

Abstract
A domino sequence has been developed between vinyldiazoacetates and racemic allyl alcohols, involving five distinct steps. The sequence generates highly functionalized cyclopentanes with four new stereogenic centers as single diastereomers in 64–92% ee. The first step is a rhodium-catalyzed oxygen ylide formation, which is then followed by a [2,3]-sigmatropic rearrangement, an oxy-Cope rearrangement, a keto/enol tautomerization, and then finally a carbonyl ene reaction. With appropriate substrates, a further silyl deprotection and a 6-exo-trig cyclization can be added to the domino process.
Introduction
Stereoselective methods for the formation of five- to seven-membered carbocycles by means of coupling acyclic fragments are important strategic reactions for the synthesis of complex targets.1 The Diels-Alder reaction is the classic transformation for the construction of cyclohexenes containing multiple stereocenters.2 Novel strategies have been developed for the formation of five- and seven-membered carbocycles, and many of these methods emulate the synthetic power of the Diels-Alder reaction.1,3 In this paper, we describe a highly diastereo- and enantioselective reaction between vinyldiazoacetates and racemic allyl alcohols for the synthesis of functionalized cyclopentanes, bearing four new stereogenic centers (Scheme 1).
Scheme 1.

Stereoselective synthesis of cyclopentanes
The discovery of the new cyclopentane synthesis originated from our studies on rhodium-carbenoid chemistry.4 The metal-catalyzed reaction of diazo compounds is an especially useful method for the generation of highly reactive metal-carbenoids, which are then capable of initiating a cascade sequence of reactions.5,6 We have been exploring the development of new cascade sequences that start from vinyldiazoacetates as substrates.7 Recently, we reported the tandem oxygen ylide formation/[2,3]-sigmatropic rearrangement between styryldiazoacetate 1 and allyl alcohols 2, which generated dienols 3 in 92–98% ee (Scheme 2).8 As the tandem oxygen ylide formation/[2,3]-sigmatropic rearrangement is broadly effective, we became intrigued with the possibility that the products of this reaction would be prone to further rearrangement. The results of a systematic study to explore the possibility of generating an extended domino sequence9 is described herein.
Scheme 2.

Tandem oxygen-ylide formation/[2,3]-sigmatropic rearrangement.
Results and discussion
Initial studies and reaction optimization
In order to determine the plausibility of a domino reaction, we examined the reactions of the styryl derivative 3 (R=Me), which is formed in 98% ee using the tandem oxygen ylide/[2,3]-sigmatropic rearrangement.8 Since 4 (Scheme 3) is a 3-hydroxy-1,5-hexadiene containing two adjacent quaternary centers, it was anticipated that this product would be liable to undergo an oxy-Cope rearrangement.10 Indeed, on heating in cyclohexane, 4 underwent a quantitative rearrangement to the enol 5, as a single diastereomer. The reaction did not proceed with complete retention of the optical purity, as the product was formed in 82% ee.
Scheme 3.

Cyclopentane synthesis from 1 and 2.
Further extension of the domino sequence was examined. Enol 5 was not expected to be a stable product, and indeed, on attempted purification by silica gel chromatography, it tautomerized to the ketone 6 as a single diastereomer in 82% ee. As the ketone in 6 is adjacent to an ester carbonyl, 6 would be anticipated to be quite reactive, and it would be reasonable to expect that it would be prone to an intramolecular carbonyl ene reaction11 to form the cyclopentane 7a. Previous examples of tandem oxy-Cope rearrangement/ene reaction sequences are known10a–b The intramolecular ene reaction was readily catalyzed by scandium(III) triflate12 at elevated temperatures, which allowed for the conversion of 6 to the cyclopentane 7a. The new stereogenic centers were formed during the concerted transformations, the oxy-Cope rearrangement and the ene reaction, and consequently, 7a was formed in a highly diastereoselective manner. The absolute configuration of 1,5-hexadiene 4 was assigned in the earlier [2,3]-sigmatropic rearrangement studies,8 and the relative configuration of cyclopentane 7a was determined by X-ray crystallography.13,14 The stereochemistry for the conversion of 4 to 7a is consistent with the oxy-Cope rearrangement of 4 to 5 proceeding via a chair transition state.10,15, Epimerization of the stereocenters in 5 would not be expected in the course of tautomerization or the ene reaction, and so, consistency in the configuration of 7a was anticipated.
