

Abstract
Psoriasis is a common, immune-mediated genetic disorder of the skin and is associated with arthritis in approximately 30% of cases. Previously, we localized PSORS2 (psoriasis susceptibility locus 2) to chromosomal region 17q25.3-qter after a genome-wide linkage scan in a family of European ancestry with multiple cases of psoriasis and psoriatic arthritis. Linkage to PSORS2 was also observed in a Taiwanese family with multiple psoriasis-affected members. In caspase recruitment domain family, member 14 (CARD14), we identified unique gain-of-function mutations that segregated with psoriasis by using genomic capture and DNA sequencing. The mutations c.349G>A (p.Gly117Ser) (in the family of European descent) and c.349+5G>A (in the Taiwanese family) altered splicing between CARD14 exons 3 and 4. A de novo CARD14 mutation, c.413A>C (p.Glu138Ala), was detected in a child with sporadic, early-onset, generalized pustular psoriasis. CARD14 activates nuclear factor kappa B (NF-kB), and compared with wild-type CARD14, the p.Gly117Ser and p.Glu138Ala substitutions were shown to lead to enhanced NF-kB activation and upregulation of a subset of psoriasis-associated genes in keratinocytes. These genes included chemokine (C-C motif) ligand 20 (CCL20) and interleukin 8 (IL8). CARD14 is localized mainly in the basal and suprabasal layers of healthy skin epidermis, whereas in lesional psoriatic skin, it is reduced in the basal layer and more diffusely upregulated in the suprabasal layers of the epidermis. We propose that, after a triggering event that can include epidermal injury, rare gain-of-function mutations in CARD14 initiate a process that includes inflammatory cell recruitment by keratinocytes. This perpetuates a vicious cycle of epidermal inflammation and regeneration, a cycle which is the hallmark of psoriasis.
Introduction
Psoriasis is a poorly understood common inflammatory disorder of the skin and other organs and affects approximately 2% of individuals of European descent.1 It can be associated with a chronic inflammatory psoriatic arthritis in ∼30% of affected individuals.2 Genome-wide association studies (GWASs) have identified over 20 susceptibility loci for psoriasis,3–11 but less than 20% of disease variance is explained.12,13 Additional low-risk loci, genetic interactions, or rare variants of large effect are thought to account for the remaining disease variance. It is difficult to identify the latter, but they are expected to be observed in rare families in which psoriasis segregates as a Mendelian trait.
Seventeen years ago, we mapped psoriasis susceptibility locus 2 (PSORS2 [MIM 602723]) to human chromosomal region 17q25-qter in a single large family (PS1) of European ancestry.14 The region of linkage spanned 3.5 Mb from D17S784 to 17qter. Affected members had plaque psoriasis, and approximately 30% also developed psoriatic arthritis. Two additional genome-wide linkage scans further implicated PSORS2. A study of 224 sibling pairs with psoriasis yielded a maximum LOD score of 2.09 with D17S802,15 and a five-generation psoriasis pedigree from Taiwan exhibited significant linkage to D17S928.16 Both of these microsatellite loci map to chromosomal region 17q25.3. In both large, multiplex families, psoriasis segregated as an autosomal-dominant Mendelian trait with high penetrance.
To identify the familial mutations, we attempted association studies and extensive resequencing of genes under the linkage peak.17–19 Recently, we included targeted and exome capture followed by NextGen sequencing of DNA from members of family PS1 and identified in caspase recruitment domain family, member 14 (CARD14 [MIM 607211])20 a mutation that segregated with psoriasis. We then identified a second CARD14 mutation in the 17q25-linked Taiwanese family, and a young child with severe generalized pustular psoriasis (PSORP [MIM 614204]) harbored a de novo mutation within CARD14. Wild-type CARD14 activates nuclear factor-kappa B (NF-kB), and the psoriasis-associated missense mutations further upregulate NF-kB activity. Compared with wild-type keratinocytes, cultured keratinocytes from affected individuals with the mutations and a keratinocyte cell line transfected with mutant CARD14 showed increased transcription of genes encoding psoriasis-associated chemokines and cytokines such as interleukin 8 (IL8 [MIM 146930]), chemokine (C-C motif) ligand 20 (CCL20 [MIM 601960]), and interleukin 36, gamma (IL36G [MIM 605542]). CARD14 peptides in the skin were almost exclusively localized in the epidermal keratinocytes, primarily in the basal layer of the epidermis. Compared with normal skin, psoriatic skin showed reduced levels of CARD14 in the basal layer but increased levels throughout the upper layers. We propose that, in the context of an inflammatory stimulus, keratinocytes harboring the CARD14 mutations can upregulate an inflammatory response via excessive activation of NF-kB-responsive genes and initiate the recruitment of the inflammatory infiltrate seen in psoriasis. Our findings resolve the identity of the elusive PSORS2 locus and identify CARD14 as a regulator of skin inflammation.
Subjects and Methods
Subjects
Family PS1 of European origin and the 17q25-linked Taiwanese psoriasis-affected family are described elsewhere.14,16 The person with pustular psoriasis (individual 2192) was a 3-year-old child from Haiti. Her father, mother, and brother were unaffected, and there was no family history of psoriasis or any other autoimmune disorder. Whole blood was obtained by venipuncture; skin samples were obtained by punch biopsy. Primary keratinocytes from family PS1 and classical-psoriasis cases were immortalized by infection with human papillomavirus E6/E7 DNA (a gift from Dr. Denise Galloway at the Fred Hutchinson Center for Cancer Research).21 Keratinocytes from individual 2192, who has pustular psoriasis, and two foreskin samples used as controls were immortalized without transformation as previously described.22 Protocols were approved by local institutional review boards (IRBs). All subjects or their parents (if the subjects were minors) provided informed consent.
Genomic Capture and Sequence Analysis
Exome capture and targeted capture of the 17q25 linkage region were performed with DNA from family PS1 and were followed by NextGen sequencing. For exome sequencing, DNA from four affected individuals was used. For targeted capture, pools of genomic DNA from 14 affected and eight unaffected samples were used. Genomic libraries were prepared with the Illumina Paired-End Genomic DNA Sample Prep Kit. Genomic capture was performed with Roche-Nimblegen SeqCap EZ Exome and Agilent SureSelect Target Enrichment kits. Captured DNA was sequenced on the Illumina Genome Analyzer II (GAIIx) for 76 cycles (one lane per sample or pool). Both capture methods significantly enriched the targeted regions (Table S1, available online).
Read Mapping and Sequence Analysis
Read mapping, sequence analysis, and variant calling were performed as described elsewhere.23 Known polymorphisms (variants in dbSNP130 and eight previously sequenced HapMap individuals) were excluded.24 Candidate mutations in the 17q25 linkage region were those observed in at least three of the four exomes sequenced and/or observed in the affected pool but not in the unaffected pool. Mutations were manually annotated for amino acid changes and were Sanger sequenced for validation and for checking segregation with disease (Table S2).
Minigene Assay
CARD14 minigenes were constructed by PCR amplification of human genomic DNA from unaffected (wild-type) and affected members of family PS1 (affected by c.349G>A) and the Taiwanese family (affected by c.349+5G>A). PCR fragments included CARD14 exon 3 and 150 bp each of the 5′ and 3′ introns. Fragments were digested by BamH1 and XbaI and were ligated into the RHCglo vector (a gift from Dr. Tom Cooper at the Baylor College of Medicine) at the corresponding digest sites. All constructs were validated with DNA sequencing. Minigene constructs were used as described previously.25 Minigenes were transfected into human embryonic kidney (HEK) 293 cells. Total RNA was harvested after 24 hr. cDNA was synthesized with the SuperScript II kit (Invitrogen) and the RTRHC oligonucleotide primer 5′-GGGCTTTGCAGCAACAGTAAC-3′. Using the TNIE4 5′-AGGTGCTGCCGCCGGGCGGTGGCTG-3′ and RSV5U 5′-CATTCACCACATTGGTGTGC-3′ primers, we sequenced the cDNA to evaluate the splicing of CARD14 exon 3 in the presence of the familial psoriasis mutations and in the presence of the reference CARD14 sequence. Sequencing primers were those described above as well as an internal exon 3 primer, 5′-ACTTGCTGGATTTGCTGAAGAC-3′.
