

Abstract
Staphylococcus aureus is an important human pathogen that can cause long-lasting persistent infections. The mechanisms by which persistent infections are maintained involve both bacterial escape strategies and modulation of the host immune response. So far, the investigations in this area have focused on strategies used by S. aureus to persist within the host. Here, we used an experimental mouse model to investigate the host response to persistent S. aureus infection. Our results demonstrated that T cells, which are critical for controlling S. aureus infection, gradually lost their ability to respond to antigenic stimulation and entered a state of anergy with the progression of infection towards persistence. The T cell hyporesponsiveness was reverted by co-stimulation with the phorbol ester PMA, an activator of protein kinase C, suggesting that a failure in the T cell receptor (TCR)-proximal signalling events underlie the hyporesponsive phenotype. The presence of these anergic antigen-specific T cells may contribute to the failure of the host immune response to promote sterilizing immunity during persistent S. aureus infection and also offers new possibilities for novel immunotherapeutic approaches.
Keywords: immunosuppressive mechanisms, persistent infection, Staphylococcus aureus, T cell anergy, T cell responses
INTRODUCTION
Staphylococcus aureus is one of the leading causes of both community-associated and nosocomial infections worldwide. As a commensal organism, S. aureus resides in the human skin and nares (Lowy, 1998). However, breaches in local protective barriers due to a skin cut, surgical wounds or implantation of medical devices provide this pathogen with the opportunity to gain access to deeper tissues and to cause life-threatening infections such as pneumonia, osteomyelitis, endocarditis and sepsis (Lowy, 1998). The host reacts to S. aureus invasion by activating innate immune mechanisms that involve the production of cytokines and chemokines that attract macrophages, neutrophils and other immune cells to the site of infection (von Köckritz-Blickwede et al, 2008). Generally, activation of adaptive immune responses ensues during which T and B cells capable of specific antigen recognition lead to the eradication of the pathogen. However, under certain circumstances S. aureus continues to thrive inside the host despite robust antimicrobial activities of the host immune defenses resulting in a persistent infection (Proctor et al, 1995; Sheehy et al, 2010). Persistent S. aureus infections such as chronic osteomyelitis or recurrent furuncolosis are difficult to eradicate. Treatment of these infections is complex and surgery is often needed to drain persistent abscesses.
In general, persistent infections result from a combination of host immune effectors that restrain the pathogen from growth and dissemination as well as from pathogen-driven counter-regulatory mechanisms that limit the effectiveness of the elicited host immune response. Like other pathogens capable of persisting within their hosts, S. aureus has evolved different strategies to interfere with the host adaptive immune response. Several studies have reported that toxic shock syndrome toxins (TSST) can alter T cell functions by targeting the T cell receptor (TCR) activation pathway (Llewelyn & Cohen, 2002). By its ability to interfere with antigen presentation mediated by MHC class II, the extracellular adherent protein (Eap) may exert an important role in subverting the adaptive immune response (Lee et al, 2002). Furthermore, staphylococcal protein A was shown to modulate B cell functions (Goodyear & Silverman, 2004). In addition, accumulating experimental evidence demonstrated that S. aureus, which is classically considered an extracellular pathogen, is capable of invading and surviving for various periods of time within a broad range of phagocytic and non-phagocytic cells (Garzoni & Kelley, 2009). Adaptation of S. aureus to the intracellular milieu is associated with a phenotype change towards small colony variants (SCVs), which resemble dormant phenotypes that favour bacterial persistence (Tuchscherr et al, 2011).
Despite substantial progress made in understanding the mechanisms employed by S. aureus to persist within the host, very few studies have addressed the host immune mechanisms evoked during a persistent staphylococcal infection. Research in this area has been hampered by the lack of feasible animal models that recapitulate the different aspects of a persistent S. aureus infection. In the study presented here, we used an experimental mouse model that sustained a long-term S. aureus infection and, therefore, enabled us to dissect the mechanisms of the host immune response during the persistent phase of infection. Our results provide unequivocal evidence that T cells are critical elements for controlling S. aureus infection. However, concomitant with the transition from acute infection to persistence, T cells became progressively hyporesponsive towards bacterial antigens and exhibited an anergic state that may explain their failure to promote sterilizing immunity. Therefore, the T cell functions seem to be tightly regulated during S. aureus infections through a complex interplay of positive (leading to responsiveness) and negative (leading to hyporesponsiveness) modulating signals. A better characterization of these modulating signals will pave the way to the development of immunotherapies aimed at stimulating protective immunity during staphylococcal infections. This is an important issue due to the increasing prevalence of S. aureus strains resistant to multiple antibiotics, especially methicillin-resistant S. aureus (MRSA). MRSA is evolving into a growing epidemic, spreading worldwide within hospitals, care facilities and the community in general and is increasingly claiming victims (Boucher & Corey, 2008). In particular, the spread of the highly virulent community-acquired MRSA clone USA300 has become a matter of concern worldwide (Deleo et al, 2010). Given this rapidly emerging epidemic and the decreased efficacy of currently available antibiotics, an immune-based antimicrobial therapy may provide a novel alternative approach to combat these infections.
RESULTS
The mouse model of persistent S. aureus infection
We previously showed that after intravenous inoculation with S. aureus, C57BL/6 mice failed to eradicate S. aureus resulting in a sustained infection (Tuchscherr et al, 2011). Here, we performed a thorough characterization of this murine model of persistent S. aureus infection. We started by quantifying the bacterial burden in the organs of C57BL/6 mice at progressive times after intravenously inoculation with 7 × 107 CFU of S. aureus. As shown in Fig 1A, the bacterial load was significantly reduced in the kidneys during the first 28 days of infection and remained at constant levels up to day 56 post-inoculation (p.i.). In contrast to the kidneys, S. aureus was progressively eliminated in the joints (B), heart (C), liver (D) and lungs (E). Infected animals demonstrated clinical signs of illness including hunched posture, ruffled fur, lethargy and weight loss mostly during the first 2 weeks of infection and started to recover and re-establish their original weight from then on (Fig 1F). As previously shown (Tuchscherr et al, 2011), persistent infection was associated with the emergence of a bacterial subpopulation that exhibited typical phenotypic features (e.g. slow growth, lack of haemolysis and pigmentation) of SCVs (Fig 1G, black arrows). Many reports support a pathogenic role for these variants in patients with persistent and/or recurrent staphylococcal infections (Proctor et al, 1995, 2006).
Figure 1. Long-term persistence of S. aureus in kidneys of C57BL/6 mice.
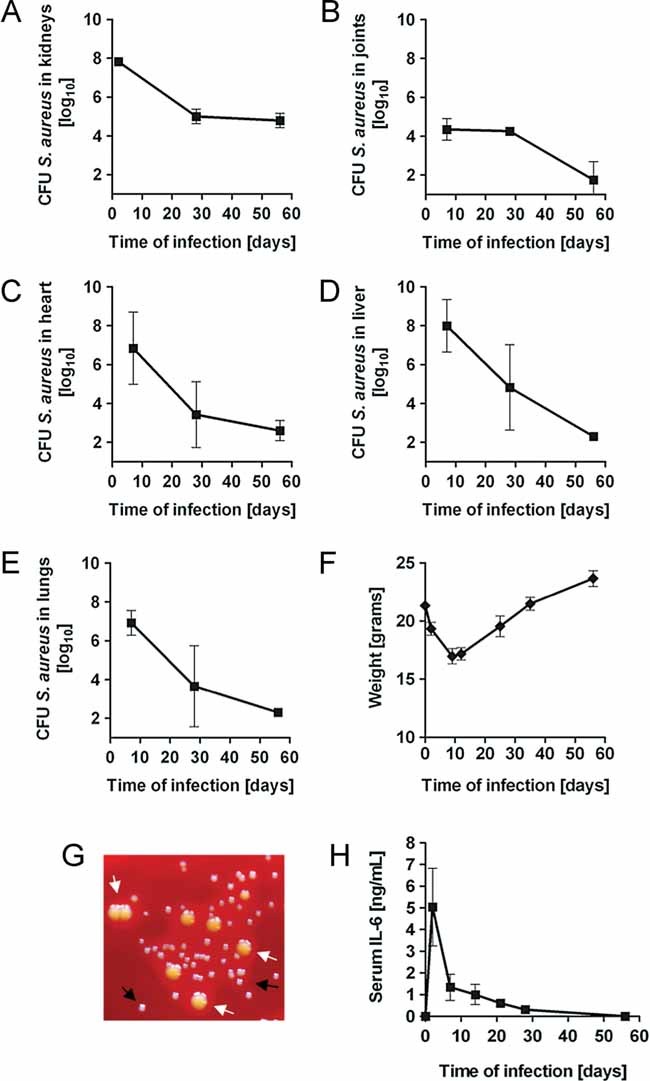
- A.–E. Bacterial burdens in the kidneys (A), joints (B), heart (C), liver (D) and lungs (E) of mice intravenously inoculated with 7 × 107 CFU of S. aureus strain SH1000. Each point represents the mean ± SD of five mice. Results are one representative of three independent experiments.
- B. Changes in body weight of S. aureus-infected mice. Results are reported as the average weight in grams ± SD of five mice.
- C. Growth of normal (white arrow) and SCVs (black arrow) S. aureus isolated from the kidneys of infected mice at day 28 after bacterial inoculation.
- D. Levels of IL-6 in serum S. aureus-infected mice at progressive times after bacterial inoculation. Each point represents the mean ± SD of five mice per group of three independent experiments.
In order to determine if the persistent S. aureus infection was associated with a systemic inflammatory response, the serum level of the pro-inflammatory cytokine interleukin (IL)-6 was measured in infected mice during the acute and persistent infection phases. Peak serum level of IL-6 (Fig 1H) was detected very early during the course of infection (day 2 p.i.) and declined thereafter. It is of interest to note that the secretion of this inflammatory cytokine was below detectable levels during the persistent phase. This suggests that the systemic inflammation quickly subsided after the acute phase and that the persistent S. aureus infection is a rather local event.
