

Abstract
The robust glow of molecular fluorescence renders even sparse molecules detectable and susceptible to analysis for concentration, mobility, chemistry, and photophysics. Correlation spectroscopy, a statistical-physics-based tool, gleans quantitative information from the spontaneously fluctuating fluorescence signals obtained from small molecular ensembles. This analytical power is available for studying molecules present at minuscule concentrations in liquid solutions (less than one nanomolar), or even on the surfaces of living cells at less than one macromolecule per square micrometer. Indeed, routines are becoming common to detect, locate, and examine individual molecules under favorable conditions.
The analytical strategy of fluorescence correlation spectroscopy (FCS) was introduced more than 25 years ago to measure chemical kinetics and the associated modulation of molecular diffusability by analysis of concentration fluctuations about the equilibrium of a small ensemble (≈103) of molecules (1). Because FCS has recently been resurrected (2) to meet several modern demands for analytical and physical sensitivity in biophysics, we discuss† some of the concepts and the modern potential of this method in chemical and biological physics.
Correlation Analysis: Gleaning Information from Noise
Local fluctuations of the intrinsic thermodynamic properties of a physical system in equilibrium are usually a source of unavoidable noise in experimental measurements, particularly in small systems. This “random noise,” however, is also a potential source of information. The dispersion (mean square amplitude) of the fluctuations around the thermodynamic mean is proportional to the number of independent accessible degrees of freedom. Moreover, temporal autocorrelation of the fluctuations is precisely governed by the dynamic parameters of the system as generally expressed by the fluctuation-dissipation theorem. In fact, according to the Onsager regression hypothesis (4), this information can be used to predict macroscopic behavior of a nonequilibrium system returning to equilibrium.
Measurements of the correlations of fluctuations about equilibrium or steady-state values have led to understanding of many physical phenomena and are particularly recognized for applications to critical phenomena in phase transformations. Optical techniques, such as quasi-elastic light scattering (QELS), provide very convenient noninvasive probes for monitoring fluctuations.‡ However, methods such as QELS had not provided signals useful for studying intramolecular transformations or homogeneous chemical kinetics. It is molecular fluorescence, employed subsequently in FCS, that has provided the appropriate sensitive molecular indicator for biomedical and chemical applications that we discuss here.
What Does Fluorescence Do for Correlation Techniques?
A useful fluorescent molecule typically emits ≥105 photons in water before photobleaching, and at rates up to ≈109 per second (at least during microsecond bursts before ground state depletion by intersystem crossing to excited triplet states). Modern photon detectors, laser excitation, and high numerical aperture microscopy optics allow collection of >3% of the emitted fluorescence photons. Sometimes a hundred photons can be detected while a single molecule in solution is diffusing (in less then a millisecond) through the focus of a laser beam tuned to excite the target fluorophore. Recognition and identification of the individual target molecules above the background fluorescence of the matrix is readily accomplished in appropriate liquid solvents. Even in the intrinsically fluorescent environment of the living biological cell surface, individual macromolecules labeled with bright fluorophores can be detected, located to a few nanometers, and tracked as they move (10, 11).
The high rate of photon detection from individual molecules and the sensitivity of fluorescence to conformational, chemical, and environmental changes can be employed by FCS for the determination of fundamental dynamical parameters. Temporal scales of microseconds to many seconds are readily accessible. Measurements of transport coefficients and chemical kinetics and recognition of aggregation are achievable. More difficult are discrimination among closely related molecular species, detection of possible photophysical differences among nominally identical molecules, and experiments in living cells and tissues.
The Renaissance in FCS
The reasons the FCS strategy is enjoying a remarkable renaissance in biophysics are twofold: (i) the current demand for sensitive analytical techniques and (ii) modern technological improvements incorporated by Rudolph Rigler, Manfred Eigen, and their coworkers as summarized by Eigen and Rigler in the Proceedings several years ago (2). Table 1 lists some of the technical improvements by many laboratories that have pushed the detection sensitivity now to single molecules. Experimentally one can realize small open volumes, often less then a femtoliter, by strong focusing of the illumination into a double cone with a waist <1 μm in diameter plus selection of the fluorescence emitted only from this region with a pinhole in the image plane (12). As in confocal laser scanning microscopy, this strategy reduces background from out-of-focus excitation (13, 14). This allows reduction of the number of observed molecules in an FCS experiment to an optimal sensitivity at about 5. These methods, including the incorporation of avalanche photodiode (APD) detection for high photon quantum efficiency, satisfy the needs even for detection of single molecules in solution (15, 16). Perhaps the most significant technological advance has been in calculating the correlations. Now commercially available digital correlator cards in a personal computer can perform autocorrelation with submicrosecond time resolution in virtually real time.