Optimization studies were then conducted such that the cascade sequence between the vinyldiazoacetate 1 and allyl alcohol 2a to form the cyclopentane 7a could be conducted as a one-pot process. It was envisioned that after the initial enantioselective rhodium-catalyzed step, the rest of the sequence should be feasible by using the appropriate combination of solvent, temperature and/or Lewis acid catalyst. Table 1 summarizes the optimization of the reaction between 1 and 2a. Prior studies had shown that the optimum temperature for the tandem oxygen ylide formation/[2,3]-sigmatropic rearrangement was 0 °C.8 After completion of the [2,3]-rearrangement, the crude product mixture was heated in the parent solvent for 20 h before treating with a catalytic amount of scandium(III) triflate for 4 h. At temperatures less than ~75 °C, only the [2,3]-product was observed in the 1H NMR of the crude reaction mixture (entry 1). By a modest increase in temperature, however, the desired product 7a was isolated in generally good yields (entries 2, 4–9). In a more polar solvent system, such as ethyl acetate, attenuated yields and enantioselectivities were observed (entry 3). The optimum conditions were found to be 1 mol % of the rhodium catalyst in heptane at 80 °C. Under these conditions 7a was formed in 95% yield and 82% ee. (entry 7). These conditions were used as the standard for the remainder of the study, although in some cases high yield of product could be obtained without the need of adding scandium(III) triflate. In these cases, however, extended reaction times were required as the ene reaction was rather sluggish. It should be noted that much lower catalyst loading can be used if desired,16 because on decreasing the catalyst loading to 0.01 mol % (entry 9) only a slight change in yield and enantioselectivity (75% yield, 79% ee) was observed.
Table 1.
Optimization of the one-pot cyclopentane synthesis.
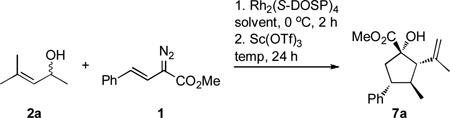 | ||||||
|---|---|---|---|---|---|---|
| entry | Rh2(S-DOSP)4 | solvent | temp (°C) |
yielda (%) |
drb | eec (%) |
| 1 | 1 mol % | n-Hex | 67 | 0 | - | - |
| 2 | 1 mol % | DCE | 83 | 43 | > 20 : 1 | 54 |
| 3 | 1 mol % | EtOAc | 77 | 18 | > 20 : 1 | 51 |
| 4 | 1 mol % | PhCH3 | 110 | 71 | > 20 : 1 | 70 |
| 5 | 1 mol % | c-Hex | 81 | 91 | > 20 : 1 | 74 |
| 6 | 1 mol % | Hep | 98 | 88 | > 20 : 1 | 80 |
| 7 | 1 mol % | Hep | 80 | 95 | > 20 : 1 | 82 |
| 8 | 0.1 mol % | Hep | 80 | 85 | > 20 : 1 | 79 |
| 9 | 0.01 mol % | Hep | 80 | 75 | > 20 : 1 | 79 |
Isolated yield.
Determined from 1H NMR of crude reaction mixture.
Determined by chiral HPLC.
Substrate scope
The next series of experiments explored the general scope of the reaction with respect to allyl alcohols bearing a 3,3´-dimethyl moiety (Table 2). The steric tolerance of the reaction was probed through substrates with various linear and branched alkyl chains (entries 1–4). All aliphatic substituents examined proved efficient substrates for the transformation, affording the cyclopentanes 7a–i in 67–95% yield. Substituents bearing a functionalized R-group, including an olefin, ketal, silyl ether, phenyl, and silyl (entries 5–9) were all compatible, indicative of the diverse functionalization one could install at the C3-position of 7. In the case of the ketal-protected alcohol 2f (entry 6), however, the product was obtained as the deprotected ketone due to the acidic reaction medium. Similarly, under the prescribed conditions, the silyl ether 2g underwent deprotection and subsequent side reactions upon exposure to scandium(III) triflate. By increasing the temperature, however, to 98 °C after the 20 h period, the corresponding cyclopentane 7g was obtained in excellent yield (65%) in the absence of Lewis acid catalyst.