To gain a deeper insight into the local infection process, we performed histopathological examination of kidney tissue obtained at 56 days p.i. As shown in Fig 2A, kidney pathology was characterized by the presence of abscesses, which are a typical pathological feature of staphylococcal infections (Cheng et al, 2009; Lowy, 1998). Renal abscesses comprised an inner accumulation of neutrophils (Fig 2B) and were surrounded by a rim of apoptotic neutrophils (Fig 2C), a peripheral fibrin wall (Fig 2A, fw) and an external layer of B and T cells (Fig 2D). Interestingly, we found collections of staphylococci within the fibrin wall (Fig 2E). This observation suggests that the initiation of abscesses probably involved the deposition of fibrin in response to infection-related inflammation and the sequestration of the staphylococci in the fibrin clot. Eventually, the abscesses resolved leaving empty spaces or caverns in the infected organ (Fig 2F, asterisks).
Figure 2. H&E-stained sections of kidney tissue collected from mice at day 56 after S. aureus inoculation.
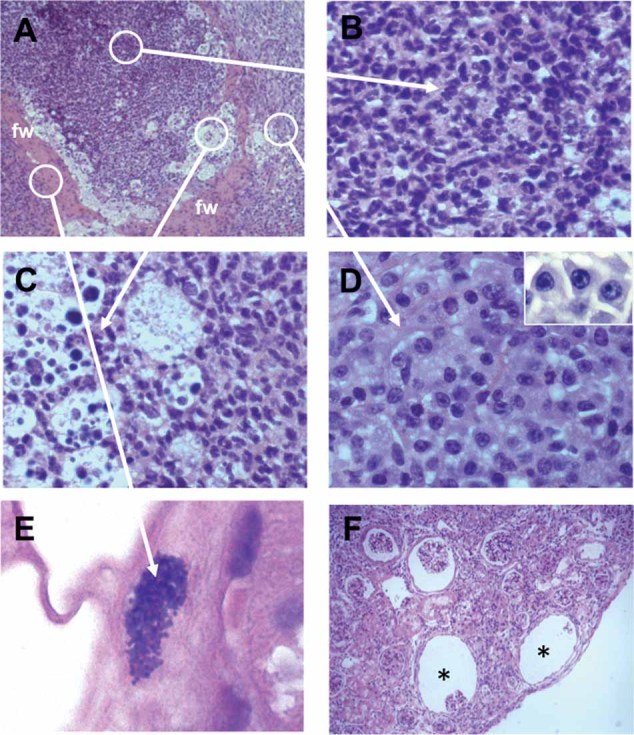
- S. aureus renal abscess with a peripheral fibrin wall (fw). Original magnification, ×20.
- Cluster of neutrophils occupying the central part of the renal abscess. Original magnification, ×63.
- Rim of apoptotic neutrophils. Original magnification, ×63.
- External layer of B and T cells. Original magnification, ×63. The inset in the upper right corner displays a high magnification (×100) photograph showing cells with the typical morphology of plasma cells.
- Collection of staphylococci can be found associated with fibrin strands. Original magnification, ×100.
- Empty spaces or caverns (asterisks) formed after the resolution of abscesses. Original magnification, ×20.
Patterns of gene expression in infected kidneys associated with temporal phases of S. aureus infection
To acquire more information regarding the local immune events in the kidneys during the different phases of a S. aureus infection, gene profiling using microarray technology was performed. Distinct gene transcription profiles were identified by comparing the relative abundance of gene transcripts at day 2 (acute) and 28 (persistent) of infection to their abundance in the kidneys of uninfected control animals. These measurements of transcript abundance may reflect both changes in gene-specific transcriptional activity as well as changes in cellular composition. We were able to assign individual clusters of genes to particular cellular and physiological categories using gene ontologies (Ashburner et al, 2000; Table S1 of Supporting information). Among the factors important for inflammation are cytokines as well as chemokines that mediate leukocyte recruitment and, thereby, orchestrate the host inflammatory response. CXCL-type chemokines (especially CXCL1/KC, CXCL2/MIP-2 and CXCL3, all chemoattractants for granulocytes; Kobayashi, 2008; Rollins, 1997) were among the highest induced chemokine genes during the acute infection (Fig 3A). The gene coding for CXCL2 was also highly upregulated during the persistent phase of infection. Most striking during the persistent phase was the upregulation of genes encoding CCL5/RANTES, CCL7/MCP-3, CCL8/MCP-2 and CXCL9/MIG, which are involved in chemoattraction of T cells (Liao et al, 1995; Loetscher et al, 1994). In accordance, the gene encoding the chemokine receptor CCR5, which is expressed by T cells (Syrbe et al, 1999), was upregulated in the kidneys principally during the persistent infection thus indicating the presence of T cells in the infected tissue at this stage of infection (Table S1 of Supporting information).
Figure 3. Expression patterns of chemokines and cytokines in the kidneys of S. aureus-infected mice.
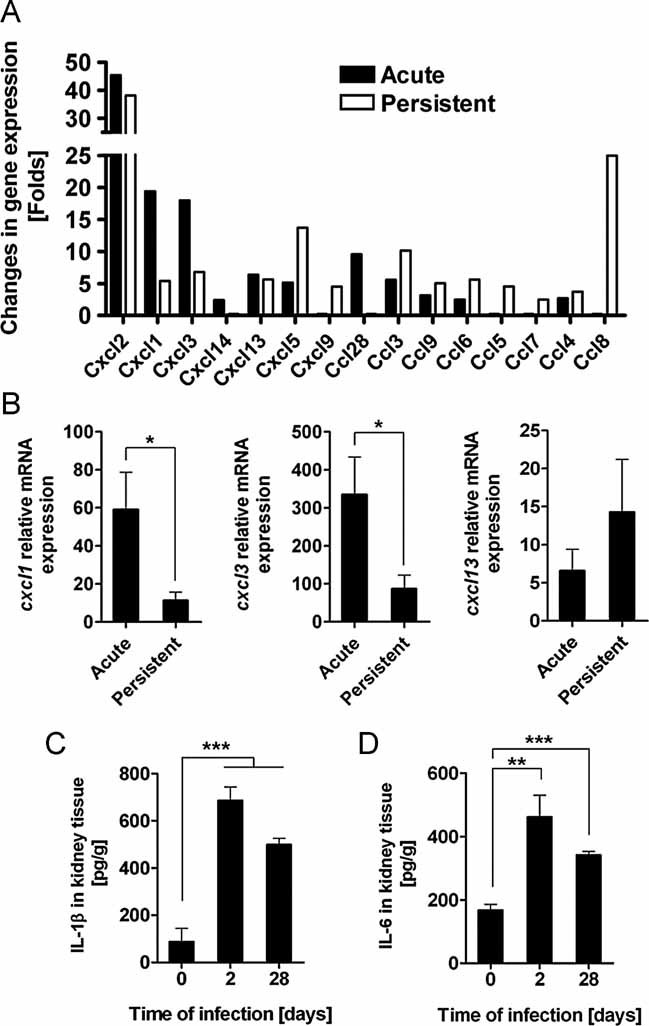
- A. Transcriptional profiles of chemokines in kidneys isolated from S. aureus-infected mice during acute (2 days of infection; black bars) and persistent (28 days of infection; white bars) infection. Bars represent average fold change in gene expression of three independent microarrays per group relative to uninfected tissue.
- B. qPCR analysis of Cxcl1 (left panel), Cxcl3 (middle panel) and Cxcl13 (right panel) gene transcription in kidneys isolated from S. aureus-infected mice during acute or persistent infection. Values were calculated using the Pfaffl equation and expressed as a ratio of mRNA expression relative to β-actin. Each column represents the mean ± SD of samples obtained from three independent experiments. *p < 0.05.
- C,D. Levels of IL-1β (C) and IL-6 (D) in kidney homogenates prepared from S. aureus-infected mice before (day 0) and 2 and 28 days after S. aureus inoculation. The levels of IL-1β and IL-6 were determined by ELISA. Each column represents the mean ± SD of triplicate samples obtained from three independent experiments. **p < 0.01 and ***p < 0.001.
To assess the validity of the microarray data, gene expression levels for selected chemokines (CXCL1, CXCL3 and CXCL13) were also measured by quantitative real-time polymerase chain reaction (PCR). The results obtained by this methodology mirrored those of the microarray analysis (Fig 3B). We observed that genes coding for the cytokines IL-6 and IL-1β were upregulated in infected kidney tissue throughout the infection (Table S1 of Supporting information). Accordingly, significantly greater levels of IL-1β (Fig 3C) and IL-6 (Fig 3D) were detected in kidney homogenates prepared from infected mice in comparison to levels in uninfected animals.
Notably, genes associated with B and T cell activation and proliferation including components of the B and TCR complexes, immunoglobulin genes (e.g. Ighg1, Igh-6, Igh-3, IgI-J2 and IgI-V1), BCR accessory molecules (e.g. CD79b), TCR chains (Tcrg-V3) and TCR accessory molecules (CD3g) were upregulated in infected kidney tissue only during persistent infection (Table S1 of Supporting information).
According to the gene expression data, the early transcriptional response to the S. aureus infection seemed to be dominated by neutrophils and macrophages while effectors of the adaptive immune system (B and T cells) dominated the transcriptional signature of the persistent phase of infection. To further prove this assumption we assessed the recruitment of B and T cells into the kidneys of S. aureus-infected mice at day 28 of infection by immunostaining. In accordance with the gene expression data, microscopic examination of stained tissue sections revealed the presence of high numbers of IgG-expressing B cells (Fig 4Ai and 4Aii) and CD3+ T cells (Fig 4Bi and 4Bii) in the kidneys of infected mice at day 28 of infection.
Figure 4. B and T cells are recruited into the kidneys of S. aureus-infected mice during the persistent phase of the infection.
