

Abstract
Targeting native progenitors with small-molecule pharmaceuticals that direct cell fate decisions is an attractive approach for regenerative medicine. Here, we show that 3,5-disubstituted isoxazoles (Isx), stem cell-modulator small-molecules originally recovered in a P19 embryonal carcinoma cell-based screen, directed cardiac muscle gene expression in vivo in target tissues of adult transgenic reporter mice. Isx also stimulated adult mouse myocardial cell cycle activity. Narrowing our focus onto one target cardiac-resident progenitor population, Isx directed muscle transcriptional programs in vivo in multipotent Notch-activated epicardium-derived cells (NECs), generating Notch-activated adult cardiomyocyte-like precursors. Myocardial infarction (MI) preemptively differentiated NECs towards fibroblast lineages, overriding Isx's cardiogenic influence in this cell population. Isx dysregulated gene expression in vivo in Notch-activated repair-fibroblasts, driving distinctive (pro-angiogenesis) gene programs, but failed to mitigate fibrosis or avert ventricular functional decline after MI. In NECs in vitro, Isx directed partial muscle differentiation, which included biosynthesis and assembly of sarcomeric α-actinin premyofibrils, beaded structures pathognomonic of early developing cardiomyocytes. Thus, although Isx small-molecules have promising in vivo efficacy at the level of cardiac muscle gene expression in native multipotent progenitors – and are first in class in this regard – a greater understanding of the dynamic interplay between fibrosis and cardiogenic small-molecule signals will be required to pharmacologically enable regenerative-repair of the heart.
Ischemic disease of the heart is almost a sine qua non of human aging and, indeed, most people will die from ischemic heart disease or its major sequelae, heart failure or arrhythmias (1). Myocardial infarction (MI) is the most catastrophic manifestation of ischemic heart disease and is associated with the nearly instantaneous loss of millions if not billions of cardiomyocytes, the working cells of the heart muscle. MI is a new problem, only seen in modern man and therefore unanticipated by evolution. Instead of rebuilding muscle, the MI-injured heart undergoes fibrosis-repair, which replaces infarcted tissue with fibroblasts and extracellular matrix, generating a tough scar that withstands elevated filling pressures but does not conduct electricity or actively contract.
Redirecting the heart's fibrosis-repair program to generate new vasculature and muscle instead of scar is one of regenerative medicine's most challenging and important clinical targets. Like many tissues, the adult mammalian heart has dynamically-regulated pools of resident stem/progenitor cells responsible for life-long tissue homeostasis (2). There is growing appreciation that cardiac-resident stem/progenitor cells, in particular epicardium-derived cells, participate in fibrosis-repair responses (3). Small-molecules targeting native heart stem/progenitor cells (or their microenvironments) that modulate fate decisions are important experimental tools and could become future pharmaceuticals for promoting regenerative heart repair (4-7).
We recently characterized a mouse model that tags Notch pathway-activated progenitors with enhanced green fluorescent protein (EGFP) (transgenic Notch reporter mice, TNR; CBF1-REx4-EGFP), and more specifically, identified NECs, CBF1-REx4-EGFP+CD31−CD45− multipotent stromal cells (MSCs), a cell population that dynamically responds to injury and participates in fibrosis-repair of the adult heart (8). The Notch-activated state in EGFP+CD31−CD45− cells, we believe, is a surrogate marker of adult multipotency and NECs are competent for multi-lineage differentiation in vivo and in vitro.
We recovered a collection of stem cell-modulator small-molecules from a high throughput screen based on activation of an Nkx2-5 luciferase knock-in bacterial artificial chromosome (BAC) integrated into the genome of P19CL6 embryonal carcinoma cells (9). Among this collection, we identified a family of 3,5-disubstituted isoxazole small-molecules that robustly activated the Nkx2-5 BAC transgene (and the native Nkx2-5 locus) in P19CL6 reporter cells (9). Collateral studies of Isx small-molecules demonstrated that this compound family induced neuronal differentiation in HCN, a clonally derived neural stem/progenitor cell line from the adult rat brain hippocampus (10). Isx small-molecules also regulated growth and differentiation in malignant astrocytoma cells (11) and promoted insulin biosynthesis in pancreatic islet β cells (12). Here, we return our attention to Isx's cardiogenic activity.
Translating in vitro-defined small-molecule bioactivities into a therapeutically beneficial in vivo functionality is one of the greatest experimental hurdles in chemical biology and medicinal chemistry. There is a myriad of reasons why small-molecules can be efficacious in vitro but not in vivo, including issues of solubility, metabolism and toxicity. Here, we explore for the first time the in vivo efficacy of our Isx small-molecules, primarily focusing on a cyclo-propyl analog obtained through in vitro structure-activity relationship studies (10). Our data show that Isx, administered to adult mice as a once daily intra-peritoneal (ip) injection, robustly activated cardiac gene programs in multipotent NECs in vivo, a promising start for a cardio-regenerative small-molecule. However, when combined with MI, NECs from Isx treated mice unexpectedly displayed decreased cardiac gene expression. Moreover, Isx failed to significantly alter the normal course of fibrosis-repair or pump functional deterioration after large MI. Thus, while our results provide proof-of-concept that Isx small-molecules target native progenitor cells in vivo and have the desired gene expression effect, they also highlight the complex challenges of developing cardio-regenerative drugs that function in a signal rich native microenvironment that dominantly promotes scar formation after serious injury.
Results and Discussion
Isx1 pharmacologically targets and regulates myocardial cells in vivo
We evaluated the in vivo efficacy of our cyclopropyl-amide analog (henceforth called Isx1) (Fig. 1a) (10); a sparingly soluble organic compound formulated in 20% 2-hydroxypropyl-β-cyclodextrin. In pharmacokinetic surveys, we detected Isx1 in the tissue of interest – heart – even though the compound's terminal half-life in plasma after single bolus ip injection was <15 minutes (data not shown). Importantly, mice treated with Isx1 daily for several weeks had no signs or symptoms of lethargy, weight loss or decreased appetite, and their serum biochemistry and hematology profiles were completely normal (data not shown). For certain experiments, we validated our results with a second structurally distinct Isx analog, Isx2, a linear aliphatic alkene chiral alcohol) (Fig. 1a).
Figure 1. Isx1 pharmacologically targets and regulates myocardial cells in vivo.
