

Abstract
Over 1 million people in the United States and 33 million individuals worldwide suffer from HIV/AIDS. Since its discovery, HIV/AIDS has been associated with an increased susceptibility to opportunistic infection due to immune dysfunction. Highly active antiretroviral therapies (HAART) restore immune function and, as a result, people infected with HIV-1 are living longer. This improved survival of HIV-1 patients has revealed a previously unrecognized risk of developing vascular complications, such as atherosclerosis and pulmonary hypertension. The mechanisms underlying these HIV-associated vascular disorders are poorly understood. However, HIV-induced elevations in reactive oxygen species, including superoxide and hydrogen peroxide, may contribute to vascular disease development and progression by altering cell function and redox-sensitive signaling pathways. In this review, we summarize the clinical and experimental evidence demonstrating HIV- and HIV antiretroviral therapy-induced alterations in reactive oxygen species (ROS) and how these effects likely contribute to vascular dysfunction and disease.
Keywords: HIV-1, HIV-1 Proteins, Oxidative Stress, Reactive Oxygen Species, Antioxidants, Pulmonary Hypertension, Atherosclerosis, Antiretroviral Therapy
Introduction
Human immunodeficiency virus type 1 (HIV-1) infection and acquired immunodeficiency syndrome (AIDS) pose one of the greatest challenges to global public health. Since the development of highly active antiretroviral therapies (HAART), mortality and the incidence of opportunistic infections in people living with HIV-1 have declined substantially [1, 2]. As HIV/AIDS patients live longer, however, serious non-AIDS events occur and are associated with a greater risk of death than opportunistic AIDS-related events [3]. As such, vascular complications including coronary heart disease, pulmonary hypertension (PH), and atherosclerosis [4–7] are some of the most widely recognized [8, 9] uncharacteristic AIDS phenomena recorded in HIV-infected patients. In addition to the increase in susceptibility, clinical data also reveal that vascular complications in HIV-1 patients progress much more rapidly than in non-infected individuals [6, 10]. The exact mechanisms by which HIV-1 promotes the development and progression of these disorders remain unknown and are likely multi-factorial. Current research has identified increased vascular oxidative stress as a contributing underlying pathway. This review summarizes the clinical and experimental data correlating HIV-1 and vascular disease, the in vitro and in vivo evidence of HIV-mediated oxidative stress, and the sources of reactive oxygen species (ROS) and antioxidants impacted by HIV-1 and HIV-1 antiretroviral agents. This review also discusses how ROS promote the development and/or progression of vascular complications and why the strategic targeting of HIV-induced ROS may be of potential therapeutic value.
HIV-associated Vascular Disorders
Noninfectious complications occur much more frequently in AIDS patients than opportunistic infections and are now associated with a greater risk of death than AIDS-related events [3]. Atherosclerosis and pulmonary hypertension (PH) are two distinct and extensively-studied examples of HIV-associated vascular disorders. Although the exact underlying mechanisms remain unknown, the pathogenesis of these diseases is strongly associated with HIV-1 infection.
Pulmonary Hypertension
Pulmonary hypertension (PH) is a persistent elevation of pulmonary artery pressure and pulmonary vascular resistance. The symptoms of PH are nonspecific and include dyspnea, syncope, fatigue, chest pain, and nonproductive cough. Chronic PH increases the load on the right ventricle (RV) causing RV hypertrophy, right heart failure, the clinical syndrome of cor pulmonale, and ultimately, death [11]. Numerous studies report an increased frequency of PH in the HIV-infected population, with a prevalence of approximately 1 case per 200 (0.5%). More recent studies suggest, however, that this number is increasing and estimate that up to 1.0% of HIV-1 patients will develop PH [12]. This increase in incidence is likely as HIV-1 positive patients are not routinely examined for PH, and PH is often misdiagnosed resulting in an inaccurate assessment of incidence among HIV-1 patients [13]. Overall, the current data suggests that more than 10,000 HIV-1-infected individuals in the U.S. alone will develop PH. This incidence of PH in the HIV-1-infected population is extremely high compared to the 1 to 2 cases per million recorded in the general population [14]. The reason HIV-infected patients develop PH at this alarming rate remains unknown. It has been suggested that the increased lifespan of HIV-1 patients on antiretroviral therapy increases the likelihood of exposure to the “multiple hits” believed to be required to develop PH [15].