Expression Plasmids
The pNFkB-luc plasmid (Clontech) contains the firefly luciferase gene and four tandem copies of the consensus NF-kB-binding sequence fused to a TATA-like promoter. The pTAL-luc plasmid (Clontech) contains an enhancerless reporter with a minimal TATA-like promoter and the firefly luciferase gene, and it was used as a negative control.
Two CARD14 cDNA clones, CARD14sh (coding for 740 amino acids; GenBank BC018142) and CARD14cl (coding for 434 amino acids; RefSeq NM_052819) were used (Capital Biosciences). A full-length (CARD14fl) construct was not available. Mutant forms of CARD14 were prepared from the CARD14sh construct (GenBank BC018142) with the QuikChange Site-Directed Mutagenesis Kit (Stratagene).
NF-kB Luciferase Reporter Assay
HEK 293 cells were cultured under standard conditions and plated at a density of 0.15 million cells per well in 12-well plates. Cells were cotransfected with (1) 0.5 μg wild-type CARD14sh or the CARD14cl expression construct, (2) 0.5 μg pTAL-luc or pNF-kB-luc plasmid, and (3) 0.025 μg pGL4.70 Renilla reporter plasmid (Promega). In total, 1.025 μg of plasmid DNA was transfected into each cell. For cotransfections, 0.5 μg of each CARD14 construct was transfected into cells. Then, 0.05 μg of pGL4.70 and 0.5 μg of reporter plasmid were added for a total of 1.55 μg plasmid DNA transfected into each cell. All transfections were performed in triplicate. For stimulation with TNF-α (tumor necrosis factor alpha [encoded by TNF (MIM 191160)]), cells were treated with 20 ng/ml TNF-α in culture medium. For TNF-α neutralization experiments, cells were treated with indicated concentrations of TNF-α neutralizing antibody (Cell Applications). Cells were harvested 24 hr after transfection, and firefly luciferase activity was determined with the Dual-Luciferase Reporter Assay System (Promega). We normalized firefly luciferase activity to Renilla luciferase activity to control for transfection efficiency. Relative NF-kB luciferase activity for each sample was then calculated by background subtraction of pTAL luciferase activity and the subsequent division of the normalized NF-kB luciferase value by the background pTAL luciferase value.
Expression Profiling
HEK 001 cells (human-papillomavirus-16-transformed keratinocytes) were transfected with wild-type CARD14sh or mutant (c.349G>A [p.Gly117Ser] or c.413A>C [p.Glu138Ala]) CARD14sh expression constructs. Cells were cultured for 24 hr, after which time total RNA was isolated. Lesional and nonlesional skin biopsies from an affected member (GEN001) of family PS1 were obtained from the Psoriasis Research Institute (Dallas, Texas), and a lesional skin biopsy from child 2192, who has severe pustular psoriasis, was obtained from the National Institute of Arthritis and Musculoskeletal and Skin Diseases. Biopsies of normal skin (n = 2) and moderate-to-severe lesional psoriasis skin (n = 2) were obtained from Rockefeller University for global expression profiling. RNA was extracted from full-thickness skin biopsies. Immortalized keratinocyte lines from uninvolved skin of affected members (K1-1 and K1-20) of family PS1 and from involved skin of classical psoriasis (K5-14, an HLA-Cw∗0602-positive case) were cultured with or without stimulation by TNF-α, and total RNA was isolated. Immortalized, nontransformed keratinocytes from individual 2192 and from two foreskin samples were cultured in medium only. For all samples described above, total RNA isolation was performed with the miRNeasy or RNeasy kit (QIAGEN). Global expression profiling was performed with HumanHT-12 v4 Expression BeadChip (Illumina). For each BeadChip (Illumina), at least 0.2 μg of total RNA per sample was reverse transcribed, amplified, and labeled. Experiments were conducted in compliance with MIAME (minimum information about a microarray experiment) guidelines. Raw and normalized expression data are deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) with accession number GSE36387.
For expression analyses, data were imported into R (v2.13.1) with the BeadArray (v2.2.0) package. Raw data were used for analysis. For single-sample-by-single-sample comparisons, probes that did not show a detection p value < 0.05 in any sample were excluded. The average signal, standard error, and number of beads were used for the computation of a two-sample, two-sided t test assuming unequal variance. Fold-changes were calculated on the basis of the average expression values. We adjusted p values with the Benjamini and Hochberg method to control for false discovery.
For group-wise comparisons, average expression values were quantile normalized and log2 transformed. Contrasts for each desired class comparison were created with Limma (v3.8.3). Probe expression values were modeled as linear models, and statistics for fold-changes and p values were calculated on these models with the limma “eBayes” function. p values were adjusted as described above.
CARD14 Transcript Isoform Analysis
cDNA Cloning and Sequencing
SuperScript II (Invitrogen) was used for the generation of cDNA from RNA derived from the involved skin of individual GEN001. PCR-based cloning of the exon 2–4 region of CARD14 was performed according to standard protocols with exon-2- and exon-4-specific oligonucleotide primers (5′-AGGAGACACTGTGGGAGATGAT-3′ for exon 2 and 5′-ATTGCTATAGTGCAGCGAGAGG-3′ for exon 4). Plasmid inserts were sequenced with the same oligonucleotide primers.
Quantitative RT-PCR
Quantitative RT-PCR (qRT-PCR) was performed in routine fashion on total RNA primed with random hexamer oligonucleotides. PCR reactions were performed with Power SYBR Green master mix (Life Technologies) on a 7900HT thermocycler (Life Technologies). Expression was normalized to 18S rRNA. Relative expression levels were calculated according to the 2-ΔΔCt method as follows: 2 × 106((Ct 18S)−(Ct CARD14)). We corrected for differences in transfection efficiency by normalizing expression levels in transfected cells to levels of FLAG. Primers for the transcripts examined by qRT-PCR are available upon request.
Immunohistochemistry
Normal skin samples (n = 3–4) and paired uninvolved and involved psoriatic samples (n = 5–6) were obtained from Rockefeller University from individuals with mild-to-severe stable plaque psoriasis. PS1 uninvolved and involved skin was collected and studied under a protocol approved by the IRB at Washington University. All frozen sections were stained with polyclonal rabbit anti-CARD14 (Sigma, 1:600). This antibody labels the internal coiled-coil domain of CARD14 and recognizes all known isoforms. Secondary biotin-labeled goat anti-rabbit antibodies (Vector Laboratories) were used. The staining signal was amplified with avidin-biotin complex (Vector Laboratories) and developed with chromogen 3-amino-9-ethylcarbazole (Sigma Life Sciences). The negative control was stained with 1% serum in lieu of the primary antibody.
Results
Exome- and Targeted-Sequencing Results
Family PS1 is of European ancestry and has multiple cases of psoriasis and psoriatic arthritis.14 DNA of family members was used for exome sequencing, targeted-capture sequencing, and NextGen sequencing. Only the following two protein-coding mutations were identified in the ∼3.5 Mb linkage region and segregated with the disease (Tables S1 and S2): c.349G>A (p.Gly117Ser) in CARD14 and c.365A>G (p.Tyr122Cys) in solute carrier family 26, member 11 (SLC26A11 [MIM 610117]) (Figures 1A and 1B and Table S2). Numbering of all CARD14 mutations in this manuscript is based on RefSeq NM_024110.3. Numbering of the SLC26A11 mutation is based on RefSeq NM_001166347.1. CARD14, also known as CARMA2, encodes a member of the CARMA (caspase recruitment domain [CARD]- and membrane-associated guanylate kinase [MAGUK]) family; this member interacts with BCL10 and activates NF-kB.20 SLC26A11 encodes a sulfate/anion transporter.26 Both mutations were predicted to be potentially damaging by PolyPhen-2.027 and SIFT.28
Figure 1.