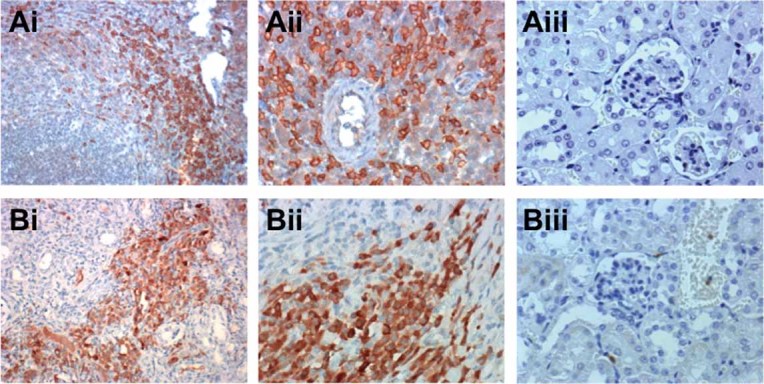
- Immunostaining of a S. aureus-infected kidney section showing the infiltration of IgG+ plasma cells (brown-stained cells in Ai and Aii). A kidney section from an uninfected animal stained for IgG+ plasma cells is shown in Aiii. Original magnification, ×20 (i) and ×40 (ii and iii).
- Immunostaining of a S. aureus-infected kidney section showing the infiltration of CD3+ T cells (brown-stained cells in Bi and Bii). A kidney section from an uninfected animal stained for CD3+ T cells is shown in (Biii). Original magnification, ×20 (i) and ×40 (ii and iii).
Adaptive immune response is required for an efficient containment of S. aureus during a persistent infection
We (von Köckritz-Blickwede et al, 2008) and others (Mölne et al, 2000; Verdrengh & Tarkowski, 1997) have shown that neutrophil recruitment is critical for effective control of bacterial growth during the acute phase of S. aureus infection. To determine their relevance for bacterial containment during the persistent phase, we depleted neutrophils in S. aureus-infected mice at day 28 p.i. by injecting anti-RB6 antibodies. The efficiency of neutrophil (∼90%) and macrophage (∼95%) depletion is shown in Figs S1 and S2 of Supporting information, respectively. In contrast to their importance during the early stage of infection (Fig S3A of Supporting information), depletion of neutrophils did not affect the capacity of C57BL/6 mice to restrain S. aureus proliferation during the persistent phase (Fig S3B of Supporting information). Similarly, depletion of macrophages after treatment with Carrageenan had a strong impact on bacterial killing during the acute phase (Fig S3C of Supporting information) but did not alter the course of infection in long-term (28 days p.i.) infected animals (Fig S3D of Supporting information). These results indicate that effectors of the innate immune defense are critical for controlling S. aureus during the acute infection but are dispensable for bacterial containment during the persistent phase.
We next investigated the relevance of the adaptive immune response for controlling S. aureus during the persistent phase of the infection using B and T cells-deficient RAG2−/− mice. The results in Fig 5A show that RAG2−/− mice exhibited a higher, more sustained bacterial burden in the kidneys during the persistent phase than immunocompentent C57BL/6 animals. Similarly, RAG2/IL-2Rγ−/− mice, which are devoid of B, T and natural killer (NK) cells, were significantly less capable of containing S. aureus during the persistent phase of infection than C57BL/6 mice (Fig S4 of Supporting information). These observations demonstrate that B and/or T cells are responsible for the superior restriction of S. aureus proliferation observed in C57BL/6 mice. Nevertheless, RAG2−/− and RAG2/IL-2Rγ−/− mice were also able to exert certain levels of control over S. aureus proliferation in the kidneys during the persistent phase that was mediated by innate immune mechanisms since depletion of neutrophils or macrophages resulted in enhanced bacterial multiplication in this organ (Fig S5 of Supporting information).
Figure 5. Adaptive immune response is required for restraining S. aureus proliferation during persistent infection.
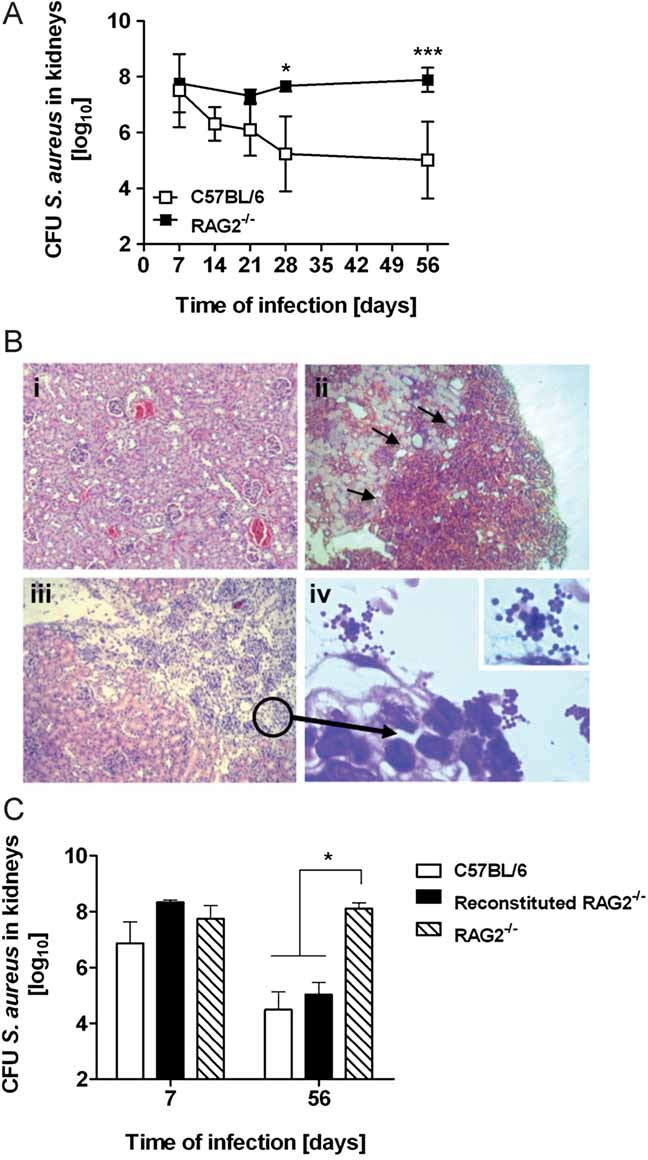
- Course of S. aureus infection in the kidneys of RAG2−/− mice after intravenous inoculation with S. aureus (black symbols). The course of S. aureus infection in the kidneys of immunocompetent C57BL/6 mice (white symbols) is included for comparison. Data points represent the mean ± SD from cohorts of five animals from three independent analyses. *p < 0.05 and ***p < 0.001.
- Histopathological appearance of H&E-stained kidney tissue of S. aureus-infected RAG2−/− mice at day 30 (ii) and 56 (iii and iv) of infection. Note the intense inflammatory infiltrate indicated by arrows (ii), the extended areas of tissue destruction (iii) and the presence of high numbers of staphylococci in (iv). Original magnifications, ×40 (i), (ii) and (iii) and ×100 (iv).
- Adoptive transfer of spleen cells from C57BL/6 donors improves the ability of RAG2−/− to control persistent S. aureus infection. RAG2−/− mice were reconstituted with splenocytes (app. 5 × 107 cells) from C57BL/6 mice and infected with S. aureus 48 h thereafter. Bacterial loads were determined in the kidneys of infected immunocompetent C57BL/6 (white bars), reconstituted mice RAG2−/− (black bars) and RAG2−/− recipient (hatched bars) at days 7 and 56 of infection. Bars represent the mean ± SD of 4–7 mice per group. One representative experiment of three independent experiments is presented. *p < 0.05.
Histological examination of kidney tissue obtained from RAG2−/− mice revealed massive inflammation at day 30 p.i. (Fig 5Bii, arrows) that degenerated into intense areas of tissue destruction (Fig 5Biii) with numerous microorganisms within the debris (Fig 5Biv) at day 56 of infection. Most interestingly, S. aureus did not lead to abscess formation in RAG2−/− mice suggesting that the development of abscesses was mediated by effector mechanisms of the adaptive immune response.
To further demonstrate the requirement for B and/or T cells in controlling S. aureus during a persistent infection, we performed cell transfer experiments. For this purpose RAG2−/− mice were reconstituted 48 h prior to S. aureus inoculation with splenocytes (appr. 5 × 107 cells) obtained from immunocompetent C57BL/6 mice. At the completion of the experiment (56 days p.i), flow cytometry analysis of spleen cell populations from reconstituted RAG2−/− mice was performed to verify the cellular reconstitution (Fig S6 of Supporting information). As shown in Fig 5C, RAG2−/− mice reconstituted with splenocytes from C57BL/6 mice exhibited a significantly improved capacity to control S. aureus at later time points than RAG2−/− mice that did not receive cells from donor animals. Furthermore, the level of resistance to S. aureus expressed by reconstituted RAG2−/− mice was comparable to those observed in immunocompetent animals (Fig 5C). These observations highlight the importance of the adaptive immune response for restricting S. aureus proliferation during the persistent phase of infection.
T cells but not B cells are critical for an efficient containment of S. aureus during the persistent infection
Because the above-described experiments strongly suggest that B and/or T cells are necessary for the S. aureus containment during the persistent infection, we next investigated the relevance of each population. Initially, we examined the dynamics of B cells in the spleen and peripheral lymph nodes of S. aureus-infected mice by combining cell counting and flow cytometric analysis. The total number of splenic B cells sharply increased (∼5-fold) during the first 30 days p.i. followed by a progressive decline (Fig 6A). Similarly, the number of B cells increased in the peripheral lymph nodes during the first 30 days of infection but was not statistically significant when compared with uninfected animals (Fig 6B). After day 30, the B cell population in the lymph nodes returned to values similar to uninfected controls (Fig 6B). During the course of infection, the B cells developed into plasma cells since sera from infected mice contained high titers of anti-S. aureus IgG antibodies (Fig S7 of Supporting information).
Figure 6. Negligible contribution of B cells to the control of S. aureus during a persistent infection.
