

Abstract
Although the prevailing view among geneticists suggests that recombination hotspots exist ubiquitously across the human genome, there is only limited experimental evidence from a few genomic regions to support the generality of this claim. A small number of true recombination hotspots are well supported experimentally, but the vast majority of hotspots have been identified on the basis of population genetic inferences from the patterns of linkage disequilibrium (LD) seen in the human population. These inferences are made assuming a particular model of human history, and one of the assumptions of that model is that the effective population size of humans has remained constant throughout our history. Our results show that relaxation of the constant population size assumption can create LD and variation patterns that are qualitatively and quantitatively similar to human populations without any need to invoke localized hotspots of recombination. In other words, apparent recombination hotspots could be an artifact of variable population size over time. Several lines of evidence suggest that the vast majority of hotspots identified on the basis of LD information are unlikely to have elevated recombination rates.
Introduction
Recombination hotspots—regions of the genome known to have much higher rates of recombination than the surrounding areas—have been characterized in yeast and bacteria.1,2 Mice have been known to have hotspots in the major histocompatibility complex (MHC) region for a while.3 Recent research has recently identified genome-wide hotspots in mice as well.4,5 The existence of hotspots in humans was first suggested in the β-globin gene cluster by looking at patterns of linkage disequilibrium in populations and the patterns of transmission in families of restriction fragment-length polymorphism (RFLP) haplotypes.6
Molecular evidence for the existence of recombination hotspots in humans comes primarily from sperm-typing analyses. Two varieties of sperm-typing exist.7 The first, single-sperm typing, relies on the DNA content of a single sperm molecule to be amplified with PCR and analyzed.8,9 The second typing technique involves pooling many sperm from a single donor before beginning the PCR amplification process.10–16 This latter technique has been particularly effective at identifying several hotspots located within the human genome.
Nevertheless, sperm typing is an expensive and challenging experiment. It is difficult to survey large fractions of the human genome. Most genome-wide surveys that have attempted to identify hotspots in humans have relied on examining patterns of linkage disequilibrium in populations.6,17 Whole-genome linkage disequilibrium data from the HapMap project18 is currently the most informative data for use in this effort. Multiple groups have published studies of this kind.19,20 These studies use the pattern of linkage disequilibrium in the genome to infer locations with an unusually large number of recombination events in their history. Required assumptions of this approach include an absence of significant natural selection in the region examined and a constant effective size of the population over time. Under these assumptions, regions of the genome with a large number of recombination events in their history are inferred to have higher rates of recombination per generation. If, however, the underlying assumptions of no natural selection and constant population size are violated, it is unclear whether regions of the genome with a large number of recombination events in their history must necessarily also have higher rates per generation, that is it is unclear whether these regions are true recombination hotspots or merely evidence for regions undergoing selection or a sign that human population size varies over time.
Several paradoxes exist around the true nature of recombination hotspots.7 Linkage disequilibrium (LD)-defined hotspots are known to be shared across populations,21 so they must be reasonably old. It has also been shown, however, that LD-defined hotspots are not shared with chimpanzees, our closest primate relative.22 This indicates that apparent hotspots cannot be older than the human-chimpanzee split. This would provide for only a very small temporal window for the origin of hotspots in the human genome.
Another question involves whether or not LD hotspots are sequence based. No motif is both necessary and sufficient to cause a hotspot, and although at least one known motif is statistically significantly associated with hotspots, it is absent from many hotspots and occurs ubiquitously throughout the genome.23 There is evidence that a zinc-finger protein, PRDM9, is both capable of binding to this motif and having a regulatory effect on hotspot usage, but there is no causal link between LD-defined hotspots and the motif itself.24–28
There are also questions related to studies attempting to verify hotspot locations in the genome. Attempts to verify hotspot locations have been done via both family and sperm-typing approaches. Family-based studies show only an imperfect correlation between recombination events and LD-defined hotspots.29,30 Sperm-typing results are consistent with some LD-defined hotspot locations but not others.15 The Jeffreys group has successfully characterized on the order of 50 hotspots by using the pooled sperm-typing technique.10–16 LDHat previously identified most of those as hotspots. Successfully picking and verifying 50 LDHat hotspots with this technique does not in any way, however, guarantee that the other 30,000 LDHat identified hotspots are all accurately described. Certainly neither approach is able to explicitly confirm a majority of LD-defined hotspots as having elevated recombination rates.
Finally, although studies have shown dramatic recombination rate differences between men and women on a broad scale,31 LD-defined hotspots have been identified in a gender neutral manner.19 It is unclear how LD-defined hotspots, theorized to be an identical set in males and females, could account for this significant difference. As a result of these and other questions surrounding LD-defined hotspots, other possible hypotheses to explain the nature of LD-defined hotspots have been examined.
One such alternative hypothesis will be detailed here. In this model we assume that recombination rates are constant throughout a region but that human population size changes over time. In particular, we assume that there was a relatively recent severe bottleneck in the human population size.32–35 One of the effects of this bottleneck was to divide the genome into regions where four or more alleles share a most-recent common ancestor that predates the bottleneck (old regions), from regions where three alleles or fewer trace their ancestor past this event (young regions). In this model, old regions have had many more recombinations in their history and have highly different patterns of linkage disequilibrium. Looking at data simulated under this model, we show that the LDHat program identifies apparent recombination hotspots even though the data were simulated with a constant recombination rate. This demographic model makes several predictions that can be used to distinguish it from the hotspot model, including, but not limited to, apparent hotspots containing more SNPs than the genome-wide average, apparent hotspots failing to cause significant breakdown in correlation between SNP counts on either side of them, and apparent hotspots being enriched on the edges of windows that recombination events have been mapped in (Figure 1).
