distance change, the His6-GS(AQ)6GS-FLAG peptide was used.
The fractional labeling fa1 = 0.99666 of this peptide by the anti-His6-XL665 conjugate was calculated from KD = 19 pm of the anti-His6-XL665 conjugate for the His6 tag (54) and the concentrations [His6-GS(AQ)6GS-FLAG] = 5 nm and [anti-His6-XL665] = 10.66 nm used in the reaction. Similarly, the fractional labeling fa2 = 0.97869 of His6-IN by the anti-His6-XL665 conjugate was calculated using the reaction concentration [His6-IN] = 10 nm.
For the His6-IN-IN-FLAG heterodimers, the donor and acceptor are on separate monomer subunits of the heterodimer; therefore it is the fractional labeling of IN-FLAG by the anti-His6-XL665/His6-IN complex that is relevant. Because the FRET efficiency is measured either in the absence of compound or in the presence of compound at the concentration that gives the peak in the dose response, the fractional labeling with the acceptor has to be calculated for each case. In the absence of compound, the fractional labeling of IN-FLAG with anti-His6-XL665 is given by fa3 in Equation 4,
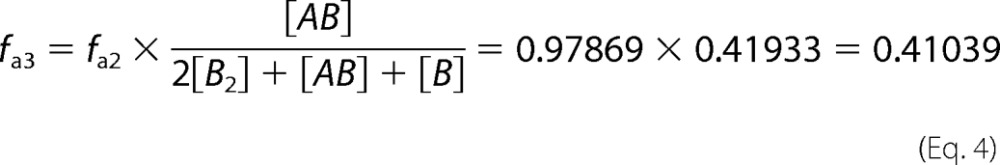 |
where A = His6-IN, B = IN-FLAG, and [AB], [B2], and [B] are equilibrium concentrations calculated from our IN dimerization model (36) after mixing together 10 nm A and 10 nm B. In the presence of compound at the concentration that gives the peak in the dose response, the fractional labeling of IN-FLAG with anti-His6-XL665 is given by fa4 in Equation 5,
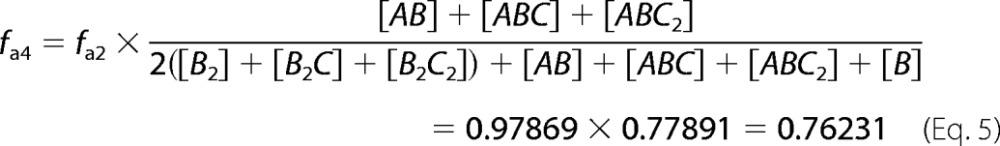 |
where A = His6-IN, B = IN-FLAG, C = compound capable of binding to the IN dimer interface, and [AB], [ABC], [ABC2], [B], [B2], [B2C], and [B2C2] are equilibrium concentrations calculated from our IN dimerization model (36) after mixing 10 nm A, 10 nm B, and the concentration of compound C that gives the dimer promotion dose-response peak. Depending on the situation, the fa in Equation 3 will be substituted for by fa1, fa3, or fa4 for the FRET efficiency calculation.
RESULTS
tBPQAs Are Potent Antiviral Compounds and Authentic Inhibitors of HIV-1 Integration
Three tBPQAs, GS-A (52), GS-B (52), and GS-C (53), displayed high antiviral potency in MT-4, MT-2, and human monocytes (peripheral blood mononuclear cells) with EC50 values ranging from 10 to 287 nm (Table 1). To assess whether these compounds were inhibitors of integration, their effects on the accumulation of 2-LTR circles (a reverse transcription side-product of nonhomologous end joining, which level can be further elevated by integration failure), and the formation of integration junctions were determined in HIV-1-infected MT-2 cells (Table 1). Similar to raltegravir and elvitegravir, but contrary to negative controls efavirenz and amprenavir, all three tBPQAs elevated the level of 2-LTR circles and decreased the number of integration junctions in HIV-1-infected cells, demonstrating that they are authentic inhibitors of HIV-1 integration.
TABLE 1.
Antiviral and anti-integration activities of tBPQAs

a Mean ± S.D. of at least three independent determinations.
b Mean ± S.D. of at least two independent determinations.
tBPQAs Selected for Drug-resistant Mutations Located Near or at the IN Dimer Interface
To identify the target of tBPQAs, HIV-1 IIIb was passaged serially in tissue culture in the presence of increasing concentrations of either GS-A or GS-B (Table 2). The antiviral potency of these two inhibitors was tested against selected viral passages to monitor the emergence of drug-resistant phenotype. GS-A selected for 34-fold resistance by passage 5 and ∼1100-fold resistance by passage 7. GS-B selected for 33-fold resistance by passage 4 and 986-fold resistance by passage 6. Each selected resistant viral pool was also cross-resistant to the other tBPQA but not to raltegravir, suggesting that the mechanism of action of tBPQAs is different from that of IN strand transfer inhibitors. Clonal sequencing of the selected viral pools revealed that GS-A selected for five mutations in IN (A128T, G134E, H171Q, T174I, and V201I), whereas GS-B selected for four mutations (Y99H, V126I, T174I and N222K) (Table 3). Under GS-A selection, passage 8 virus underwent a purifying selection for the double mutant A128T/T174I with the total disappearance of the other three mutations. Under GS-B selection, passage 6 was enriched to 83.3% of viruses containing both V126I and T174I mutations. With the exception of N222K, located in the CTD of IN, all the selected mutations are located in the CCD, in or near the LEDGF binding pocket at the IN dimer interface (Fig. 1).
TABLE 2.
Phenotype of viral pools selected with tBPQAs
The -fold change is calculated from the ratio of EC50 of the selected viral pool over the EC50 of wild-type HIV-1 IIIb (shown in parentheses). The data represent the mean of two independent determinations. EFV, efavirenz; APV, amprenavir; RAL, raltegravir.
| Selected virus | Duration of selection | [Drug] reached | -Fold change |
||||
|---|---|---|---|---|---|---|---|
| EFV | APV | GS-A | GS-B | RAL | |||
| day | nm | ||||||
| HIV-1 IIIb | 1 (0.8 nm) | 1 (62 nm) | 1 (55 nm) | 1 (14 nm) | 1 (7.2 nm) | ||
| EFV P2 | 17 | 2 | 1.2 | 1.2 | 1.0 | 1.1 | 1.3 |
| EFV P5 | 43 | 16 | 90 | 0.8 | 0.8 | 0.8 | 1.1 |
| EFV P7 | 57 | 64 | 181 | 0.7 | 0.7 | 0.6 | 1.2 |
| APV P2 | 17 | 100 | 1.0 | 1.5 | 1.2 | 1.2 | 1.3 |
| GS-A P2 | 17 | 160 | 1.7 | 1.1 | 14 | 2.1 | 1.4 |
| GS-A P3 | 23 | 320 | 2.4 | 1.0 | 10 | 0.7 | 1.2 |
| GS-A P5 | 42 | 1,280 | 1.3 | 0.6 | 34 | 20 | 1.1 |
| GS-A P6 | 50 | 2,560 | 1.1 | 1.1 | 583 | 1,321 | 1.2 |
| GS-A P7 | 60 | 5,120 | 1.5 | 2.0 | 1,127 | 822 | 2.5 |
| GS-A P8 | 69 | 10,240 | 0.9 | 1.0 | 1,061 | 599 | 1.0 |
| GS-B P3 | 37 | 60 | 2.5 | 1.0 | 65 | 30 | 1.5 |
| GS-B P4 | 43 | 120 | 2.1 | 1.2 | 81 | 33 | 1.6 |
| GS-B P5 | 49 | 240 | 2.8 | 1.1 | 93 | 277 | 1.7 |
| GS-B P6 | 59 | 480 | 1.7 | 0.6 | 1,081 | 986 | 1.4 |
TABLE 3.