Recent studies investigating HIV-PH suggest that HAART fails to prevent the development of HIV-PH or improve the hemodynamic parameters in HIV-PH patients [16]. In addition, although HAART regulates viral replication and improves survival, patients with well-controlled HIV infection still develop PH. These observations underscore the severity of this disorder and also the need for further investigation of this disease.
Coronary Heart Disease and Atherosclerosis
HIV-1 positive patients also have a higher prevalence of atherosclerotic lesions [17–20], and elevated markers of subclinical atherosclerosis including increased carotid artery intima-media thickness [6, 21–28], increased arterial stiffness [29, 30] and endothelial dysfunction [31–34]. Clinical studies examining cardiovascular disease in HIV-1-positive people prior to the era of HAART are relatively few, yet there is evidence of serious cardiovascular anomalies in these patients. Seminal work by Joshi revealed coronary arteriopathy in 3 of 6 HIV-1-infected children at autopsy; it also described vasculitis and perivasculitis with infiltration of lymphocytes and mononuclear cells in vessel walls [35]. Other post-mortem analyses described major atherosclerotic lesions in proximal coronary arteries in 6 out of 8 HIV-infected patients who were 23–32 years of age [36]. The high frequency of abnormalities in these early studies is striking considering that cardiovascular pathologies are normally rare in these age groups. Vasculitis in small blood vessels [37, 38], aneurysms in medium or large arteries [39], and significantly lower levels of high density lipoprotein cholesterol (HDLc) in the bloodstream [40] of untreated HIV-1-positive individuals indicate that HIV-1 infection increases cardiovascular complications. These findings provide initial evidence of vascular dysfunction in HIV-1 patients, and support the premise that HIV-1 viral proteins have a role in the development of cardiovascular disease in this population.
Antiretroviral-naïve HIV-1-positive patients are found to have markers of endothelial activation including elevated plasma levels of von Willebrand factor, plasminogen activator inhibitor-1 antigen, and tissue-type plasminogen activator [41, 42]. These elevations in markers of endothelial dysfunction were found to correlate with anti-p24 antibodies and disease severity [41]. Antiretroviral-naïve HIV-1-positive people have also been found to have higher levels of soluble vascular cell adhesion molecule-1 (VCAM-1) [43], intracellular adhesion molecule-1 (ICAM-1) [44], and E-selectin [42, 44, 45] compared to healthy controls. This up-regulation of cell adhesion markers suggests that HIV-1 increases endothelial cell activation and dysregulation. These derangements may contribute to the increased incidence of pulmonary and systemic vascular disease.
There is also indirect clinical evidence showing that the presence of the HIV-1 virus potentiates cardiovascular risk. An ongoing retrospective analysis by the Kaiser Permanente Medical Care Program of Northern California has determined hospitalization rates for coronary heart disease and myocardial infarction in 4159 HIV-1-positive male members [46]. The authors did not find a correlation between antiretroviral therapies (ARTs) and hospitalization rates in the HIV-1 positive group after a 4 year follow-up. However, they demonstrated significantly higher hospitalization rates in the infected group when comparing them to age- and sex-matched HIV-1-negative controls during this same timeframe. They were unable to establish correlations between the increase in hospitalization rate with other known risk factors (i.e. smoking, hypertension, diabetes, and hyperlipidemia) in the HIV-1-positive group, thus postulating that HIV-1 infection itself increases the hospitalization rate for coronary heart disease and myocardial infarction. This conclusion has been supported by other clinical studies as well. A 2007 study examined acute myocardial infarction in patients (3851 HIV-1-positive and 1,044,589 HIV-1-negative) at 2 large Massachusetts hospitals [47]. The authors found a significantly increased risk for heart attack in the HIV-1-positive population at all ages examined, even when adjusted for the presence of other traditional risk factors in this group. Interestingly, the risk for myocardial infarction was roughly tripled in HIV-1-positive women compared to uninfected women, a group generally considered to be at lower risk of developing cardiovascular disease compared to men. However, the impact of HAART on this observation could not be adjusted for due to insufficient data. Another long-term multi-institution analysis, the Strategies for Management of Antiretroviral Therapy (SMART) Study Group, concluded that cessation of antiretroviral therapy (ART) in HIV-1-positive patients increases their short-term risk of developing cardiovascular disease [48]. Because prolonged ART has been associated with major metabolic and cardiovascular disorders, the authors of the study had hoped to evaluate the effectiveness of episodic ART in 2,720 HIV-1-positive patients using a treatment paradigm that administered HAART to maintain CD4+ lymphocyte levels. Unfortunately, interruption of antiretroviral therapy did not benefit this cohort and actually increased the incidence of major cardiovascular events.