Figure 1.
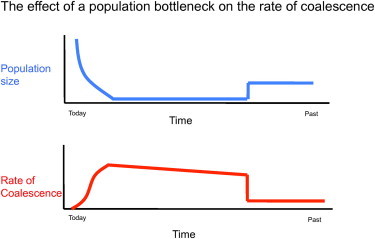
The Coalescent Process
The rate of coalescence is directly related to the size of the population. Going backward in time, the small bottleneck population has sequences that are rapidly coalescing. Any sequence that does not coalesce during or before the bottleneck takes much longer to eventually coalesce. These are the regions of the genome that we term “old.”
Materials and Methods
A Wright-Fisher coalescent simulator generates the simulated human population.36,37 A sample of 60 individuals is generated to match the available HapMap data. Each simulation consists of 300 replications of the history of a 1 Mb region of the genome in all 60 individuals. Varying sets of input parameters are used, consisting of the initial population size in individuals, the bottleneck size in individuals, the number of generations ago the bottleneck begins, the duration of the bottleneck in generations, and the duration of the exponential growth phase in generations. Each set of parameters is simulated under two models, one that does not censor the SNPs and one that is censored first to dbSNP levels of variation and second to the HapMap 5 kb windows.36 The uncensored simulation permits analysis of population statistics, whereas the censored simulation matches HapMap data.
To generate simulations with LD patterns that mimic hotspots, we created a fine-tuned demographic history, consisting of a large initial equilibrium population, a sharp bottleneck, an extended holding period, and a short exponential growth phase to modern population levels. This final model has an initial equilibrium population size of 75,000 individuals that is assumed to have been stable long enough to reach equilibrium. It collapses into a bottleneck of 150 individuals approximately 1,575 generations ago. This bottleneck persists for 575 generations, at which time exponential growth occurs for 1,000 generations to reach the current population size of 6 billion individuals. In all simulations the mutation rate is held constant at 2 × 10−8 mutation events per base pair per generation. For each 1 Mb simulation, the recombination rate is constant over the entire window. Different 1 Mb regions were simulated with recombination rates between 1 × 10−10 and 3.6 × 10−8 recombination events per base pair per generation. The final model utilizes a tiling pattern based on a rough 5 Mb sex-averaged map generated from published data30 to accurately replicate the human genome (Table 1). The genome-wide average recombination rate is 1 × 10−8 after all of the tiled 1 Mb windows are averaged together.
Table 1.
Broad Scale Recombination Rate Variation in Our Simulated Human Genomes
| Recombination Rate | Number of Megabases |
|---|---|
| 1.0 × 10−10 | 53.19 |
| 1.0 × 10−9 | 79.79 |
| 2.0 × 10−9 | 42.55 |
| 3.0 × 10−9 | 101.06 |
| 4.0 × 10−9 | 117.02 |
| 5.0 × 10−9 | 138.30 |
| 6.0 × 10−9 | 212.77 |
| 7.0 × 10−9 | 303.19 |
| 8.0 × 10−9 | 196.81 |
| 9.0 × 10−9 | 202.13 |
| 1.0 × 10−8 | 228.72 |
| 1.1 × 10−8 | 117.02 |
| 1.2 × 10−8 | 164.89 |
| 1.3 × 10−8 | 117.02 |
| 1.4 × 10−8 | 148.94 |
| 1.5 × 10−8 | 101.06 |
| 1.6 × 10−8 | 74.47 |
| 1.7 × 10−8 | 74.47 |
| 1.8 × 10−8 | 95.74 |
| 1.9 × 10−8 | 63.83 |
| 2.0 × 10−8 | 47.87 |
| 2.1 × 10−8 | 47.87 |
| 2.2 × 10−8 | 26.60 |
| 2.3 × 10−8 | 42.55 |
| 2.4 × 10−8 | 15.96 |
| 2.5 × 10−8 | 26.60 |
| 2.6 × 10−8 | 37.23 |
| 2.7 × 10−8 | 21.28 |
| 2.8 × 10−8 | 21.28 |
| 2.9 × 10−8 | 21.28 |
| 3.0 × 10−8 | 15.96 |
| 3.1 × 10−8 | 15.96 |
| 3.2 × 10−8 | 15.96 |
| 3.3 × 10−8 | 0.00 |
| 3.4 × 10−8 | 0.00 |
| 3.5 × 10−8 | 5.32 |
| 3.6 × 10−8 | 5.32 |
| Total Mb | 3,000 |
| Genome-wide average recombination rate | 1.1 × 10−8 |
Each recombination rate is present in the simulated genome for the number of megabases listed.
The fine-tuning of these parameters was done to match known human population characteristics as closely as possible. The goals included achieving an overall human nucleotide diversity (Watterson's θS)38 that is approximately .001,18 as well as an overall nucleotide heterozygosity (Tajima's θπ)39 of .0008. θS is the estimate of 4Nu based on the number of segregating sites in the population, whereas θπ is the estimate of 4Nu derived from nucleotide heterozygosity. Tajima's D, the statistic that compares the two values of θ, is known to be negative in humans, indicating a recent rapid population expansion.40–42 Additionally, the fraction of recombination occurring in 10%–15% of the genome has been estimated to be 50% in humans, and the fraction of recombination occurring in 20% of the genome has been estimated to be 60%.19
The final selection of parameters attempts to match these results as closely as possible but is in no way the only set of parameters that is broadly consistent with this pattern of variation. Additionally, we should be clear that this set of parameters is not intended to imply that we believe that we have accurately or completely described the full and complex demographic and selective history of the human population. The true human history is a complex one, with multiple expansions, possible contractions, intermittent waves of migration, and almost surely significant natural selection at some loci. The full richness of this history is beyond any simple model. The goal here is modest. It is to determine whether a simple model of expansion and contraction can explain the broad patterns of diversity and linkage disequilibrium. The results of the optimal set of simulations are a population with Watterson's estimate of θ ∼.000794 and a slightly negative Tajima's D of −.000583. Although this is not a perfect match to the human population, it is reasonably close, and its differences might very well be due to the lack of migration, selection, and complexity to the demographic changes.