Genotype of HIV-1 variants selected in vitro in the presence of tBPQAs
| Selected virusa | Duration of selection | Compound concentration reached | Integrase mutations identifiedb | Integrase mutationsc |
|---|---|---|---|---|
| day | nm | |||
| HIV-1 IIIbd | Y99H (0%), V126I (0%), A128T (0%), G134E (0%), H171Q (0%), T174I (0%), V201I (0%), N222K (0%) | |||
| GS-A P2 | 17 | 160 | A128T, G134E | A128T (83.3%), G134E (12.5%) |
| GS-A P5 | 42 | 1,280 | A128T, G134E, H171Q, V201I | A128T/H171Q/V201I (33.3%), G134E/H171Q/V201I (62.5%), G134E/H171Q (4.2%) |
| GS-A P6 | 50 | 2,560 | A128T, G134E, H171Q, T174I, V201I | A128T/H171Q/V201I (20.8%), A128T/H171Q (4.2%), A128T/T174I (70.8%), G134E/H171Q/V201I (4.2%), G134E/H171Q (4.2%) |
| GS-A P8d | 69 | 10,240 | A128T, T174I | A128T/T174I (100%) |
| GS-B P3 | 37 | 60 | Y99H, N222K | Y99H/N222K (100%) |
| GS-B P5 | 49 | 240 | Y99H, V126I, T174I, N222K | Y99H/N222K (83.3%), Y99H/T174I/N222K (4.2%), V126I/T174I (8.3%), V126I/T174I/N222K (4.2%), |
| GS-B P6d | 59 | 480 | Y99H, V126I, T174I, N222K | Y99H/N222K (4.2%), V126I/T174I (33.3%), V126I/N222K (4.2%), V126I/T174I/N222K (50%) |
a Twenty-four clones from each viral passage were sequenced using two sequencing primers.
b A128T was not selected with GS-B, but three new mutations were selected: Y99H, V126I, and N222K.
c Percentage of total clones analyzed is shown in parentheses.
d HIV-1 IIIb contains 42% T124/T125 and 58% A124/T125. By passage 8 of selection with GS-A, 100% of the clones are T124/T125. By passage 6 of selection with GS-B, 100% of the clones are A124/T125.
FIGURE 1.

Crystal structure of GS-A bound to IN CCD dimer. A, view of the IN CCD dimer complexed with two molecules of GS-A colored in orange. Each individual CCD monomer is colored in either green or tan. The active site residues are highlighted in red. B, view of GS-A (orange) and GS-B (gray) in the binding pocket at the CCD dimer interface. Key residues in the binding pockets are identified. Resistance mutations at positions 201 and 222 are not near the tBPQA binding site. Val-201 is at the CCD dimer interface but near the C-terminal end of CCD, and Asn-222 is in the C-terminal domain.
Crystal Structure of IN CCD and tBPQAs Confirmed Compound Interaction at the IN Dimer Interface
Apocrystals of IN CCD were soaked with either GS-A or GS-B and their x-ray structures determined (Table 4). In both structures, electron density consistent with GS-A and GS-B was found at the CCD dimer interface. The refined structures show clear density for both inhibitors without ambiguity (Fig. 1). Approximately half of the binding pocket is formed by each monomer subunit. The acid group forms two hydrogen bonds to the backbone nitrogen atoms of residues Glu-170 and His-171 and another hydrogen bond to the side-chain oxygen of Thr-174 (Fig. 1). The tert-butyl group is positioned into a pocket lined with residues Tyr-99, Gln-95, and Thr-174. The Gln-95 side chain lies over the top of the tert-butyl group, partially sequestering it from solvent. The quinoline core is positioned against one face of the pocket primarily formed by residues Ala-124 and Thr-125. Ala-128 is also positioned near the quinoline core. The functional group attached at position 4 of the quinoline core (R4 group) is further positioned along the dimer interface cleft and points toward Trp-131. This part of the pocket is also lined by residues Leu-102, Ala-128, Ala-129, Thr-174, Gln-168, Ala-169, and Met-178.
A majority of the amino acids found to be mutated during resistance selection with GS-A are part of the binding pocket. Ala-128 is located in close proximity to the quinoline core, and Thr-174 forms significant contacts with the acid, tert-butyl, and R4 moieties. GS-B selected mutations at amino acids Val-126 and Thr-174. Val-126 is not directed into the pocket but is within the helix that forms a significant part of the pocket including residues Ala-124, Thr-125, and Ala-128.
tBPQAs Compete with LEDGF for Binding to IN, Promote IN Dimer Formation, and Inhibit IN Dimer Subunit Exchange
With the knowledge that tBPQAs bind to the same pocket at the IN dimer interface as the IBD of LEDGF (43), we tested the ability of tBPQAs to compete with LEDGF for binding to IN dimers using the IN-LEDGF interaction assay we described previously (Fig. 2A) (54). GS-A, GS-B, and GS-C showed IC50 values ranging from 19 to 228 nm (Fig. 2B). Previously, we had shown, using an IN dimerization assay, that LEDGF-derived peptides binding to the IN dimer interface can promote IN dimer formation and inhibit its subunit exchange (36). tBPQAs were tested in this assay and displayed the characteristic biphasic dose-response curves with an ascending phase reflecting dimer promotion and a descending phase reflecting inhibition of subunit exchange at the higher tested concentrations (Fig. 2C). The EC50 values of dimer promotion determined from the ascending phase range from 22 to 249 nm for the three compounds shown in Fig. 2B. As with the LEDGF-derived peptides, the dose-response curves of the tBPQAs displayed different peak heights that are beyond the peak height of ∼183% predicted by the law of mass action, suggesting possible FRET distance change upon compound binding (36).
FIGURE 2.

tBPQA competition binding against LEDGF and promotion of IN dimer formation correlate with their antiviral potency. A, diagram of the experimental setup for the IN-LEDGF interaction assay. B, competition dose response of tBPQAs using the IN-LEDGF interaction assay. Data represent the means of two independent experiments done in quadruplicate with standard deviations shown as error bars. C, dimer promotion dose response of tBPQAs. Data represent the means of four independent experiments done in quadruplicate with standard deviations shown as error bars. D, correlation plot between the EC50 of dimer promotion and the IC50 of competition binding against LEDGF for a diverse set of 11 tBPQAs. E, correlation plot between the EC50 of dimer promotion and the antiviral EC50 in MT-4 cells for the same set of 11 tBPQAs.