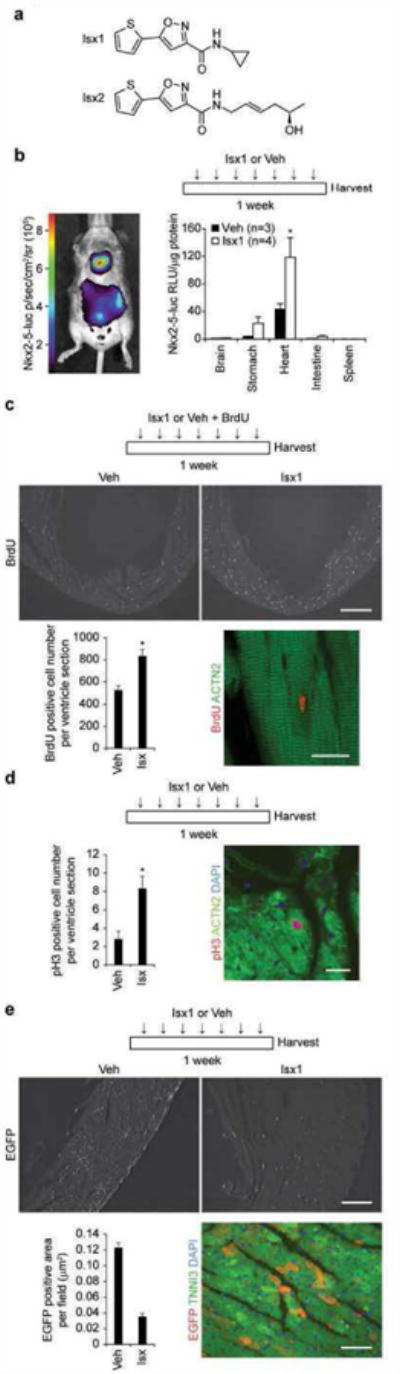
(a) Chemical structures of Isx1 (N-cyclopropyl-5-(thiophen-2-yl)isoxazole-3-carboxamide) and Isx2 ((R,E)-N-(5-hydroxyhex-2-enyl)-5-(thiophen-2-yl)isoxazole-3-carboxamide) compounds. (b) Time map of Isx1 in vivo treatment paradigm. Bioluminescence imaging of Nkx2-5-luc-BAC mouse demonstrating cardiac and stomach (a known extra-cardiac target) restricted luciferase activity after Isx injection in vivo. Quantitative evaluation of Isx-induced Nkx2-5-luc-BAC activity normalized per μg protein in tissue extracts from reporter mice injected ip once daily for 7 days with cyclodextrin (Veh, n=3) or Isx1 at 16 mg/kg (n=4). (c) Time map of Isx1 or Veh plus BrdU in vivo treatment paradigm. BrdU immunohistochemistry in uninjured adult mouse heart injected with vehicle or Isx (top panels, scale bar = 500μm). Quantitative evaluation of BrdU+ cells per ventricle section (9 sections from 3 animals were counted for each group, * = p<0.005). Immunohistochemistry localizing a BrdU+ nucleus (red) to an ACTN2+ cardiomyocyte (green) in an uninjured adult mouse injected with Isx (bottom right panel, Scale bar = 20μm). (d) Time map of Isx1 in vivo treatment paradigm. Quantitative evaluation of pH3+ cells per ventricle section in uninjured adult mice injected with Veh or Isx1 for 7 days (6 sections from 3 animals were counted for each group, * = p<0.05). Phospho-histone H3+ nucleus (red) localizing to an ACTN2+ cardiomyocyte (green), co-stained with DAPI (blue), in an uninjured adult mouse injected with Isx (right hand panel, Scale bar = 30μm). (e) Time map of Isx1 in vivo treatment paradigm. EGFP immunohistochemistry in uninjured adult mouse heart injected with Veh or Isx1 (top panels, scale bar = 100μm). Quantitative evaluation of EGFP+ area per field (18 fields per group) in uninjured adult mice treated with Veh or Isx1 for 7 days. EGFP+ (red) immunohistochemistry localized to TNNI3+ (green) cardiomyocytes, co-stained with DAPI (blue) (bottom right panel, scale bar = 30μm).
First, we probed Isx1's in vivo bioactivity in our Nkx2-5-luc-BAC transgenic reporter mice, whole animal counterparts to the Nkx2-5-luc-BAC P19CL6 cells used in the original high throughput chemical screen (Fig. 1b) (9). After one week of daily ip injection, Isx1 increased steady state luc activity (per microgram of tissue protein) in target tissues (heart and stomach, a known extra-cardiac Nkx2-5 expression site), but not in spleen, brain or intestine (Fig. 1b). Thus, systemically administered Isx1 specifically activated the original target reporter gene in expected target tissues, a promising in vivo starting point, demonstrating this compound's potential in vivo cardiogenic function.
Pharmacologically targeting the heart in vivo, Isx demonstrated several additional biological activities, including increased myocardial cell cycle and Notch pathway activity. For example, we observed increased cell cycle activity in myocardial cells of adult mice treated with Isx1 (Fig. 1c). We co-injected adult mice with Isx1 or vehicle and the DNA synthesis tracer, bromodeoxyuridine (BrdU), daily for one week and then immunostained histological sections of perfusion fixed hearts for BrdU incorporated into myocardial cellular DNA (Fig. 1c, upper panels). We counted the total number of BrdU+ myocardial cells in serial sections from replicate Isx1 or vehicle treated mice. When compared with vehicle, Isx1 significantly increased the number of BrdU+ myocardial cells (non-myocytes and cardiomyocytes) by almost 50% (Fig. 1c, bottom left hand bar graph). Although the majority of BrdU+ myocardial cells were non-myocytes, we also observed BrdU+ cardiomyocytes – extremely rarely in vehicle treated mice and significantly more frequently in Isx1 treated mice (Fig. 1c, bottom right hand panel and Supplemental Fig. 1).
To independently corroborate these results, we evaluated serial heart sections by phosphohistone H3 (pH3) immunohistochemistry. As expected, we detected far fewer pH3+ (a snapshot) as compared to BrdU+ myocardial cells (a weeklong pulse), but once again Isx1 significantly increased pH3+ cell number positivity, almost 4-fold (Fig. 1d, bottom left hand bar graph). Once again, we observed rare pH3+ cardiomyocytes essentially only in Isx1 treated mouse hearts (Fig. 1d, bottom right hand panel and Supplemental Figure 2). Although additional studies will be required to identify and enumerate the exact myocardial cell types involved and characterize the functional consequences, these BdrU and pH3 data independently confirmed that Isx1 small-molecules – administered systemically – significantly increased cell cycle activity in the adult mouse heart.
Reporting activity of a complex signaling cascade (at the downstream level of target gene activation) involved in cardiomyogenesis, the TNR mouse presents a unique opportunity to examine how pathophysiologic states or pharmacological agents regulate Notch pathway signaling in vivo. Treating TNR with Isx1 daily for 7 days resulted in overall decreased myocardial CBF1-REx4-EGFP detected by immunohistochemistry (Fig. 1e, upper panels). We evaluated the total number of EGFP+ myocardial cells in serial sections from replicate Isx1 or vehicle treated adult TNR mice using an indirect computer-based scanning method that counted the EGFP+ area per field (Fig. 1e). When compared with vehicle, Isx1 markedly decreased the EGFP+ area per field, more than 4-fold (Fig. 1e, bottom left hand bar graph). Although the majority of EGFP+ myocardial cells were non-myocytes, we readily confirmed EGFP+ cardiomyocytes in Isx treated mice (Fig. 1e, bottom right hand panel). Although we only observed a single EGFP+ cardiomyocyte in all of the sections studied from vehicle treated mice, EGFP+ cardiomyocytes were numerous in sections from Isx treated mice, and these cardiomyocytes often localized in clusters and pairs (Fig. 1e, bottom right hand panel and Supplemental Fig. 3). Thus, although Isx1 decreased Notch pathway activity in myocardial cells overall, it markedly increased EGFP localization in cardiomyocytes of the adult TNR mouse heart.