The censored simulation output is then run through the LDHat software. The output comes in the form of estimated recombination rates between each SNP. Within each simulation there is no variation in recombination anywhere. Despite this, the presence of the bottleneck causes LDHat to infer significant rate variation. LDHat believes that 38% of the recombination occurs in 10% of the genome, and 50% of the recombination occurs in 20% of the genome (Figure 2). This is the crucial metric. In simulations that are otherwise identical in parameters but do not contain a bottleneck, LDHat identifies ∼50% of the recombination as occurring in ∼50% of the genome. The bottleneck causes LDHat to misinterpret a region with an even recombination rate as one with dramatic recombination rate changes.
Figure 2.
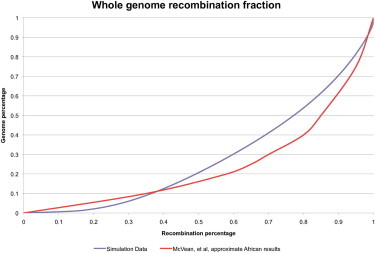
Genome-wide Recombination Rate Comparison
The similarity between the fraction of recombination per fraction of the genome identified by LDHat in an African population and in our simulated population.
Additional analyses are then performed to roughly determine the number of hotspots that have been identified by LDHat. This is complicated by the fact that the simulations are physically small, 1 megabase each, whereas the actual analysis that was used to identify hotspots employed much larger contiguous regions. Therefore, we have elected to find hotspots in as simple a manner as possible, meaning that we do not attempt to mimic the previously reported complex approach.19 Any contiguous region of a simulation with an LDHat estimated recombination rate significantly higher than the LDHat estimated mean rate for the entire simulation is called a hotspot. Using this simplified approach, our model generates approximately three to four hotspots per megabase or ∼10,000–12,000 per genome, which is somewhat lower than the estimate of ∼32,00043 previously identified. The intent here is not to attempt to match the number of individual hotspots identified by LDHat, but instead simply to confirm that there are multiple recombination peaks in any given simulation. The other possibility, that there is a single massive rate change in each simulation, has been ruled out.
Additionally, the X chromosome is simulated independently, as it is assumed to have a population size that is 3/4 that of the total population throughout history. The X also is assumed to have 1/2 the global recombination rate of the rest of the genome. Simulations of the X chromosome show that it has the same basic properties as the rest of our simulated genome; 50% of its recombination occurs in 20% of its length. As expected, however, it has a lower estimated value for θ, ∼.00035, and a slightly more negative Tajima's D of −.0092.
Results
Human demographic history can generate LD patterns that are largely indistinguishable from those created by true recombination hotspots. To demonstrate this, a simulated population is generated that matches a wide range of human population metrics. This simulated population, although not representative of the vastly complex history of human demography, provides a model in which it is evident that the presence of population bottlenecks can drive the inference of LD-defined recombination hotspots. LD-defined hotspots do not have elevated rates of recombination in this model but are instead regions of the genome that are very old. These regions did not coalesce until after the bottleneck, giving them ample time to collect additional SNPs and recombination events that neighboring regions did not. LDHat, by assuming a constant population size, misinterprets the increase in the number of recombination events as an increased recombination rate in these regions, tagging them as hotspots. Two competing models for regions identified as LD-defined hotspots now exist, and the next step is to identify which model better explains the available data.
Number of SNPs in a Hotspot
The demographic model predicts that LD-defined apparent hotspots are older than the surrounding regions and therefore have had more recombination events in their history. They should also have accumulated more mutations in their history. Therefore, LD-defined apparent hotspots should contain more SNPs than similarly matched control regions, if the demographic hypothesis is correct. Using the April 2009 Pilot Data release from the 1000 Genomes Project,44 we find that apparent hotspots have 7.5 SNPs per kb. Random regions of the genome, matched to apparent hotspots for length and GC content have 6.5 SNPs per kb. Because of the enormous number of hotspots, this result is significantly different at p < 10−300. This is consistent with the argument that LD-defined apparent hotspots are older regions of the genome. If the recombination process were highly mutagenic, this could provide another explanation for our results. We note, however, that the 1000 Genomes Project Consortium finds no such increase in divergence around the PRDM9 motif believed to be associated with recombination.44
Correlation Analysis
Recombination can be thought of as having at least two interrelated, but easily separable, effects on variation. First and foremost, recombination creates four-gametes among diallelic markers, that is if we label the two alleles at two loci 0 and 1, in the absence of recombination 00, 01, and 10 gametes are possible. Only recombination (or recurrent mutation) can create the fourth, 11, gamete.45 Recombination's effect of creating the fourth gamete is the primary signal used by LDHat and others20,46 to estimate underlying recombination rates.
However, this is not the only signal that can be used. Recombination also de-couples neighboring coalescent trees.37 Increased recombination decreases the correlation between neighboring coalescent trees, which in turn decreases the correlation between the number of SNPs in neighboring regions.47 This particular signal of recombination is not used at all by programs such as LDHat and can thus be viewed as an independent method of estimating recombination rates.