Correlation among Activities of tBPQAs in Dimer Promotion, IN-LEDGF Competition Binding, and Antiviral Assays
To examine whether there is any correlation among dimer promotion, IN-LEDGF competition binding, and the antiviral activities of tBPQAs, the EC50 and IC50 values of eight additional tBPQAs were determined in these three assays. Our results showed that the EC50 values of dimer promotion correlated well with the IC50 values of IN-LEDGF competition binding with an R2 = 0.9393. This result indicates that competition with LEDGF and IN dimer promotion are two manifestations of the same compound interaction with the LEDGF binding pocket at the IN dimer interface (Fig. 2D). In addition, the EC50 values of dimer promotion also correlated with the antiviral EC50 values with an R2 = 0.8470, suggesting that the interaction of tBPQAs in the LEDGF pocket at the IN dimer interface may underlie the antiviral effect of this class of compounds (Fig. 2E).
tBPQAs Interfere with IN Catalytic Activity
To assess whether tBPQAs could affect IN catalytic activity, we tested them in both strand transfer and 3′-processing assays (Table 5). Because non-processed donor DNA was used in the strand transfer assay and compound was added to IN before the DNA substrates, the inhibition of signal generating strand transfer product formation could result from the inhibition of IN-donor assembly, 3′-processing, or strand transfer steps. In the 3′-processing assay, the inhibition of signal decrease resulting from cleavage of the 3′-dinucleotide could come from either inhibition of IN-donor assembly or 3′-processing. Efavirenz served as a negative control, as it does not inhibit IN catalytic activities. Raltegravir, which specifically inhibits the strand transfer activity but is much less effective at inhibiting the 3′-processing activity (∼40-fold less), was used as a positive control (Table 5). Unlike raltegravir, the three tested tBPQAs as well as LEDGF(354–378) inhibited IN activity with similar IC50 values in both assays (∼2-fold difference between the two assays). These results suggest that tBPQAs could also inhibit IN activity at a step that differs from that of raltegravir and that this inhibition could occur either at the 3′-processing step or at the IN-donor assembly step.
TABLE 5.
Inhibitory activities of tBPQAs in strand transfer and 3′-processing assays
| Compound | IC50 Strand transfera,b | IC50 3′-Processingb,c |
|---|---|---|
| nm | ||
| GS-A | 55 ± 3 | 106 ± 5 |
| GS-B | 67 ± 4 | 151 ± 12 |
| Raltegravir | 15 ± 1 | 585 ± 42 |
| LEDGF(354–378) | 7,061 ± 2,842 | 7,297 ± 1,532 |
| Efavirenz | >25,000 | >25,000 |
a IN + compound was preincubated for 5 min at 22 °C, and then non-processed donor DNA and target DNA were added and the reaction was incubated for 2 h at 37 °C.
b The final concentration (nm) of the components are: 250 IN, 12.5 non-processed donor, 5 target DNA, and 12.5 3′-processing substrate.
c IN + compound was preincubated for 5 min at 22 °C, and then 3′-processing substrate DNA was added and the reaction was incubated for 2 h at 37 °C.
IN-Donor DNA Assembly Is Target of Inhibition by tBPQAs
To identify which of the three steps of the strand transfer reaction is targeted by tBPQAs, the strand transfer assay was modified to either remove or isolate a particular step (Fig. 3, schemes A–F). Schemes A and B, show the standard strand transfer assay and the 3′-processing assay schemes used in Table 5. Schemes C and E, used 3′-processed donor DNA, whereas schemes D and F used unprocessed donor DNA. The effect of 3′-end preprocessing on the inhibitory activity of a compound was assessed by comparing the results of schemes C and D and E and F. In schemes C and D the compound was present during the assembly/3′-processing steps, whereas in schemes E and F, IN and donor DNA were allowed to preassemble in the absence of compound for 45 min. A comparison of schemes C and E or D and F (Fig. 3) allowed for the assessment of the effect of preassembly on the inhibitory activity of the compound. The preprocessing of the donor DNA 3′-end did not affect the inhibitory activity of the three tBPQAs and raltegravir (Fig. 4, superimposable red and blue curves and superimposable brown and green curves). These results indicated that like raltegravir, tBPQAs do not target the 3′-processing step. Preassembly of IN and donor DNA had no effect on the maximum level of inhibition by raltegravir (Fig. 4A, all four curves plateau to the same level) because the final strand transfer step is the target of raltegravir. In contrast, preassembly rendered the preassembled IN-donor DNA complex refractory to inhibition by tBPQAs (Fig. 4, B and C, brown and green curves displayed a 22–25% decreased maximum level of inhibition compared with the red and blue curves). These results indicated that tBPQAs do not target the strand transfer step but the correct assembly of donor DNA on IN. To further show that the inhibitory activity of tBPQAs is dependent on the duration of IN-donor DNA assembly, GS-A was added with target DNA to the reaction after various times of IN-donor DNA preassembly (Fig. 4, D and E). When GS-A was present during IN-donor DNA assembly, strand transfer activity was at its minimum irrespective of the duration of IN-donor DNA assembly (Fig. 4D). In contrast, when GS-A was added after an increasing duration of IN-donor DNA assembly, strand transfer activity was gradually recovered, suggesting that an increasing amount of preassembled IN-donor DNA complex became refractory to inhibition (Fig. 4E).
FIGURE 3.

Strand transfer and 3′-processing assay schemes. A, standard strand transfer assay. In this assay, IN, compound, donor, and target DNAs were all present at the start of the reaction. Because non-processed donor DNA (i.e. Blunt Donor) was used, all three steps, IN-donor DNA assembly, 3′-processing, and strand transfer, had to be functional for the generation of the final strand transfer product. B, 3′-processing assay. IN, compound, and donor DNA were all present at the start of the reaction. In this assay, both steps, IN-donor DNA assembly and 3′-processing, had to be functional for the generation of the final 3′-processed donor DNA product. C and D, IN, compound, and donor DNAs (either 3′-processed or non-processed) were all present at the start, but target DNA was added after assembly/3′-processing was allowed to proceed for 45 min. Strand transfer was allowed to proceed for 30 min after the addition of target DNA. E and F, only IN and donor DNAs (either 3′-processed or non-processed) were present at the start. Assembly/3′-processing was allowed to proceed for 45 min in the absence of compound after which compound and target DNA were added and strand transfer was allowed to proceed for 30 min.
FIGURE 4.

Effect of pre-3′-processing and preassembly on the inhibitory activity of compounds. A (raltegravir), B (GS-A), and C (GS-B). Red and blue curves (see Fig. 3, schemes C and D), compound was added with donor DNA (either 3′-processed or non-processed). Brown and green curves (see Fig. 3, schemes E and F), compound was added with target DNA after assembly/3′-processing was allowed to proceed for 45 min. The dose-response data from A–C represent the mean of at least two independent experiments done in triplicate with the standard deviation shown as error bars. D, preassembly of IN-donor DNA was allowed to proceed for various lengths of time in the presence of GS-A before the addition of target DNA. E, preassembly of IN-donor DNA was allowed to proceed for various lengths of time in the absence of compound before the addition of GS-A and target DNA. Blue curve, IN alone. Red curve, IN + compound. The time course data from D and E represent the mean of at least three independent experiments done in triplicate with the standard deviation shown as error bars.
Binding of tBPQAs to IN Dimer Interface Induced Detectable Movement of One IN Monomer Relative to the Other within the Dimer
Because the binding of tBPQAs to the IN dimer interface gave a dimer promotion dose-response peak height larger than the 183% predicted by the law of mass action (Fig. 2B) (36), we hypothesized that this FRET signal enhancement could have resulted from the relative movement between the monomer subunits of the IN dimer, leading to the shortening of the distance between fluorophores (i.e. europium cryptate and XL665) on individual monomers. To verify whether there is a shortening of the FRET distance, we measured the FRET efficiency of the donor-acceptor pair in the absence of a tBPQA and in its presence at the concentration that gives the dose-response peak (Fig. 5). The experimental setup for the measurement of donor fluorescence intensity in the absence of acceptor, FD, and donor fluorescence intensity in the presence of acceptor, FDA, is depicted in Fig. 5A. The FRET distance of the donor-acceptor pair on the negative control peptide, His6-GS(AQ)6GS-FLAG, by itself is 113.7 Å (Fig. 5B). The addition of tBPQAs or LEDGF(354–378), which do not interact with either the control peptide or the antibody conjugates, had no effect on the FRET distance of the donor-acceptor pair on this control peptide (Fig. 5B). The FRET distance of the donor-acceptor pair on IN dimer by itself is 120.7 Å (Fig. 5C). The binding of GS-B and GS-C to the IN dimer shortened the FRET distance between donor and acceptor by 14.7 and 17.4 Å, respectively, whereas the binding of the LEDGF-derived peptide LEDGF(354–378) left the FRET distance roughly unaltered (Fig. 5C). These results showed that binding of tBPQAs to the IN dimer induced the movement of one monomer subunit relative to the other in the dimer, preventing the correct assembly of IN with donor DNA.