Table 1.
Experimental advances in detection, identification, and characterization of sparse and single molecules in dilute solution by FCS
| Utilization of a high-speed dedicated digital correlator for nearly real-time highly efficient determination of correlation functions |
| Reduction of the dark-current noise and improvement of the signal/shot noise with small area and high quantum efficiency avalanche photodiode (APD) detector |
| Reduction of the in-focus background by tight focusing to a small probe volume and marker wavelength selection |
| Reduction of out-of-focus background and improved probe volume definition by confocal detection |
| Elimination of the out of focus background and photo bleaching by two-photon excitation (TPE) |
| Avoiding Raman and Rayleigh scattering by TPE to reduce in-focus background |
How Does FCS Work?
In an FCS experiment, the fluorescence emitted from a small, optically well-defined open volume element of a solution in equilibrium is monitored as a function of time (Fig. 1a). The recorded fluorescence emission signal is proportional to the number of fluorescent molecules in the probe volume. This number fluctuates about its equilibrium value as molecules diffuse in and out of the volume and as fluorescent molecules chemically transform to and from nonfluorescent forms. The temporal autocorrelation of the fluorescence signal fluctuation (Fig. 1b Lower) yields the time scale of such dynamics and its variance yields the average number of independent fluorophores (〈N〉) in the probe volume. The correlation functions therefore contain information about chemical reaction kinetics, coefficients of diffusion, and the equilibrium chemical concentrations.
Figure 1.

FCS. (a) Fluorescence is collected from the molecules (circles) in a small well defined volume (red) near the focus of a laser beam (pink) within an equilibrated solution. The number of fluorescent molecules (yellow) in this volume fluctuates due to diffusion/flow in and out of this volume and due to chemical transformation to and from a nonfluorescent (black) species. The fluorescence fluctuates (b Upper) in proportion to the number of fluorescent molecules in the volume, and its autocorrelation (b Lower) yields the dynamic parameters.
Mathematically, the normalized autocorrelation function G(τ) is calculated as the time average (〈〉) of the product of the fluctuations of the detected fluorescence [δF(t)] at every time t and the fluctuations at the delayed times t + τ, normalized by the squared time average of the fluorescence emission [F(t)] (17)—i.e.,
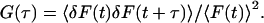 |
1 |
The zero time correlation G(0) = 〈δF(t)2〉/〈F(t)〉2 is the normalized variance of F(t): it represents the relative magnitude of fluctuations. The temporal variation of δF(t) is proportional to δN(t), the fluctuations of the number of independent fluorophores in the probe volume. For a dilute homogeneous dye solution G(0) = 1/〈N〉, where 〈N〉 is the average number of fluorescent molecules in the probe volume (18). One can think of the statistical physics of the system as a grand canonical ensemble with very few particles and correspondingly large particle density fluctuations about a thermodynamic average. The small mean occupation numbers 〈N〉 make the fluctuations large in the sampling volume so the method works theoretically and experimentally.
In actual experiments, δF(t) remains locally proportional to δN(t), but the proportionality constant φ (the “detectivity”) is a function of the position vector r and is determined by the optical system [i.e., δF(r, t) = φ(r)δN(r, t) = φ(r)δC(r, t) dr, where δC(r, t) is the concentration fluctuation]. Thus it follows from Eq. 1:
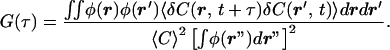 |
2 |
The limiting value as τ → 0 yields the time-independent quantity G(0) that determines the effective average number of molecules 〈N〉 in the focal volume:
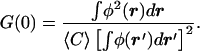 |
3 |
Because φ(r) is a smooth function of r, no sharply delineated focal volume exists. However, the effective average number of molecules 〈N〉 [=1/G(0)], does still provide a consistent way of defining an effective volume 〈V〉 (19, 20) by the relation 〈V〉 = 〈N〉/〈C〉 = 1/[G(0)〈C〉].