Taken together, these results established that Isx1 targeted myocardium and tissue-specifically activated a transgenic Nkx2-5 reporter gene. Isx1 also increased cell cycle activity in myocardial cells, including a small subset of cardiomyocytes, in vivo. Moreover, although Isx decreased overall myocardial Notch pathway activity, new EGFP+ cardiomyocytes appeared in significant numbers only after Isx1 treatment. Individually, although each of these in vivo pharmacological actions of Isx1 warrants further mechanistic exploration, collectively, they provided conclusive evidence that Isx1 regulates myocardial biology at cellular and molecular levels. The next question was whether Isx1 had cardiomyogenic efficacy in vivo specifically in NECs, our chosen candidate native heart stem/progenitor cell.
Isx activates cardiomyogenic transcriptional programs in NECs in vivo
Next, we sharpened our focus onto NECs, a dynamically regulated adult mouse cardiac-resident MSC-like stem/progenitor population involved in fibrosis-repair after MI and pressure overload injury (8). NECs are exceedingly rare cells, only 10-20,000 cells/normal adult TNR mouse heart, restricting us to transcriptome analysis of freshly purified primary cells from individual hearts. Here, we examined the effects of Isx small-molecules on NEC number per heart and on gene expression profiles of NECs freshly isolated from mice treated with Isx1 and Isx2 compared to control.
The NEC pool is dynamically upregulated with distinctive amplification kinetics depending upon the type of heart injury (8). Isx1 conversely contracted the size of the NEC pool (Fig. 2a). Treating TNR mice with daily Isx1 for one week decreased the heart's NEC pool by ∼50% and we observed an even more dramatic decrement in NEC number with Isx2 (Fig. 2a, FACS scattergrams and graph).
Figure 2. Isx chemically programs cardiomyogenic gene expression in NECs in vivo.
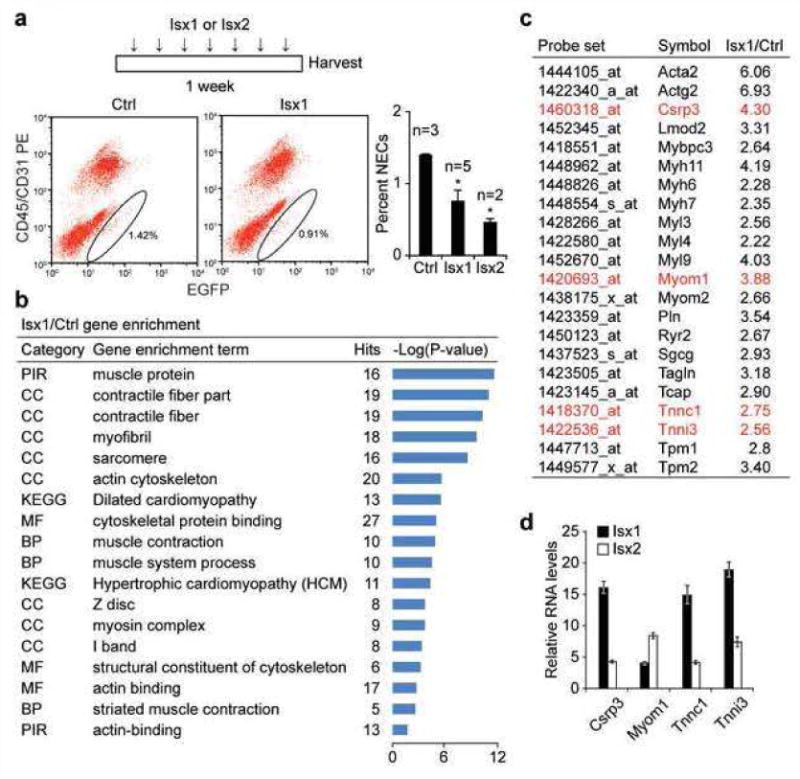
(a) Time map of Isx1, Isx2 or Veh in vivo treatment paradigm. FACS profiles and graph demonstrating dynamic regulation of NEC (Notch/CBF1-REx4-EGFP+CD45−CD31− cell) number per adult age-matched male TNR mouse hearts, after treating mice with Isx1 or Isx2 (n = 3 for Ctrl; n = 5 for Isx1; n = 2 for Isx2; * = p<0.05 comparing NECs from experimental groups to Ctrl; using a two-tailed unpaired Student's t-test). (b) Microarray gene enrichment analysis of NECs from Isx1 versus Ctrl TNR mice demonstrating significant enrichment of cardiac muscle terms in NECs from Isx1 treated mice. (c) A representative list of genes from the gene ontology groups in (b), comparing relative transcript levels in NECs from Isx1 versus Ctrl TNR hearts. (d) Independent confirmation of microarray cardiac gene activation (genes highlighted in red in (c)) by Isx1 in NECs in vivo compared with Isx2 by QPCR.
The in vivo gene expression profile of NECs reflects the sum of fate-specifying endogenous and/or exogenous signals or cues converging on native multipotent progenitors at the time of cell harvest from the heart. Conceptually, NECs are “transition state” cells, a type of MSC poised for differentiation towards lineages like fibroblasts, smooth muscle cells and, more controversially, cardiomyocytes. Thus, we reasoned that the gene expression phenotype of NECs could serve as an in vivo biosensor reporting the cell fate instructive influences of pharmacological or pathophysiological stimuli.
With this concept in mind, we FACS-purified NECs from experimentally manipulated TNR mice, and performed Affymetrix microarray gene expression analysis on purified RNA. We compared the transcriptomes of NECs from control and Isx1 treated mice side-by-side for gene enrichment using the GO (Gene Ontology) database. Unbiased transcriptome analysis revealed highly statistically significant enrichment (P value >10-10) of muscle gene categories like “muscle protein,” “contractile fiber part,” “myofibril” and “sarcomere” in primary NECs isolated from Isx1 treated versus control TNR mouse hearts (Fig. 2b). Importantly, our NEC purification protocol mechanically excludes the possibility of isolating adult cardiomyocytes, as previously detailed (8). These data provided compelling bioinformatics evidence that injecting Isx1 cardiomyogenic small-molecules into normal adult mice directly influenced the NEC gene expression program, quite dramatically.