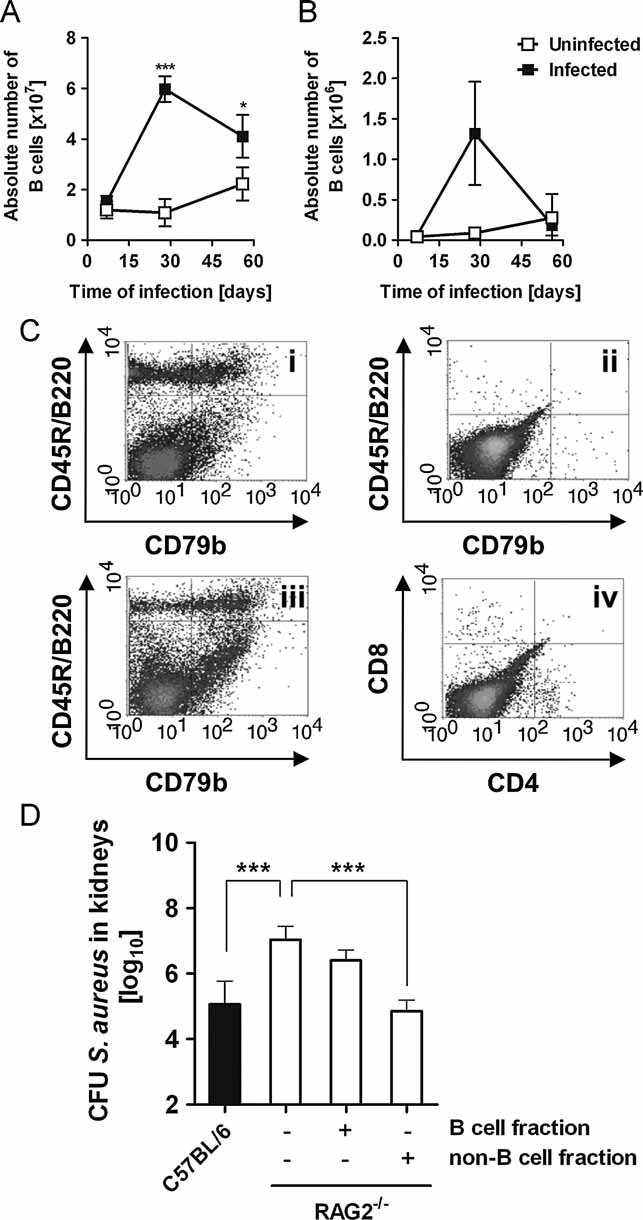
- A,B. Dynamics of B cells in the spleen (A) and peripheral lymph nodes (B) of uninfected (white symbols) or S. aureus-infected C57BL/6 mice (black symbols) at progressive times after bacterial inoculation as determined by flow cytometry analysis of CD45R/B220+ cells. Each symbol represents the mean ± SD of the absolute number of B cells determined in three animals. *p < 0.05 and ***p < 0.001.
- C. Dot plot flow cytometry analysis of splenic B cells (CD45R/B220+) from RAG2−/− mice prior (ii) and 56 days after (iii) reconstitution with app. 107 purified B cells isolated from the spleen of C57BL/6 donor (i) mice. Dot plot analysis of splenic CD4+ and CD8+ T cells from B cells-reconstituted RAG−/− mice is shown in (iv).
- D. Bacterial loads in the kidneys of C57BL/6 mice (black bar) and different groups of RAG2−/− mice (white bars), non-reconstituted or reconstituted with either purified B cells or with the non-B cell fraction at 56 days after intravenous inoculation with S. aureus. Bars represent the mean ± SD of four mice per group. One representative experiment of three independent experiments is presented. ***p < 0.001.
To determine the relevance of B cells for the containment of S. aureus during the persistent infection, we transferred B cells from C57BL/6 donor mice (Fig 6Ci) into RAG2−/− recipient mice (Fig 6Cii). The purity of the transferred B cells was ∼97% (Fig S8 of Supporting information). The efficiency of long-term B cell reconstitution was ∼50% and was determined by assessing the expression of CD45R/B220 and CD79b (component of the BCR highly expressed on mature B cells) on splenocytes of reconstituted RAG2−/− mice after the completion of the experiment (day 56 p.i.; Fig 6Ciii). The carryover of CD4+ or CD8+ T cells were <1% in the B cell-reconstituted RAG2−/− mice (Fig 6iv). The transfer of purified B cells, however, did not improve their capacity to restrain S. aureus proliferation during persistent infection (Fig 6D). Similar results were obtained when primed B cells isolated from S. aureus-infected mice were used for the reconstitution of the RAG2−/− mice (data not shown). Conversely, RAG−/− mice adoptively transferred with the non-B cell splenocyte fraction were as effective as immunocompetent C57BL/6 mice at controlling S. aureus during the persistent infection (Fig 6D) suggesting that T cells rather than B cells were responsible for this effect. To confirm this assumption, purified T cells (CD4+ plus CD8+ cells) from C57BL/6 mice (Fig 7Ai) were adoptively transferred into RAG2−/− mice (Fig 7A ii) prior to S. aureus infection (Fig 7). The purity of the transferred T cells was ∼98% (Fig S9 of Supporting information). The efficiency of reconstitution was ∼60% for CD4+ and ∼50% for CD8+ T cells (determined at day 56 p.i.; Fig 7Aiii). No B cells were detected in the T cells-reconstituted RAG2−/− mice (Fig 7Aiv). Adoptive transfer of T cells significantly improved the capacity of RAG2−/− mice to control S. aureus during persistent infection to levels seen in immunocompetent C57BL/6 mice (Fig 7B). In contrast, adoptive transfer of the non-T cell splenocyte fraction into RAG−/− had no beneficial effect in the ability of recipient RAG2−/− mice to control the staphylococcal infection (Fig 7B). These observations support the critical role of T cells for limiting bacterial proliferation during persistent infection.
Figure 7. T cells are critical for the control of S. aureus proliferation during a persistent infection.
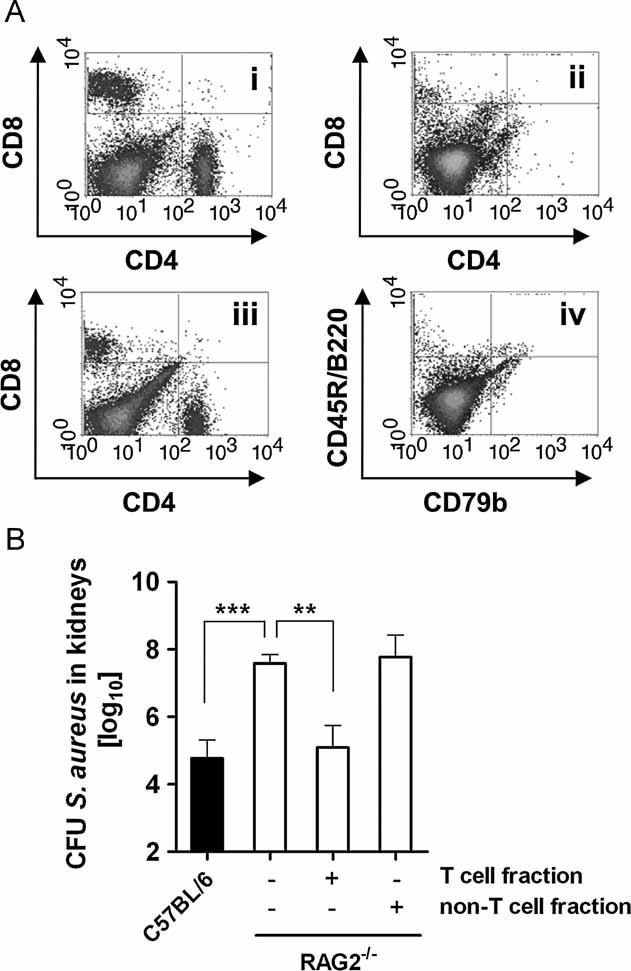
- Dot plot flow cytometry analysis of splenic CD4+ and CD8+ T cells from RAG2−/− mice prior (ii) and 56 days after (iii) reconstitution with purified T cells (5 × 105) isolated from the spleen of C57BL/6 donor mice (i). Dot plot analysis of splenic B cells from T cells-reconstituted RAG−/− mice is shown in (iv).
- Bacterial loads in the kidneys of C57BL/6 mice (black bar) and groups of RAG2−/− mice (white bars), non-reconstituted or reconstituted with either purified T cells or with the non-T cell fraction at 56 days after intravenous inoculation with S. aureus. Bars represent the mean ± SD of four mice per group. One representative experiment of three independent experiments is presented. **p < 0.01 and ***p < 0.001.
To obtain further insights into the dynamics of T cells during the progress of infection, quantitative and phenotypic analysis was performed in CD4+ and CD8+ T cell subsets. The numbers of CD4+ T cells increased approximately fivefold in the spleen (Fig 8A) and about threefold in the peripheral lymph nodes (Fig 8B) during the first 30 days of infection. The expansion in the CD8+ T cell compartment was not as pronounced as the CD4+ T cells in the spleen (∼3-fold increase; Fig 8C) but similar in the lymph nodes (Fig 8D). Phenotypic analysis of the CD4+ and CD8+ T cells revealed an increase in the percentage of activated (CD44high) and memory (CD62Llow) cells during the persistent phase of infection (Fig 8E).
Figure 8. Dynamic and phenotype of T cells in S. aureus-infected C57BL/6 mice.