Various optical schemes of FCS essentially differ only in the detectivity profile represented by φ(r). For an infinite homogeneous dilute dye solution, for any given excitation and emission wavelengths, φ(r) is proportional to the product of the normalized excitation [ɛ(r)] and collection [χ(r)] profiles—i.e., φ(r) ∝ ɛ(r)χ(r). For a conventional FCS experiment with single-photon excitation and confocal detection, the excitation profile ɛ(r) = I(r) [I(r) = the illumination intensity profile]. The detection profile χ(r) is determined by the experimental geometry (e.g., optical characteristics of the objective lens, the size of the confocal aperture, etc.), yielding φ(r) ∝ I(r)χ(r). For two-photon excited fluorescence without a confocal aperture (see later) the probe volume is entirely determined by the profile of the excitation illumination—i.e., ɛ(r) = I2(r), with full-field detection [χ(r) = 1], yielding φ(r) ∝ I2(r). Measurement of G(0) for known 〈C〉 yields information on φ(r) and determines V, which provides experimental calibration to determine unknown values of 〈C〉.
In chemically equilibrated solutions, values of dynamical parameters for each species j [e.g., diffusion coefficients (Dj) and chemical kinetic rate constants (Tjk)] can be obtained from the measured time evolution of G(τ). Changes in G(τ) originate from the time dependence of δCj(τ), which is governed by (1):
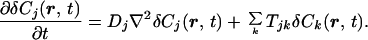 |
4 |
The resultant form of G(τ) must be evaluated in terms of the solutions of Eq. 4, for a given φ(r). The case with only one species and no chemical kinetics (Dj = D, Tjk = 0) and where φ(r) is a three-dimensional Gaussian function with half axes of r and l (which approximates a one-photon excited confocal FCS experiment) provides an illustrative example (2):
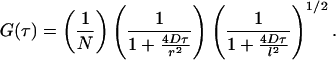 |
5 |
Fig. 2 shows an example of an actual FCS experiment, where only a few minutes of data collection provides an accurate measure of concentrations, diffusion coefficient, and the rate of singlet ↔ triplet interconversion of a fluorophore in solution.
Figure 2.

Fluorescence autocorrelation functions as a function of time. Data were obtained from several different concentrations of rhodamine 6G (R6G) molecules in 70% sucrose aqueous solution. The concentrations used are (green curves, from the bottom): 20 nM, 10 nM, 5 nM, 2.5 nM, 1.25 nM, and 0.62 nM. The autocorrelation decays at a time scale of ≈100 ms due to diffusion of molecules in and out of the excitation volume. (Inset) Average number 〈N〉 of molecules in the focal volume (as determined by the inverse of the value of the autocorrelation function extrapolated to time zero [1/G(0)]) as a function of concentration. The topmost curve (red) is also obtained from 0.62 nM R6G, but at 70 times higher excitation power. Lowering of the molecular ground state population by long-lived triplet excited state formation is evident in the higher G(0), while the triplet state lifetime manifests itself in the sub-millisecond decay of the extra part of the autocorrelation.
Early Applications
In the first FCS experiment, the power of the technique was demonstrated by measuring the diffusion and binding kinetics of the small fluorescent drug ethidium to DNA. This early research formulated the mathematical framework of the general FCS problem in terms of the eigenvalues and eigenvectors of the coupled diffusion and chemical kinetic rate equations, so that it still provides the foundations for analysis (1, 12, 21–24). The original experiments showed how binding of the small drug molecules to larger DNA molecules effectively slows the drug diffusion (1). The ability of FCS to measure the absolute fluorophore concentration was subsequently utilized for determining the number densities of macromolecules in solution (25, 26), in membranes (27), and on surfaces (28). The same concepts have been applied also for determining molecular weights and molecular aggregation (29). The temporal decay of the autocorrelation function has been used to measure diffusion coefficients [both translational (22, 30) and rotational (31, 32)] and chemical kinetic rate constants (1, 33, 34) of fluorescent molecules in chemical and biological systems. Measurements of fluorescence decay times by autocorrelation on a nanosecond time scale have been reported (35). Scanning of the specimen on a sub-diffusion time scale improves the signal-to-noise ratio (25, 36–40) of fluctuation measurements and can measure spatial heterogeneity in a static system. Alternatively, flow of fluids can sample large volumes for FCS to provide a good measure of convective transport and improve sampling statistics for detection of sparse molecules (17).