It is suggested that HIV-1 infection elicits endothelial dysfunction in patients, as measured by flow-mediated dilation (FMD) of the brachial artery. A controlled case-study of 4 HIV-1-positive patients suggested that viral load inversely correlated with endothelium-dependent FMD without any relation to antiretroviral regimens [49]. Solages monitored FMD in 75 HIV-1-positive and 223 control subjects, and found significantly impaired endothelial function in the infected population. This study also found that viral load was a significant predictor of FMD [50]. The authors did not observe an association between endothelial dysfunction and the use of HAART, which could potentially be explained by the small sample size and unrepresentative demographic characteristics of this specific population. A smaller study in patients from 3.5–19.5 years of age also showed that HIV-1-infected children had significantly reduced FMD, increased wall stiffness, and lower cross-sectional compliance and distensibility of the carotid artery than non-infected children [51]. Interestingly, no differences in these parameters were observed when comparing HIV-1-positive children on HAART with those who were HAART-naïve, suggesting that the HIV-1 viral infection increased endothelial dysfunction. Other data also suggest that HIV, independent of HAART, can induce vascular dysfunction [32, 52]. However, a 2007 study published by Lorenz concluded that HIV-1 infection and HAART are both independent risk factors for the development of atherosclerosis in adults [53]. They found that intima media thickness of the carotid bifurcation, a predictor of subclinical atherosclerosis, was 24.8% higher in an HIV-1-positive/antiretroviral-naïve group compared to an uninfected control group. They also observed significantly greater IMT of the carotid bifurcation and the common carotid artery associated with HAART treatment in HIV-1-positive individuals. This effect of HIV-1 proteins was confirmed in a 2009 study which demonstrated that HIV-1 infection is independently associated with carotid intima media thickening, a measure of sub-clinical atherosclerosis [54].
HIV-1 Infection and Elevated ROS
Clinical Evidence
HIV-1 patients consistently demonstrate marked increases in ROS production as well as significant reductions in antioxidant availability and activity. In a study with treated and untreated HIV-infected subjects, the oxidative stress marker, d-ROM (derivatives of reactive oxygen metabolites) was shown to be greater in serum of HIV-1 patients than that of healthy controls [55]. Malondialdehyde (MDA), an index of lipid peroxidation, was also significantly elevated in the serum from both symptomatic and asymptomatic HIV-1 patients [56–58]. The marked increases in ROS biomarkers, d-ROM and MDA in HIV-1 patients demonstrate a HIV-induced dysregulation of ROS. These alterations are likely attributable to numerous mediators. However, several studies suggest that increased ROS production in HIV-1 patients results from diminished antioxidant expression and activity (Figure 1). For example, a dramatic attenuation in the total antioxidant capacity including vitamin A and C serum concentrations has been noted in HIV-1 seropositive patients [56, 59, 60]. Glutathione (GSH), the predominant antioxidant in the lung, is also significantly altered in HIV-1 patients. Studies demonstrate that total and reduced GSH in the epithelial lining fluid of symptom-free HIV-seropositive individuals was 60% less than those in normal subjects [61]. GSH levels are also reduced in the blood of HIV-1 patients [62–64]. Plasma of HIV-infected patients displays a 30% reduction in glutathione when compared to healthy controls. There was no difference in glutathione levels in the untreated HIV-infected group when compared to subjects on ART regimens [65]. Interestingly, however, HIV-1 produces a contradictory effect in the antioxidant, thioredoxin (Trx) which is significantly elevated in the plasma of HIV-infected healthy volunteers [66]. Studies show that Trx functions as an antioxidant in both the cytosol and mitochondria and contributes to cell growth, DNA repair and transcription factor regulation [67]. The HIV-induced increase in Trx may, therefore, function as a cellular compensatory mechanism or an attempt to normalize antioxidant capacity.
Figure 1. Effects of HIV-1 and Antiretroviral Therapies on ROS Sources and Scavengers.