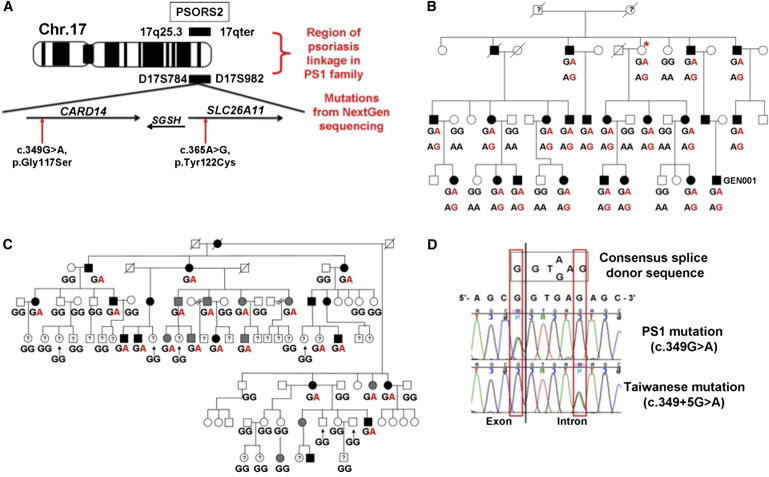
Positional Cloning of PSORS2 and Mutations Segregating in Linked Families
(A) Location of PSORS2 showing polymorphic microsatellites D17S784 and D17S982 and the locations of CARD14 and SLC26A11 and familial mutations. The following abbreviation is used: SGSH, N-sulfoglucosamine sulfohydrolase (MIM 605270).
(B) The pedigree of family PS1 shows genotypes at mutated loci (phased as displayed) of CARD14 (c.349G>A; upper) and SLC26A11 (c.365A>G; lower). The unaffected woman with an asterisk recently developed psoriasis at 83 years of age. The affected male labeled GEN001 provided skin biopsies of uninvolved and involved (psoriatic plaque) skin used for this study.
(C) The pedigree of 17q25-linked Taiwanese psoriasis-affected family shows genotypes of the c.349+5G>A mutation in family members. Black-filled symbols indicate classical psoriasis. Gray-filled symbols indicate mild skin manifestation of psoriasis. Question marks indicate unknown disease status.
(D) Both familial CARD14 mutations are located within the consensus splice donor sequence of exon 3. Chromatograms obtained after resequencing of this splice donor sequence in DNA of an affected member of each family are shown. The exon-intron junction is indicated by the vertical black line. The two mutations are boxed in red.
Further Evidence of CARD14 Mutations Predisposing to Psoriasis
Coding exons and flanking intronic DNA of CARD14 and SLC26A11 were resequenced in affected members of the Taiwanese psoriasis-affected family.16 This revealed a c.349+5G>A mutation in CARD14 but none in SLC26A11. Resequencing of 54 members of this family (21 affected, 20 unaffected, and 13 of unknown disease status) demonstrated that this mutation segregated with psoriasis (Figure 1C). This family had previously been sequenced for all genes under the linkage peak, and only one other possible disease-causing variant, a mutation upstream of the transcriptional start site of zinc finger protein 750 (ZNF750 [MIM 610226]), had been identified (discussed further below).19
We also identified a de novo germline mutation (c.413A>C [p.Glu138Ala]) in exon 4 of CARD14 in a 3-year-old Haitian child (individual 2192) who had severe pustular psoriasis since the age of 6 months (Figures 2A–2C). The mutation was predicted to be damaging by PolyPhen-2.027 and SIFT.28 The child had no family history of psoriasis or any other inflammatory disorder. She did not harbor any mutation in SLC26A11 nor the interleukin 36 receptor antagonist (IL36RN [MIM 605507]), a gene recently described to be mutated in pustular psoriasis.32,33 Neither the multiplex families nor the child with pustular psoriasis harbored the PSORS1 risk variant, HLA-Cw∗0602,34 suggesting that CARD14 mutations are sufficient to lead to disease. None of these mutations were detected in the sequences of the 1,000 Genomes Project,35 dbSNP130, or in eight previously exome-sequenced HapMap individuals.24 They were also shown to be present in the psoriatic population at very low frequencies (<0.1% of cases) and to be even less frequent or absent in controls (see the accompanying paper36 in this issue of AJHG).
Figure 2.
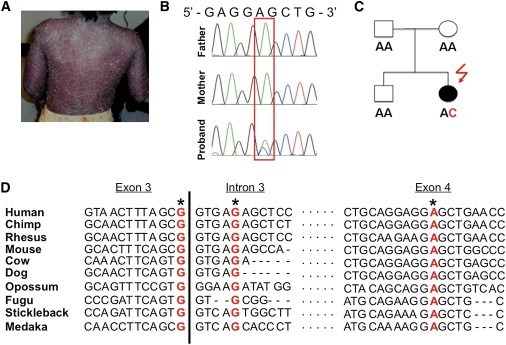
Detection of a De Novo CARD14 Mutation in an Individual with Pediatric Pustular Psoriasis and Conservation of Mutated Nucleotides
(A) Child 2192, who has pustular psoriasis, before treatment. Most of the body is covered with lesions.
(B) Sequence traces for 2192 and her parents revealing a de novo CARD14 mutation, c.413A>C (p.Glu138Ala).
(C) The pedigree of individual 2192 shows parental genotypes for de novo mutation c.413A>C (p.Glu138Ala).
(D) Alignment of DNA sequences from the indicated species is shown for the segments of CARD14 exon 3, intron 3, and exon 4 harboring the CARD14 mutations described here. The mutations are shown in red and are marked with asterisks. DNA and protein sequences were downloaded from the UCSC Genome Browser29 and were aligned with ClustalW2.30,31 The genome builds used for each species are as follows: hg19 (human), panTro2 (chimp), rheMac2 (rhesus), mm9 (mouse), bosTau4 (cow), canFam2 (dog), monDom5 (opossum), fr2 (fugu), gasAcu1 (stickleback), and oryLat2 (medaka).
Familial Psoriasis Mutations Alter Splicing of Exon 3
The CARD14 nucleotides mutated in the two families and in the pustular case were highly conserved in vertebrates (Figure 2D). The amino acids altered in family PS1 and individual 2192 with pustular psoriasis were also highly conserved (Figure S1). Although the p.Gly117Ser substitution was a missense alteration, the fact that both familial mutations lie in the splice donor sequence of exon 3 suggests that they might affect splicing (Figure 1D). We investigated this with in vitro splicing assays (Figures 3A and 3B),25 which revealed that both familial mutations lead to the use of a cryptic splice donor site 66 bp from the start of intron 3 (AG/GTGCCC, Figures 3B and 3C). This caused an insertion of 22 amino acids into the CARD14 peptide between exons 3 and 4 (Figure 3D).
Figure 3.
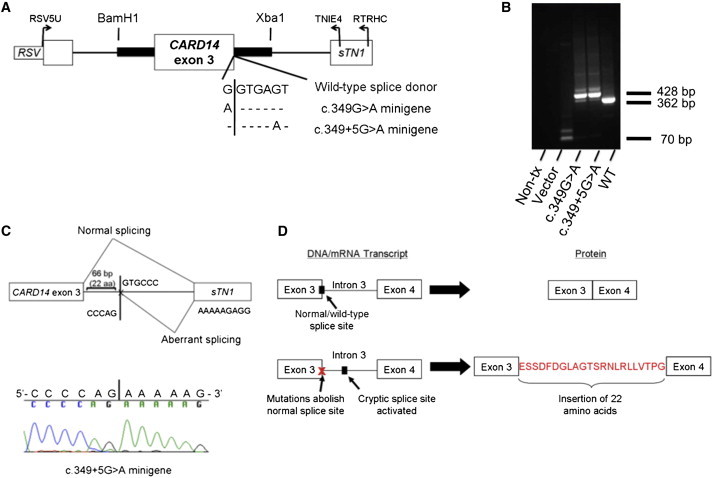
Familial CARD14 Mutations Alter Splicing of Exon 3
(A) The CARD14 exon 3 minigene constructs are wild-type, c.349G>A (mutation in the northern European PS1 family), and c.349+5G>A (mutation in the Taiwanese psoriasis-affected family). The vertical line indicates the junction of exon 3 and the intron of CARD14. The following abbreviation is used: RSV, Rous sarcoma virus promoter.