Focusing on these enrichment terms, we selected a number of candidate muscle genes and directly compared microarray-determined transcript levels in NECs from Isx1 treated versus control mice (Fig. 2c). Among these, we found significant upregulation of transcripts encoding cysteine and glycine rich protein-3 (Csrp3; cardiac LIM protein; upregulated 4.3-fold by Isx in vivo) and the cardiac myofibrillar proteins myomesin-1 (Myom1; upregulated 3.88-fold by Isx in vivo), cardiac troponin C (Tnnc1; upregulated 2.75-fold by Isx in vivo) and cardiac troponin I (Tnni3; upregulated 2.56-fold by Isx in vivo) (Fig. 2c). Even further, we treated additional mice with Isx1 or Isx2, isolated NECs and used quantitative polymerase chain reaction (QPCR) to confirm that all four of these genes – Csrp3, Myom1, Tnnc1 and Tnni3 – were significantly upregulated by Isx1 or Isx2 in vivo (Fig. 2d). Indeed, by QPCR, troponin C and I mRNAs were increased by Isx1 in NECs in vivo by more than 15-fold (Fig. 2d). The pharmacological and physiological significance of quantitative distinctions between Isx1 and Isx2 in these in vivo experiments remain to be established. Nonetheless, these results – confirmed and validated in multiple small-molecule treated TNR mice, using two independent mRNA detection techniques (microarray and QPCR), and further independently confirmed with two structurally distinct Isx analogs – provided compelling evidence that week-long daily injection of Isx small-molecules radically changed the in vivo gene expression phenotype of NECs, a native multipotent heart progenitor population involved in injury-repair. The robust and comprehensive nature of the transcriptional program triggered by Isx1 and Isx2 in NECs in vivo allowed us to designate these FACS-isolated cells as bona fide cardiomyocyte-like precursors (from the adult mouse heart), at least by gene expression criteria. Importantly, despite these microscopic and molecular changes under physiological conditions, we noted no significant change in heart gross structure or function. Therefore, these results led to the next important question: how does Isx affect the ischemically injured or infarcted myocardium already engaged in life-preserving repair process?
Fibrosis overrides Isx1's cardiomyogenic influence in vivo
To evaluate whether Isx1 induced cardiac muscle differentiation in NECs after MI, we injected Isx1 daily for the first 7 days following MI, isolated NECs and evaluated their transcriptomes, as described above. First of all, as before (Fig. 2a), Isx1 decreased the number of NECs in the TNR mouse heart, completely counteracting the NEC number boosting effect normally observed after MI (Fig. 3a, scattergrams and bar graph). This result confirmed that, indeed, Isx1 targeted NECs after MI. Analyzing the gene expression response of these cells, we focused on the “heart development” gene group and observed a striking difference between NEC transcriptomes from control and Isx1-alone versus MI-alone and (MI + Isx1) treated mice: basically, all MI mice, irrespective of Isx1 treatment, had markedly reduced “heart development” transcript levels (blue zone on heat map, Fig. 3b). Indeed, compared to control NECs in vivo, sarcomeric α-myosin heavy chain mRNA (Myh6) was downregulated >5-fold in NECs from MI-alone and (MI + Isx1) treated mice (Fig. 3b). The most likely explanation for the downregulation of “heart development” transcripts including Myh6 following MI was that muscle-related mRNAs were diluted by fibrosis and cell proliferation gene transcripts, which were massively upregulated in NECs after MI, as we previously reported (8). Most critically, Isx1's demonstrated ability to activate cardiac muscle genes failed to penetrate through the pro-fibrosis gene profile of MI-activated NECs.
Figure 3. MI switches NEC's Isx-transcriptional response from cardiomyogenic to fibrosis and angiogenesis genes.
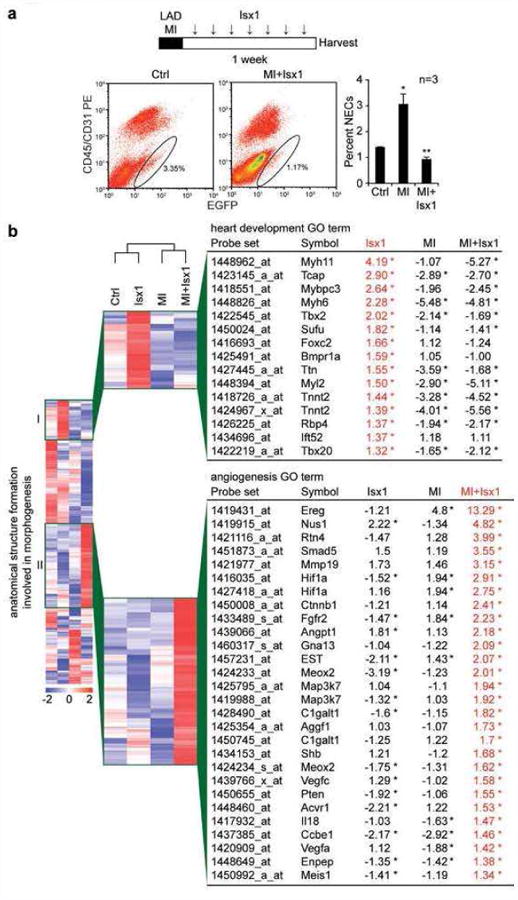
(a) Time map of LAD-MI and Isx1 in vivo treatment paradigm. FACS profiles and graph demonstrating dynamic regulation of NEC (Notch/CBF1-REx4-EGFP+CD45−CD31− cell) number in adult age-matched male TNR mouse hearts after LAD-MI and weeklong treatment with Isx1 (n=3 for Ctrl, MI, and MI+Isx1; *=p<0.05 comparing NECs from experimental groups to Ctrl; ** = p<0.01 comparing NECs from MI+Isx1 to MI alone using a two-tailed unpaired Student's t-test). (b) Gene enrichment analysis of Affymetrix microarray data comparing gene expression in NECs isolated from Ctrl, Isx1, MI and MI+Isx1 treated TNR mice. Heat map of the “anatomical structure formation involved in morphogenesis” gene ontology group highlighting an enlarged section of a unique gene cluster with high expression after Isx1 treatment and a list of genes in the “heart development” ontology group (cluster I, top) and a second unique gene cluster with high expression after MI+Isx1 treatment and a list of genes in the “angiogenesis” ontology group (cluster II, bottom), identified by enrichment analysis of the cluster, with fold change of transcript levels for Isx1, MI and MI+Isx1 versus Ctrl (red=high expression; blue=low expression; the asterisk indicates transcripts that are “present” by Affymetrix chip criteria in Ctrl and the indicated samples, and whose transcript levels after Isx1, MI or MI+Isx1 differed from Ctrl, up or down, in a statistically significant manner (see M & M for details).
On the other hand, Isx1 dysregulated numerous other genes (either up or down) in vivo in NEC-fibroblasts after MI, confirming that there was indeed a major pharmacological effect of this small-molecule after MI. Although we do not yet understand the significance of the gene expression changes induced by Isx1 in post-MI NEC-fibroblasts, MI-alone versus (MI + Isx1) had distinctive transcriptome heat maps for the GO term, “anatomical structure formation involved in morphogenesis,” and indeed we identified a comprehensive “angiogenesis” transcriptional program (group II) specifically upregulated by Isx1 in NECs after MI (Fig. 3b). Thus, although the small-molecule “worked” in vivo after MI, the cardiac muscle gene-inducing activity disappeared once NEC target cells had been costimulated by MI.