LDHat predicts that approximately 47.8% of all recombination occurs in their predicted hotspots. Because these hotspots correspond to ∼6% of the genome, LDHat predicts that these hotspots have an average recombination rate ∼7.5 times the genome average. Regions with these recombination rates ought to see dramatic decorrelation in the number of SNPs found on either side of a hotspot. In particular, we know that correlation between the number of SNPs found in a sample of size two is
| (Equation 1) |
where κA and κB are the observed number of SNPs found in regions A and B, ΘA and ΘB are the expected number of SNPs in those regions, and c is the scaled recombination rate. This gives c = 4Nerb, where Ne is the effective population size, r is the recombination rate per base in the region between A and B, and b is the number of bases between regions A and B.47
The expectation herein is that if hotspots truly have elevated recombination rates, the correlation in number of SNPs found in a sample of size two will break down across an LDHat-inferred hotspot. Given an average hotspot intensity of 7.5-fold over the local recombination rate, we expect the correlation to drop by a factor of almost six across a real hotspot when measured at a distance of 10 kb47 (Figures 3A–3E). If on the other hand, the demographic model is correct, the correlations across LD-defined hotspot locations will not be dramatically lower than the correlation predicted by the genome-wide average recombination rate of 1x10−8 per base.
Figure 3.

The Correlation between Number of SNPs on Either Side of LD-Defined Hotspots in Individual Genomes
(A–E) Identified LD-based hotspots, shown in dark blue, show correlation levels nearly equal to what would be expected if they had genome-wide average recombination rates, shown in light blue. This differs dramatically from the expected correlation for regions of the genome that have a five-fold increased recombination rate above the genome-wide average, shown in light green. (A) The Venter Genome,50 (B) the Korean SJK genome,51 (C) the Chinese YH genome,52 (D) the African NA18507 genome on Illumina sequencing technology,53 (E) the African NA18507 genome on SOLiD sequencing technology.54
The recent sequencing of individual genomes has made the optimal analysis of this prediction possible. The assumptions made in this analysis require that SNPs come from a sample of size two. If only heterozygous SNPs from individual genomes are used, they are a perfect data set. For each LD-defined hotspot, we form two 10 kb windows on either side of the hotspot. The distance between these two windows is varied from 10 kb (5 kb each from the center of the hotspot), to 20 kb, 30 kb, etc. Within each window we count the number of heterozygotes seen in a single individual and measure the correlation between those counts across all hotspots. That correlation is plotted in Figures 3A–3E. The correlation across a hotspot is nearly identical to the predicted correlation from the genome-wide average recombination rate. More importantly, the correlation across LD-defined hotspots does not come anywhere close to the expected correlation for 5× or 10× hotter LD-defined hotspots. At a distance of 10 kb, the LD-defined hotspots have a correlation that is five times higher than would be predicted for 7.5× hotter hotspots. When the observed correlation and Equation 1 are used, the average recombination rate across hotspots appears to be approximately 1 × 10−8 per base per generation. This is approximately the genome-wide average recombination rate.31
Comparing LD-Defined Hotspots to Known Recombination Events in Families
The demographic model predicts that LD-predicted hotspots are regions of the genome that have unusually high levels of variation and have an unusually large number of recombination events in their histories. The hotspot model predicts that LD-defined hotspots have unusually high recombination rates. These two predictions can be distinguished by looking at recombination events prospectively. The demographic model predicts that future recombination events are no more likely to happen in LD-defined hotspots than would be predicted based on the size of the hotspot and the genome-wide average recombination rate. The hotspot model predicts that future recombination events are more likely to occur in hotspots on average than elsewhere in the genome.
Excellent data30 are available to test these predictions. These data were generated by mapping recombination events in a large Hutterite pedigree with SNPs on the Affymetrix GeneChip Mapping 500k Array Set. The key limitation to inferring the position of recombination events in pedigrees involves informative markers. A recombination event occurs at some position. In order to detect that event, an informative marker (a marker heterozygous in the parents) must exist on either side of the event. Thus, the precise event is not detectable, but instead a window that contains the event is found. That window includes the precise position of the recombination event, and extends 5′ and 3′ away from that event until an informative marker is detected. Using a novel phasing algorithm, we mapped 24,095 crossovers to windows; 12,278 (51%) mapped to windows of less than 100 kb, and 4,854 (20%) mapped to windows of less than 30 kb. Seventy-two percent of the recombination events that could be mapped to windows of 30 kb or less overlap with an LD-defined recombination hotspot.30 This is far more than expected by chance and would seem to strongly support the notion that LD-defined hotspots do in fact have higher than average recombination rates. There is, however, a further detail of the analysis that must not be overlooked. In our model, LD-defined hotspots are the oldest regions of the genome and as such have more SNPs. Additionally, those SNPs are, on average, at higher intermediate frequency and are thus more likely to be informative. This would make LD-defined hotspots likely end points for mapping windows and create an apparent excess of hotspots on the edges of recombination mapping windows. On the other hand, one expects the position of the actual recombination event to be on average near the center of windows, assuming the distance to the nearest 5′ informative marker is on average the same as the distance to the nearest 3′ informative marker. If the LD-defined hotspots are real, therefore, one would expect to see hotspots enriched in the centers of recombination event windows (Figures 4A and 4B).
Figure 4.
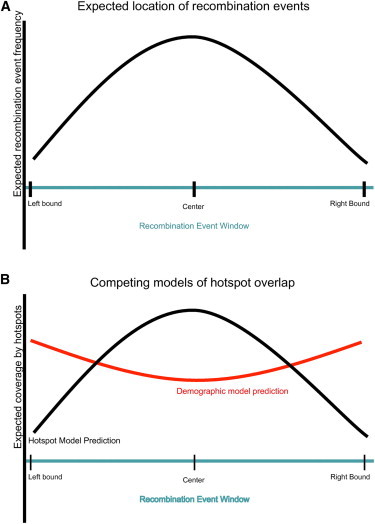
Analysis of Recombination Mapping Windows
(A) Expected location of average recombination event within mapping windows. In recombination mapping windows30 the expectation is that, averaged across all windows, the position of the true recombination event will be in the center of the window on average.