Table 2.
Scope of the one-pot cyclopentane synthesis with alcohols 2.
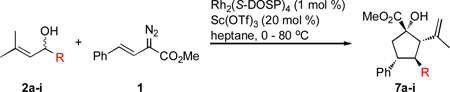 | |||||
|---|---|---|---|---|---|
| entry | comp. | R | yielda (%) |
drb | eec (%) |
| 1 | a | Me | 95 | > 20 : 1 | 82 |
| 2 | b | i-Pr | 67 | > 20 : 1 | 80 |
| 3 | c | i-Bu | 73 | > 20 : 1 | 80 |
| 4 | d | n-Hex | 80 | > 20 : 1 | 78 |
| 5 | e | 86 | > 20 : 1 | 76 | |
| 6 | f | 45d | > 20 : 1 | 90 (96)e | |
| 7 | g | 65f | > 20 : 1 | 78 | |
| 8 | h | Bn | 42 | > 20 : 1 | 87 |
| 9 | i | 59 | > 20 : 1 | 84 | |
Isolated yield.
Determined from 1H NMR of crude reaction mixture.
Determined by chiral HPLC.
Yield of the deprotected ketone.
Number in parentheses indicates ee of recrystallized product.
Reaction conducted in refluxing heptane in the absence of Sc(OTf)3.
The reaction of various vinyldiazoacetates 8 was then explored, which introduced different functionality at the C4-position of the cyclopentane 9 (Table 3). The effect of substituents on the aryl ring was probed (entries 1–3). The best result was obtained for the electron rich 4-methoxy group in 8b; however, substrates 8a and 8c with electron poor groups (entries 1 and 3) also afforded the desired products. Moreover, for the p-methoxy and p-bromostyryldiazoacetates, 8b and 8c, an improvement in enantioselectivity was observed. The relatively low yield for 8c was associated with competing O—H insertion versus the [2,3]-sigmatropic rearrangement.8 The E-methyl hexenoate 8d (entry 4) was an efficient substrate; however, the enantioselectivity of the [2,3]-rearrangement in this case was modest, and this translated to low enantioselectvity (64% ee) in the corresponding cyclopentane 9d. The absolute configuration of 9c was determined by X-ray crystallography.13 Since equivalent transitions states are presumed to be involved in the formation of 7a–i, 9a, 9b, and 9d, the absolute configuration of those products was assigned by analogy.
Table 3.
Scope of the one-pot cyclopentane synthesis with diazoacetates 8.
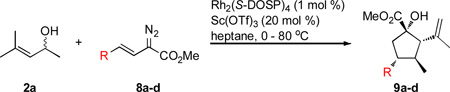 | |||||
|---|---|---|---|---|---|
| entry | comp. | R | yielda (%) |
drb | eec (%) |
| 1 | a | p-CF3C6H4 | 63 | > 20 : 1 | 78 |
| 2 | b | p-(OMe)C6H4 | 94 | > 20 : 1 | 87 |
| 3 | c | p-BrC6H4 | 48 | > 20 : 1 | 92 |
| 4 | d | Et | 63 | > 20 : 1 | 64 |
Isolated yield.
Determined from 1H NMR of crude reaction mixture.
Determined by chiral HPLC.
Further examples
The domino sequence could be extended to include two additional steps when the mono-silyl protected diol 10 was used as substrate. (Scheme 4). When the standard reaction was conducted, but now using an equivalent of scandium(III) triflate in refluxing heptane, the reaction did not stop at the cyclopentane 11. Instead, the fused pyran 12 was isolated as a single diastereomer in 80% ee. The formation of 12 is likely to proceed by silyl deprotection and a 6-exo-trig cyclization. The order of the last two steps in not known. The relative configuration of the trans-fused ring system was determined by X-ray crystallography.12,13
Scheme 4.