To conclude this section, Isx small-molecules pharmacologically targeted multipotent NECs, prominently upregulating cardiac muscle genes and generating a new class of Notch-activated cardiomyocyte-like precursors, at least by gene expression criteria. Yet, in the context of MI, when cardiomyocyte-generating pharmacological activity would presumably be most desirable in NECs, Isx1's cardiac muscle gene-activating effects were overpowered by injury-triggered activation of fibrosis genes. After MI, Isx1 induced a distinctive transcriptional program in NEC-fibroblasts, and this program included angiogenesis genes but excluded muscle genes.
Isx1 fails to mitigate scar formation or ventricular functional decline after MI
Thus far, our in silico results demonstrated that Isx1 activated cardiac muscle gene programs in multipoint NECs in vivo. However, after MI, Isx1's cardiogenic effect was “washed-out” in the heart's robust fibrosis-repair transcriptional response, as NECs differentiated towards fibroblasts. On the other hand, Isx1 strongly dysregulated NEC-fibroblast gene expression after MI, and in this regard, Isx1 might still affect post-MI fibrosis, remodeling or ventricular function, either positively or negatively. We therefore evaluated the effects of Isx therapy on post-MI cardiac function and structure.
First, we did MI surgery in age-matched wild type adult male mice and selected the most severely injured animals, documented by echocardiography on post-operative day 3 (∼50% decline in fractional shortening), for studies of Isx1's effects on ventricular function (Fig. 4a). We randomized this cohort into two groups: an Isx1 treatment arm (9 mice) and a cyclodextrin vehicle-control arm (8 mice). We treated mice with Isx1 (once daily ip injection) or vehicle starting on day 3 after MI and continued for a total of 7 days (Fig. 4a). We re-evaluated ventricular function by echocardiography (with operator blinded to treatment arm) on day 7 post-MI (half-way through the Isx treatment course) and found significantly improved ventricular function and decreased dimensions in Isx-treated MI mice (Fig. 4a). However, after cessation of treatment, the Isx-mediated improvement in ventricular function was not sustained and on day 21 after MI, the day of final echocardiography, all animals had roughly equivalent (and severely depressed) systolic contractile function (Fig. 4a). Although not powered as an efficacy study, this pilot demonstrated a tantalizing early positive signal – a trend towards improved ventricular function; yet, a short course of Isx1 therapy did not have a durable positive effect on heart function.
Figure 4. Isx improves post-MI ventricular function at week one without gross structural changes.
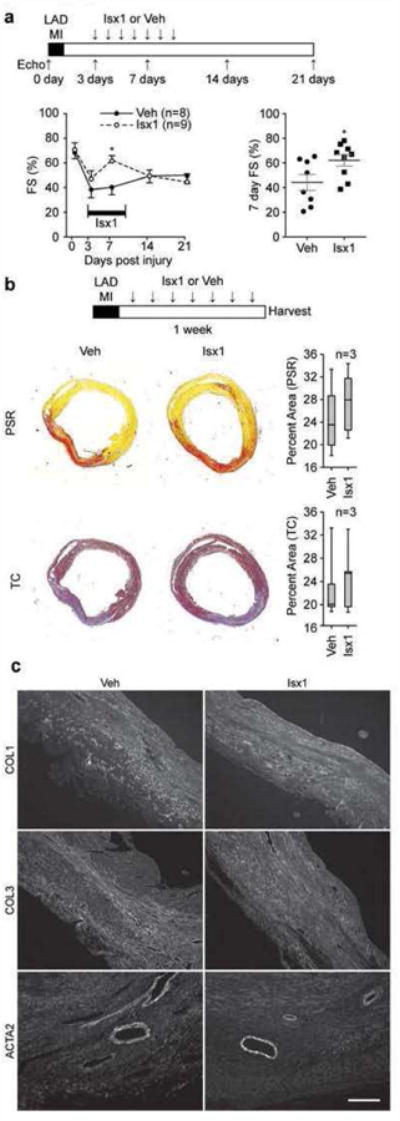
(a) Time map of LAD-MI, Isx1 or Veh in vivo treatment and echocardiography paradigm. Isx1 significantly improved ventricular function (fractional shortening) evaluated on post-MI day 7 during Isx1 treatment (time course graph on the left and day 7 post-MI scatterplot on the right; n=8 mice for Veh; n=9 mice for Isx1; * = p<0.05 comparing Isx1 and Ctrl using a two-tailed unpaired Student's t test). (b) Time map of LAD-MI, Isx1 or Veh in vivo treatment paradigm. At week one post-MI, Isx1 treatment did not change scar size (measured by quantitating Picrosirius Red (PSR) or Masson's Trichrome (TC) staining area of myocardial cross-sections) or (c) composition (evaluated by immunohistochemical staining for collagens-1 (COL1) and -3 (COL3) and smooth muscle α–actin (ACTA2) (Scale bar = 200μm).
To correlate this functional result with possible Isx-mediated changes in cardiac structure, we evaluated the effect of Isx on post-MI scar size and histology, this time starting Isx therapy on the day of MI and continuing for 7 days. Hearts were perfusion-fixed and paraffin-embedded, and histological sections stained with Picrosirius Red (PSR), demonstrating collagen, or Masson's trichrome (TC), demonstrating connective tissue (Fig. 4b). Comparing hearts from Isx versus vehicle treated mice at day 7 after MI, at multiple cross-sectional levels, we determined that there was no significant difference in scar area, measured by PSR or TC (Fig. 4b). Even further, we confirmed by immunohistochemistry for collagen 1 and 3 (COL1 and COL3) and smooth muscle α-actin muscle (ACTA2) that irrespective of study arm, scar zones were indistinguishable (Fig. 4c).
In summary, Isx1 significantly improved ventricular function early after MI, but this improvement was not durable and had completely disappeared by day 21 post-MI. Additionally, Isx1 therapy did not alter early scar histology. We tentatively conclude, with the important caveat that we have thus far only evaluated one short-term Isx1 treatment regimen, that the heart's robust fibrosis-repair response is a formidable barrier. Although this is an important launching point and proof-of-concept, higher efficacy Isx small-molecules will likely be needed to drive regenerative-repair of the adult heart.
Isx induces cardiomyogenic differentiation of NECs in vitro
Our results thus far have shown that injury-activated fibrosis obscured Isx1's muscle gene inducing effects at the level of NECs in vivo and, therefore not surprisingly, post-MI fibrosis-repair and remodeling proceeded unabated despite the presence of Isx1. Although numerous cell types, including monocytes, macrophages and other inflammatory cells, producing numerous proinflammatory and profibrotic cytokines, target NECs in vivo and influence their growth and differentiation, we evaluated the effects of Isx1 on isolated NECs in vitro. Importantly, despite clonality, NEC cultures demonstrated marked cellular heterogeneity, a reassuring functional hallmark of MSCs (8).