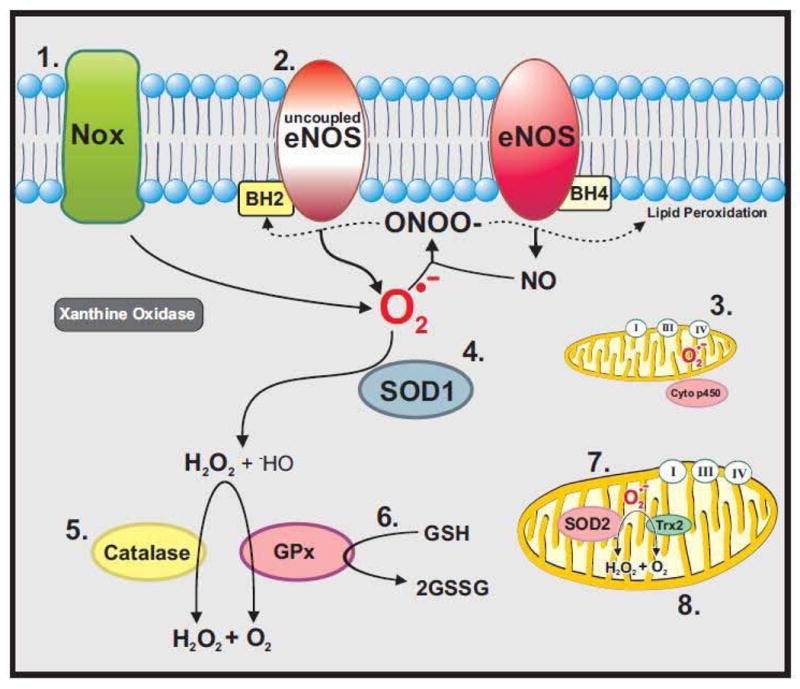
1. NADPH Oxidases (Noxes), the primary producer of ROS in vascular cells, are dramatically up-regulated by HIV-1 [69, 92, 96] and ART [133]. 2. Uncoupled eNOS produces superoxide, instead of nitric oxide. Superoxide may then couple with NO to generate the highly reactive radical peroxynitrite (ONOO−) [173], which oxidizes tetrahydrobiopterin and causes lipid peroxidation [174]. HIV-1 [56, 68] and ART [158] stimulate elevations in lipid peroxidation markers such as MDA and nitrotyrosine. 3. HIV-1 [77, 89] and ART [131, 137–139] promote ROS release by inducing mitochondrial dysfunction. 4. HIV [71] and ART [153, 293] reduce SOD expression and activity. 5. HIV-1 negatively modulates catalase expression and activity. 6. GSH levels are significantly decreased in HIV-1 patients [65, 128]. In addition, HIV-1 proteins alter GSH release [86] and regulation [88, 116]. ART also deplete cellular GSH [131, 158]. 7. Mitochondrial antioxidant, SOD2 is decreased in HIV-1 [80, 89, 293].
These studies demonstrate an obvious association between HIV-1 and altered ROS production and antioxidant availability. These alterations are noted in numerous tissues as elevated ROS biomarkers and decreased antioxidant levels are observed in both serum and epithelial lining fluid from HIV-1 patients. The attenuation in cellular antioxidant systems provide a likely explanation for the considerable elevation of ROS documented in HIV-1 patients. However, while clinical studies are unable to determine the cellular sources of ROS, experimental HIV models have shed more light on specific mediators of HIV-induced ROS.
Experimental Models
In vitro models of HIV-1 infection mirror clinical studies demonstrating both increases in ROS and declines in antioxidant activity. HIV-1 infection of human primary macrophages produces a 6-fold increase in malondialdehyde (MDA) [68]. This finding implicates HIV-1 infection as the principal cause for elevated macrophage ROS levels. However, it remains controversial whether HIV-1 infection or HIV-1-induced mediators contribute to the increased ROS production and antioxidant depletion seen in infected patients. Data from several recent studies strongly suggest that HIV-induced mediators, independent of HIV-1 infection, are sufficient to alter cellular ROS generation. In an in vitro model of HIV-induced oxidative stress, podocytes expressing the NL4-3 HIV-1 construct with a deleted gag/pol region exhibit a marked increase in ROS generation over a 3-hour interval of HIV exposure. The NL4-3-induced increase in podocyte ROS generation was attenuated by diphenyleneiodonium (DPI) administration, implicating flavin-containing enzymes such as NADPH oxidase as the primary source for ROS increases [69]. This study suggests that virus infectivity due to gag and pol function is not necessary for HIV-induced ROS release.