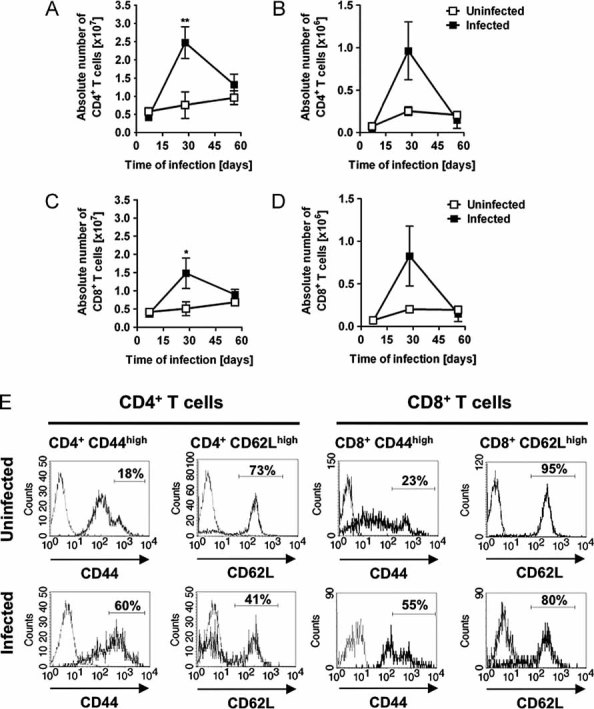
- A.–D. Absolute numbers of CD4+ T cells in the spleen (A) and peripheral lymph nodes (B) as well as CD8+ T cells in the spleen (C) and lymph nodes (D) of uninfected (white symbols) or S. aureus-infected (black symbols) C57BL/6 mice at progressive times after bacterial inoculation. Each symbol represents the mean ± SD of the absolute number of cells determined in three animals. *p < 0.05 and **p < 0.01.
- E. Gated CD4+ or CD8+ splenocytes from uninfected mice (upper panels) and from mice infected with S. aureus for 56 days (lower panels) were analysed for the expression of CD44 (left panels, thick lines) or CD62L (right panels, thick lines) by flow cytometry. Cells stained with isotype control antibodies (thin line) are included in each histogram. Numbers within histograms indicate mean % of CD4+ or CD8+ T cells expressing high levels of the corresponding marker.
T cells undergo in vivo anergy during persistent S. aureus infection
Although our data above demonstrate the importance of T cells in the control of S. aureus infection, these cells did not succeed in clearing the infection. Therefore, we further investigated the mechanisms underlying the incomplete expression of T cell immunity. Under certain circumstances, sustained antigenic stimulation of T cells can lead to the development of a hyporesponsive state (Schwartz, 2003). Hence, we hypothesized that the permanent activation of T cells by the continuous presence of staphylococcal antigens during persistent infection may lead to immunological hyporesponsiveness. To corroborate this hypothesis, we determined the proliferative response of spleen cells isolated from mice at progressive times after bacterial inoculation to in vitro antigenic re-stimulation. Our results show that antigen-specific spleen T cells obtained from mice at day 7 p.i. responded to in vitro antigenic re-stimulation by active proliferation (Fig 9A). On the other hand, antigen-specific spleen T cells obtained from mice at >14 days p.i. became unresponsive to antigenic re-stimulation (Fig 9A). Spleen T cells isolated from S. aureus-infected mice during the persistent phase of infection were also hyporesponsive to stimulation with anti-CD3 along with the costimulatory signal provided by the anti-CD28 antibody (Fig 9B) or Concanavalin A (Con A; Fig 9C), which activate T cells in a TCR-mediated and antigen-presenting-cells-dependent manner. These results indicate that, with the transition from the acute to the persistent phase of staphylococcal infection, T cells entered a state of anergy.
Figure 9. T cells become hyporesponsive to antigen-specific stimulation during a persistent S. aureus infection.
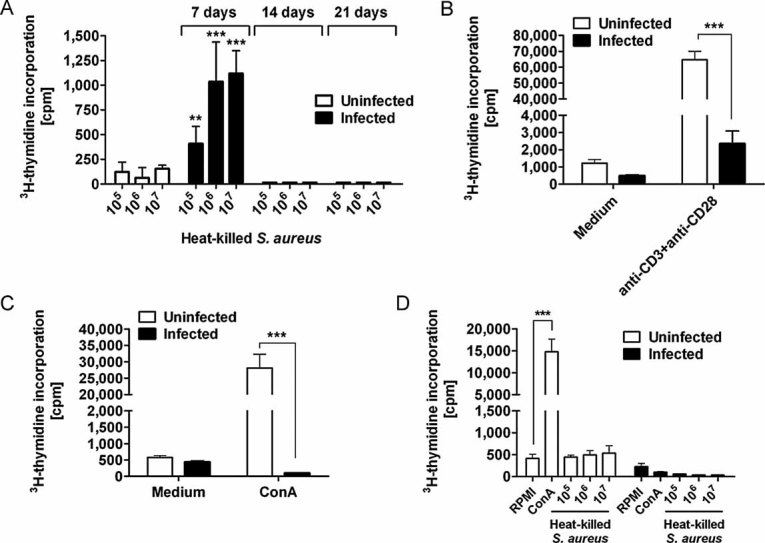
- Proliferative response of spleen cells isolated from uninfected (white bars) or from S. aureus-infected mice (black bars) at days 7, 14 and 21 of infection to in vitro re-stimulation with increasing concentrations of heat-killed S. aureus. Results are expressed as mean cpm minus background cpm ± SD of three individual experiments. **p < 0.01 and ***p < 0.001.
- Proliferative responses of spleen cells isolated from uninfected (white bars) or from S. aureus-infected mice (black bars) at day 21 of infection to in vitro re-stimulation with anti-CD3 plus anti-CD28. Results are expressed as mean cpm ± SD of three individual experiments. ***p < 0.001.
- Proliferate response of lymphocytes isolated from either uninfected (white bars) or S. aureus-infected (black bars) mice at day and 21 of infection to in vitro stimulation with Con A (5 µg/ml). Results are expressed as mean cpm ± SD of three individual experiments. ***p < 0.001
- Proliferative response of spleen cells isolated from uninfected (white bars) or from S. aureus-infected (black bars) mice at day 21 of infection to in vitro re-stimulation with increasing concentrations of heat-killed S. aureus or Con A in the presence of 100 pg/ml of rIL-2. Results are expressed as mean cpm ± SD of three individual experiments. ***p < 0.001.
Two categories of T cell anergy can be distinguished namely ‘clonal anergy’ and ‘in vivo anergy’. Clonal anergy represents a state of growth arrest associated with impaired production of selective cytokines (e.g. IL-2) in response to TCR stimulation, does not require antigen to maintain the anergic state and can be reversed by stimulation with IL-2 (Essery et al, 1988). In contrast, in vivo anergy or adaptive tolerance represents a more generalized inhibition of effector functions and develops after prolonged in vivo exposure to antigens (Choi & Schwartz, 2007; Schwartz, 2003). In our experimental settings, we observed that spleen cells isolated from long-term (>21 days) S. aureus-infected mice failed to produce IL-2 after antigen-specific or TCR stimulation (data not shown) and addition of recombinant (r)IL-2 did not restore their ability to proliferate (Fig 9D). These observations suggest that T cells undergo in vivo anergy during S. aureus persistent infection.
The anergic state is generally the result of a failure in the proximal TCR-mediated signal transduction pathways leading to a perturbation in cell signalling of downstream pathway(s) (Choi & Schwartz, 2007). The earliest step in intracellular signalling following TCR ligation is the activation of PTKs leading to phosphorylation of the immunoreceptor tyrosine-based activation motif (ITAMs) followed by recruitment of zeta-chain-associated protein kinase 70 (ZAP-70). ZAP-70 targets are the transmembrane adapter protein linker for the activation of T cells (LAT) and the cytosolic adapter protein src homology 2 (SH2) domain-containing leukocyte phosphoprotein of 76 kDa (SLP-76). The later is an essential component of the TCR-proximal signalling cascade, serving as the docking site for Vav1. The assembly and activation of these adapters ultimately trigger PKCθ activation leading to the translocation of transcription factors such as NF-κB and the production of cytokines (Smith-Garvin et al, 2009).
To determine if the hyporesponsiveness of T cells from long-term infected mice to antigenic stimulation originated from a blockade in the proximal TCR signal transduction pathway, we assessed the proliferative response of these splenocytes to TCR activation after stimulation with anti-CD3 in the presence of the phorbol ester PMA, an activator of PKC. The results in Fig 10A show that activation of PKC by PMA fully rescued the hyporesponsive phenotype of splenic T cells to TCR stimulation. Similarly, stimulation of T cells in an antigen-specific manner in the presence of PMA restored their proliferative capacity (Fig 10B) as well as their ability to produce IL-2 (Fig 10C). Together, these observations suggest that a blockage in the TCR-proximal signalling event may likely be responsible for the hyporesponsive phenotype of spleen cells from long-term infected mice.
Figure 10. Hyporesponsiveness of T cells to TCR stimulation can be reversed by co-stimulation with PMA.
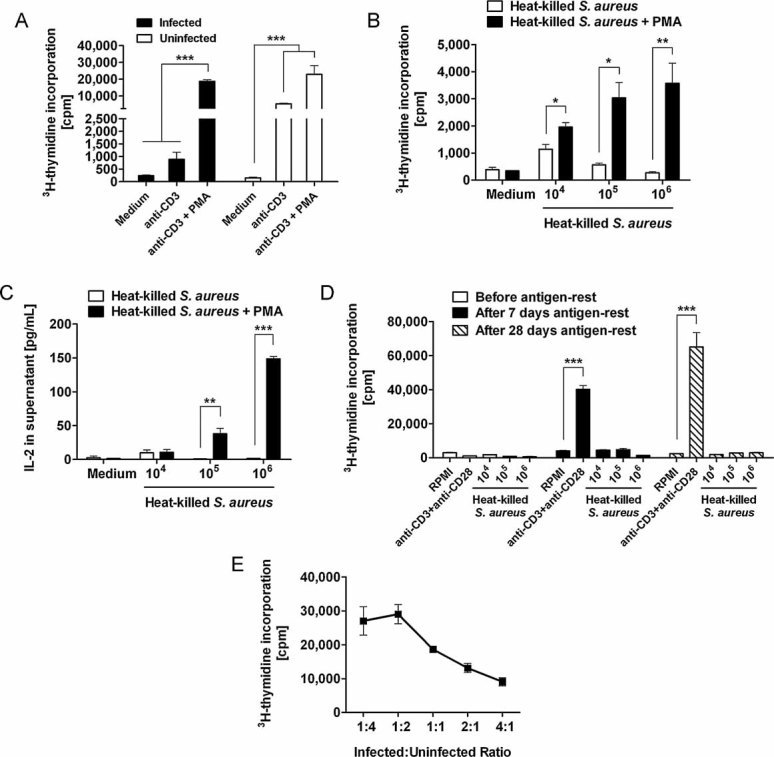
- Proliferative response of lymphocytes isolated from uninfected (white bars) or from S. aureus-infected (black bars) mice at day 21 of infection to in vitro re-stimulation with anti-CD3 in the presence or absence of the PKC activator PMA. ***p < 0.001.