(B) Results of agarose gel electrophoresis after transfection of minigenes and isolation of cDNA. Mutant minigene PCR products were 60–70 bp larger than those of the wild-type construct. The following abbreviations are used: Non-tx, nontransfected; Vector, RHCglo25 vector alone; and WT, wild-type minigene. c.349G>A and c.349+5G>A are the mutant minigenes.
(C) Results of sequencing minigenes. Minigene c.349+5G>A is shown. Both mutations altered splicing of exon 3 and led to the addition of 66 bp from intron 3 and splicing to exon 4 at a cryptic splice donor site (AG/GTGCCC). The following abbreviation is used: sTN1, chicken skeletal troponin I (RHCglo23 downstream gene fragment).
(D) Consequence of altered splicing on CARD14.
We were also interested in evaluating the effects of these mutations in vivo. Although RNA and cDNA from the skin of affected members of the Taiwanese family were unavailable, we obtained a skin biopsy and extracted cDNA from the skin of an affected member (GEN001) of family PS1. cDNA sequencing with primers amplifying transcripts between exons 2 and 4 of CARD14 from skin of this individual revealed the presence of low levels of a correctly spliced mutant c.349A allele transcript (Figure S2A) and an isoform with splicing directly from exon 2 to exon 4 (e.g., skipping of exon 3; Figure S2B).
We performed further analyses of wild-type and mutant CARD14 transcripts from the exon 2–4 region by using RNA-Seq employing RNA from involved skin from GEN001. This revealed that 47% of CARD14 transcripts harbored the wild-type c.349G allele and that 12% of transcripts harbored the c.349A allele with correct splicing between exons 3 and 4. 9% of transcripts harbored the 66 bp intronic insertion (predicted by the minigene assay) between exons 3 and 4. We also identified 17% of the transcripts that were due to the skipping of exon 3 (presumably as a result of the c.349A mutation). Similar ratios of wild-type and mutant transcripts of each type were seen after PCR-based cloning and sequencing of cDNA spanning exons 2–4 (Figure S3 and Table S3).
However, RNA-Seq revealed an additional RNA species that could not be identified with PCR-based amplification of cDNA. This species extended from exon 3 into the intron and terminated 838 bp downstream (Figure S3 and Table S3). This mRNA isoform was confirmed to be from the sense strand of CARD14 (Figure S4). It was represented by ∼15% of all transcripts and would encode a peptide of an additional 59 amino acids after exon 3 and a 3′UTR of 656 bp. This transcript would be predicted to encode only the CARD domain. This peptide could exhibit constitutive CARD14-induced activation of NF-kB or exert a dominant-negative effect on wild-type CARD14. Additional studies are needed if researchers are to determine its effect as well as the contribution of the other mutant species to psoriasis pathogenesis. Nevertheless, additional functional studies (described below) indicated that the c.349A canonically spliced transcript itself has gain-of-function activity and is likely to contribute to disease. RNA-Seq with RNA derived from normal skin or classical (nonfamilial) psoriatic skin only revealed correctly spliced CARD14 transcripts between exons 3 and 4 (Figures S3 and S4 and Table S3).
Effect of Mutations on CARD14 Function In Vitro
CARD14 encodes a 1,004 amino acid protein that activates NF-kB20 and is represented by several isoforms. Full-length CARD14 (CARD14fl, also known as CARD14 isoform 1) encodes both an N-terminal CARD domain necessary for activation of NF-kB and a C-terminal tripartite MAGUK domain (PDZ/SH3/GUK).18 A shorter isoform, CARD14sh, encodes the CARD domain but not the tripartite domain. Another isoform, CARDcl, lacks the CARD domain and the tripartite domain (these latter isoforms lacking the tripartite domain are both known as isoform 2; Figure S5). Interrogation of a human tissue panel revealed that CARD14sh was the most abundant isoform in all tissues, including skin, in which it was expressed (Table S4).
We used luciferase reporter assays to test the effects of the familial and pustular-psoriasis substitutions on the NF-kB-inducing activity of the abundant CARD14sh protein isoform. Expression constructs with the p.Gly117Ser and p.Gly138Ala substitutions were generated, and an expression construct encoding CARD14cl, which lacks the CARD domain, served as a negative control for NF-kB activation. A construct encoding the full-length CARD14fl protein isoform was not available for subcloning into the expression vector.
The NF-kB assay revealed that compared with wild-type CARD14sh, both substitutions lead to increased levels of the luciferase reporter. The pustular-psoriasis substitution (p.Glu138Ala) led to a 7- to 9-fold increase in levels, and the PS1 p.Gly117Ser substitution led to a 3- to 4-fold increase in levels (Figure 4B). CARD14cl failed to activate NF-kB, in agreement with previous studies.20,37 We also investigated the dominant nature of the substitutions by cotransfecting wild-type and either p.Glu138Ala or p.Gly117Ser CARD14sh constructs and performing the NF-kB luciferase reporter assay. The resultant NF-kB activity was similar to that seen with the p.Glu138Ala and p.Gly117Ser substitutions alone (Figure 4B), providing additional evidence of their gain-of function activity. Cotransfection of CARD14cl and CARD14sh caused NF-kB activity to be reduced by approximately 50% as a result of either a dilution of CARD14sh or a dominant-negative effect of CARD14cl (Figure 4B).
Figure 4.

Location of CARD14 Alterations and Their Effect on Transcriptional Activation
(A) The location of the familial alterations and the de novo substitution in CARD14 are shown relative to the key protein domains. The two familial alterations are shown by blue triangles; the pustular-psoriasis alteration is shown by the purple triangle.
(B) NF-kB activation levels measured in HEK 293 cells transfected with one of four options: (1) CARD14sh alone, (2) the same construct harboring one of the rare variants shown, (3) CARD14cl, or (4) a combination of two constructs from options 1–3. The change in NF-kB activity relative to the background vector was determined for each variant (y axis) (see Subjects and Methods). Every data point represents the average value of three replicates. Asterisks represent results from a two-tailed, unpaired student's t test between the indicated construct and CARD14sh. The NF-kB activity induced by cotransfection of the p.Gly117Ser substitution and wild-type CARD14sh was not statistically significant from the NF-kB activity induced by CARD14sh alone (p = 0.1) or p.Gly117Ser alone (p = 0.7).
(C) HEK 001 cells were transfected with wild-type or altered (c.349G>A [p.Gly117Ser] or c.413A>C [p.Glu138Ala]) CARD14sh, and qRT-PCR was performed so that the upregulation of CCL20, IL8, SOD2, and IL36G identified by global expression profiling could be confirmed. Asterisks represent results from a two-tailed, unpaired student's t test between the indicated construct and CARD14sh.
(D) Upregulation of those same transcripts was confirmed in primary keratinocytes of individual 2192 with pustular psoriasis (c.413A>C [p.Glu138Ala]). Expression in these primary keratinocytes was compared with that in two human foreskin keratinocyte samples (control 1 and control 2). For all qRT-PCR, expression levels were normalized to 18S by the 2-ΔΔCt method. For the transformation of expression levels to nondecimal integers, all expression levels were multiplied by 100,000 before being plotted on graphs. Asterisks represent results from a two-tailed, unpaired student's t test between the indicated control and the mutant (p.Glu138Ala). For (B–D), error bars represent the standard deviation of replicates, and ∗p ≤ 0.05, ∗∗p ≤ 0.01, and ∗∗∗p ≤ 0.001. The following abbreviations are used: NT, nontransfected; G117S, p.Gly117Ser; E138A, p.Glu138Ala; and Ctrl, control.
Because TNF-α activates NF-kB pathways, it was important to ascertain whether the increase in NF-kB activity seen in some of our assays was due to TNF-α secreted by transfected cells. The addition of a neutralizing TNF antibody did not have an effect on NF-kB reporter activity (Figure S6). These results support the disease-causing potential of the familial and de novo pustular-psoriasis substitutions through activation of the NF-kB pathway.