FIGURE 5.

FRET distance measurement between donor and acceptor labels. A, diagram of the experimental setup for FRET distance measurement. Donor fluorescence intensity in the absence of acceptor, FD was measured by adding only the europium cryptate conjugate to either the IN heterodimer mixture or the negative control peptide in the presence or absence of compound. Donor fluorescence intensity in the presence of acceptor, FDA, was measured by adding the europium cryptate conjugate and the XL665 conjugate to either the IN heterodimer mixture or the negative control peptide in the presence or absence of compound. B, FRET distance measurement between europium cryptate and XL665 from the ends of a negative control peptide. C, FRET distance measurement between europium cryptate and XL665 from two monomers of a heterodimer. The mean FRET efficiency was calculated from three independent experiments done in octuplicates.
tBPQAs Decreased the Frequency of Deletions at the 2-LTR Junctions
When the two long terminal repeats of double-stranded HIV DNA are joined together to form a circular DNA, the junction is referred to as the 2-LTR junction. To determine whether evidence of inhibition of IN-donor DNA assembly or 3′-processing by tBPQAs could be identified in HIV-1-infected cells, we examined the sequences at the 2-LTR junctions of 2-LTR circles isolated from HIV-1-infected MT-2 cells that were either untreated or treated with strand transfer inhibitors or tBPQAs. We hypothesized that inhibitors of the 3′-processing and strand transfer steps could leave different signatures at the 2-LTR junction sequence of 2-LTR circles, which level can be increased by failed integrations, and that these signatures could provide clues to the mechanism by which integration was inhibited. This rationale is summarized in Fig. 6. The preintegration complexes can be theoretically subdivided into two pools, one containing 3′-processed viral DNA ends and the other containing unprocessed blunt-ended viral DNA or viral DNA with various lengths of primer binding site attached to the U5 end or polypurine tract attached to the U3 end (60). In the absence of drug treatment, the unprocessed viral DNA fail to integrate and form 1-LTR circles and 2-LTR circles with a wild-type sequence or insertions at the 2-LTR junctions. The 3′-processed viral DNA will either integrate to form the provirus or fail to integrate and circularize. Because of 3′-processing, circularization will require some trimming of the non-base-paired overhangs at the viral DNA ends, leading to deletions of 2-LTR circle junctions in the absence of drug (60) (Fig. 6A). In the presence of strand transfer inhibitors such as raltegravir and elvitegravir, the compound will bind to the IN-viral DNA complex at the pocket formed between the IN active site and the 3′-processed viral DNA end, targeting this 3′-processed viral DNA pool for circularization and increasing the proportion of 2-LTR circles containing deletions at the 2-LTR junctions (Fig. 6B). In contrast, in the presence of an inhibitor of IN-viral DNA assembly or 3′-processing, the size of the unprocessed viral DNA pool increases, resulting in a reduced proportion of 2-LTR circles with deletions at the 2-LTR junctions and an increased proportion with wild-type sequences or insertions at the 2-LTR junctions (Fig. 6C). The result of this study is shown in Table 6. The concentration of each compound used in this study corresponds to the lowest concentration that gives the maximum 2-LTR circle accumulation. The proportion of clones with deletions at the 2-LTR junction is 65.5 and 42.5% for raltegravir and elvitegravir treatments, respectively (Table 6). For the two tBPQA-treated controls, this proportion significantly decreased to 14.8 and 23.2% for GS-A and GS-B, respectively (Table 6). This decreased frequency of deletions at the 2-LTR junctions is consistent with the inhibition of IN-donor DNA assembly or 3′-processing by tBPQAs.
FIGURE 6.

A working model of decreased frequency in deletions at the 2-LTR junctions consistent with inhibition of IN-donor DNA assembly or 3′-processing. A, under conditions of no drug treatment, the preintegration complexes (PIC) containing unprocessed donor DNA ends form the circularized viral DNA pool with either a wild-type 2-LTR junction or those with insertion. The majority of the preintegration complexes containing 3′-processed viral DNA ends are integrated, whereas a minority of this pool circularizes giving rise to 2-LTR junctions with deletions. B, under conditions of strand transfer inhibitor treatment, preintegration complexes containing 3′-processed viral DNA ends are inhibitor-bound and are not available for integration. The viral DNA in this inhibitor-bound pool will eventually circularize and increase the proportions of 2-LTR circles with deletions at the 2-LTR junctions. C, under conditions of treatment with an IN-donor assembly or 3′-processing inhibitor, the preintegration complex pool containing 3′-processed viral DNA decreases considerably, resulting in a smaller proportion of 2-LTR circles with deletions at the 2-LTR junction.
TABLE 6.
Frequencies of deletions and insertions at the 2-LTR junctions from 2-LTR circles isolated from drug-treated HIV-1–infected cells
| 2-LTR junction types | Frequencya |
||||
|---|---|---|---|---|---|
| No drug | 240 nm raltegravir | 120 nm elvitegravir | 1 μm GS-B | 10 μm GS-A | |
| % total clones analyzed | |||||
| Wild type | 34.2 | 43.7 | 56.3 | 43.0 | 42.0 |
| Insertions | 40.5 | 4.6 | 9.2 | 44.2 | 50.6 |
| Left deletionsb | 26.6 | 44.8 | 32.2 | 15.1 | 7.4 |
| Right deletionsb | 21.5 | 20.7 | 10.3 | 8.1 | 7.4 |
a Ninety-six clones of 2-LTR junctions from each drug treatment condition were sequenced. The percentages of the various types of 2-LTR junctions for a given drug treatment condition do not necessarily add up to 100%, because deletions and insertions may occur simultaneously and left and right deletions may also occur simultaneously.
b t tests were conducted for the deletion values between the no drug control and each of the drug treatments: no drug/raltegravir, p = 0.2883; no drug/elvitegravir, p = 0.6884; no drug/GS-B, p = 0.0076; no drug/GS-A, p = 0.0003.
DISCUSSION
tBPQAs are a new class of putative IN inhibitors, originally designed by Boehringer Ingelheim Pharmaceuticals Inc. (51–53) and subsequently licensed by Gilead Sciences, Inc. for further development. They are structurally distinct from INSTIs but are close structural analogs of LEDGINs, a new class of allosteric inhibitors of IN shown to disrupt the IN-LEDGF interaction (50). In this report, we have systematically characterized the tBPQAs in order to elucidate their mechanism of action. The tested tBPQAs are potent inhibitors of HIV-1 replication with EC50 values of 10–20 nm in a variety of infected cells, including primary peripheral blood mononuclear cells. Quantification of 2-LTR circles and late-RT product by PCR showed that tBPQAs do not inhibit reverse transcription, as the level of neither 2-LTR circles nor late-RT products decreased in the presence of the compound. Instead, tBPQAs were shown to elevate the level of 2-LTR circles and decrease integration junctions as quantified by Alu-PCR, indicating their direct effect on the integration of proviral DNA.