- Proliferative responses of lymphocytes isolated from S. aureus-infected mice at day 21 of infection to in vitro re-stimulation with increasing concentrations of heat-killed S. aureus in the presence (black bars) or absence (white bars) of PMA. Results are expressed as the mean cpm ± SD as determined from three independent experiments. *p < 0.05 and **p < 0.01.
- Levels of IL-2 in the culture supernatant of spleen cells isolated from S. aureus-infected mice at day 21 of infection and in vitro re-stimulated with increasing concentrations of heat-killed S. aureus in the presence (black bars) or absence (white bars) of PMA. The level of IL-2 was assessed by ELISA and each bar represents the mean value ± SD of three independent experiments. **p < 0.01 and ***p < 0.001.
- Proliferative responses of lymphocytes isolated from S. aureus-infected mice at day 21 of infection to in vitro re-stimulation with increasing concentrations of heat-killed S. aureus or anti-CD3/anti-CD28 before (white bars) or after 7 days (black bars) or 28 days (hatched bars) antigen-rest. Each bar represents the mean value ± SD of three independent experiments. ***p < 0.001.
- Suppressive effect of lymphocytes isolated from S. aureus-infected mice at day 21 of infection in the proliferation of naive T cells. Splenocytes from infected mice were co-cultured with splenocytes from naive mice at different ratios and stimulated with anti-CD3/anti-CD28. Proliferation was measured by 3H-thymidine incorporation, and the data represents the average of three independent experiments.
If the loss of T cell function is antigen-driven it can be proposed that the removal of the stimulating antigens might reverse T cell anergy. To address this issue, spleen cells isolated from 30-days-infected C57BL/6 mice were transferred into uninfected RAG2−/− mice. Spleen cells were harvested from the recipient mice after 7 and 28 days of rest and re-stimulated in vitro with different concentrations of heat-killed S. aureus or anti-CD3 plus anti-CD28. As shown in Fig 10D, the proliferative response to anti-CD3 plus anti-CD28 but not to antigen-specific stimulation was recovered after 7 and 28 days of antigen-rest. This data suggests that the anergic state of antigen-specific T cells seems to be irreversible and cannot be recovered after antigenic resting. In contrast, the global proliferative response of T cells to TCR stimulation could be recovered, at least in part, in the absence of persistent antigens.
We also investigated whether suppressive mechanisms were generated during persistent S. aureus infection. For this purpose, spleen cells isolated from infected mice during the persistent phase of infection (21 days p.i.) were co-cultured at different ratios with naive spleen cells in the presence of anti-CD3 plus anti-CD28. As shown in Fig 10E, proliferation of naive splenic T cells is inhibited by the addition of splenocytes from infected mice in a dose-dependent manner. These results indicate the presence of suppressive mechanisms in the spleen of persistently infected mice.
DISCUSSION
Exposure to S. aureus generally leads to the generation of antigen-specific humoral and cellular immune responses that culminate in the elimination of the pathogen. However, under certain circumstances S. aureus survives inside the host and induces persistent relapsing diseases (Proctor et al, 1995; Sheehy et al, 2010). Considering the large numbers of studies addressing virulence factors and survival strategies employed by staphylococci to persist within the host, surprisingly large gaps remain in our understanding of the host immune response evoked during persistent staphylococcal infections. In this study, we used an experimental mouse model of persistent staphylococcal infection to define the host immune mechanisms that selectively govern S. aureus persistence.
Using different experimental approaches, we provide unequivocal evidence that the adaptive immune system, specifically the T cell-mediated immune response, is critical for the containment of S. aureus proliferation during the persistent phase of infection. Initially, we used a whole genome microarray approach combined with histology and flow cytometry to provide a global and comprehensive assessment of the complexity of the immune networks associated with S. aureus persistent infection. The transcription profile of kidney tissue isolated during the acute (2 days p.i.) or persistent phases of infection (28 days p.i.) showed a specific signature. Associated with the persistent infection were genes coding for chemokines involved in T (CCL7/MCP-3, CCL8/MCP-2 and CXCL9/MIG) and B (CXCL13) cell trafficking (Gunn et al, 1998; Liao et al, 1995; Loetscher et al, 1994) suggesting that these cells were recruited into the infected tissue during the persistent phase of infection. This postulation was confirmed by immunohistological staining detecting B cells (Fig 4A) and T cells (Fig 4B) in kidney sections of long-term infected mice. Furthermore, B and T cell-deficient RAG2−/− mice were less capable of restraining bacterial growth during the persistent phase of infection than immunocompetent mice, confirming the necessity of acquired immune mechanisms for bacterial containment. Most importantly, reconstitution with T but not with B lymphocytes significantly improved the capacity of RAG2−/− mice to control S. aureus during persistence indicating that T cells rather than B cells were involved in this process. Our data is consistent with that of Spellberg et al (Spellberg et al, 2008) who found that mice deficient in T but not in B cells were significantly more susceptible to lethal challenge with S. aureus than wild-type animals. Furthermore, patients with B cell deficiency were shown not to be at greater risk of S. aureus infections or more severe staphylococcal diseases than normal individuals (Rich et al, 2001). However, the importance of T cells for controlling S. aureus infection in humans is clearly evidenced by the high incidence of recurrent S. aureus infections in HIV-infected individuals (Popovich et al, 2010).
Our results also demonstrate that S. aureus was able to survive within infected animals in the presence of high levels of antigen-specific antibodies generated during the course of infection. This is in line with the observation that despite increased antibody levels against S. aureus detected in the sera from convalescing patients, the rate of recurrence of staphylococcal infections in those patients may range from 25 to 45% (Huang & Platt, 2003). Often caused by the same S. aureus strain (Huang et al, 2008), such recurrent infections emphasize that the humoral immune response does not protect against re-infection.
In contrast to the well-defined abscesses present in the kidneys of infected immunocompetent mice (Fig 2), histopathological examination of kidney tissue from infected RAG−/− mice indicated an impaired capacity to develop abscesses (Fig 5B). Similar to our data, Schmaler et al (2011) reported that kidney abscesses in S. aureus-infected CD3−/− and Rag2−/− mice lacked necrotic zones and were less organized than in immunocompetent mice. Therefore, structural abscess formation seems to be critical for effective containment of S. aureus replication and appears to be dependent on the effective recruitment of T cells as previously reported for murine models of S. aureus intra-abdominal (Chung et al, 2003; Tzianabos et al, 2000) and subcutaneous (McLoughlin et al, 2006) abscess formation.
Despite the requirement for T cells to restrict S. aureus proliferation during persistence they did not succeed in clearing the actual infection. These results are consistent with the recent findings that activation of T and B cells by vaccination of mice with heat-killed bacteria did not result in the eradication of S. aureus (Schmaler et al, 2011). The authors argued that vaccination skewed the cytokine pattern towards immunosuppression rather than towards bacterial clearance (Schmaler et al, 2011). Understanding why the immune system failed to eradicate S. aureus will greatly aid in the design of immunomodulatory interventions for patients affected by persistent staphylococcal infections.
The long-standing interaction between S. aureus and the host during persistent infections can have severe consequences for an ongoing immune response. In this regard, it was proposed that the induction and maintenance of T cell responses could be substantially altered when the pathogen persists. Several studies have developed the view that the pathogenesis of various persistent bacterial, viral or parasitic infections is the result of T cell anergy (Anderson et al, 2006; Klenerman & Hill, 2005; Stadecker, 1992). In vivo anergy is a form of T cell hyporesponsiveness that develops after prolonged exposure to antigens (Schwartz, 2003). This type of anergy requires antigenic persistence in order to maintain the hyporesponsive state and is not recovered after addition of IL-2 (Schwartz, 2003). In this study, we provide evidence that T cells entered an anergic state during persistent staphylococcal infection since they failed to proliferate after in vitro receptor-mediated stimulation and their proliferative response could not be rescued by addition of exogenous IL-2.
Furthermore, we also demonstrated that the anergic state of antigen-specific T cells isolated from persistently infected mice was irreversible in the absence of antigen and could not be recovered after antigenic resting. In contrast, the global proliferative response of T cells to TCR stimulation could be recovered, at least in part, in the absence of persistent antigens. These observations indicate that cell-intrinsic factors control antigen-specific T cell anergy in vivo. In this regard, it has been reported that T cells undergoing in vivo anergy often entails a blockage at the level of TCR-proximal signalling events which in turn results in diminished activation of PKCθ and translocation of NF-κB (Chiodetti et al, 2006). We found that the hyporesposiveness of antigen-specific spleen cells obtained from long-term infected mice to antigenic stimulation could be fully reversed by co-stimulation with PMA, an activator of PKC. These observations suggest that the anergic status may be indeed caused by a flaw in the proximal TCR signalling events upstream of PKCθ.
Interestingly, we also showed that proliferation of naive splenic T cells was inhibited by the addition of splenocytes from infected mice in a dose-dependent manner. These results suggest the presence of extrinsic immunosuppressive mechanisms in long-term infected mice. Several extrinsic mechanisms have been shown to be responsible for the negative regulation of immune responses in a persistent antigen environment including the production of suppressive cytokines, tolerogenic dendritic cells and the development of regulatory T cells (Jin et al, 2011). We are currently investigating the specific mechanism responsible for T cell immunosuppression during persistent S. aureus infection.