(B) Competing predictions on the locations where hotspots will overlap recombination mapping windows. The hotspot model predicts that LD-defined hotspots would occur more frequently than expected by chance in the center of recombination windows. Our demographic model predicts that LD-defined hotspots will overlap disproportionately on the edges of recombination mapping windows.
To analyze this, each recombination event window is broken into ten equal fragments. For each of the ten fragments, the percentage of each hotspot that overlapped the fragment is counted. Results are then combined for all mapped recombination event windows. The result of this analysis is striking. Edges of recombination event windows show marked enrichment for LD-defined hotspots. The centers of event windows, however, show no more LD-defined hotspot coverage than the genome-wide average (Figure 5). This matches the hypothesis of our demographic model and is not easily explainable in the context of the hotspot model.
Figure 5.
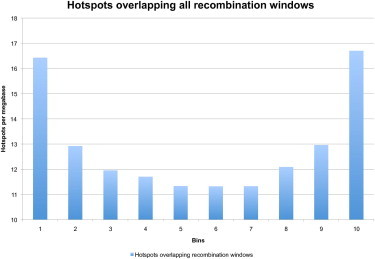
Overlap Pattern of Hotspots onto Mapping Windows
Each recombination mapping window is broken into ten bins. The overlap of hotspots is summed for each bin across all windows. LD-defined hotspots overlap greatly on the edges of recombination mapping windows. The centers of the windows show approximately the genome-wide average number of hotspots, i.e., no enrichment for hotspots.
Dissecting the LD-Defined Hotspots
It is possible that there are significant differences among the 33,000 LD-identified hotspots. To find out, we first sorted the LD-defined hotspots by the number of SNPs per base they contain. The top 10% of LD-defined hotspots have 13.16 SNPs per kb. One can call these SNP-rich LD-defined hotspots. The bottom 10% of hotspots have 3.69 SNPs per kb. One can call these SNP-poor LD-defined hotspots. SNP-poor LD-defined hotspots have slightly more than half the number of SNPs per kb that random spots in the genome have. SNP-rich LD-defined hotspots, however, have over twice the genome-wide average the number of SNPs.
When the LD-identified hotspots are sorted in this manner, SNP-rich LD-defined hotspots have an average LDHat identified recombination rate of 1.362 × 10−7 recombination events per base. The SNP-poor LD-defined hotspots have an average recombination rate of 7.02 × 10−8 recombination events per base. Clearly the number of recombination events being identified by LDHat is significantly greater in the SNP-rich LD-identified hotspots. The natural interpretation is that SNP-rich regions are older.
When this sorting is applied to the analysis of recombination windows, there is a clear difference between the two groups of hotspots. First, windows contain SNP-rich LD-defined hotspots preferentially, five times more than the genome-wide average. These SNP-rich regions fall significantly more often on the edges of recombination windows, even in windows that were mapped to less than 30 kb (Figures 6A and 6B). Very few of SNP-poor LD-defined hotspots are found in recombination windows, and among those that are, the distribution is nearly uniform and is roughly the same rate at which they appear in the genome (Figures 7A and 7B).
Figure 6.
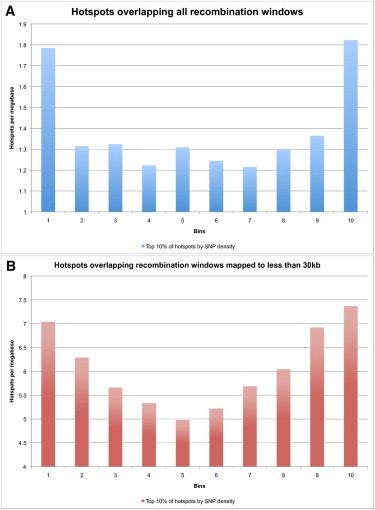
Overlap Pattern for top 10% of Hotspots by SNP Density onto Mapping Windows
(A–B) The top 10% of hotspots by SNP density were selected. All recombination mapping windows (A) and those windows mapped to less than 30 kb (B) were again broken into ten bins each and the overlap of this set of hotspots was summed for each bin across all windows in the set. (A) SNP-rich LD-defined hotspots are dramatically overrepresented on the edges of all recombination mapping windows. (B) SNP-rich LD-defined hotspots are dramatically overrepresented on the edges of recombination events that can be mapped to windows less than 30 kb across.
Figure 7.
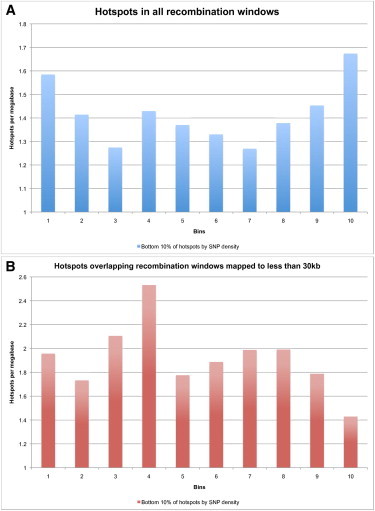
Overlap Pattern for Bottom 10% of Hotspots by SNP Density onto Mapping Windows
The bottom 10% of hotspots by SNP density were selected. All recombination mapping windows (A) and those windows mapped to less than 30 kb (B) were again broken into ten bins each and the overlap of this set of hotspots was summed for each bin across all windows in the set. (A) SNP-poor LD-defined hotspots are distributed nearly uniformly and at near the genome-wide average in recombination mapping windows. (B) SNP-poor LD-defined hotspots are distributed at near the genome-wide average in recombination events that can be mapped to windows less than 30 kb. There might be a slight humping effect in the center of these windows suggesting the possibility that some fraction of them might be actual recombination hotspots.