Using serial passaging of HIV-1 in the presence of increasing concentrations of tBPQAs, mutations were selected in IN at the dimer interface that forms the binding pocket for the LEDGF IBD. Notably, GS-A selected for HIV-1 variants containing the double mutant A128T/T174I, whereas GS-B selected for a majority of variants containing the double mutant V126I/T174I (Table 3). The IN CCD dimer was co-crystallized with GS-A and GS-B, showing that these compounds bind in the LEDGF binding pocket of the IN dimer (Fig. 1 and Table 4). In particular, the carboxylic acid of the compounds forms a hydrogen bond to the side-chain oxygen of Thr-174 and two hydrogen bonds to the backbone nitrogen atoms of residues Glu-170 and His-171. Thr-174 also forms significant contacts with the tert-butyl and R4 groups. H171Q was selected in this study using GS-A, and A128T and E170G where selected previously by growing HIV-1 in a cell line overexpressing the IBD of LEDGF (61). The reason that GS-B selected for V126I instead of A128T became clear when the compounds were tested against viral clones containing either V126I or A128T with a wild-type background (data not shown). The A128T virus was 5-fold more sensitive to GS-B than wild type but 6-fold less sensitive to GS-A than wild type. V126I in contrast was 5–9-fold more resistant to both GS-A and GS-B than wild type. This selection pattern is consistent with the fact that Val- 126, in contrast to Ala-128, is not directed into the pocket but is within the helix that forms a significant part of the pocket, and therefore, V126I is selected only when selection of the alternative A128T is not possible. The quinoline core is positioned against one face of the pocket primarily formed by residues Ala-124 and Thr-125. Positions 124 and 125 are highly polymorphic in the IN sequences of HIV-1 clinical isolates (62), and the potency of individual compounds can be differentially affected by a particular combination of polymorphic variants at residues 124 and 125. In fact, this differential effect was observed during our resistance selection (Table 3). Our starting virus was HIV-1 IIIb, which is a mixture of 42% Thr-124/Thr-125 and 58% Ala-124/Thr-125. Whereas selection with GS-A purified the population by keeping the Thr-124/Thr-125 variant, selection with GS-B purified the population by keeping the Ala-124/Thr-125 variant. Taken together, these results show that the LEDGF binding pocket located at the IN dimer interface is the target of the tBPQAs.
With the knowledge of the protein target of tBPQAs, we showed by competition-binding dose response that tBPQAs are able to compete with LEDGF for binding to IN, with IC50 values ranging from 19 to 228 nm using a previously described IN-LEDGF interaction assay (54). Because ligands binding to the IN dimer interface were shown previously to promote IN dimer formation and inhibit its subunit exchange (33, 35–36), we also tested the tBPQAs in the IN dimer promotion dose-response assay. The tBPQAs produced the characteristic biphasic dose-response curves with an ascending phase reflecting dimer promotion and a descending phase reflecting inhibition of subunit exchange and gave EC50 values of dimer promotion ranging from 22 to 249 nm as determined from the ascending phase. Our results showed that there is good correlation between the EC50 values of dimer promotion and the IC50 values of IN-LEDGF competition binding with an R2 = 0.9393, indicating that displacement of LEDGF and stabilization of IN dimers are two manifestations of the same interaction between the compound and IN dimer interface. In addition, a good correlation was observed between the EC50 of dimer promotion and the antiviral EC50 with an R2 = 0.8470, suggesting that the interaction of tBPQAs with the LEDGF pocket at the IN dimer interface is tied directly to the antiviral effect of this class of compounds.
Using a combination of 3′-processing and strand transfer assays, we showed initially that tBPQAs are also capable of inhibiting IN catalytic activity at a step that differs from that of raltegravir and that this inhibition could occur either at the 3′-processing step or at the IN-donor assembly step (Table 5). By modifying the strand transfer assay, which incorporates the IN-donor DNA assembly, 3′-processing, and strand transfer steps, we were able to identify IN-donor DNA assembly as the step that is targeted directly by tBPQAs. The 3′-processing step was ruled out as a target of inhibition because tBPQAs effectively inhibited the IN strand transfer reaction even when 3′-preprocessed donor DNA was used (Fig. 4, A–C). The strand-transfer step was also ruled out as a target because tBPQAs had no effect on the strand transfer activity of preassembled IN-donor DNA complexes (Fig. 4, A–C). In addition, the level of preassembled IN-donor DNA complex refractory to inhibition can be gradually increased by prolongation of the time of preassembly in the absence of tBPQAs (Fig. 4, D and E). When the time of preassembly data in Fig. 4E was analyzed with a single exponential function, it yielded a rate constant of 0.0365 min−1 or a doubling time of 19.0 min. Using an HTRF-based IN-donor DNA assembly assay (data not shown), the rate constant of assembly was determined independently to be 0.0324 min−1 or a doubling time of 21.4 min. These results further confirmed that tBPQAs interfere at the assembly step. In the raltegravir control (Fig. 4A), when preassembly of IN-donor DNA and 3′-processing in the absence of drug were allowed to proceed for 45 min before drug addition, the dose-response curves were shifted to the right (Fig. 4A, brown and green curves) but still plateaued at the same level as the controls with drug added from the start (red and blue curves). This shift in the dose-response curve could be due to the delay for raltegravir to reach binding equilibrium with the binding pocket created during the previous 45 min of IN-donor DNA assembly and 3′-processing. When raltegravir was added together with IN and donor DNA from the start, the binding equilibrium was established as soon as new binding pockets became available, and thus would not incur a delay in inhibition.
With the IN-donor DNA assembly step identified as the target of tBPQAs, we sought to understand the mechanism of this inhibition. Using an HTRF-based IN-donor DNA assembly assay, we showed that tBPQAs were not able to compete with donor DNA for binding to IN (IC50 > 300 μm, data not shown), suggesting that tBPQAs do not inhibit by disrupting IN-donor DNA binding. This result is not surprising, as DNA binds to extensive surfaces on IN. Our first clue as to a plausible mechanism of inhibition of IN-donor DNA assembly came from the observation that the peak heights (>420%) of the IN dimer promotion dose-response curves of tBPQAs were much larger than predicted by the law of mass action (∼183%). These larger peak heights suggested an increase in FRET efficiency upon compound binding due to a conformational change in the IN dimer that decreased the distance between the donor-acceptor pair. Our measurement of FRET efficiency in the absence and presence of tBPQAs confirmed a shortening of the distance between donor and acceptor by 14.7 to 17.4 Å upon binding of tBPQAs to IN dimers. Taken together, these results indicated that the binding of tBPQAs to the LEDGF binding pocket at the IN dimer interface induced a conformational change that rigidified the naturally flexible IN dimer and prevented the correct assembly of donor DNA onto IN. As a result, the donor DNA end was incorrectly positioned in the enzyme active site. There is a parallel between the effect of tBPQAs on IN dimers described here and a recent observation showing that LEDGF/IBD can stabilize an IN tetramer conformation that differs from that found in the stable synaptic complex (63). Although the binding of tBPQAs to the IN dimer changed its conformation and interfered with correct IN-donor DNA assembly, LEDGF/IBD binding to the IN tetramer induced a tetramer conformation incapable of stable synaptic complex formation in the presence of viral DNA. There is however a difference. Whereas LEDGF/IBD-bound IN can still interact with viral DNA to carry out 3′-processing (35), binding of tBPQAs to IN prevents correct assembly of IN with viral DNA and therefore 3′-processing.