Tissue-Specific Localization of CARD14
To examine the localization of CARD14 in healthy and psoriatic skin, we stained normal and psoriatic skin samples with a polyclonal antibody specific to the coiled-coil domain of CARD14 (Figure 5). Staining of basal keratinocytes was observed in normal and uninvolved skin, and decreased expression was seen in the upper layers of the skin, including the granular layer. This contrasted with involved skin, in which there was loss of labeling of basal keratinocytes but labeling throughout the remainder of the epidermis and of some dermal cells. There was reduced, patchy epidermal labeling in involved skin of GEN001 (Figures 5A and 5B and Figure S7). Although there might be a subset of dermal immune cells that express CARD14, keratinocytes are clearly a major source of CARD14.
Figure 5.
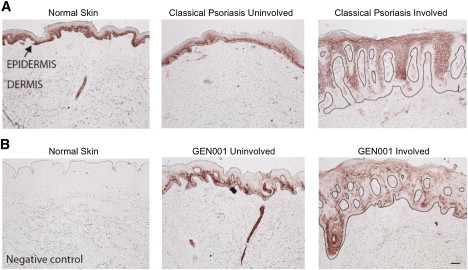
Distribution of CARD14 in Normal and Psoriatic Skin
Representative images of normal and psoriatic (uninvolved and involved) skin labeled with a polyclonal antibody to the internal coiled-coil CARD14 domain that is shared by all known isoforms.
(A) Normal skin and classical uninvolved and involved psoriasis skin.
(B) A normal-skin negative control and uninvolved and involved affected skin from individual GEN001 from family PS1. The epidermis is the darker-stained upper band (black arrow), and the dermis is the paler region below. The black line denotes the dermoepidermal junction; lesional skin is cut on a slight cross section, and the dermis is evident as islands projecting upwards into the epidermis. The scale bar represents 100 μm.
Previous mRNA-expression studies have reported elevated levels (a 2.7-fold increase) of CARD14 in involved psoriatic skin versus uninvolved skin.38 We have confirmed this with qRT-PCR (not shown). However, our data indicate that this apparent increase in gene expression might be due to an increase in the ratio of CARD14-positive keratinocytes to CARD14-negative keratinocytes in involved skin rather than an increase in the number of CARD14 transcripts at the cellular level.
Effect of CARD14 Substitutions on Keratinocyte Gene Expression
CARD14 localizes strongly to epidermal keratinocytes (see above), suggesting that the effect of the substitutions is mediated at least in part through this cell type. We therefore investigated the effect of wild-type and altered CARD14 (p.Gly117Ser and p.Glu138Ala) on the transcriptome of psoriatic skin and keratinocytes separately. We performed global expression profiling of classical psoriatic skin, skin from an affected member (GEN001) of family PS1, and skin from individual 2192 with pustular psoriasis, and we compared transcript levels with those from normal skin. We also profiled HEK 001 immortalized keratinocytes that had been transfected with a wild-type CARD14sh construct or a CARD14sh construct encoding one of the missense substitutions (p.Gly117Ser or p.Glu138Ala). We then looked for genes that were upregulated in psoriatic skin and that were expressed at significantly higher levels in the p.Glu138Ala and p.Gly117Ser transfectants than in the wild-type-CARD14sh transfectants (Table S5).
A number of transcripts consistently showed increased expression in psoriatic skin, upregulation after keratinocytes were transfected with CARD14sh, and further upregulation in the presence of the p.Glu138Ala and p.Gly117Ser substitutions. This was confirmed with qRT-PCR (Figure 4C). For example, in the presence of the p.Glu138Ala and p.Gly117Ser substitutions, CCL20 was upregulated 7.2× and 1.9× more, respectively, and IL8 was upregulated 4.6× and 1.6× more, respectively, than in the presence of wild-type CARD14sh. The p.Glu138Ala substitution increased expression of superoxide dismutase 2, mitochondrial (SOD2 [MIM 147460]) and IL36G 4.1× and 1.8× more, respectively, than did wild-type CARD14sh (Table S5 and Figure 4C). We also confirmed upregulation of these transcripts in primary keratinocytes from individual 2192 with pustular psoriasis (Figure 4D; primary keratinocytes from family PS1 were not available).
As a complement to these studies, we also profiled three immortalized keratinocyte lines before and after treatment with TNF-α. Two lines were from uninvolved skin of affected members (K1-1 and K1-20) of family PS1, and the other was from involved skin of a classical-psoriasis-affected individual (K5-14) who harbors the HLA-Cw∗0602 risk allele. In all three immortalized keratinocyte lines, TNF-α stimulation led to upregulation of the same transcripts described above. All of this provided evidence that after an inflammatory trigger, these transcripts are induced in keratinocytes of affected members of family PS1 and of classical-psoriasis-affected individuals (Table S5).
It is also relevant that compared with those of normal skin, the transcriptomes of involved skin from the familial, pustular-psoriasis, and classical-psoriasis samples exhibited very similar differences. This suggests that pathways downstream of CARD14 can be similarly altered in classical, pustular, and familial psoriasis. Table S6 provides fold changes of transcripts that most strongly differentiate classical psoriasis from normal skin, as well as fold changes of the same transcripts in involved skin from GEN001 (PS1) and the child, 2192, with pustular psoriasis.
Discussion
Here, rare, highly penetrant mutations in CARD14 have been shown to cause psoriasis, thus concluding a 17 year search for PSORS2.14 We identified two disease-causing CARD14 mutations in two multiplex psoriasis-affected families (c.349G>A [p.Gly117Ser] in family PS1 of northern European ancestry and c.349+5G>A in a family from Taiwan). In the European family, approximately 30% of affected members also had psoriatic arthritis, indicating that the c.349G>A (p.Gly117Ser) mutation might also contribute to inflammatory joint disease. Both of the familial mutations disrupted splicing of CARD14 and lead to an in-frame insertion of 22 amino acids in vitro. The c.349G>A mutation was confirmed in vivo to lead to multiple splice forms, including the 22 amino acid insertion predicted from the minigene assay, another with skipping of exon 3, and a third with the addition of 59 amino acids from intron 3 and a subsequent termination signal and 661 bp 3′UTR. The latter is predicted to result in a truncated peptide with only the CARD domain. A de novo mutation (c.413A>C [p.Glu138Ala]) in exon 4 of CARD14 in a child with sporadic, severe, early-onset pustular psoriasis and no family history or susceptibility factors for psoriasis independently confirmed the disease-causing potential of these mutations. Interestingly, exon 4 encodes part of the coiled-coil domain of the protein. A recessive mouse mutation in the coiled-coil domain of a structurally related scaffold protein, CARD11, leads to an inflammatory skin phenotype resembling atopic dermatitis. This suggests that the coiled-coil domains of CARD14 and related family members are important in regulating inflammation.39
The identification of a CARD14 mutation in the Taiwanese family settles a paradox that existed between the two multiplex psoriasis-affected families. The Taiwanese family harbors a variant (c.625A>C in the promoter of ZNF750) that was thought to cause psoriasis.19 However, we identified no disease-causing mutation in ZNF750 in the PS1 family, and we found the ZNF750 c.625A>C variant in only 2 out of 172 healthy Asian controls. Hence, the CARD14 mutation rather than the ZNF750 variant is probably causative for psoriasis in this family.
It is also worth noting that one PS1 family member who harbored the c.349G>A (p.Gly117Ser) mutation was initially unaffected when ascertained in 199314 but developed psoriasis 17 years later at 83 years old (indicated by the asterisk in Figure 1B). It is unknown which precipitating factors contribute to this variable age of onset, but genetic background40 and environmental factors are likely to play a role. Determining whether the SLC26A11 variant that cosegregated with psoriasis in family PS1 contributes to psoriasis or psoriatic arthritis independently needs further investigation.