In vivo models studying the effect of HIV-1 proteins on oxidative stress reach similar conclusions. Mice expressing the HIV-1 Tat protein (HIV-1 Tat+) exhibit a significant reduction in total intracellular GSH content in both the liver and erythrocytes. Additionally, glutathione synthetase activity in HIV-1 Tat+ mouse liver was decreased to 73% of control levels [70]. Similarly, studies from our group employing an HIV-1 transgenic rat model indicate that the expression of HIV-1 proteins is sufficient to augment ROS production and alter antioxidant expression. These animals express a HIV-1 provirus that encodes for the viral genes env, tat, nef, rev, vif, vpu, and vpr. However, due to the deletion of the gag and pol regions, the HIV-1 transgene is both nonreplicative and noninfectious. Studies using this model show that HIV-1 transgenic (Tg) rat aortas display significant increases in superoxide and 3-nitrotyrosine levels compared to wild-type controls. HIV-1 Tg rats also exhibit marked decreases in circulating nitric oxide (NO) and total GSH as well as reductions in aortic SOD1 expression and activity [71]. HIV-1 transgene expression also induces marked elevations in rat lung superoxide, hydrogen peroxide (H2O2), and NO metabolite levels as well as concomitant decreases in lung lavage fluid GSH when compared to wild-type rats [72, 73].
Altogether, these studies highlight the ability of HIV-1 proteins to independently alter ROS release and antioxidant activity. Indeed, details regarding whether HIV-1 protein concentrations in these model are physiologically relevant are needed. Additionally, further investigation to ascertain if these proteins potentiate the effect of HIV-1 infection is warranted. Nonetheless, the contribution of HIV-1 proteins to altered ROS production and regulation is clear.
Increased ROS – Contribution of HIV-1 Proteins
Considerable research indicates that HIV-1 significantly alters the cellular oxidant/antioxidant balance. However, research utilizing HIV- and Tat-Tg animal models argues that virus-induced mediators such as HIV-1 proteins may serve as sufficient inducers of oxidative stress. As a result, the investigation of HIV-1 proteins and their effects on ROS release and regulation has increased substantially. This research sheds light on the imbalance between oxidants and antioxidants, and strongly suggests that HIV-1 proteins contribute to both increased production of ROS and diminished antioxidant activity.
HIV-1 proteins are encoded by 9 genes located within the virion capsid. Three of these genes, gag, pol, and env, are found in all retroviruses and are vital to the structure of HIV-1. For example, the gag and pol regions encode for the reverse transcriptase and integrase enzymes necessary for efficient HIV-infection and replication. The env gene encodes for gp160, the precursor for the envelope proteins gp120 and gp41, which are necessary for virus entry into cells. The 6 remaining “accessory” genes are unique to HIV-1. Two of these, tat and rev, perform a regulatory function and are essential for viral replication [74]. However, the roles of HIV-1 genes, vpr, vpu, vif, and nef are less fully understood [75]. The following section will focus on the role of HIV-1 proteins that have been shown to affect ROS levels – Tat, Nef, gp120, and Vpr.
Tat
Of all the HIV-1 proteins, the early viral protein, Tat is the most widely studied. Composed of 86–101 amino acids, Tat serves as a transcriptional transactivator of viral gene expression by binding to a transactivation-responsive region in the HIV long terminal repeat (LTR). The expression of Tat is critical for productive HIV infection, as Tat-deficient viruses are non-infectious. In HIV-1 patients, Tat can be secreted from infected T cells and monocytes [76] and following its release, circulates in the bloodstream. From the circulation, Tat enters uninfected cells [76–79] and alters cellular physiology by positively or negatively affecting gene expression. In 1995, Westendorp et al reported plasma Tat levels between 1–3 ng/mL in HIV-infected patients [80]. More recently, however, Tat serum levels in HIV-1-infected patients were estimated to fall between 2 and 40 ng/mL [81]. It is also suggested that Tat concentrations are higher around HIV-infected perivascular cells and in the proximity of endothelial cells [82]. This effect is thought to occur because macrophages and monocytes act as viral reservoirs and secrete Tat as well as cytokines and oxidants near endothelial cells.