Recent Applications
The power of FCS for counting sparse molecules has recently been innovatively utilized by La Clair (3) for the study of disfavored reaction pathways, which have higher activation energy barriers than the pathways to major products. This study required the detection of the rare product (10−11 M) among an abundance of the favored product (10−7 M). In a chosen spectral window, individual molecules of the sparse disfavored product are much brighter compared with each of their more abundant counterparts. For problems with these characteristics, FCS is the logical choice. It works this way: On the occasions when a bright sparse molecule diffuses through the focal sampling volume a transient fluorescence fluctuation burst is recorded with a time scale determined by the diffusion time τD ≈ w2/4D, where w is the radius of the focal volume and D is the diffusion constant. These bursts make a large contribution to the variance of the fluorescence fluctuations and hence to the experimentally determined zero-time autocorrelation function G(0). The background fluorescence, although possibly quite bright due to the large concentration of dimly fluorescent molecules, contributes minimally to G(0). In a molecular mixture, the relative contribution of each species i to G(0) is proportional to the product of the number of molecules ni and the square of the detectivity φi (for simplicity, φi is assumed to be independent of position within the focal volume): G(0) = Σ φi2Ni/(Σ φiNi)2. Thus in a two-component mixture with N2 = 104 N1 and φ2 = 10−3 φ1, species 2 contributes 91% of the total fluorescence, but only 1% of the G(0). This facilitates an accurate concentration measurement of the rare but bright species by means of FCS, as in the present case.
La Clair’s experiment is one of several recent results suggesting that FCS is poised to become a versatile tool in the arsenal of the chemist and the biologist. As the limits of time resolution and sensitivity have improved, many innovative applications have been conceived. For example, FCS has recently been utilized for studying the dynamics of intersystem crossing among excited states of fluorophores where the occupancy of the triplet excited state blocks out emission for microsecond intervals (41, 42).
The ability of FCS to study dilute solutions has been carried to the limit where, on average, less than one molecule is present in the probe volume (13, 20). The principal barrier to detecting these small concentrations seems to be the experimental time required to record enough “events” (an event is a single molecule passing through the probe volume) to obtain a statistically meaningful autocorrelation function, which in turn determines the diffusion coefficient. One remedy for this sampling problem is transport of the solution through a set of sampling volumes and analysis by cross-correlation of the fluorescence signals from each volume (43). One intriguing and widely discussed possibility for application of this technique is gene sequencing. A DNA strand synthesized completely from fluorescence-labeled nucleotides is immobilized in the flow by attachment at one end and sequentially shortened one base at a time at the other end by action of an exonuclease. The individual nucleic acids are then detected in sequence as they flow through a first illuminated band and are verified by time-shifted cross-correlation as they pass through the second band downstream. There are also programs underway to develop strongly fluorescent nucleotide analogs or methods of utilizing active nucleotide fluorescence for this approach to rapid sequencing.
Some of the most interesting new applications use FCS in combination with gene amplification techniques [e.g., NASBA (nucleic acid sequence-based amplification)] to multiply sparse genetic material into a multitude. Eigen and co-workers have demonstrated the identification of particular RNA and DNA strands with the help of fluorescent primers in the context of viral pathogen analysis (44). Before the symptomatic stage, the virus is present at a very low level (≈10−18 M) in blood plasma, and a high level of chemical amplification is required for detection. The problem is brought under the purview of FCS by attaching a fluorophore with a DNA probe that hybridizes with a specific part of the viral RNA, which in turn changes the diffusion constant of the fluorophore. The sensitivity of FCS affords detection after amplification to 1 nM, more than 100-fold lower compared with conventional techniques, thereby making the diagnosis faster and more reliable. It remains to explore the practical problems of measurement in unpurified blood plasma. Eigen and Rigler have also outlined the application of FCS to molecular evolution studies (2).
Measurements of chemical kinetics by FCS have recently been advanced by applying cross-correlation between two distinguishable fluorescent markers on the reagents, which then become perfectly cross-correlated when the reactive pairs bind to each other (45). Another exciting possibility is the study of the kinetics of protein folding (46). FCS offers an appealing method to follow the dynamics of folding at a time scale spanning from hundreds of nanoseconds to hundreds of milliseconds, all in an equilibrium solution without the need for perturbation to provide a temporal trigger for a transient response.