A substantial proportion of individuals with psoriasis have no family history of the disease. This has been thought to be due to a requirement for environmental triggers and/or inheritance of multiple low-risk genetic factors. Until now, evidence implicating de novo mutations as being causative in psoriasis or other common disease for which there is no family history has been lacking. However, the early age of onset of severe generalized pustular psoriasis in a child with a de novo mutation indicates that such rare variants can account for some of these cases. The frequency of de novo mutations contributing to common diseases such as psoriasis is of interest, and genes harboring such variants are expected to be identified at an increasing rate now that global sequencing of DNA from parents and affected children is feasible.
The role of CARD14 in health and disease remains to be fully determined. It encodes a 1,004 amino acid protein that activates the transcription factor NF-kB20 and inhibits apoptosis.37 It has not been implicated previously in psoriasis pathogenesis. CARD14 is expressed in the placenta and mucosal surfaces,20,37,41 and, as demonstrated here, in the keratinocytes of normal and inflamed skin. The localization pattern of CARD14 differs between normal and involved lesional skin. We show that it is localized in the basal layer of the skin and is absent from the upper layers, including the granular layer of normal skin, but the opposite is true of psoriatic skin. In psoriatic skin, CARD14 is absent from the basal layer and is present in all suprabasal layers. This difference in expression pattern cannot be easily explained. However, it highlights intrinsic differences between keratinocytes of healthy and psoriatic skin.
Although variants in CARD14 have not been described previously in psoriasis, genetic alterations in components of the NF-kB pathway are implicated from GWASs.5–11 Moreover, basal-level constitutively active phosphorylated NF-kB/RelA is found in uninvolved skin and is upregulated in psoriasis plaques.42 Uncontrolled NF-kB activation has also been suggested as the inflammatory pathway in a recently published monogenic form of pustular psoriasis caused by mutations in IL36RN.32,33 Our findings suggest that in psoriasis, NF-kB-pathway alterations implicated from GWASs can occur at the level of the keratinocyte as well as at the level of the cells of the hematopoietic system.
Both of the missense substitutions reported here significantly increased NF-kB activation and led to upregulation of psoriasis-associated transcripts such as IL8 and CCL20. The pustular-psoriasis substitution also led to upregulation of IL36G and SOD2. TNF-α stimulation of epidermal keratinocytes has previously been reported to lead to upregulation of many genes, including IL8, CCL20, and SOD2, harboring NF-kB binding sites in their promoters.43–45 These transcripts are also notable because of the functions of the proteins they encode. IL8 is a well-known neutrophil chemotaxin,46 and CCL20 is a chemotactic factor for CCR6+ immature dendritic cells and T cells. IL36G is a proinflammatory cytokine, and SOD2 is a potential antinecroptosis gene.47 All of these molecules have been shown to be upregulated in keratinocytes in response to injury,48 suggesting that CARD14 mediates an important conserved pathway that is triggered in response to disrupted homeostasis (due to injury, infection, etc.) of the skin. The effect of the pustular mutation on gene expression was generally greater than that of the family PS1 mutation in transfected keratinocytes. This might reflect an increased severity in the presence of certain mutations such as c.413G>A (p.Glu138Ala). However, it might be due in part to the fact that we were unable to assay the combined effect of the c.349G>A (p.Gly117Ser) mutation with all of the in-frame, altered splice forms that occur as a result of this mutation (and that are also predicted as a consequence of the Taiwanese psoriasis mutation). Indeed, the relative frequency of the splice forms might influence the severity of disease as well as the effect of the missense p.Gly117Ser substitution on NF-kB activation.
The expression alterations seen in keratinocytes with the PS1 and pustular mutations allow us to propose a model of psoriasis pathogenesis. After an inflammatory trigger that might include infection and/or epidermal injury,48 a subset of CARD14 substitutions that are found in psoriasis patients induce enhanced activation of NF-kB. This leads to pathologic levels of transcripts of some target genes, including key chemokines implicated in psoriasis, such as IL8 and CCL20. We suggest that the release of these inflammatory mediators leads to the recruitment and differentiation of inflammatory cells. Activated dendritic cells then produce IL-23, a critical factor for Th17 development, and T cells produce IL17 and IL22, leading to further keratinocyte activation and epidermal hyperplasia.49 The activation of keratinocytes and expression of IL36G also induce the activation of the NF-kB pathway in inflammatory cells and the further production of cytokines and chemokines. This creates a vicious cycle of inflammation and acanthosis that characterizes psoriasis (Figure 6). Notably, GWASs have identified psoriasis-associated alterations at other loci harboring genes encoding members of the NF-kB pathway. For example, variants near TNFAIP3 (tumor necrosis factor, alpha-induced protein 3, [TNFAIP3 (MIM 191163)]) are associated with psoriasis. That gene encodes the protein A20, which negatively regulates NF-kB activation by the CARD14-related peptides CARD11 and CARD10.50 TNIP1 (TNFAIP3-interacting protein 1, [TNIP1 (MIM 607714)]) might also be involved in this process because it acts in concert with A20 and is downregulated in psoriatic skin.8 Although future studies are needed for the dissection of the interaction between CARD14 and the NF-kB pathway, we know that CARD14-induced activation of NF-kB is dependent on TRAF2 and might also require TRAF3 or TRAF6.37 Thus, one can hypothesize that activation of the CARD14-NF-kB pathway might be downstream of Act1, encoded by TRAF3IP2 (TRAF3-interacting protein 2, [TRAF3IP2 (MIM 607043)]), which harbors a variant associated with psoriasis and psoriatic arthritis.5,6,9 Alternatively, CARD14 might be part of a parallel pathway leading to NF-kB activation. Loss-of-function amino acid substitutions in IL36RN were recently identified in some cases of pustular psoriasis.32,33 This protein directly opposes the activity of IL36γ and also intersects with the NF-kB pathway. Determining whether and where activation pathways for CARD14 and IL36RN intersect requires further evaluation.
Figure 6.
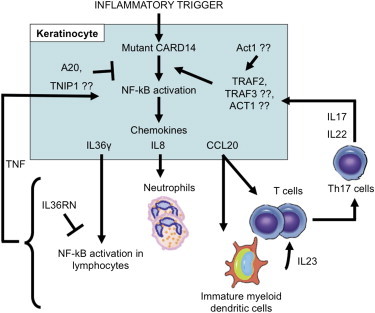
Proposed Model of Psoriasis Pathogenesis in the Presence of Psoriasis-Specific CARD14 Alterations
After an inflammatory trigger, alterations in CARD14 induce activation of NF-kB. This leads to the transcription of many genes, including key chemokines such as CCL20, IL8, and IL36G, implicated in psoriasis. The proteins encoded by these transcripts can recruit immune cells involved in disease pathogenesis. Those cells, in turn, produce cytokines and chemokines that cause inflammation and lead to further keratinocyte activation and epidermal hyperplasia (see Discussion). Other proteins in the NF-kB pathway have been previously implicated in psoriasis and might be important in CARD14 signaling to NF-kB. These proteins include A20,50 TNIP1,8 and Act1,5,6,9 and these, in turn, implicate TRAF2, TRAF3, and TRAF6.37 IL36RN, recently found to be mutated in pustular psoriasis, might also play a role in this pathway by inhibiting IL36γ-induced immune cell activation.32,33 All of these events contribute to this vicious cycle of inflammation and acanthosis seen in psoriasis.
Finally, our findings highlight the shortcomings of traditional schemes for classifying human disease on the basis of clinical observations and underscore the utility of adapting molecular classification schemes based on the physiology that is dysregulated in disease. In this instance, common plaque psoriasis, pustular psoriasis, and, possibly, psoriatic arthritis, represent different parts of a disease severity spectrum and might all be ascribed to mutations in the same gene (CARD14). Our findings also pave the way for novel therapeutic interventions for these diseases.