Domino ylide formation/[2,3]-sigmatropic rearrangement/Oxy-Cope rearrangement/tautomerization/Ene reaction/silyl deprotection/6-exo-trig cyclization.
The synthetic utility of the domino sequence was showcased in the reaction with the readily available diterpenoid, (−)-pulegol (13). Two diastereomeric hydrindane products 14 and 15 are possible, containing five stereogenic centers, two of which are quaternary. We have already demonstrated that the diastereoselectivity of the ylide formation/2,3-sigmatropic rearrangement with (−)-pulegol (13) is controlled by the chiral catalyst, with Rh2(R-DOSP)4 as catalyst giving a 10 : 1 diastereomeric mixture and Rh2(S-DOSP)4 giving a 4 : 1 diastereomeric mixture, favoring the other diastereomer.8 In contrast, the enantiomers of the catalyst show very different levels of diastereocontrol in the domino sequence. In the Rh2(R-DOSP)4-catalyzed reaction between 13 and 1, the single diastereomer 14 of the hydrindane was formed in 69% yield. In the Rh2(S-DOSP)4-catalyzed reaction between 13 and 1, a 1 : 2 mixture of the diastereomers 14 and 15 was formed.17 The reason for the difference in diastereocontrol will be discussed later.
Stereochemical rationale
One of the most impressive features of the domino sequence for the synthesis of cyclopentanes is the high level of diastereocontrol possible in the sequence. Four new stereogenic centers are generated and the monocyclic products are produced as single diastereomers with 64–92% ee. The enantiomeric purity of the cyclopentanes is slightly degraded compared to the enantiomeric purity of the initial 2,3-sigmatropic rearrangement products. The enantioselectivity of the tandem ylide formation/2,3-sigmatropic rearrangement is already established.8 The highly stereoselective nature of the oxy-Cope and the anionic oxy-Cope has been documented in detail.15,18 In general, in the absence of significant 1,3-diaxial interactions, a chair-like transition state is strongly favored.18 Two additional stereogenic centers are generated in the ene reaction, in which the relative stereochemistry is presumably controlled by the already established stereocenters.
From the exploratory studies in Scheme 3, it is clear that the slight loss of enantiomeric purity occurs during the oxy-Cope rearrangement. This can be explained by considering the two possible chair-like transition states TS-C1 and TS-C2 for the [3,3]-rearrangement portrayed in Figure 1. Enantiocontrol will depend on the axial/equatorial preference of the hydroxyl or the carbomethoxy groups. The cyclohexane A-values of the hydroxyl and carbomethoxy substituents are 0.60–1.04 and 1.2–1.3 kcal mol−1, respectively.19 Therefore the TS-C1, with the hydroxyl group in an axial position should be favored, forming (4S,5R)-5a as the predominant product, but the difference in A-values is not sufficient to eliminate some of the product formed via TS-C2. Both transition states generate the same diastereomer but opposite enantiomers of the product.
Figure 1.

Transition states for the oxy-Cope rearrangement.
For the reaction with (−)-pulegol, the major diastereomer produced in the Rh2(R-DOSP)4-catalyzed reaction with 1 has been shown to be the [2,3]-sigmatropic rearrangement product 16.8 Compound 16 is ideally suited to undergo a stereoselective oxy-Cope rearrangement as illustrated in Figure 2. In the chair transition state TS-C3, the less sterically demanding hydroxyl group is adopting the axial position and the remote C9-methyl group is oriented away from the site of bond formation, minimizing any unfavorable steric interactions. Transition-state TS-C3 leads to the formation of the observed diastereomer 14. In the case of the major diastereomer of the [2,3]-sigmatropic rearrangement product from the Rh2(S-DOSP)4-catalyzed reaction, neither of the two possible chair forms is strongly favored because one would have the ester group in the axial position and the other would have bond formation occurring on the same face as the C9-methyl group.
Figure 2.

Oxy-Cope transition state for the reaction with (−)-pulogol.