Taken together, the data indicates that the hyporesponsiveness of T cells during persistent S. aureus infection involves a combination of both anergy and immunosuppression. A similar situation has been observed in T cells of mice immunized repeatedly with bacterial superantigens like staphylococcal enterotoxins. In these animals, the anergic state observed at the T cell-population level has been shown to be a combination of both intrinsic unresponsiveness to antigenic stimulation and cytokine-mediated immunosuppression probably produced by regulatory T cells (Miller et al, 1999).
In conclusion, the results of our study show that during persistent S. aureus infection an intricate network of regulatory mechanisms exists that limits the necessary immune responses required for pathogen clearance. Nonetheless, the T cells still retain some level of residual function during the persistent infection and exert control over bacterial replication. This was most apparent in the situation of RAG2−/− mice wherein the lack of T cells led to increased bacterial titers. It is therefore plausible that the immune balance during S. aureus infection is tightly regulated by the complex interplay of immune-modulating factors responsible for the suppression or sustainment of T cell responses. Variations in the level of these immune-modulating factors between individuals may explain inherent differences in the ability of each person to control these infections. This paradox is exemplified by an increased risk of persistent S. aureus carriers to acquire S. aureus infections despite having developed an antigen-specific immune response against this pathogen (Kluytmans et al, 1991). On the other hand, this immune response still confers certain levels of protection since the risk of death from staphylococcal bacteremia is significantly lower in carriers than in non-carriers (Wertheim et al, 2004).
Understanding the complex regulation of T cell responses during S. aureus infection will provide valuable information for the development of immunotherapeutic strategies to overcome immune suppression and stimulate optimal T cell responses to eliminate the persistent pathogen.
MATERIALS AND METHODS
Bioethics statement
All experiments involving animals were performed in strict accordance with the guidelines of the European Convention for the Protection of Vertebrate Animals used for Experimental and other Scientific Purposes. All experiments were approved by the ethical board Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit, Oldenburg in Germany (Az. 33.42502/07-04.05).
The paper explained
PROBLEM
Exposure to S. aureus generally leads to the generation of antigen-specific humoral and cellular immune responses that culminate in the elimination of the pathogen. However, under certain circumstances, S. aureus continues to thrive inside the host despite robust antimicrobial activities of the host immune defenses resulting in a persistent infection. Despite substantial progress made in understanding the mechanisms employed by S. aureus to persist within the host, very few studies have addressed the host immune mechanisms evoked during persistent infection.
RESULTS
Using an experimental murine model of staphylococcal persistent infection, we demonstrate that T cells are critical elements for controlling S. aureus growth during the persistent phase. However, concomitant with the transition from acute infection to persistence, T cells became progressively hyporesponsive towards bacterial antigens and exhibited an anergic state that may explain their failure to promote sterilizing immunity.
IMPACT
Our findings indicate that restoring T cell functionality during a persistent staphylococcal infection may provide a novel alternative approach to achieve eradication of the persistent pathogen.
Bacterial strains
The rsbU+ derivative of strain 8325-4 named SH1000 (Jonsson et al, 2004) was used in this study. Bacteria were grown to the Mid-Log phase at 37°C with shaking (150 rpm) in brain heart infusion (BHI)-medium, collected by centrifugation for 10 min at 4000 rpm, washed twice with sterile PBS and adjusted to 5 × 108 CFU/ml.
Mice
Pathogen-free C57BL/6 (8–12-week-old female) mice were purchased from Harlan-Winkelmann (Borchen, Deutschland). The B and T cell-deficient RAG2−/− and the B, T and NK cell-deficient RAG2/IL-2Rγ−/− (C57BL/6 background) were kindly provided by S. Weiss (HZI, Braunschweig, Germany).
Infection model
The mice were inoculated with 7 × 107 CFU of S. aureus in 0.2 ml of PBS via a lateral tail vein. Infected mice were killed by CO2 asphyxiation at different times of infection and the amount of bacteria determined by preparing organ homogenates in 5 ml of PBS and plating 10-fold serial dilutions on blood agar (Invitrogen, Karlsruhe, Germany).
Cytokine determination
Cytokine levels were determined in serum or kidney homogenates by ELISA according to the recommendations of the manufacturer (BD Pharmingen, San Diego, CA, USA) using matched antibody pairs and recombinant cytokines as standards.
Histopathological studies
Tissue samples from kidneys were placed in 10% neutral-buffered formalin and subsequently embedded in paraffin. 0.5 µm sections were stained with haematoxylin and eosin (H&E) and then examined microscopically using an Olympus BX51 microscope for pathological changes.
RNA extraction
Total RNA was extracted from the kidneys at day 0 (uninfected), days 2 and 28 of infection using the RNeasy Midi kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The quality and quantity of the RNA was checked using the Agilent 2100 bioanalyser (Agilent Technologies, Waldbronn, Germany).
Gene array analysis
Total RNA (500 ng) was used for biotin labelling according to the 3′ IVT Express Kit (Affymetrix, Santa Clara, CA). Ten microgram of biotinylated cRNA from each sample was fragmented and placed in a hybridization cocktail containing four biotinylated hybridization controls (BioB, BioC, BioD and Cre) as recommended by the manufacturer. Samples were hybridized to an identical lot of Affymetrix GeneChip MOE430 2.0 for 16 h at 45°C.
Steps for washing and SA-PE staining were processed on the fluidics station 450 using the EukGEws2v5_FS450 protocol (Affymetrix). Image Analysis was performed on GCS3000 Scanner and GCOS1.2 Software Suite (Affymetrix). Data analysis was performed using RMA normalized data sets (GeneSpring GX11, Agilent Technologies). Differentially expressed genes were determined by ANOVA with a cut-off for the p-value of 0.05 and a mean fold change of more than twofold. The array data can be found under the GEO accession number GSE25244 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE25244).
Real-time RT-PCR
Total RNA was isolated as described above and cDNA synthesis was performed using a reverse transcription-polymerase chain reaction (RT-PCR) kit (Invitrogen) following the manufacturer's instructions. A detailed description is provided in the Supporting information section.
Adoptive transfer experiments
CD45R/B220+ B cells and T cells (CD4+ plus CD8+ T cells) were purified from the spleen of C57BL/6 donor mice using PE-conjugated antibodies, anti-PE microbeads and MACS MS columns (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer's protocol. Purified B (>95% purity; Fig S7 of Supporting information) or T cells (>95% purity; Fig S8 of Supporting information) were adoptively transferred intravenously into recipient RAG2−/− mice. Animals were challenged with S. aureus 48 h after adoptive cell transfer.
Determination of anti-S. aureus specific serum antibodies
Anti-S. aureus specific IgG antibodies in serum samples were measured by ELISA using microtiter plates coated with 100 µl/well of S. aureus Wood 46 cell lysates. A detailed description is provided in the Supporting information section.
Proliferation assay
At the specified times, spleens from either uninfected or S. aureus infected mice were transformed into a single cell suspension and stimulated with different concentrations of heat-inactivated S. aureus, 5 µg/ml Con A type IV (Sigma), 50 ng/ml PMA (Sigma), 0.5 µg/ml anti-mouse CD3ε mAb (eBioscience), 1 µg/ml anti-mouse CD28 mAb or 100 pg/ml recombinant mouse IL-2 protein (eBioscience). After 3 days of incubation, the cells were pulsed with 1 µCi of 3H-thymidine (Amersham, Buchler, Germany) and 16–18 h later the cells were harvested on Filtermats A (Wallac, Freiburg, Germany) using a cell harvester (Inotech, Wohlen, Switzerland). The amount of 3H-thymidine incorporation was measured in a gamma scintillation counter (Wallac 1450, MicroTrilux).
Statistical analyses
All data was analysed in Excel 2000 (Microsoft Office, Microsoft, Redmond, WA) or GraphPad Prism 4.0 (GraphPad Software, San Diego, CA). Comparisons between groups were performed by use of a two-tailed t-test. p-Values <0.05 were considered as significant.
Acknowledgments
We would like to thank S. Weiss for providing the RAG2−/− and the Rag2/IL-2Rγ−/− mice. This work was supported by the Bundesministerium für Bildung und Forschung (SkinStaph-2; 01KI1009B) and by the Ministry for Science and Culture in Lower Saxony through a scholarship from the Center for Infection Biology (ZIB) at Hannover Medical School and the Helmholtz International Research School for Infection Biology (HIRSIB).
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author contributions
CZ, GP and EM conceived and designed the experiments; CZ and OG performed the experiments; CZ, OG, RG and EM analysed the data; EH and RG contributed reagents/materials/analysis tools; CZ and EM wrote the paper; OG and GP gave comments on the manuscript.