Acknowledgments
This research was supported by the following grants from the National Institutes of Health: AR050266 and 5RC1AR058681 (A.M.B.), T32AR007279 (E.D.O.R.), T32HL083822 and T32GM07200 (C.T.J), AR060222 (M.A.L and K.C.P), and T32HG000045 (C.E.J). R.G.M., Y.L., and Y.C. are supported by the Intramural Research Programs of the National Institute of Arthritis and Musculoskeletal and Skin Diseases. A.A.M. was supported by the Intramural Research Programs of the National Institute of Allergy and Infectious Diseases. Additional funding came from the Academia Sinica and National Science Council (National Clinical Core, National Genotyping Core) of Taiwan. The authors thank the many individuals with psoriasis and the controls who participated in this study. Mike Lovett provided helpful comments on the manuscript. The authors are indebted to the National Psoriasis Foundation for continuing support during the course of this study.
Supplemental Data
Web Resources
The URLs for data presented here are as follows:
ClustalW2, http://www.ebi.ac.uk/Tools/msa/clustalw2/
Microarray Gene Expression Data Society (MIAME), http://www.mged.org/Workgroups/MIAME_checklist.html
NCBI Gene Expression Omnibus (GEO), http://www.ncbi.nlm.nih.gov/geo/
NCBI Reference Sequence (RefSeq), http://www.ncbi.nlm.nih.gov/RefSeq/
Online Mendelian Inheritance in Man (OMIM), http://omim.org
PolyPhen-2.0, http://genetics.bwh.harvard.edu/pph2/
SIFT, http://sift.jcvi.org/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Lowes M.A., Bowcock A.M., Krueger J.G. Pathogenesis and therapy of psoriasis. Nature. 2007;445:866–873. doi: 10.1038/nature05663. [DOI] [PubMed] [Google Scholar]
- 2.Nograles K.E., Brasington R.D., Bowcock A.M. New insights into the pathogenesis and genetics of psoriatic arthritis. Nat. Clin. Pract. Rheumatol. 2009;5:83–91. doi: 10.1038/ncprheum0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Capon F., Bijlmakers M.J., Wolf N., Quaranta M., Huffmeier U., Allen M., Timms K., Abkevich V., Gutin A., Smith R. Identification of ZNF313/RNF114 as a novel psoriasis susceptibility gene. Hum. Mol. Genet. 2008;17:1938–1945. doi: 10.1093/hmg/ddn091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cargill M., Schrodi S.J., Chang M., Garcia V.E., Brandon R., Callis K.P., Matsunami N., Ardlie K.G., Civello D., Catanese J.J. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am. J. Hum. Genet. 2007;80:273–290. doi: 10.1086/511051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ellinghaus E., Ellinghaus D., Stuart P.E., Nair R.P., Debrus S., Raelson J.V., Belouchi M., Fournier H., Reinhard C., Ding J. Genome-wide association study identifies a psoriasis susceptibility locus at TRAF3IP2. Nat. Genet. 2010;42:991–995. doi: 10.1038/ng.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hüffmeier U., Uebe S., Ekici A.B., Bowes J., Giardina E., Korendowych E., Juneblad K., Apel M., McManus R., Ho P. Common variants at TRAF3IP2 are associated with susceptibility to psoriatic arthritis and psoriasis. Nat. Genet. 2010;42:996–999. doi: 10.1038/ng.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Y., Helms C., Liao W., Zaba L.C., Duan S., Gardner J., Wise C., Miner A., Malloy M.J., Pullinger C.R. A genome-wide association study of psoriasis and psoriatic arthritis identifies new disease loci. PLoS Genet. 2008;4:e1000041. doi: 10.1371/journal.pgen.1000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nair R.P., Duffin K.C., Helms C., Ding J., Stuart P.E., Goldgar D., Gudjonsson J.E., Li Y., Tejasvi T., Feng B.J., Collaborative Association Study of Psoriasis Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat. Genet. 2009;41:199–204. doi: 10.1038/ng.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strange A., Capon F., Spencer C.C., Knight J., Weale M.E., Allen M.H., Barton A., Band G., Bellenguez C., Bergboer J.G., Genetic Analysis of Psoriasis Consortium & the Wellcome Trust Case Control Consortium 2 A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat. Genet. 2010;42:985–990. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stuart P.E., Nair R.P., Ellinghaus E., Ding J., Tejasvi T., Gudjonsson J.E., Li Y., Weidinger S., Eberlein B., Gieger C. Genome-wide association analysis identifies three psoriasis susceptibility loci. Nat. Genet. 2010;42:1000–1004. doi: 10.1038/ng.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun L.D., Cheng H., Wang Z.X., Zhang A.P., Wang P.G., Xu J.H., Zhu Q.X., Zhou H.S., Ellinghaus E., Zhang F.R. Association analyses identify six new psoriasis susceptibility loci in the Chinese population. Nat. Genet. 2010;42:1005–1009. doi: 10.1038/ng.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vineis P., E Pearce N. Genome-wide association studies may be misinterpreted: Genes versus heritability. Carcinogenesis. 2011;32:1295–1298. doi: 10.1093/carcin/bgr087. [DOI] [PubMed] [Google Scholar]
- 13.Chen H., Poon A., Yeung C., Helms C., Pons J., Bowcock A.M., Kwok P.Y., Liao W. A genetic risk score combining ten psoriasis risk loci improves disease prediction. PLoS ONE. 2011;6:e19454. doi: 10.1371/journal.pone.0019454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tomfohrde J., Silverman A., Barnes R., Fernandez-Vina M.A., Young M., Lory D., Morris L., Wuepper K.D., Stastny P., Menter A. Gene for familial psoriasis susceptibility mapped to the distal end of human chromosome 17q. Science. 1994;264:1141–1145. doi: 10.1126/science.8178173. [DOI] [PubMed] [Google Scholar]
- 15.Nair R.P., Henseler T., Jenisch S., Stuart P., Bichakjian C.K., Lenk W., Westphal E., Guo S.W., Christophers E., Voorhees J.J., Elder J.T. Evidence for two psoriasis susceptibility loci (HLA and 17q) and two novel candidate regions (16q and 20p) by genome-wide scan. Hum. Mol. Genet. 1997;6:1349–1356. doi: 10.1093/hmg/6.8.1349. [DOI] [PubMed] [Google Scholar]
- 16.Hwu W.L., Yang C.F., Fann C.S., Chen C.L., Tsai T.F., Chien Y.H., Chiang S.C., Chen C.H., Hung S.I., Wu J.Y., Chen Y.T. Mapping of psoriasis to 17q terminus. J. Med. Genet. 2005;42:152–158. doi: 10.1136/jmg.2004.018564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Helms C., Cao L., Krueger J.G., Wijsman E.M., Chamian F., Gordon D., Heffernan M., Daw J.A., Robarge J., Ott J. A putative RUNX1 binding site variant between SLC9A3R1 and NAT9 is associated with susceptibility to psoriasis. Nat. Genet. 2003;35:349–356. doi: 10.1038/ng1268. [DOI] [PubMed] [Google Scholar]
- 18.Speckman R.A., Wright Daw J.A., Helms C., Duan S., Cao L., Taillon-Miller P., Kwok P.Y., Menter A., Bowcock A.M. Novel immunoglobulin superfamily gene cluster, mapping to a region of human chromosome 17q25, linked to psoriasis susceptibility. Hum. Genet. 2003;112:34–41. doi: 10.1007/s00439-002-0851-y. [DOI] [PubMed] [Google Scholar]
- 19.Yang C.F., Hwu W.L., Yang L.C., Chung W.H., Chien Y.H., Hung C.F., Chen H.C., Tsai P.J., Fann C.S., Liao F., Chen Y.T. A promoter sequence variant of ZNF750 is linked with familial psoriasis. J. Invest. Dermatol. 2008;128:1662–1668. doi: 10.1038/jid.2008.1. [DOI] [PubMed] [Google Scholar]
- 20.Bertin J., Wang L., Guo Y., Jacobson M.D., Poyet J.L., Srinivasula S.M., Merriam S., DiStefano P.S., Alnemri E.S. CARD11 and CARD14 are novel caspase recruitment domain (CARD)/membrane-associated guanylate kinase (MAGUK) family members that interact with BCL10 and activate NF-kappa B. J. Biol. Chem. 2001;276:11877–11882. doi: 10.1074/jbc.M010512200. [DOI] [PubMed] [Google Scholar]