A stereochemical analysis of the highly diastereoselective carbonyl ene reaction is shown in Figure 3. The stereoelectronic requirement for transposition of the ene and enophile for the carbonyl ene reaction limits the possible transition states to the two boat-like TS-B1 would be expected to be strongly favored, because TS-B2 would have substantial pseudo 1,3-diaxial interactions between the phenyl and carbomethoxy moieties.
Figure 3.

Transition states for the carbonyl ene reaction.
Conclusions
In summary, we have developed a novel, multi-step, domino sequence for the asymmetric synthesis of highly decorated cyclopentanes. The domino sequence proceeds in high yield and with well-defined stereochemistry. These studies demonstrate the synthetic utility of highly reactive metal-carbenoids to initiate an elaborate sequence of reactions, resulting in the rapid generation of synthetic complexity.
Supplementary Material
Scheme 5.

Derivitization of (−)-pulogeol 13.
Acknowledgements
This material is based on work supported by the National Institutes of Health (GM080337). We thank Dr. Ken Hardcastle for the X-ray crystallographic structural determination.
Footnotes
Electronic Supplementary Information (ESI) available: [Full experimental data and spectral data]. See DOI: 10.1039/b000000x/
General Procedure for One-Pot Cyclopentane Synthesis. An oven-dried, 25 mL round-bottomed flask, equipped with a stir bar, was capped with a rubber septum and placed under a dry argon atmosphere. The reaction vessel was charged with Rh2(S-DOSP)4 (19 mg, 0.01 mmol, 0.01 equiv) and the allyl alcohol (1.0 mmol, 1.0 equiv) in heptane (1.0 mL). The solution was cooled to 0 °C in an ice bath before adding a heptane solution (10 mL) of the diazo compound (1.1 mmol, 1.1 equiv) drop-wise over 30 min. Following addition, the reaction was stirred at 0 °C for 2 h before warming to rt for 30 min. The rubber septum was removed and the reaction flask was fixed with a reflux condenser and heated to 80 °C for 24 h or until TLC indicated complete conversion of the [2,3]-rearrangement product to a mixture of oxy-Cope and ene products. Scandium triflate (98 mg, 0.20 mmol, 0.20 equiv) was then added in a single portion and the reaction was heated for an addition 2 h or until TLC indicated complete conversion of the oxy-Cope product to the cyclopentane. The reaction was then cooled to ambient temperature and concentrated in vacuo. The product was purified by flash chromatography.
Notes and references