Multiphoton Fluorescence Excitation (MPE) in FCS
In a new and promising experimental scheme, simultaneous molecular absorption of two or more infrared photons from a focused mode-locked laser beam energizes visible wavelength fluorescence emission. The excitation occurs in a well defined subfemtoliter focal volume where the focused intensity is high enough to allow simultaneous (<10−15 s) absorption of two or more photons by a molecule. Because excitation can occur only in the high-intensity focal volume, there is no out-of-focus fluorescence background and confocal detection is not required for FCS. Thus, as noted in a previous section, the probe volume of FCS is precisely defined by the illumination optics.§ MPE provides for higher penetration depth in biological tissue, lower phototoxicity, and the ability to excite and image native UV chromophores in living cells (49). All of these advantages may be important for biological applications of FCS. Furthermore, MPE at a single excitation wavelength can excite a range of fluorophores across the spectrum of emission colors (50, 51), thereby providing an easy way of performing simultaneous auto- or cross-correlation spectroscopy of multiple species. Two-photon excitation (TPE) of fluorescence has an additional advantage: usually the principal background source in conventional fluorescence excitation is Raman and Rayleigh scattering by the solvent molecules. With TPE the corresponding hyper-Rayleigh and hyper-Raman scattering of water is very weak and is shiftable within the broad MPE spectra to harmless wavelengths (52). TPE has been utilized for several diffusion measurements by FCS (46, 53). One apparent drawback of this scheme, however, is an extra limitation on the maximum number of fluorescence photons available per molecule per unit time to the frequency (usually ≈80 MHz) of pulses of the mode-locked lasers used for MPE. Increasing the laser pulse frequency or dividing the laser beam into multiple optically delayed beams optimizes the signal.
Future Possibilities
The applications mentioned above are all generally still nascent, so their applicability remains to be seen as they mature. We think that one of the most promising is the combination of FCS with genetic amplification technologies for detection of sparse genetic fragments such as those of the HIV virus in human plasma. Similarly, cross-correlation among fluorescent markers (45) of receptors and drug molecules may provide a useful tool in the search for effective pharmaceuticals. We hope that our laboratory will be able to make FCS a useful tool for measurement of the dynamics of the folding of proteins and nucleic acids. Perhaps the most productive functions of FCS will, however, come about by extensions of the original simple concept. One can expect FCS to spawn a variety of new techniques driven by the current interest in detecting and analyzing sparse and single molecules.
There is another capability of FCS that we think is valuable. That is the minute spatial scale of the sampling volume. Research in cell biology requires measurements of the molecular transport and chemical kinetics within cellular interiors, and the complex compartmental geometry of tissues is a challenge. FCS measurements in principle could provide prized information. Whether the anticipated technical difficulties of autofluorescence, photodamage, insertion of fluorescent markers, and interference from cellular activity can be overcome is not clear. The only published intracellular FCS experiments of which we are aware observed microinjected fluorescent latex spheres that are bright enough to overwhelm the background autofluorescence (53), but it did prove the feasibility of intracellular FCS. The potential value of answers to outstanding questions provides the motivation to advance to the molecular and organelle level inside and among living cells.
Acknowledgments
This work was carried out in the Developmental Resource for Biophysical Imaging and Optoelectronics with funding provided by the National Science Foundation (Grant DIR 88002787) and the National Institutes of Health (Grants RR07719 and RR04224). U.H. was supported by an Otto Hahn Grant from the Max Planck Society.
ABBREVIATIONS
- FCS
fluorescence correlation spectroscopy
- MPE
multiphoton fluorescence excitation
Footnotes
This article was originally solicited by the editors of the Proceedings as an invited commentary on a paper by James J. La Clair (3), but the authors preferred to expand their comments to a broader Perspective.
For example, in our own laboratory, quasi-elastic light scattering (QELS) revealed the two-dimensional thermal excitation of critical fluid interfaces (5), and the coupling of thermal and concentration fluctuations in tricritical isotopic mixtures of 3He and 4He at 0.7 K (6); thermal fluctuations of phospholipid membranes measured their interfacial energies (7) and cross-correlation of mechanical and electrical fluctuations of the mechanosensory receptors of the inner ear established fundamental limits (8) and mechanisms of mechanoelectrical transduction in hearing (9).