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Anderson K, Czinn S, Redline R, Blanchard T. Induction of CTLA-4 mediated anergy contributes to persistent colonization in the murine model of gastric Helicobacter pylori infection. J Immunol. 2006;176:5306–5313. doi: 10.4049/jimmunol.176.9.5306. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher HW, Corey GR. Epidemiology of methicillin-resistant Staphylococcus aureus. Clin Infect Dis. 2008;46:S344–S349. doi: 10.1086/533590. [DOI] [PubMed] [Google Scholar]
- Cheng AG, Kim HK, Burts ML, Krausz T, Schneewind O, Missiakas DM. Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J. 2009;23:3393–3404. doi: 10.1096/fj.09-135467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiodetti L, Choi S, Barber DL, Schwartz RH. Adaptive tolerance and clonal anergy are distinct biochemical states. J Immunol. 2006;176:2279–2291. doi: 10.4049/jimmunol.176.4.2279. [DOI] [PubMed] [Google Scholar]
- Choi S, Schwartz RH. Molecular mechanisms for adaptive tolerance and other T cell anergy models. Semin Immunol. 2007;19:140–152. doi: 10.1016/j.smim.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung DR, Kasper DL, Panzo RJ, Chitnis T, Grusby MJ, Sayegh MH, Tzianabos AO. CD4+ T cells mediate abscess formation in intra-abdominal sepsis by an IL-17-dependent mechanism. J Immunol. 2003;170:1958–1963. doi: 10.4049/jimmunol.170.4.1958. [DOI] [PubMed] [Google Scholar]
- Deleo FR, Otto M, Kreiswirth BN, Chambers HF. Community-associated meticillin-resistant Staphylococcus aureus. Lancet. 2010;375:1557–1568. doi: 10.1016/S0140-6736(09)61999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essery G, Feldmann M, Lamb JR. Interleukin-2 can prevent and reverse antigen-induced unresponsiveness in cloned human T lymphocytes. Immunology. 1988;64:413–417. [PMC free article] [PubMed] [Google Scholar]
- Garzoni C, Kelley WL. Staphylococcus aureus: new evidence for intracellular persistence. Trends Microbiol. 2009;17:59–65. doi: 10.1016/j.tim.2008.11.005. [DOI] [PubMed] [Google Scholar]
- Goodyear CS, Silverman GJ. Staphylococcal toxin induced preferential and prolonged in vivo deletion of innate-like B lymphocytes. Proc Natl Acad Sci USA. 2004;101:11392–11397. doi: 10.1073/pnas.0404382101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn MD, Ngo VN, Ansel KM, Ekland EH, Cyster JG, Williams LT. A B-cell-homing chemokine made in lymphoid follicles activates Burkitt's lymphoma receptor-1. Nature. 1998;391:799–803. doi: 10.1038/35876. [DOI] [PubMed] [Google Scholar]
- Huang SS, Platt R. Risk of methicillin-resistant Staphylococcus aureus infection after previous infection or colonization. Clin Infect Dis. 2003;36:281–285. doi: 10.1086/345955. [DOI] [PubMed] [Google Scholar]
- Huang SS, Diekema DJ, Warren DK, Zuccotti G, Winokur PL, Tendolkar S, Boyken L, Datta R, Jones RM, Ward MA, et al. Strain-relatedness of methicillin-resistant Staphylococcus aureus isolates recovered from patients with repeated infection. Clin Infect Dis. 2008;46:1241–1247. doi: 10.1086/529381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin HT, Jeong YH, Park HJ, Ha SJ. Mechanism of T cell exhaustion in a chronic environment. BMB Rep. 2011;44:217–231. doi: 10.5483/BMBRep.2011.44.4.217. [DOI] [PubMed] [Google Scholar]
- Jonsson IM, Arvidson S, Foster S, Tarkowski A. Sigma factor B and Rsbu are required for virulence in Staphylococcus aureus induced arthritis and sepsis. Infect Immun. 2004;72:6106–6111. doi: 10.1128/IAI.72.10.6106-6111.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenerman P, Hill A. T cells and viral persistence: lessons from diverse infections. Nat Immunol. 2005;6:873–879. doi: 10.1038/ni1241. [DOI] [PubMed] [Google Scholar]
- Kluytmans J, van Belkum A, Verbrugh H. Nasal carriage of Staphylococcus aureus: epidemiology, underlying mechanisms, and associated risks. Clin Microbiol Rev. 1991;10:505–520. doi: 10.1128/cmr.10.3.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi Y. The role of chemokines in neutrophil biology. Front Biosci. 2008;13:2400–2407. doi: 10.2741/2853. [DOI] [PubMed] [Google Scholar]
- Lee LY, Miyamoto YJ, McIntyre BW, Höök M, McCrea KW, McDevitt D, Brown EL. The Staphylococcus aureus Map protein is an immunomodulator that interferes with T cell-mediated responses. J Clin Invest. 2002;110:1461–1471. doi: 10.1172/JCI16318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao F, Rabin RL, Yannelli JR, Koniaris LG, Vanguri P, Farber JM. Human Mig chemokine: biochemical and functional characterization. J Exp Med. 1995;182:1301–1314. doi: 10.1084/jem.182.5.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llewelyn M, Cohen J. Superantigens: microbial agents that corrupt immunity. Lancet Infect Dis. 2002;2:156–162. doi: 10.1016/s1473-3099(02)00222-0. [DOI] [PubMed] [Google Scholar]
- Loetscher P, Seitz M, Clark-Lewis I, Baggiolini M, Moser B. Monocyte chemotactic proteins MCP-1, MCP-2, and MCP-3 are major attractants for human CD4+ and CD8+ T lymphocytes. FASEB J. 1994;8:1055–1060. doi: 10.1096/fasebj.8.13.7926371. [DOI] [PubMed] [Google Scholar]
- Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- McLoughlin RM, Solinga RM, Rich J, Zaleski KJ, Cocchiaro JL, Risley A, Tzianabos AO, Lee JC. CD4+ T cells and CXC chemokines modulate the pathogenesis of Staphylococcus aureus wound infections. Proc Natl Acad Sci USA. 2006;103:10408–10413. doi: 10.1073/pnas.0508961103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C, Ragheb JA, Schwartz RH. Anergy and cytokine-mediated suppression as distinct superantigen-induced tolerance mechanisms in vivo. J Exp Med. 1999;190:53–64. doi: 10.1084/jem.190.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mölne L, Verdrengh M, Tarkowski A. Role of neutrophil leukocytes in cutaneous infection caused by Staphylococcus aureus. Infect Immun. 2000;68:6162–6167. doi: 10.1128/iai.68.11.6162-6167.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovich KJ, Weinstein RA, Aroutcheva A, Rice T, Hota B. Community-associated methicillin-resistant Staphylococcus aureus and HIV: intersecting epidemics. Clin Infect Dis. 2010;50:979–987. doi: 10.1086/651076. [DOI] [PubMed] [Google Scholar]
- Proctor RA, van Langevelde P, Kristjansson M, Maslow JN, Arbeit RD. Persistent and relapsing infections associated with small colony variants of Staphylococcus aureus. Clin Infect Dis. 1995;20:95–102. doi: 10.1093/clinids/20.1.95. [DOI] [PubMed] [Google Scholar]
- Proctor RA, von Eiff C, Kahl BC, Becker K, McNamara P, Herrmann M, Peters G. Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat Rev Microbiol. 2006;4:295–305. doi: 10.1038/nrmicro1384. [DOI] [PubMed] [Google Scholar]
- Rich RR, Fleisher TA, Shearer WT, Kotzin BL, Schroeder HWJ, editors. Clinical Immunology: Prinicples and Practice. 2nd ed. New York: Mosby; 2001. [Google Scholar]
- Rollins BJ. Chemokines. Blood. 1997;90:909–928. [PubMed] [Google Scholar]
- Schmaler M, Jann NJ, Ferracin F, Landmann R. T and B cells are not required for clearing Staphylococcus aureus in systemic infection despite a strong TLR2-MyD88-dependent T cell activation. J Immunol. 2011;186:443–452. doi: 10.4049/jimmunol.1001407. [DOI] [PubMed] [Google Scholar]
- Schwartz RH. T cell anergy. Annu Rev Immunol. 2003;21:305–334. doi: 10.1146/annurev.immunol.21.120601.141110. [DOI] [PubMed] [Google Scholar]
- Sheehy SH, Atkins BA, Bejon P, Byren I, Wyllie D, Athanasou NA, Berendt AR, McNally MA. The microbiology of chronic osteomyelitis: prevalence of resistance to common empirical anti-microbial regimens. J Infect. 2010;60:338–343. doi: 10.1016/j.jinf.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellberg B, Ibrahim AS, Yeaman MR, Lin L, Fu Y, Avanesian V, Bayer AS, Filler SG, Lipke P, Otoo H,s, et al. The antifungal vaccine derived from the recombinant N terminus of Als3p protects mice against the bacterium Staphylococcus aureus. Infect Immun. 2008;76:4574–4580. doi: 10.1128/IAI.00700-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadecker MJ. The role of T-cell anergy in the immunomodulation of schistosomiasis. Parasitol Today. 1992;8:199–204. doi: 10.1016/0169-4758(92)90264-3. [DOI] [PubMed] [Google Scholar]
- Syrbe U, Siveke J, Hamann A. Th1/Th2 subsets: Distinct differences in homing and chemokine receptor expression. Springer Semin Immunopathol. 1999;21:263–285. doi: 10.1007/BF00812257. [DOI] [PubMed] [Google Scholar]
- Tuchscherr L, Medina E, Hussain M, Völker W, Heitmann V, Niemann S, Holzinger D, Roth J, Proctor RA, Becker K, et al. Staphylococcus aureus phenotype switching: an effective bacterial strategy to escape host immune response and establish a chronic infection. EMBO Mol Med. 2011;3:129–141. doi: 10.1002/emmm.201000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzianabos AO, Chandraker A, Kalka-Moll W, Stingele F, Dong VM, Finberg RW, Peach R, Sayegh MH. Bacterial pathogens induce abscess formation by CD4(+) T-cell activation via the CD28-B7-2 costimulatory pathway. Infect Immun. 2000;68:6650–6655. doi: 10.1128/iai.68.12.6650-6655.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdrengh M, Tarkowski A. Role of neutrophils in experimental septicemia and septic arthritis induced by Staphylococcus aureus. Infect Immun. 1997;65:2517–2521. doi: 10.1128/iai.65.7.2517-2521.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Köckritz-Blickwede M, Rohde M, Oehmcke S, Miller LS, Cheung AL, Herwald H, Foster S, Medina E. Immunological mechanisms underlying the genetic predisposition to severe Staphylococcus aureus infection in the mouse model. Am J Pathol. 2008;173:1657–1668. doi: 10.2353/ajpath.2008.080337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertheim HF, Vos MC, Ott A, van Belkum A, Voss A, Kluytmans JA, van Keulen PH, Vandenbroucke-Grauls CM, Meester MH, Verbrugh HA. Risk and outcome of nosocomial Staphylococcus aureus bacteraemia in nasal carriers versus non-carriers. Lancet. 2004;364:703–705. doi: 10.1016/S0140-6736(04)16897-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.