- 1.(a) Carreira EM, Kvaerno L. Classics in Stereoselective Synthesis. Weinheim: Wiley-VCH Verlag GmbH & Co.; 2009. [Google Scholar]; (b) Takao K-I, Munakata R, Tadano K-I. Chem. Rev. 2005;105:4779. doi: 10.1021/cr040632u. [DOI] [PubMed] [Google Scholar]; (c) Crimmins MT, Ellis JM. J. Org. Chem. 2008;73:1649. doi: 10.1021/jo0712695. [DOI] [PubMed] [Google Scholar]; (d) Kobayashi S, Jørgensen KA. Cycloaddition Reactions in Organic Synthesis. Weinheim: Wiley-VCH Verlag GmbH & Co.; 2002. [Google Scholar]; (e) Bach T, Hehn JP. Angew. Chem., Int. Ed. 2011;50:1000. doi: 10.1002/anie.201002845. [DOI] [PubMed] [Google Scholar]; (f) Harmata M. Chem. Commun. 2010;46:8904. doi: 10.1039/c0cc03621h. [DOI] [PubMed] [Google Scholar]; (g) Buchanan GS, Feltenberger JB, Hsung RP. Current Organic Synthesis. 2010;7:363. doi: 10.2174/157017910791414490. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Taylor MS, Jacobsen EN. Angew. Chem., Int. Ed. 2006;45:1520. doi: 10.1002/anie.200503132. [DOI] [PubMed] [Google Scholar]
- 2.(a) Kagan HB, Riant O. Chem. Rev. 1992;92:1007. [Google Scholar]; (b) Wessig P, Müller G. Chem. Rev. 2008;108:2051. doi: 10.1021/cr0783986. [DOI] [PubMed] [Google Scholar]; (c) Brieger G, Bennett JN. Chem. Rev. 1980;80:63. [Google Scholar]; (d) Martin JG, Hill RK. Chem. Rev. 1961;61:537. [Google Scholar]; (e) Whiting A. Asymmetric Diels-Alder Reactions. In: Stephenson GR, editor. Advanced Asymmetric Synthesis. 1st ed. London: Blackie Academic & Professional; 1996. p. 126. [Google Scholar]; (f) Oppolzer W. Angew. Chem., Int. Ed. 1984;23:840. [Google Scholar]
- 3.(a) Pellissier H. Adv. Synth. Catal. 2011;353:189. [Google Scholar]; (b) Butenschoen H. Angew. Chem., Int. Ed. 2008;47:5287. doi: 10.1002/anie.200801738. [DOI] [PubMed] [Google Scholar]; (c) Le Marquand P, Tam W. Angew. Chem., Int. Ed. 2008;47:2926. doi: 10.1002/anie.200705481. [DOI] [PubMed] [Google Scholar]; (d) Widenhoefer RA. Angew. Chem., Int. Ed. 2009;48:6950. doi: 10.1002/anie.200902404. [DOI] [PubMed] [Google Scholar]; (e) Davies HML, Denton JR. Chem. Soc. Rev. 2009;38:3061. doi: 10.1039/b901170f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Harmata M. Adv. Synth. Catal. 2006;348:2297. [Google Scholar]
- 4.(a) Davies HML, Beckwith REJ. Chem. Rev. 2003;103:2861. doi: 10.1021/cr0200217. [DOI] [PubMed] [Google Scholar]; (b) Davies HML, Morton D. Chem. Soc. Rev. 2011;40:1857. doi: 10.1039/c0cs00217h. [DOI] [PubMed] [Google Scholar]
- 5.For recent reviews see: Davies HML, Dai X. Total Synthesis of Natural Products using the Combined C-H Activation/Cope Rearrangement as the Key Step. In: Harmata M, editor. Strategies and Tactics in Organic Synthesis. 1st ed. Vol. 7. Hungary: Elsevier, Ltd.; 2008. p. 383. Doyle MP, McKervy MA, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides. New York: John Wiley & Sons, Inc.; 1998. Padwa A. J. Org. Chem. 2009;74:6421. doi: 10.1021/jo901300x. Padwa A. Chem. Soc. Rev. 2009;38:3072. doi: 10.1039/b816701j.
- 6.For recent examples see: Hu W, Xu X, Zhou J, Liu W-J, Huang H, Hu J, Yang L, Gong L-Z. J. Am. Chem. Soc. 2008;130:7782. doi: 10.1021/ja801755z. Yan M, Jacobsen N, Hu W, Gronenberg SS, Doyle MP, Coyler JT, Bykowski D. Angew. Chem., Int. Ed. 2004;43:6713. doi: 10.1002/anie.200461722. Prein M, Padwa A. Tetrahedron Lett. 1996;37:6981.