We treated NEC cultures with Isx1 (20 μM) or control in serum-rich media for four days. During this period, control cells continued to grow until reaching confluence (Fig. 5a, upper left hand panel). Isx1 treated NECs, on the other hand, rapidly flattened-out, dramatically loosing contrast by phase microscopy, and stopped growing, arresting at sub-confluent cell density; at 20 μM, there was little, if any, Isx1-mediated cell death in these cultures (Fig. 5a, upper right hand panel). We characterized the phenotype of NEC cultures by staining for COL1 and ACTA2, markers of fibroblast and smooth (or early cardiac) muscle cells, respectively (Fig. 5b, lower panels). Compared to control cultures (Fig. 5a, lower left hand panel), Isx1 treated NECs were phenotypically shifted towards muscle lineages (Fig. 5a, lower right hand panel). We quantitated these results in replicate cultures and present them here in pie chart format demonstrating the preponderance of COL1+ cells in the control cultures (Fig. 5a, bottom left) versus ACTA2+ cells in Isx1 treated cultures (Fig. 5a, bottom right). As further confirmation, we demonstrated both qualitative and quantitative changes in COL1 and ACTA2 gene expression in Isx1 treated NEC cultures by protein blot (Fig. 5b). Thus, Isx1 induced a fibroblast-to-muscle gene expression switch in multipotent NEC cultures in vitro.
Figure 5. Isx induces cardiomyogenic differentiation of NECs in vitro.
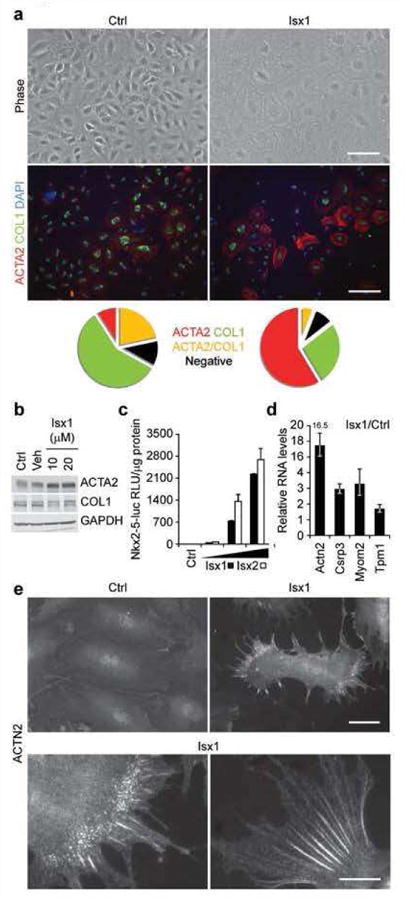
(a) Phase contrast images (top panels, scale bar = 100μm) of clonally-derived NECs cultured for 4 days in media alone (Ctrl) or treated with Isx1 (20 μM), and double-immunostained for ACTA2 (red) and COL1 (green) with DAPI (blue) counter stain (bottom panels, scale bar = 200μm). Pie charts represent the percentage of total cells that were ACTA2+ (red), COL1+ (green), ACTA2+/COL1+ (yellow) or double negative (black). (b) Protein blotting for ACTA2 and COL1 in NECs cultured for 4 days in media alone (Ctrl), in the presence of DMSO (Veh) or increasing amounts of Isx1 with GAPDH to control for loading. (c) Dose-dependent activation of Nkx2-5-luc-BAC transgene in NEC reporter cell line (3G8) by Isx1 and 1sx2. (d) Upregulation of candidate cardiac genes in NECs cultured for 4 days in media alone or in the presence of 20 μM Isx1 by QPCR. (e) ACTN2 immunohistochemistry in NECs cultured for 4 days in media alone (Ctrl-top left panel) or 20 μM Isx1 (top right panel) (Scale bar = 50μm). Bottom panel are higher magnification images of ACTN2 immunohistochemistry in Isx1 treated NECs (Scale bar = 20μm).
To focus more specifically on activation of cardiomyogenic genes by Isx small-molecules in vitro, we studied an Nkx2-5-luc-BAC NEC double transgenic reporter cell line, generated by crossbreeding TNR (CBF1-REx4-EGFP) and transgenic Nkx2-5-luc-BAC reporter mice. Here, we compared Isx1 and Isx2, which both strongly and dose-dependently activated the genomically integrated Nkx2-5-luc-BAC reporter gene in these cells (Fig. 5c). These results confirmed that Isx1 and Isx2 targeted the Nkx2-5-luc-BAC, the original target gene that identified Isx in the P19CL6 chemical screen, in adult mouse heart-derived cells in vitro.
We also surveyed a number of genes by quantitative polymerase chain reaction (QPCR) in Isx1 treated NECs, focusing here on sarcomeric α-actinin (Actn2), Csrp3 and the cardiac myofibrillar proteins myomesin-2 (Myom2) and tropomyosin-1 (Tpm1), gene candidates cherry-picked from our Isx in vivo microarray data (Fig. 2c). Notably, after four days, Isx1 increased sarcomeric α-actinin (Actn2) mRNA levels more than 16-fold (compared with control) in cultured NECs (Fig. 5d). Even further, although control NECs had only background immunocytochemical staining (Fig. 5e, upper left hand panel), Isx1 treated NECs had highly organized “beaded” sarcomeric α-actinin structures, premyofibrils pathognomonic of early developing cardiomyocytes (Fig. 5e, remaining panels) (13). Sarcomeric α-actinin premyofibrils conclusively demonstrated Isx1's pharmacological action, inducing cardiomyogenic differentiation in primary NECs isolated from the adult heart. Taken together, these molecular and cellular data provided compelling evidence that Isx robustly activated a cardiomyogenic gene expression and differentiation program in NECs in vitro, corroborating the in vivo results in the uninjured heart presented above. Additional studies will be required to develop strategies for overcoming or circumventing the profibrosis signals that seem to thwart Isx1's cardiomyogenic effects in NECs after MI, when they are most critically needed.
Conclusion
Our goal is promoting regenerative-repair of the injured adult mouse heart by targeting native progenitors with cardiogenic stem cell-modulator small-molecules (9). We previously characterized a native fibrosis-repair program driven by NECs – fluorescent protein-tagged, molecularly defined and dynamically regulated multipotent heart progenitors derived from the injury-activated epicardial mesothelium (8). Here, we evaluated the efficacy of Isx small-molecules – cardiac gene-activating compounds identified in a previous cell-based chemical screen (9) – for modulating gene expression in NECs in vivo. Indeed, weeklong daily injection of Isx small-molecules (either of two structurally distinct analogs) robustly drove steady-state cardiac muscle gene expression, defined by microarray and confirmed by Q-PCR, in NECs in vivo. Activating muscle gene programs in NECs, a rare and quiescent reserve cell population, did not alter normal cardiac structure or function, positively or negatively. Nonetheless, demonstrating in vivo efficacy, even if limited to changes in gene expression in one particular cell population, marked an important milestone in our cardiogenic stem cell-modulator small-molecule program. We identified two mechanistic roadblocks that will need to be overcome or circumvented before we can hope to successfully re-engineer fibrosis into muscle-rebuilding repair using Isx small-molecules.