Two-photon excitation of fluorescence was first understood by Maria Goeppert-Mayer in 1931 (47), but its experimental realization had to wait more than 30 years for the invention of pulsed lasers in the 1960s. Multiphoton excitation was adopted for two-photon laser scanning fluorescence microscopy and for spatially resolved photochemical activation of caged bioactive molecules by Denk et al. in 1990 (48).
References
- 1.Magde D, Elson E, Webb W W. Phys Rev Lett. 1972;29:705–708. [Google Scholar]
- 2.Eigen M, Rigler R. Proc Natl Acad Sci USA. 1994;91:5740–5747. doi: 10.1073/pnas.91.13.5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.La Clair J J. Proc Natl Acad Sci USA. 1997;94:1623–1628. doi: 10.1073/pnas.94.5.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Onsager L. Phys Rev. 1931;37:405–426. [Google Scholar]
- 5.Huang J S, Webb W W. Phys Rev Lett. 1969;23:160–163. [Google Scholar]
- 6.Leiderer P, Nelson D R, Watts D R, Webb W W. Phys Rev Lett. 1975;34:1080–1083. [Google Scholar]
- 7.Schneider M B, Jenkins J T, Webb W W. J Phys (Paris) 1984;45:1457–1472. [Google Scholar]
- 8.Mantese J V, Webb W W. Phys Rev Lett. 1985;55:2212–2215. doi: 10.1103/PhysRevLett.55.2212. [DOI] [PubMed] [Google Scholar]
- 9.Denk W, Keolian R M, Webb W W. J Neurophysiol. 1992;68:927–932. doi: 10.1152/jn.1992.68.3.927. [DOI] [PubMed] [Google Scholar]
- 10.Ghosh R N, Webb W W. Biophys J. 1994;66:1301–1318. doi: 10.1016/S0006-3495(94)80939-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feder T J, Chang E Y, Holowka D, Webb W W. J Cell Phys. 1994;158:7–16. doi: 10.1002/jcp.1041580103. [DOI] [PubMed] [Google Scholar]
- 12.Koppel D E, Axelrod D, Schlessinger J, Elson E L, Webb W W. Biophys J. 1976;16:1315–1329. doi: 10.1016/S0006-3495(76)85776-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rigler R, Widengren J, Mets U. In: Fluorescence Spectroscopy: New Methods and Applications. Wolfbeis O S, editor. Berlin: Springer; 1992. pp. 13–24. [Google Scholar]
- 14.Qian H, Elson E L. Appl Opt. 1991;30:1185–1195. doi: 10.1364/AO.30.001185. [DOI] [PubMed] [Google Scholar]
- 15.Edman L, Mets U, Rigler R. Proc Natl Acad Sci USA. 1996;93:6710–6715. doi: 10.1073/pnas.93.13.6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiu D T, Zare R N. J Am Chem Soc. 1996;118:6512–6513. [Google Scholar]
- 17.Magde D, Webb W W, Elson E. Biopolymers. 1978;17:361–376. doi: 10.1002/bip.1974.360130103. [DOI] [PubMed] [Google Scholar]
- 18.Reif F. Fundamentals of Statistical and Thermal Physics. New York: McGraw–Hill; 1965. [Google Scholar]
- 19.Palmer A G, Thompson N L. Appl Opt. 1989;28:1214–1220. doi: 10.1364/AO.28.001214. [DOI] [PubMed] [Google Scholar]
- 20.Mertz J, Xu C, Webb W W. Opt Lett. 1995;20:2532–2534. doi: 10.1364/ol.20.002532. [DOI] [PubMed] [Google Scholar]
- 21.Elson E L, Webb W W. Annu Rev Biophys Bioeng. 1975;4:311–334. doi: 10.1146/annurev.bb.04.060175.001523. [DOI] [PubMed] [Google Scholar]
- 22.Magde D, Elson E L, Webb W W. Biopolymers. 1974;13:29–61. doi: 10.1002/bip.1974.360130103. [DOI] [PubMed] [Google Scholar]
- 23.Elson E L, Magde D. Biopolymers. 1974;13:1–27. doi: 10.1002/bip.1974.360130103. [DOI] [PubMed] [Google Scholar]
- 24.Koppel D E. Phys Rev A. 1974;10:1938–1945. [Google Scholar]
- 25.Weissman M, Schindler H, Feher G. Proc Natl Acad Sci USA. 1976;73:2776–2780. doi: 10.1073/pnas.73.8.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sorscher S M, Klein M P. Rev Sci Instrum. 1980;51:98–102. [Google Scholar]