To determine whether any evidence of inhibition of IN-donor DNA assembly or 3′-processing by tBPQAs could be identified in HIV-1-infected cells, we compared the sequences at the 2-LTR junctions of 2-LTR circles isolated from HIV-1-infected cells that were either untreated or treated with INSTIs or tBPQAs. The proportion of clones with deletions at the 2-LTR junction was found to be 1.8–4.4-fold higher for raltegravir and elvitegravir treatments than for tBPQAs treatments (Table 6). This decreased frequency of deletions at the 2-LTR junctions is consistent with the antiviral mechanism involving the inhibition of IN-donor DNA assembly as concluded from extensive biochemical characterization.
In conclusion, the mechanism of action of the tBPQAs was investigated systematically to demonstrate that this new class of compounds can inhibit HIV-1 integration through binding to the IN dimer interface. This interface also constitutes a functionally important pocket for interaction with the host integration cofactor, LEDGF. We have shown that the binding of tBPQAs to IN can have a dual effect on the process of HIV proviral integration: (i) it induces a conformational change in the IN dimer with loss of flexibility, which prevents the correct assembly of viral DNA-IN complex; and (ii) it inhibits the interaction of IN with LEDGF, which could block the tethering of the preintegration complex to host chromatin. We note that a separate study demonstrating the dual mode of action of a class of IN inhibitors similar to tBPQAs was published after the submission of our manuscript (64). Further detailed studies are in progress to determine how these two distinct effects contribute to the overall antiviral activity of this new class of potent antiretrovirals. This potential for a dual effect on the HIV integration process in infected cells makes tBPQAs attractive candidates for further development as novel HIV therapeutics.
The atomic coordinates and structure factors (codes 4E1M and 4E1N) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- IN
- integrase
- LEDGF
- lens epithelium-derived growth factor
- 2-LTR
- two long terminal repeat
- INSTI
- IN strand transfer inhibitor
- NTD
- N-terminal domain
- CTD
- C-terminal domain
- tBPQA
- tert-butoxy-(4-phenyl-quinolin-3-yl)-acetic acid
- CCD
- catalytic core domain
- HTRF
- homogeneous time-resolved FRET
- DMSO
- dimethyl sulfoxide
- IBD
- integrase binding domain.
REFERENCES
- 1. Bushman F. D., Craigie R. (1991) Activities of human immunodeficiency virus (HIV) integration protein in vitro: specific cleavage and integration of HIV DNA. Proc. Natl. Acad. Sci. U.S.A. 88, 1339–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Craigie R., Mizuuchi K., Bushman F. D., Engelman A. (1991) A rapid in vitro assay for HIV DNA integration. Nucleic Acids Res. 19, 2729–2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hazuda D. J., Hastings J. C., Wolfe A. L., Emini E. A. (1994) A novel assay for the DNA strand-transfer reaction of HIV-1 integrase. Nucleic Acids Res. 22, 1121–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Savarino A. (2006) A historical sketch of the discovery and development of HIV-1 integrase inhibitors. Expert Opin. Investig. Drugs 15, 1507–1522 [DOI] [PubMed] [Google Scholar]
- 5. Al-Mawsawi L. Q., Al-Safi R. I., Neamati N. (2008) Anti-infectives: clinical progress of HIV-1 integrase inhibitors. Expert Opin. Emerg. Drugs 13, 213–225 [DOI] [PubMed] [Google Scholar]
- 6. Cooper D. A., Gatell J., Rockstroh J., Katlama C., Yeni P., Lazzarin A., Xu X., Isaacs R., Teppler H., Nguyen B. Y., and the BENCHMRK-1 Study Group (2008) 15th Conference on Retroviruses and Opportunistic Infections, Boston, February 3–6, 2008, Abstr. 788, p. 357, CROI and UC San Diego School of Medicine, Alexandria, VA [Google Scholar]
- 7. Steigbigel R., Kumar P., Eron J., Schechter M., Markowitz M., Loutfy M., Zhao J., Isaacs R., Nguyen B. Y., Teppler H., and the BENCHMRK-2 Study Group (2008) 15th Conference on Retroviruses and Opportunistic Infections, Boston, February 3–6, 2008, Abstr. 789, p. 357, CROI and UC San Diego School of Medicine, Alexandria, VA [Google Scholar]
- 8. Markowitz M., Nguyen B. Y., Gotuzzo E., Mendo F., Ratanasuwan W., Kovacs C., Prada G., Morales-Ramirez J. O., Crumpacker C. S., Isaacs R. D., Campbell H., Strohmaier K. M., Wan H., Danovich R. M., Teppler H., and Protocol 004 Part II Study Team (2009) Sustained antiretroviral effect of raltegravir after 96 weeks of combination therapy in treatment-naive patients with HIV-1 infection. J. Acquir. Immune Defic. Syndr. 52, 350–356 [DOI] [PubMed] [Google Scholar]
- 9. Zolopa A. R., Berger D. S., Lampiris H., Zhong L., Chuck S. L., Enejosa J. V., Kearney B. P., Cheng A. K. (2010) Activity of elvitegravir, a once-daily integrase inhibitor, against resistant HIV type 1: results of a phase 2, randomized, controlled, dose-ranging clinical trial. J. Infect. Dis. 201, 814–822 [DOI] [PubMed] [Google Scholar]
- 10. Shimura K., Kodama E. N. (2009) Elvitegravir: a new HIV integrase inhibitor. Antivir. Chem Chemother. 20, 79–85 [DOI] [PubMed] [Google Scholar]
- 11. Mathias A., Lee M., Callebaut C., Xu L., Tsai L., Murray B., Liu H., Yale K., Warren D., Kearney. B. (2009) 16th Conference on Retroviruses and Opportunistic Infections, Montreal, February 8–11, 2009, Abstr. 40, p. 79, CROI and UC San Diego School of Medicine, Alexandria, VA [Google Scholar]
- 12. Min S., Song I., Borland J., Chen S., Lou Y., Fujiwara T., Piscitelli S. C. (2010) Pharmacokinetics and safety of S/GSK1349572, a next-generation HIV integrase inhibitor, in healthy volunteers. Antimicrob. Agents Chemother. 54, 254–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kobayashi M., Yoshinaga T., Seki T., Wakasa-Morimoto C., Brown K. W., Ferris R., Foster S. A., Hazen R. J., Miki S., Suyama-Kagitani A., Kawauchi-Miki S., Taishi T., Kawasuji T., Johns B. A., Underwood M. R., Garvey E. P., Sato A., Fujiwara T. (2011) In vitro antiretroviral properties of S/GSK1349572, a next-generation HIV integrase inhibitor. Antimicrob. Agents Chemother. 55, 813–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. DeJesus E., Cohen C., Elion R., Ortiz R., Maroldo L., Franson S., et al. (2007) 4th International AIDS Society Conference on HIV Pathogenesis, Treatment, and Prevention, Sydney, July 22–25, 2007, Abstr. TUPEB032, International AIDS Society [Google Scholar]
- 15. Wai J., Fisher T., Embrey M., Egbertson M., Vacca J., Hazuda D., Miller M., Witmer M., Gabryelski L., Lyle T. (2007) 14th Conference on Retroviruses and Opportunistic Infections, Los Angeles, February 25–28, 2007, Abstr. 87, p. 96, CROI and UC San Diego School of Medicine, Alexandria, VA [Google Scholar]