- 7.(a) Lian Y, Miller LC, Born S, Sarpong R, Davies HML. J. Am. Chem. Soc. 2010;132:12422. doi: 10.1021/ja103916t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Davies HML, Dai X. J. Am. Chem. Soc. 2006;128:2485. doi: 10.1021/ja056877l. [DOI] [PubMed] [Google Scholar]; (c) Schwartz BD, Denton JR, Lian Y, Davies HML, Williams CM. J. Am. Chem. Soc. 2009;131:8329. doi: 10.1021/ja9019484. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Davies HML, Jin Q. J. Am. Chem. Soc. 2004;126:10862. doi: 10.1021/ja047185k. [DOI] [PubMed] [Google Scholar]
- 8.Li Z, Davies HML. J. Am. Chem. Soc. 2010;132:396. doi: 10.1021/ja9075293. [DOI] [PubMed] [Google Scholar]
- 9.Nicolaou KC, Edmonds DJ, Bulger PG. Angew. Chem., Int. Ed. 2006;45:7134. doi: 10.1002/anie.200601872. [DOI] [PubMed] [Google Scholar]
- 10.(a) Warrington JM, Yap GPA, Barriault L. Org. Lett. 2000;2:663. doi: 10.1021/ol005502w. [DOI] [PubMed] [Google Scholar]; (b) Arns S, Barriault L. Chem. Commun. 2007:2211. doi: 10.1039/b700054p. [DOI] [PubMed] [Google Scholar]; (c) Nubbemeyer U. Synthesis. 2003:961. [Google Scholar]; (d) Mehta G, Reddy KS. Synlett. 1996:625. [Google Scholar]; (e) Jacobi PA, Selnick HG. J. Org. Chem. 1990;55:202. [Google Scholar]; (e) Ladouceur G, Paquette LA. J. Org. Chem. 1989;54:4278. [Google Scholar]; (g) Janardhanam S, Rajagopalan K. J. Chem. Soc., Perkin Trans. 1. 1992:2727. [Google Scholar]
- 11.(a) Grachan ML, Tudge MT, Jacobsen EN. Angew. Chem., Int. Ed. 2008;47:1469. doi: 10.1002/anie.200704439. [DOI] [PubMed] [Google Scholar]; (b) Bigot A, Breuninger D, Breit B. Org. Lett. 2008;10:5321. doi: 10.1021/ol8016148. [DOI] [PubMed] [Google Scholar]; (c) Barbero A, Castreño P, Garc a C, Pulido FJ. J. Org. Chem. 2001;66:7723. doi: 10.1021/jo0158736. [DOI] [PubMed] [Google Scholar]; (d) Williams JT, Bahia PS, Snaith JS. Org. Lett. 2002;4:3727. doi: 10.1021/ol0266929. [DOI] [PubMed] [Google Scholar]; (e) Hoye TR, Kyle JB, Caruso AJ, Dellaria JF. J. Org. Chem. 1980;45:4287. [Google Scholar]; (f) Song Z, Beak P. J. Am. Chem. Soc. 1990;112:8126. [Google Scholar]
- 12.Yang D, Yang M, Zhu N-Y. Org. Lett. 2003;5:3749. doi: 10.1021/ol035486d. [DOI] [PubMed] [Google Scholar]
- 13.On attempted recrystallization of enantioenriched 7a and 12, the crystalline material that was obtained was the racemate, and so, the X-ray determination from these materials, only gave the relative configuration of 7a and 12.
- 14.The crystal structures of 7a, 9c, and 12 have been deposited at the Cambridge Crystallographic Data Centre under deposition numbers CCDC 827543, 827544, and 827545, respectively.
- 15.(a) Paquette LA, Teleha CA, Taylor RT, Maynard GD, Rogers RD, Gallucci JC, Springer JP. J. Am. Chem. Soc. 1990;112:265. [Google Scholar]; (b) Paquette LA, Maynard GD. J. Am. Chem. Soc. 1992;114:3010. [Google Scholar]; (c) Lee E, Shin I-J, Kim T-S. J. Am. Chem. Soc. 1990;112:260. [Google Scholar]; (d) Koreeda M, Tanaka Y, Schwartz A. J. Org. Chem. 1980;45:1172. [Google Scholar]
- 16.Pelphrey P, Hansen J, Davies HML. Chem. Sci. 2010;1:254. [Google Scholar]
- 17.The configuration of 14 was determined by 1D-NOE and COSY NMR studies.
- 18.Evans DA, Baillargeon DJ, Nelson JV. J. Am. Chem. Soc. 1978;100:2242. [Google Scholar]
- 19.Anslyn EV, Dougherty DA. Modern Physical Organic Chemistry. United States of America: University Science Books; 2006. [Google Scholar]
- 20.(a) Oppolzer W, Snieckus V. Angew. Chem., Int. Ed. 1978;17:476. [Google Scholar]; (b) Hoffmann HMR. Angew. Chem., Int. Ed. 1969;8:556. [Google Scholar]; (c) Zhao Y-J, Li B, Tan L-JS, Shen Z-L, Loh T-P. J. Am. Chem. Soc. 2010;132:10242. doi: 10.1021/ja104119j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.