- 21.Kaur P., McDougall J.K., Cone R. Immortalization of primary human epithelial cells by cloned cervical carcinoma DNA containing human papillomavirus type 16 E6/E7 open reading frames. J. Gen. Virol. 1989;70:1261–1266. doi: 10.1099/0022-1317-70-5-1261. [DOI] [PubMed] [Google Scholar]
- 22.Chapman S., Liu X., Meyers C., Schlegel R., McBride A.A. Human keratinocytes are efficiently immortalized by a Rho kinase inhibitor. J. Clin. Invest. 2010;120:2619–2626. doi: 10.1172/JCI42297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harbour J.W., Onken M.D., Roberson E.D., Duan S., Cao L., Worley L.A., Council M.L., Matatall K.A., Helms C., Bowcock A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330:1410–1413. doi: 10.1126/science.1194472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ng S.B., Turner E.H., Robertson P.D., Flygare S.D., Bigham A.W., Lee C., Shaffer T., Wong M., Bhattacharjee A., Eichler E.E. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh G., Cooper T.A. Minigene reporter for identification and analysis of cis elements and trans factors affecting pre-mRNA splicing. Biotechniques. 2006;41:177–181. doi: 10.2144/000112208. [DOI] [PubMed] [Google Scholar]
- 26.Vincourt J.B., Jullien D., Amalric F., Girard J.P. Molecular and functional characterization of SLC26A11, a sodium-independent sulfate transporter from high endothelial venules. FASEB J. 2003;17:890–892. doi: 10.1096/fj.02-0787fje. [DOI] [PubMed] [Google Scholar]
- 27.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 29.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chenna R., Sugawara H., Koike T., Lopez R., Gibson T.J., Higgins D.G., Thompson J.D. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 32.Marrakchi S., Guigue P., Renshaw B.R., Puel A., Pei X.Y., Fraitag S., Zribi J., Bal E., Cluzeau C., Chrabieh M. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N. Engl. J. Med. 2011;365:620–628. doi: 10.1056/NEJMoa1013068. [DOI] [PubMed] [Google Scholar]
- 33.Onoufriadis A., Simpson M.A., Pink A.E., Di Meglio P., Smith C.H., Pullabhatla V., Knight J., Spain S.L., Nestle F.O., Burden A.D. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am. J. Hum. Genet. 2011;89:432–437. doi: 10.1016/j.ajhg.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elder J.T., Cluster 17 Collaboration Fine mapping of the psoriasis susceptibility gene PSORS1: A reassessment of risk associated with a putative risk haplotype lacking HLA-Cw6. J. Invest. Dermatol. 2005;124:921–930. doi: 10.1111/j.0022-202X.2005.23729.x. [DOI] [PubMed] [Google Scholar]
- 35.1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jordan C.T., Cao L., Roberson E.D.O., Duan S., Helms C.A., Nair R.P., Duffin K.C., Stuart P.E., Goldgar D., Hayashi G. Rare and Common Variants in CARD14, Encoding an Epidermal Regulator of NF-kappaB, in Psoriasis. Am. J. Hum. Genet. 2012;90 doi: 10.1016/j.ajhg.2012.03.013. in press. Published online April 19, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scudiero I., Zotti T., Ferravante A., Vessichelli M., Vito P., Stilo R. Alternative splicing of CARMA2/CARD14 transcripts generates protein variants with differential effect on NF-κB activation and endoplasmic reticulum stress-induced cell death. J. Cell. Physiol. 2011;226:3121–3131. doi: 10.1002/jcp.22667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suárez-Fariñas M., Lowes M.A., Zaba L.C., Krueger J.G. Evaluation of the psoriasis transcriptome across different studies by gene set enrichment analysis (GSEA) PLoS ONE. 2010;5:e10247. doi: 10.1371/journal.pone.0010247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jun J.E., Wilson L.E., Vinuesa C.G., Lesage S., Blery M., Miosge L.A., Cook M.C., Kucharska E.M., Hara H., Penninger J.M. Identifying the MAGUK protein Carma-1 as a central regulator of humoral immune responses and atopy by genome-wide mouse mutagenesis. Immunity. 2003;18:751–762. doi: 10.1016/s1074-7613(03)00141-9. [DOI] [PubMed] [Google Scholar]
- 40.Prots I., Skapenko A., Wendler J., Mattyasovszky S., Yoné C.L., Spriewald B., Burkhardt H., Rau R., Kalden J.R., Lipsky P.E., Schulze-Koops H. Association of the IL4R single-nucleotide polymorphism I50V with rapidly erosive rheumatoid arthritis. Arthritis Rheum. 2006;54:1491–1500. doi: 10.1002/art.21832. [DOI] [PubMed] [Google Scholar]
- 41.Blonska M., Lin X. NF-κB signaling pathways regulated by CARMA family of scaffold proteins. Cell Res. 2011;21:55–70. doi: 10.1038/cr.2010.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lizzul P.F., Aphale A., Malaviya R., Sun Y., Masud S., Dombrovskiy V., Gottlieb A.B. Differential expression of phosphorylated NF-kappaB/RelA in normal and psoriatic epidermis and downregulation of NF-kappaB in response to treatment with etanercept. J. Invest. Dermatol. 2005;124:1275–1283. doi: 10.1111/j.0022-202X.2005.23735.x. [DOI] [PubMed] [Google Scholar]
- 43.Stein B., Yang M.X. Repression of the interleukin-6 promoter by estrogen receptor is mediated by NF-kappa B and C/EBP beta. Mol. Cell. Biol. 1995;15:4971–4979. doi: 10.1128/mcb.15.9.4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Banno T., Gazel A., Blumenberg M. Pathway-specific profiling identifies the NF-kappa B-dependent tumor necrosis factor alpha-regulated genes in epidermal keratinocytes. J. Biol. Chem. 2005;280:18973–18980. doi: 10.1074/jbc.M411758200. [DOI] [PubMed] [Google Scholar]
- 45.Banno T., Gazel A., Blumenberg M. Effects of tumor necrosis factor-alpha (TNF alpha) in epidermal keratinocytes revealed using global transcriptional profiling. J. Biol. Chem. 2004;279:32633–32642. doi: 10.1074/jbc.M400642200. [DOI] [PubMed] [Google Scholar]
- 46.Sticherling M., Sautier W., Schröder J.M., Christophers E. Interleukin-8 plays its role at local level in psoriasis vulgaris. Acta Derm. Venereol. 1999;79:4–8. doi: 10.1080/000155599750011606. [DOI] [PubMed] [Google Scholar]
- 47.Thapa R.J., Basagoudanavar S.H., Nogusa S., Irrinki K., Mallilankaraman K., Slifker M.J., Beg A.A., Madesh M., Balachandran S. NF-kappaB protects cells from gamma interferon-induced RIP1-dependent necroptosis. Mol. Cell. Biol. 2011;31:2934–2946. doi: 10.1128/MCB.05445-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kennedy-Crispin M., Billick E., Mitsui H., Gulati N., Fujita H., Gilleaudeau P., Sullivan-Whalen M., Johnson-Huang L.M., Suarez-Farinas M., Krueger J.G. Human Keratinocytes' Response to Injury Upregulates CCL20 and Other Genes Linking Innate and Adaptive Immunity. J Invest Dermatol. 2012;132:105–113. doi: 10.1038/jid.2011.262. Published online September 1, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnston A., Xing X., Guzman A.M., Riblett M., Loyd C.M., Ward N.L., Wohn C., Prens E.P., Wang F., Maier L.E. IL-1F5, -F6, -F8, and -F9: a novel IL-1 family signaling system that is active in psoriasis and promotes keratinocyte antimicrobial peptide expression. J. Immunol. 2011;186:2613–2622. doi: 10.4049/jimmunol.1003162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stilo R., Varricchio E., Liguoro D., Leonardi A., Vito P. A20 is a negative regulator of BCL10- and CARMA3-mediated activation of NF-kappaB. J. Cell Sci. 2008;121:1165–1171. doi: 10.1242/jcs.021105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.