The first roadblock is the “resistance” to Isx1's in vitro cardiogenic function in NECs triggered by injury/inflammatory signals unleashed by MI. Numerous paracrine growth factors and cytokines, coupled with systemic mediators, converge to control the growth, differentiation and function of NECs and other repair cells, ultimately producing the scar. Hypoxia has recently been shown to have anti-cardiomyogenic effects in cultured human fetal heart progenitors, promoting an angiogenic program instead, paralleling our in vivo observations (14). Although we anticipated that Isx1 would activate cardiac muscle genes in NECs after MI, post-MI NECs displayed a strong fibro-proliferative gene expression signature instead. NEC-fibroblasts responded to Isx1 with significant gene expression changes, including activation of an angiogenesis program, but other than a modest echocardiographic signal in the Isx1 group early after MI, which may arise from a variety of pharmacological actions of the injected molecules, we have yet to demonstrate the histological or functional consequences of these Isx1 induced gene expression changes. We conclude that, in terms of Isx small-molecules, NECs in the injured heart are ‘moving targets,’ preemptively differentiating into fibroblasts, a competing cell fate that diminishes or excludes cardiac muscle gene activation by Isx1.
The second roadblock is the arrest of cell cycle progression triggered by Isx1 in differentiating progenitors. In principle, the net effect of Isx1's negative regulation of cell cycling in vivo would be to diminish the total number of muscle lineage differentiating cells available for regenerative repair. This is a numbers game. Typically, millions or billions of new cardiomyocytes would be needed to repair an MI, and this is several orders of magnitude beyond what our data suggest might be possible with Isx small-molecules and NECs. In this regard, Isx's growth and differentiation effects might, paradoxically, be counterproductive for repair and necessitate first boosting progenitor number with agents like thymosin-β4, as recently suggested (15). These results stress the dynamic nature of tissue repair processes in vivo.
Fibrosis is a clearly a formidable opponent of regenerative heart repair. Our small-molecules were selected for cardiogenic activity in multipotent progenitors not fibroblasts, although, in principle, small-molecules that trans-differentiate fibroblasts into cardiac muscle cells might be identified in appropriately designed screens. Alternatively, Isx small-molecules might ‘beat’ NECs to cardiac muscle instead of fibroblast differentiation, if pro-fibrosis signals were concomitantly blocked or attenuated, e.g., by non-steroidal anti-inflammatory drugs or steroids. Additionally, the lack of functional efficacy of Isx small-molecules in the injured heart in vivo might be a problem of timing, e.g., administering Isx therapy immediately after MI (or pretreating/‘priming’ with Isx) might enhance Isx's in vivo efficacy for defeating fibrosis and enhancing muscle rebuilding. Conversely, Isx might have a greater effect on long term remodeling rather than acute phase repair, and prolonged continuous therapy (several months) might be required to observe a structural, functional or survival benefit. One final possibility is that the nature of current experimental model, a massive acute MI, is not suited for this approach. Isx, even if structurally and pharmacologically optimized, will be no match for the fibrotic, lifesaving, response triggered by catastrophic tissue injury like MI. Other less-severe injury models may be more approachable pharmacologically.
In summary, although short term Isx1 therapy falls short of providing definitive proof-of-principle for therapeutic efficacy in heart repair, this small organic molecule does exactly what it was asked to do in the original in vitro chemical screen, in vivo, namely, activate cardiac muscle genes in native progenitors. Optimistically, further optimization of this important small-molecule bioactivity will, we believe, lead to the first cardio-regenerative pharmaceutical.
Material and methods
Cell culture
NECs were isolated and cultured as previously described (8). NECs were treated with 20μM Isx1 or Isx2 or the (vol:vol) equivalent of DMSO for 4 days, unless otherwise specified.
Animals
The UTSW IACUC approved all animal protocols. Adult ICR wild type or TNR transgenic male mice were injected IP once daily for 7 days with small-molecules (16 mg/kg/day) formulated in 30% w/v 2-hydroxypropyl-β-cyclodextrin (Sigma). Animals were observed daily and sacrificed 7 days post-injury. In vivo BLI (bioluminescence imaging) was performed on anesthetized animals after a SC injection of D-Luciferin (450 mg/kg in PBS in a total volume of 250 μl). The bioluminescence signal was collected for one minute.
Myocardial injuries
We subjected mice to experimental MI by permanent ligation of the left anterior descending coronary artery, as previously described (8).
Echocardiography
Transthoracic echocardiograms (General Electric Vivid-7 Pro machine equipped with a 12-mHz transducer) were performed in unsedated mice at baseline, 3, 7, 14 and 21 days post-MI. Motion mode (M-mode) images were obtained in the parasternal short-axis view. Fractional shortening was calculated from M-mode images as the left ventricular end-diastolic dimension (LVEDD) minus the left ventricular end-systolic dimension (LVESD) divided by LVEDD.
Immunoblot analysis
Protein extracts were prepared in 1× CLB buffer (Cell Signaling) containing 1μM PMSF in the presence of a phosphatase inhibitor cocktail (Roche). Blots were probed with the following antibodies: mouse anti-α smooth muscle actin/ACTA2 (1A4) (Sigma), rabbit anti-type I Collagen (Rockland) and mouse anti-GAPDH (BD Bioscience Pharmingen).
RNA extraction/QPCR
Total RNA was isolated with Trizol (Invitrogen) and reverse transcription performed using the Superscript First-Stand Synthesis System (Invitrogen). QPCR was performed using SYBR Green QPCR or Taqman Mastermix (ABI), and relative transcript levels calculated using the comparative method (ΔΔCT) with Gapdh as the endogenous control and specific control calibrator. Neonatal heart mRNA was used as the positive control. Primer sequences are available upon request.
Immunocytochemistry/histochemistry/histology
Cells were fixed for 10 minutes with 4% PFA in PBS, washed and stained with the following antibodies: mouse anti-α smooth muscle actin/ACTA2 (1A4) (Sigma), goat anti-Collagen Type I (COL1) (SouthernBiotech) and mouse anti-α sarcomeric actinin/ACTN2 (clone EA-53) (Sigma). Paraffin-embedded sections from PFA fixed adult mouse hearts were deparaffinized, treated with pronase (Sigma) for antigen retrieval and stained with rabbit anti-Collagen Type I (Rockland), rabbit anti-Collagen Type III (Rockland), mouse anti-α smooth muscle actin/ACTA2 (1A4) (Sigma), mouse anti-α sarcomeric actinin/ACTN2 (clone EA-53) (Sigma), rat anti-BrdU (Axyll), rabbit anti-phospho-histone H3 (Ser10)/pH3 (Millipore), rabbit anti-Troponin I (H-170)/TNNI3 (Santa Cruz), and mouse anti-GFP (Invitrogen). The Tyramide signal amplification kit (TSA) (Invitrogen) was used for EGFP immunostaining per manufacturer's protocol. Quantification of EGFP was performed using ImageJ software to calculate average area per field.