- 16. Malet I., Delelis O., Valantin M. A., Montes B., Soulie C., Wirden M., Tchertanov L., Peytavin G., Reynes J., Mouscadet J. F., Katlama C., Calvez V., Marcelin A. G. (2008) Mutations associated with failure of raltegravir treatment affect integrase sensitivity to the inhibitor in vitro. Antimicrob. Agents Chemother. 52, 1351–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Métifiot M., Marchand C., Maddali K., Pommier Y. (2010) Resistance to integrase inhibitors. Viruses 2, 1347–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Al-Mawsawi L. Q., Neamati N. (2011) Allosteric inhibitor development targeting HIV-1 integrase. Chem. Med. Chem. 6, 228–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Engelman A., Englund G., Orenstein J. M., Martin M. A., Craigie R. (1995) Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication. J. Virol. 69, 2729–2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zheng R., Jenkins T. M., Craigie R. (1996) Zinc folds the N-terminal domain of HIV-1 integrase, promotes multimerization, and enhances catalytic activity. Proc. Natl. Acad. Sci. U.S.A. 93, 13659–13664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cai M., Zheng R., Caffrey M., Craigie R., Clore G. M., Gronenborn A. M. (1997) Solution structure of the N-terminal zinc binding domain of HIV-1 integrase. Nat. Struct. Biol. 4, 567–577 [DOI] [PubMed] [Google Scholar]
- 22. Wu X., Liu H., Xiao H., Conway J. A., Hehl E., Kalpana G. V., Prasad V., Kappes J. C. (1999) Human immunodeficiency virus type 1 integrase protein promotes reverse transcription through specific interactions with the nucleoprotein reverse transcription complex. J. Virol. 73, 2126–2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carayon K., Leh H., Henry E., Simon F., Mouscadet J. F., Deprez E. (2010) A cooperative and specific DNA-binding mode of HIV-1 integrase depends on the nature of the metallic cofactor and involves the zinc-containing N-terminal domain. Nucleic Acids Res. 38, 3692–3708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Du L., Shen L., Yu Z., Chen J., Guo Y., Tang Y., Shen X., Jiang H. (2008) Hyrtiosal, from the marine sponge Hyrtios erectus, inhibits HIV-1 integrase binding to viral DNA by a new inhibitor binding site. Chem. Med. Chem. 3, 173–180 [DOI] [PubMed] [Google Scholar]
- 25. Lodi P. J., Ernst J. A., Kuszewski J., Hickman A. B., Engelman A., Craigie R., Clore G. M., Gronenborn A. M. (1995) Solution structure of the DNA binding domain of HIV-1 integrase. Biochemistry 34, 9826–9833 [DOI] [PubMed] [Google Scholar]
- 26. Lutzke R. A., Vink C., Plasterk R. H. (1994) Characterization of the minimal DNA-binding domain of the HIV integrase protein. Nucleic Acids Res. 22, 4125–4131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Williams K. L., Zhang Y., Shkriabai N., Karki R. G., Nicklaus M. C., Kotrikadze N., Hess S., Le Grice S. F., Craigie R., Pathak V. K., Kvaratskhelia M. (2005) Mass spectrometric analysis of the HIV-1 integrase-pyridoxal 5′-phosphate complex reveals a new binding site for a nucleotide inhibitor. J. Biol. Chem. 280, 7949–7955 [DOI] [PubMed] [Google Scholar]
- 28. Maroun R. G., Gayet S., Benleulmi M. S., Porumb H., Zargarian L., Merad H., Leh H., Mouscadet J. F., Troalen F., Fermandjian S. (2001) Peptide inhibitors of HIV-1 integrase dissociate the enzyme oligomers. Biochemistry 40, 13840–13848 [DOI] [PubMed] [Google Scholar]
- 29. Zhao L., O'Reilly M. K., Shultz M. D., Chmielewski J. (2003) Interfacial peptide inhibitors of HIV-1 integrase activity and dimerization. Bioorg. Med. Chem. Lett. 13, 1175–1177 [DOI] [PubMed] [Google Scholar]
- 30. Li H. Y., Zawahir Z., Song L. D., Long Y. Q., Neamati N. (2006) Sequence-based design and discovery of peptide inhibitors of HIV-1 integrase: insight into the binding mode of the enzyme. J. Med. Chem. 49, 4477–4486 [DOI] [PubMed] [Google Scholar]
- 31. Molteni V., Greenwald J., Rhodes D., Hwang Y., Kwiatkowski W., Bushman F. D., Siegel J. S., Choe S. (2001) Acta Crystallogr. D Biol. Crystallogr. 57, 536–544 [DOI] [PubMed] [Google Scholar]
- 32. Al-Mawsawi L. Q., Fikkert V., Dayam R., Witvrouw M., Burke T. R., Jr., Borchers C. H., Neamati N. (2006) Discovery of a small-molecule HIV-1 integrase inhibitor-binding site. Proc. Natl. Acad. Sci. U.S.A. 103, 10080–10085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kessl J. J., Eidahl J. O., Shkriabai N., Zhao Z., McKee C. J., Hess S., Burke T. R., Jr., Kvaratskhelia M. (2009) An allosteric mechanism for inhibiting HIV-1 integrase with a small molecule. Mol. Pharmacol. 76, 824–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Du L., Zhao Y. X., Yang L. M., Zheng Y. T., Tang Y., Shen X., Jiang H. L. (2008) Symmetrical 1-pyrrolidineacetamide showing anti-HIV activity through a new binding site on HIV-1 integrase. Acta Pharmacol. Sin. 29, 1261–1267 [DOI] [PubMed] [Google Scholar]
- 35. McKee C. J., Kessl J. J., Shkriabai N., Dar M. J., Engelman A., Kvaratskhelia M. (2008) Dynamic modulation of HIV-1 integrase structure and function by cellular lens epithelium-derived growth factor (LEDGF) protein. J. Biol. Chem. 283, 31802–31812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tsiang M., Jones G. S., Hung M., Samuel D., Novikov N., Mukund S., Brendza K. M., Niedziela-Majka A., Jin D., Liu X., Mitchell M., Sakowicz R., Geleziunas R. (2011) Dithiothreitol causes HIV-1 integrase dimer dissociation while agents interacting with the integrase dimer interface promote dimer formation. Biochemistry 50, 1567–1581 [DOI] [PubMed] [Google Scholar]
- 37. Cherepanov P., Maertens G., Proost P., Devreese B., Van Beeumen J., Engelborghs Y., De Clercq E., Debyser Z. (2003) HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem. 278, 372–381 [DOI] [PubMed] [Google Scholar]
- 38. Maertens G., Cherepanov P., Pluymers W., Busschots K., De Clercq E., Debyser Z., Engelborghs Y. (2003) LEDGF/p75 is essential for nuclear and chromosomal targeting of HIV-1 integrase in human cells. J. Biol. Chem. 278, 33528–33539 [DOI] [PubMed] [Google Scholar]
- 39. Cherepanov P., Devroe E., Silver P. A., Engelman A. (2004) Identification of an evolutionarily conserved domain in human lens epithelium-derived growth factor/transcriptional co-activator p75 (LEDGF/p75) that binds HIV-1 integrase. J. Biol. Chem. 279, 48883–48892 [DOI] [PubMed] [Google Scholar]