This leads to a possibility that LD-identified hotspots comprise two different groups. One, consisting of most of the LD-identified hotspots, contains regions that are misidentified as hotspots because of their older age. The second, consisting of SNP-poor regions, might contain some actual recombination hotspots that truly have elevated recombination rates. Properly separating these two groups would be of significant benefit.
Discussion
Regions of the genome with unusually low linkage disequilibrium have had more recombination events in their history than surrounding regions. Such regions have been identified,19 and we refer to them as LD-defined recombination hotspots. However, evidence that LD-defined hotspots will have more recombination events in the future (i.e., have higher recombination rates) has always been limited. We have proposed a model of human demographic history in an attempt to offer a different hypothesis to explain the nature of LD-defined hotspots. This hypothesis suggests that LD-defined hotspots are simply regions of the genome that have a significantly increased time to common ancestor. Conceptually, these regions are older than their neighbors.
It is important to note that this analysis does not preclude the existence of hotspots in the genome. There are very clearly some regions of the genome, such as the MHC region,11 that appear to have hotspots in single-sperm-typing experiments. Other molecularly identified hotspots also exist.16 Whether such hotspots exist on the scale surmised by LD-block analysis is as yet unknown.
Current research indicates that the PRDM9 protein is likely to play a role in the recombination process.24–28 Part of that role might, in fact, mediate the initiation of recombination events in the genome. It is useful to note, however, that the putative binding site for PRDM9 is ubiquitous throughout the genome with nearly 300,000 copies, distributed nearly uniformly, with an average distance of approximately 10 kb between them. Thus, the number of PRDM9 motifs is are an order of magnitude greater than the number of LD-defined hotspots, and 89% of those motifs do not occur in LD-defined hotspots. If we assume for the moment that a PRDM9 binding site is both necessary and sufficient to initiate a recombination event, we would conclude from the distribution of PRDM9 binding sites that recombination is nearly uniform throughout the genome and that LD-defined hotspots have on average approximately 1.5× times the recombination rate of genome average, because they are enriched for PRDM9 motifs by about 50% over genome-wide average (i.e., nothing like the 7.5-fold increase hypothesized elsewhere). Also, unlike LD-defined hotspots, regions surrounding PRDM9 binding sites are not enriched for SNP density,44 suggesting that if PRDM9 is responsible for the initiation of recombination events, those recombination events are not causing the elevated SNP density seen in LD-defined hotspots.
Genetic variation at the PRDM9 locus might further complicate this picture. Admixture mapping in African Americans48 confirms a distinct correlation between a specific PRDM9 allele and apparent recombination hotspots in African populations.49 In addition to suggesting that PRDM9 variants are likely to play a role in the location of recombination events, this also confirms that identified recombination hotspots can differ dramatically across populations over relatively short timescales. Whatever role PRDM9 does play, the research surmises that the mutability of the PRDM9 protein might allow the PRDM9 protein to bind to new motifs even as the recombination process destroys the original motif in the genome. If this is the case, 300,000 current PRDM9 motifs might only be the tip of the iceberg, and historically an even larger fraction of the genome might have been targets for the initiation of recombination events, all of which argues in favor of a relatively uniform recombination rate over evolutionary timescales.
Analysis of LD patterns, such as that performed by LDHat, usually assumes that every region of the genome has the same average time to a common ancestor. Our model, on the other hand, creates a genome that is a mixture of regions that share a common ancestor long before a bottleneck (old regions) and regions that share an ancestor after a bottleneck (young regions). The older regions of the genome, in our model, have had many more recombination events in their histories. As a result, LDHat infers that because more recombinations have happened in these regions they must have a higher recombination rate. Our hypothesis is that these regions have approximately the same recombination rate, but a much older age. In every analysis for which pertinent data were available, our demographic model was the better-supported model. In several analyses, our model was the only one supported. This allows us to conclude that, although some LD-defined hotspots might in fact be true hotspots, most are not.
Acknowledgments
We thank A. Locke, Y. Jakubek, and M. Zwick for informative discussions and F. Spencer, A. McCallion, A. Chakravarti, and J. Mendell for helpful suggestions. We thank S. Warren for critical reading of the manuscript. This work was supported by a grant (5R01HG003461-05) from the National Institutes of Health to D. Cutler.