- 27.Fahey P F, Koppel D E, Barak L S, Wolf D E, Elson E L, Webb W W. Science. 1977;195:305–306. doi: 10.1126/science.831279. [DOI] [PubMed] [Google Scholar]
- 28.Thompson N L, Axelrod D. Biophys J. 1983;43:103–114. doi: 10.1016/S0006-3495(83)84328-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palmer A G, Thompson N L. Proc Natl Acad Sci USA. 1989;86:6148–6152. doi: 10.1073/pnas.86.16.6148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rigler R, Grasselli P, Ehrenberg M. Physica Scripta. 1979;19:486–490. [Google Scholar]
- 31.Ehrenberg M, Rigler R. Q Rev Biophys. 1976;9:69–81. doi: 10.1017/s003358350000216x. [DOI] [PubMed] [Google Scholar]
- 32.Kask P, Piksarv P, Mets U, Pooga M, Lippmaa E. Eur Biophys J. 1987;14:257–261. doi: 10.1007/BF00256359. [DOI] [PubMed] [Google Scholar]
- 33.Icenogle R D, Elson E L. Biopolymers. 1983;22:1919–1948. doi: 10.1002/bip.360220808. [DOI] [PubMed] [Google Scholar]
- 34.Sorscher S M, Bartholomew J C, Klein M P. Biochim Biophys Acta. 1980;610:28–46. doi: 10.1016/0005-2787(80)90053-2. [DOI] [PubMed] [Google Scholar]
- 35.Kask P, Piksarv P, Mets U. Eur Biophys J. 1985;12:163–166. doi: 10.1007/BF00256359. [DOI] [PubMed] [Google Scholar]
- 36.Koppel D E, Morgan F, Cowan A E, Carson J H. Biophys J. 1994;66:502–507. doi: 10.1016/s0006-3495(94)80801-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Srivastava M, Petersen N O. Methods Cell Sci. 1996;18:47–54. [Google Scholar]
- 38.Berland K M, So P T C, Chen Y, Mantulin W W, Gratton E. Biophys J. 1996;71:410–420. doi: 10.1016/S0006-3495(96)79242-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petersen N O, Johnson D C, Schlesinger M J. Biophys J. 1986;49:817–820. doi: 10.1016/S0006-3495(86)83710-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petersen N O. Biophys J. 1986;49:809–815. doi: 10.1016/S0006-3495(86)83709-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Widengren J, Rigler R, Mets U. J Fluorescence. 1994;4:255–258. doi: 10.1007/BF01878460. [DOI] [PubMed] [Google Scholar]
- 42.Widengren J, Mets Ulo, Rigler R. J Phys Chem. 1995;99:13368–13379. [Google Scholar]
- 43.Brinkmeier, M. (1996) Ph.D. dissertation (Tech. Univ. Braunschweig, Germany).
- 44.Oehlenschlager F, Schwille P, Eigen M. Proc Natl Acad Sci USA. 1996;93:12811–12816. doi: 10.1073/pnas.93.23.12811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schwille P, Meyer-Almes F J, Rigler R. Biophys J. 1997;72:1878–1886. doi: 10.1016/S0006-3495(97)78833-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maiti S, Webb W W. Biophys J. 1996;70:A262–A262. [Google Scholar]
- 47.Goeppert-Mayer M. Ann Phys. 1931;9:273–295. [Google Scholar]
- 48.Denk W, Strickler J H, Webb W W. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- 49.Maiti S, Shear J B, Williams R M, Zipfel W R, Webb W W. Science. 1997;275:530–532. doi: 10.1126/science.275.5299.530. [DOI] [PubMed] [Google Scholar]
- 50.Xu C, Zipfel W, Shear J B, Williams R M, Webb W W. Proc Natl Acad Sci USA. 1996;93:10763–10768. doi: 10.1073/pnas.93.20.10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu C, Webb W W. J Opt Soc Am B. 1996;13:481–491. [Google Scholar]
- 52.Xu C, Shear J B, Webb W W. Anal Chem. 1997;68:1285–1287. doi: 10.1021/ac961185o. [DOI] [PubMed] [Google Scholar]
- 53.Berland K M, So P T C, Gratton E. Biophys J. 1995;68:694–701. doi: 10.1016/S0006-3495(95)80230-4. [DOI] [PMC free article] [PubMed] [Google Scholar]