- 40. Emiliani S., Mousnier A., Busschots K., Maroun M., Van Maele B., Tempé D., Vandekerckhove L., Moisant F., Ben-Slama L., Witvrouw M., Christ F., Rain J. C., Dargemont C., Debyser Z., Benarous R. (2005) Integrase mutants defective for interaction with LEDGF/p75 are impaired in chromosome tethering and HIV-1 replication. J. Biol. Chem. 280, 25517–25523 [DOI] [PubMed] [Google Scholar]
- 41. Ciuffi A., Llano M., Poeschla E., Hoffmann C., Leipzig J., Shinn P., Ecker J. R., Bushman F. (2005) A role for LEDGF/p75 in targeting HIV DNA integration. Nature Medicine 11, 1287–1289 [DOI] [PubMed] [Google Scholar]
- 42. Cherepanov P., Sun Z. Y., Rahman S., Maertens G., Wagner G., Engelman A. (2005) Solution structure of the HIV-1 integrase-binding domain in LEDGF/p75. Nat. Struct. Mol. Biol. 12, 526–532 [DOI] [PubMed] [Google Scholar]
- 43. Cherepanov P., Ambrosio A. L., Rahman S., Ellenberger T., Engelman A. (2005) Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc. Natl. Acad. Sci. U.S.A. 102, 17308–17313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vandekerckhove L., Christ F., Van Maele B., De Rijck J., Gijsbers R., Van den Haute C., Witvrouw M., Debyser Z. (2006) Transient and stable knockdown of the integrase cofactor LEDGF/p75 reveals its role in the replication cycle of human immunodeficiency virus. J. Virol. 80, 1886–1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Llano M., Saenz D. T., Meehan A., Wongthida P., Peretz M., Walker W. H., Teo W., Poeschla E. M. (2006) An essential role for LEDGF/p75 in HIV integration. Science 314, 461–464 [DOI] [PubMed] [Google Scholar]
- 46. De Rijck J., Vandekerckhove L., Gijsbers R., Hombrouck A., Hendrix J., Vercammen J., Engelborghs Y., Christ F., Debyser Z. (2006) Overexpression of the lens epithelium-derived growth factor/p75 integrase binding domain inhibits human immunodeficiency virus replication. J. Virol. 80, 11498–11509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hou Y., McGuinness D. E., Prongay A. J., Feld B., Ingravallo P., Ogert R. A., Lunn C. A., Howe J. A. (2008) Screening for antiviral inhibitors of the HIV integrase-LEDGF/p75 interaction using the AlphaScreen luminescent proximity assay. J. Biomol. Screen 13, 406–414 [DOI] [PubMed] [Google Scholar]
- 48. Du L., Zhao Y., Chen J., Yang L., Zheng Y., Tang Y., Shen X., Jiang H. (2008) D77, one benzoic acid derivative, functions as a novel anti-HIV-1 inhibitor targeting the interaction between integrase and cellular LEDGF/p75. Biochem. Biophys. Res. Commun. 375, 139–144 [DOI] [PubMed] [Google Scholar]
- 49. De Luca L., Barreca M. L., Ferro S., Christ F., Iraci N., Gitto R., Monforte A. M., Debyser Z., Chimirri A. (2009) Pharmacophore-based discovery of small-molecule inhibitors of protein-protein interactions between HIV-1 integrase and cellular cofactor LEDGF/p75. Chem. Med. Chem. 4, 1311–1316 [DOI] [PubMed] [Google Scholar]
- 50. Christ F., Voet A., Marchand A., Nicolet S., Desimmie B. A., Marchand D., Bardiot D., Van der Veken N. J., Van Remoortel B., Strelkov S. V., De Maeyer M., Chaltin P., Debyser Z. (2010) Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 6, 442–448 [DOI] [PubMed] [Google Scholar]
- 51. Tsantrizos Y. S., Boes M., Brochu C., Fenwick C., Malenfant E., Mason S., Pesant M. (November 22, 2007) Inhibitors of Human Immunodeficiency Virus Replication. International patent application WO 2007/131350 A1
- 52. Tsantrizos Y. S., Bailey M. D., Bilodeau F., Carson R. J., Coulombe R., Fader L., Halmos T., Kawai S., Landry S., Laplante S., Morin S., Parisien M., Poupart M. A., Simoneau B. (May 22, 2009) Inhibitors of Human Immunodeficiency Virus Replication. International patent application WO 2009/062285 A1
- 53. Tsantrizos Y. S., Bailey M. D., Bilodeau F., Carson R. J., Fader L., Halmos T., Kawai S., Landry S., Laplante S., Simoneau B. (May 22, 2009) Inhibitors of Human Immunodeficiency Virus Replication. International patent application WO 2009/062289 A1
- 54. Tsiang M., Jones G. S., Hung M., Mukund S., Han B., Liu X., Babaoglu K., Lansdon E., Chen X., Todd J., Cai T., Pagratis N., Sakowicz R., Geleziunas R. (2009) Affinities between the binding partners of the HIV-1 integrase dimer-lens epithelium-derived growth factor (IN dimer-LEDGF) complex. J. Biol. Chem. 284, 33580–33599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jones G. S., Yu F., Zeynalzadegan A., Hesselgesser J., Chen X., Chen J., Jin H., Kim C. U., Wright M., Geleziunas R., Tsiang M. (2009) Preclinical evaluation of GS-9160, a novel inhibitor of human immunodeficiency virus type 1 integrase. Antimicrob. Agents Chemother. 53, 1194–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Otwinowski Z. M., Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 57. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 59. Wang Y., Klock H., Yin H., Wolff K., Bieza K., Niswonger K., Matzen J., Gunderson D., Hale J., Lesley S., Kuhen K., Caldwell J., Brinker A. (2005) Homogeneous high-throughput screening assays for HIV-1 integrase 3β-processing and strand transfer activities. J. Biomol. Screen 10, 456–462 [DOI] [PubMed] [Google Scholar]
- 60. Svarovskaia E. S., Barr R., Zhang X., Pais G. C., Marchand C., Pommier Y., Burke T. R., Jr., Pathak V. K. (2004) Azido-containing diketo acid derivatives inhibit human immunodeficiency virus type 1 integrase in vivo and influence the frequency of deletions at two long-terminal-repeat-circle junctions. J. Virol. 78, 3210–3222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hombrouck A., De Rijck J., Hendrix J., Vandekerckhove L., Voet A., De Maeyer M., Witvrouw M., Engelborghs Y., Christ F., Gijsbers R., Debyser Z. (2007) Virus evolution reveals an exclusive role for LEDGF/p75 in chromosomal tethering of HIV. PLoS Pathog. 3, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lataillade M., Chiarella J., Kozal M. J. (2007) Natural polymorphism of the HIV-1 integrase gene and mutations associated with integrase inhibitor resistance. Antiviral Ther. 12, 563–570 [PubMed] [Google Scholar]
- 63. Kessl J. J., Li M., Ignatov M., Shkriabai N., Eidahl J. O., Feng L., Musier-Forsyth K., Craigie R., Kvaratskhelia M. (2011) FRET analysis reveals distinct conformations of IN tetramers in the presence of viral DNA or LEDGF/p75. Nucleic Acids Res. 39, 9009–9022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kessl J. ., Jena N., Koh Y., Taskent-Sezgin H., Slaughter A., Feng L., de Silva S., Wu L., Le Grice S. F., Engelman A., Fuchs J. R., Kvaratskhelia M. (March 21, 2012) Multimode, cooperative mechanism of action of allosteric HIV-1 integrase inhibitors. J. Biol. Chem. 10.1074/jbc.M112.354373 [DOI] [PMC free article] [PubMed] [Google Scholar]