Microarray/gene-enrichment analysis
All samples were prepared as previously described (16). Briefly, total RNA was extracted with Trizol (Invitrogen) and amplified and labeled with MessageAMP II-Biotin kit (Ambion) and submitted to UT Southwestern Microarray Core for processing, hybridization and scanning. Samples were normalized and compared using dChip software 2010 as described (8). Change was specified as significant if the lower 90% confidence bound of fold change was greater than or equal to 2 and less than or equal to -2 and the absolute difference of the mean expression was greater than 100. Gene-enrichment analysis of upregulated genes was determined by a modified Fisher Exact P-Value and presented as the -Log(P-value) (17). Unsupervised hierarchical clustering was performed with dChip software using default options. The master gene list for cluster analysis consisted of all “present” probe sets with a signal intensity of ≥ 100.
Flow cytometry
Antibodies: PE anti-CD45 (30-F11), PE anti-CD31 (MEC 13.3) (BD). All samples were stained as recommended by the manufacturer. After dissociation, cells were sorted and collected with the MoFlo sorter (Beckman-Coulter).
Luciferase assays
Tissues where harvested, snap frozen in liquid nitrogen and homogenized in cell lysis buffer (Cell Signaling). Luciferase activity of lysates was assayed using BrightGlo (Promega) and normalized to protein concentration determined by the Pierce BCA protein assay kit (Thermo Scientific).
Quantification of fibrosis
Transverse sections were made of hearts fixed in 4% paraformaldehyde in PBS and stained with Picrosirius Red or Masson's Trichrome. Quantification of interstitial fibrosis was performed using digital microscopic analysis using ImageJ software using three sections taken at 500 μm intervals beginning from the level of the suture from 3 mice.
Supplementary Material
Acknowledgments
The authors wish to acknowledge scientific contributions by H. May, W. Tan, J. Hill, E. Olson and M. Xu. We thank A. Chang and S. Ludwig for reading the manuscript and the following funding sources: American Heart Association-Jon Holden DeHaan Cardiac Myogenesis Research Center Network, NIH/NHLBI Progenitor Cell Biology Consortium 1U01HL100401-01, Cancer Prevention and Research Institute of Texas Multi-Investigator Research Award and Sponsored Research Agreement from LoneStar Heart, Inc. (UTSW BioCenter).
Footnotes
Supporting Information Available: This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Tsao L, Gibson CM. Heart failure: an epidemic of the 21st century. Crit Pathw Cardiol. 2004;3:194–204. doi: 10.1097/01.hpc.0000146867.90558.ca. [DOI] [PubMed] [Google Scholar]
- 2.Daley GQ, Scadden DT. Prospects for stem cell-based therapy. Cell. 2008;132:544–548. doi: 10.1016/j.cell.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 3.Zhou B, Honor LB, He H, Ma Q, Oh JH, Butterfield C, Lin RZ, Melero-Martin JM, Dolmatova E, Duffy HS, Gise A, Zhou P, Hu YW, Wang G, Zhang B, Wang L, Hall JL, Moses MA, McGowan FX, Pu WT. Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. J Clin Invest. 121:1894–1904. doi: 10.1172/JCI45529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grayson P, Mendlein J, Thies S, Yingling J. Stem cell modulators (SCMs): a small molecule and biologic approach to stem cell therapeutics. Drug Discovery Today: Therapeutic Strategies. 2009;6:141–145. [Google Scholar]
- 5.Adams GB, Martin RP, Alley IR, Chabner KT, Cohen KS, Calvi LM, Kronenberg HM, Scadden DT. Therapeutic targeting of a stem cell niche. Nature biotechnology. 2007;25:238–243. doi: 10.1038/nbt1281. [DOI] [PubMed] [Google Scholar]
- 6.Mukherjee S, Raje N, Schoonmaker JA, Liu JC, Hideshima T, Wein MN, Jones DC, Vallet S, Bouxsein ML, Pozzi S, Chhetri S, Seo YD, Aronson JP, Patel C, Fulciniti M, Purton LE, Glimcher LH, Lian JB, Stein G, Anderson KC, Scadden DT. Pharmacologic targeting of a stem/progenitor population in vivo is associated with enhanced bone regeneration in mice. J Clin Invest. 2008;118:491–504. doi: 10.1172/JCI33102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agrawal S, Schaffer DV. In situ stem cell therapy: novel targets, familiar challenges. Trends Biotechnol. 2005;23:78–83. doi: 10.1016/j.tibtech.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 8.Russell JL, Goetsch SC, Gaiano NR, Hill JA, Olson EN, Schneider JW. A dynamic notch injury response activates epicardium and contributes to fibrosis repair. Circulation research. 2011;108:51–59. doi: 10.1161/CIRCRESAHA.110.233262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sadek H, Hannack B, Choe E, Wang J, Latif S, Garry MG, Garry DJ, Longgood J, Frantz DE, Olson EN, Hsieh J, Schneider JW. Cardiogenic small molecules that enhance myocardial repair by stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:6063–6068. doi: 10.1073/pnas.0711507105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schneider JW, Gao Z, Li S, Farooqi M, Tang TS, Bezprozvanny I, Frantz DE, Hsieh J. Small-molecule activation of neuronal cell fate. Nature chemical biology. 2008;4:408–410. doi: 10.1038/nchembio.95. [DOI] [PubMed] [Google Scholar]
- 11.Zhang L, Li P, Hsu T, Aguilar HR, Frantz DE, Schneider JW, Bachoo RM, Hsieh J. Small-molecule blocks malignant astrocyte proliferation and induces neuronal gene expression. Differentiation. 2011;81:233–242. doi: 10.1016/j.diff.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dioum EM, Osborne JK, Goetsch S, Russell J, Schneider JW, Cobb MH. A small molecule differentiation inducer increases insulin production by pancreatic beta cells. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:20713–20718. doi: 10.1073/pnas.1118526109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanger JW, Ayoob JC, Chowrashi P, Zurawski D, Sanger JM. Assembly of myofibrils in cardiac muscle cells. Advances in experimental medicine and biology. 2000;481:89–102. doi: 10.1007/978-1-4615-4267-4_6. discussion 103-105. [DOI] [PubMed] [Google Scholar]
- 14.van Oorschot AA, Smits AM, Pardali E, Doevendans PA, Goumans MJ. Low oxygen tension positively influences cardiomyocyte progenitor cell function. Journal of cellular and molecular medicine. 2011;15:2723–2734. doi: 10.1111/j.1582-4934.2011.01270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smart N, Bollini S, Dube KN, Vieira JM, Zhou B, Davidson S, Yellon D, Riegler J, Price AN, Lythgoe MF, Pu WT, Riley PR. De novo cardiomyocytes from within the activated adult heart after injury. Nature. 474:640–644. doi: 10.1038/nature10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goetsch SC, Hawke TJ, Gallardo TD, Richardson JA, Garry DJ. Transcriptional profiling and regulation of the extracellular matrix during muscle regeneration. Physiol Genomics. 2003;14:261–271. doi: 10.1152/physiolgenomics.00056.2003. [DOI] [PubMed] [Google Scholar]
- 17.Hosack DA, Dennis G, Jr, Sherman BT, Lane HC, Lempicki RA. Identifying biological themes within lists of genes with EASE. Genome Biol. 2003;4:R70. doi: 10.1186/gb-2003-4-10-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.