Web Resources
The URL for data presented herein are as follows:
Wright-Fischer coalescent simulator, http://genome.emory.edu/faculty/dcutler/
References
- 1.de Massy B. Distribution of meiotic recombination sites. Trends Genet. 2003;19:514–522. doi: 10.1016/S0168-9525(03)00201-4. [DOI] [PubMed] [Google Scholar]
- 2.Eggleston A.K., Mitchell A.H., West S.C. In vitro reconstitution of the late steps of genetic recombination in E. coli. Cell. 1997;89:607–617. doi: 10.1016/s0092-8674(00)80242-1. [DOI] [PubMed] [Google Scholar]
- 3.Steinmetz M., Stephan D., Fischer Lindahl K. Gene organization and recombinational hotspots in the murine major histocompatibility complex. Cell. 1986;44:895–904. doi: 10.1016/0092-8674(86)90012-7. [DOI] [PubMed] [Google Scholar]
- 4.Smagulova F., Gregoretti I.V., Brick K., Khil P., Camerini-Otero R.D., Petukhova G.V. Genome-wide analysis reveals novel molecular features of mouse recombination hotspots. Nature. 2011;472:375–378. doi: 10.1038/nature09869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paigen K., Szatkiewicz J.P., Sawyer K., Leahy N., Parvanov E.D., Ng S.H.S., Graber J.H., Broman K.W., Petkov P.M. The recombinational anatomy of a mouse chromosome. PLoS Genet. 2008;4:e1000119. doi: 10.1371/journal.pgen.1000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chakravarti A., Buetow K.H., Antonarakis S.E., Waber P.G., Boehm C.D., Kazazian H.H. Nonuniform recombination within the human beta-globin gene cluster. Am. J. Hum. Genet. 1984;36:1239–1258. [PMC free article] [PubMed] [Google Scholar]
- 7.Arnheim N., Calabrese P., Tiemann-Boege I. Mammalian meiotic recombination hot spots. Annu. Rev. Genet. 2007;41:369–399. doi: 10.1146/annurev.genet.41.110306.130301. [DOI] [PubMed] [Google Scholar]
- 8.Cullen M., Perfetto S.P., Klitz W., Nelson G., Carrington M. High-resolution patterns of meiotic recombination across the human major histocompatibility complex. Am. J. Hum. Genet. 2002;71:759–776. doi: 10.1086/342973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schneider J.A., Peto T.E.A., Boone R.A., Boyce A.J., Clegg J.B. Direct measurement of the male recombination fraction in the human beta-globin hot spot. Hum. Mol. Genet. 2002;11:207–215. doi: 10.1093/hmg/11.3.207. [DOI] [PubMed] [Google Scholar]
- 10.Jeffreys A.J., Holloway J.K., Kauppi L., May C.A., Neumann R., Slingsby M.T., Webb A.J. Meiotic recombination hot spots and human DNA diversity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004;359:141–152. doi: 10.1098/rstb.2003.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jeffreys A.J., Kauppi L., Neumann R. Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nat. Genet. 2001;29:217–222. doi: 10.1038/ng1001-217. [DOI] [PubMed] [Google Scholar]
- 12.Jeffreys A.J., Neumann R. Reciprocal crossover asymmetry and meiotic drive in a human recombination hot spot. Nat. Genet. 2002;31:267–271. doi: 10.1038/ng910. [DOI] [PubMed] [Google Scholar]
- 13.Jeffreys A.J., Neumann R. Factors influencing recombination frequency and distribution in a human meiotic crossover hotspot. Hum. Mol. Genet. 2005;14:2277–2287. doi: 10.1093/hmg/ddi232. [DOI] [PubMed] [Google Scholar]
- 14.Jeffreys A.J., Neumann R., Panayi M., Myers S., Donnelly P. Human recombination hot spots hidden in regions of strong marker association. Nat. Genet. 2005;37:601–606. doi: 10.1038/ng1565. [DOI] [PubMed] [Google Scholar]
- 15.Neumann R., Jeffreys A.J. Polymorphism in the activity of human crossover hotspots independent of local DNA sequence variation. Hum. Mol. Genet. 2006;15:1401–1411. doi: 10.1093/hmg/ddl063. [DOI] [PubMed] [Google Scholar]
- 16.Jeffreys A.J., Murray J., Neumann R. High-resolution mapping of crossovers in human sperm defines a minisatellite-associated recombination hotspot. Mol. Cell. 1998;2:267–273. doi: 10.1016/s1097-2765(00)80138-0. [DOI] [PubMed] [Google Scholar]
- 17.Hudson R.R. Two-locus sampling distributions and their application. Genetics. 2001;159:1805–1817. doi: 10.1093/genetics/159.4.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Consortium T.I.H., International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McVean G.A.T., Myers S.R., Hunt S., Deloukas P., Bentley D.R., Donnelly P. The fine-scale structure of recombination rate variation in the human genome. Science. 2004;304:581–584. doi: 10.1126/science.1092500. [DOI] [PubMed] [Google Scholar]
- 20.Myers S., Bottolo L., Freeman C., McVean G., Donnelly P. A fine-scale map of recombination rates and hotspots across the human genome. Science. 2005;310:321–324. doi: 10.1126/science.1117196. [DOI] [PubMed] [Google Scholar]
- 21.Laayouni H., Montanucci L., Sikora M., Melé M., Dall'Olio G.M., Lorente-Galdos B., McGee K.M., Graffelman J., Awadalla P., Bosch E. Similarity in recombination rate estimates highly correlates with genetic differentiation in humans. PLoS ONE. 2011;6:e17913. doi: 10.1371/journal.pone.0017913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winckler W., Myers S.R., Richter D.J., Onofrio R.C., McDonald G.J., Bontrop R.E., McVean G.A.T., Gabriel S.B., Reich D., Donnelly P., Altshuler D. Comparison of fine-scale recombination rates in humans and chimpanzees. Science. 2005;308:107–111. doi: 10.1126/science.1105322. [DOI] [PubMed] [Google Scholar]
- 23.Myers S., Freeman C., Auton A., Donnelly P., McVean G. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat. Genet. 2008;40:1124–1129. doi: 10.1038/ng.213. [DOI] [PubMed] [Google Scholar]
- 24.Baudat F., Buard J., Grey C., Fledel-Alon A., Ober C., Przeworski M., Coop G., de Massy B. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science. 2010;327:836–840. doi: 10.1126/science.1183439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berg I.L., Neumann R., Lam K.-W.G., Sarbajna S., Odenthal-Hesse L., May C.A., Jeffreys A.J. PRDM9 variation strongly influences recombination hot-spot activity and meiotic instability in humans. Nat. Genet. 2010;42:859–863. doi: 10.1038/ng.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McVean G., Myers S. PRDM9 marks the spot. Nat. Genet. 2010;42:821–822. doi: 10.1038/ng1010-821. [DOI] [PubMed] [Google Scholar]
- 27.Myers S., Bowden R., Tumian A., Bontrop R.E., Freeman C., MacFie T.S., McVean G., Donnelly P. Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science. 2010;327:876–879. doi: 10.1126/science.1182363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parvanov E.D., Petkov P.M., Paigen K. Prdm9 controls activation of mammalian recombination hotspots. Science. 2010;327:835. doi: 10.1126/science.1181495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khil P.P., Camerini-Otero R.D. Genetic crossovers are predicted accurately by the computed human recombination map. PLoS Genet. 2010;6:e1000831. doi: 10.1371/journal.pgen.1000831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coop G., Wen X., Ober C., Pritchard J.K., Przeworski M. High-resolution mapping of crossovers reveals extensive variation in fine-scale recombination patterns among humans. Science. 2008;319:1395–1398. doi: 10.1126/science.1151851. [DOI] [PubMed] [Google Scholar]
- 31.Broman K.W., Murray J.C., Sheffield V.C., White R.L., Weber J.L. Comprehensive human genetic maps: individual and sex-specific variation in recombination. Am. J. Hum. Genet. 1998;63:861–869. doi: 10.1086/302011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harpending H., Sherry S., Rogers A. The Genetic Structure of Ancient Human Populations. Curr. Anthropol. 1993;34:483–496. [Google Scholar]
- 33.Hawks J., Hunley K., Lee S.H., Wolpoff M. Population bottlenecks and Pleistocene human evolution. Mol. Biol. Evol. 2000;17:2–22. doi: 10.1093/oxfordjournals.molbev.a026233. [DOI] [PubMed] [Google Scholar]
- 34.Rampino M. Climate-volcanism feedback and the Toba eruption of 74,000 years ago. Quaternary Research. 1993;40:269–280. [Google Scholar]
- 35.Rogers A.R., Jorde L.B. Genetic evidence on modern human origins. Hum. Biol. 1995;67:1–36. [PubMed] [Google Scholar]
- 36.Lin S., Chakravarti A., Cutler D.J. Exhaustive allelic transmission disequilibrium tests as a new approach to genome-wide association studies. Nat. Genet. 2004;36:1181–1188. doi: 10.1038/ng1457. [DOI] [PubMed] [Google Scholar]
- 37.Hudson R.R. Properties of a neutral allele model with intragenic recombination. Theor. Popul. Biol. 1983;23:183–201. doi: 10.1016/0040-5809(83)90013-8. [DOI] [PubMed] [Google Scholar]
- 38.Watterson G.A. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- 39.Tajima F. Evolutionary relationship of DNA sequences in finite populations. Genetics. 1983;105:437–460. doi: 10.1093/genetics/105.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ptak S.E., Przeworski M. Evidence for population growth in humans is confounded by fine-scale population structure. Trends Genet. 2002;18:559–563. doi: 10.1016/s0168-9525(02)02781-6. [DOI] [PubMed] [Google Scholar]
- 41.Tajima F. The effect of change in population size on DNA polymorphism. Genetics. 1989;123:597–601. doi: 10.1093/genetics/123.3.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M., International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hudson R.R., Kaplan N.L. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics. 1985;111:147–164. doi: 10.1093/genetics/111.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crawford D.C., Bhangale T., Li N., Hellenthal G., Rieder M.J., Nickerson D.A., Stephens M. Evidence for substantial fine-scale variation in recombination rates across the human genome. Nat. Genet. 2004;36:700–706. doi: 10.1038/ng1376. [DOI] [PubMed] [Google Scholar]
- 47.Griffiths R. Neutral two-locus multiple allele models with recombination. Theor. Popul. Biol. 1981;19:169–186. [Google Scholar]
- 48.Wegmann D., Kessner D.E., Veeramah K.R., Mathias R.A., Nicolae D.L., Yanek L.R., Sun Y.V., Torgerson D.G., Rafaels N., Mosley T. Recombination rates in admixed individuals identified by ancestry-based inference. Nat. Genet. 2011;43:847–853. doi: 10.1038/ng.894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hinch A.G., Tandon A., Patterson N., Song Y., Rohland N., Palmer C.D., Chen G.K., Wang K., Buxbaum S.G., Akylbekova E.L. The landscape of recombination in African Americans. Nature. 2011;476:170–175. doi: 10.1038/nature10336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Levy S., Sutton G., Ng P.C., Feuk L., Halpern A.L., Walenz B.P., Axelrod N., Huang J., Kirkness E.F., Denisov G. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahn S.M., Kim T.H., Lee S., Kim D., Ghang H., Kim D.S., Kim B.C., Kim S.Y., Kim W.Y., Kim C. The first Korean genome sequence and analysis: full genome sequencing for a socio-ethnic group. Genome Res. 2009;19:1622–1629. doi: 10.1101/gr.092197.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang J., Wang W., Li R., Li Y., Tian G., Goodman L., Fan W., Zhang J., Li J., Zhang J. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–65. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bentley D.R., Balasubramanian S., Swerdlow H.P., Smith G.P., Milton J., Brown C.G., Hall K.P., Evers D.J., Barnes C.L., Bignell H.R. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McKernan K.J., Peckham H.E., Costa G.L., McLaughlin S.F., Fu Y., Tsung E.F., Clouser C.R., Duncan C., Ichikawa J.K., Lee C.C. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res. 2009;19:1527–1541. doi: 10.1101/gr.091868.109. [DOI] [PMC free article] [PubMed] [Google Scholar]