Extensive research demonstrates that HIV-1 Tat increases ROS levels and decreases antioxidant levels. For example, Tat causes a dose-dependent increase of ROS in cultured brain microvascular cells [83] and significantly induces ROS production and lipid peroxidation in rat brain endothelial cells [84]. In the HIV indicator (HeLa-CD4-LTR-B-gal), or MAGI cells, transfection with a Tat-expressing plasmid for 48 hours significantly increases ROS levels and reduces intracellular GSH levels by 50%. This study also showed that the Tat-induced alterations are reversed by pretreatment with the antioxidant, N-acetylcysteine (NAC) [85]. Murine fibroblasts expressing the full-length HIV-1 Tat protein exhibit a similar 50% decrease in cellular GSH concentrations [86]. Additionally, in vivo studies demonstrate that the intravenous injection of Tat protein decreases mouse brain GSH levels by 85% [87]. Tat is also shown to affect other cellular antioxidant enzymes as Tat over-expression in HeLa cells results in a 3-fold reduction in the glutathione peroxidase (GPx) mRNA ratio as well as a 2.5 fold decrease in GPx activity [88]. Moreover, HeLa cells stably producing the Tat protein express 48% less SOD2 compared to control cells [80, 89], which may be caused by Tat-induced disruption of Sp1 and Sp3 binding in the SOD2 basal promoter [90]
These studies demonstrate that Tat alters cellular ROS and antioxidant regulation. Yet, the exact mechanism and source of Tat-induced oxidative stress remain unclear. Recent studies, however, have demonstrated Tat-induced activation of several ROS-producing enzymes. For example, Gu et al showed that Tat acutely increases intracellular oxidant levels in ECV-304 cells. This Tat-induced oxidant activity is decreased by pretreatment with two NADPH oxidase inhibitors, DPI and apocynin [91]. Co-culture of human umbilical vein endothelial cells with HeLa-Tat cells also significantly induces endothelial H2O2 production via Nox 4 activation [92]. These studies implicate NADPH oxidases as potential mediators of Tat-induced ROS. However, other oxidases may contribute to HIV-induced ROS release as DPI and apocynin are somewhat nonspecific inhibitors.
These studies provide remarkable evidence of the independent effects of Tat on ROS levels and antioxidant availability. Recent studies examining novel anti-AIDS therapies have attempted to target Tat activity and binding. However, the available data regarding the effectiveness of these agents is controversial. Although further investigation is needed to better understand the mechanism underlying its effects, Tat clearly alters cellular function and may likely contributes to the vascular dysfunction and disease associated with HIV-1 infection.
Nef
Several studies implicate the HIV-1 protein Nef as a potential mediator in HIV-induced oxidative stress and vascular disease. Nef, “the negative factor” is an HIV viral accessory protein with a molecular weight ranging between 27–34 kDa. Although normally found within the cytoplasm, Nef associates with the cellular membrane upon activation via myristoylation. Nef expression has been shown to down-regulate the cell-surface levels of both CD4 and MHC-1 molecules. It also interferes with numerous intracellular pathways, leading to the dysregulation of cellular signaling and activation [93]. In vitro studies indicate that Nef influences HIV-1 pathogenesis through its ability to increase viral replication and infectivity in primary lymphocytes and macrophages. In addition, in vivo studies show that Nef is essential for high virus replication and disease progression to AIDS in HIV-infected individuals [94].
Research investigating ROS elevations in response to Nef is limited. In 2002, it was shown that Nef protein expression does not independently induce microglial NADPH oxidase [95]. However, Nef significantly enhanced superoxide release by NADPH oxidase following challenge with the calcium ionophore, formyl peptide or lipopolysaccharide (LPS) [95]. Other studies reveal that Nef regulates superoxide production in a biphasic manner. Research by Olivetta et al demonstrates that human monoblastic cells (U937) stably transfected with a vector expressing a Nef-ER fusion protein produce greater ROS than controls at 1- and 4- hours post-transfection. However, Nef-expressing cells exhibit a complete ablation in ROS production at later time points. In more recent studies, exposure of neutrophils from healthy donors to Nef for one hour increased superoxide production. DPI administration significantly reduced the Nef-induced superoxide production, implicating activation of a flavin-containing enzyme. Also, studies performed with neutrophil cellular lysates demonstrate that Nef associates with p22-phox, but not any other NADPH oxidase subunits [96]. Similarly, in ex vivo studies, exposure of porcine pulmonary arteries or pulmonary artery endothelial cells (HPAEC) to Nef markedly increases superoxide release by 54% and 70%, respectively. In addition to these effects on ROS, Nef also concomitantly decreased eNOS expression and NO production in porcine arterial rings and HPAEC [97].
Collectively, these reports implicate Nef as a mediator of HIV-induced ROS and vascular dysfunction. Although in vitro data suggest a potential cell-type dependent effect, ex vivo studies underscore the potential physiological relevance of Nef in HIV-related vascular disease. Moreover, studies performed by the Flores group demonstrate that HIV-1 Nef contributes to HIV-associated PH by promoting vascular remodeling [98–100]. Altogether, HIV-1 Nef may contribute to the vascular dysfunction documented in the HIV-1 population via increased ROS production and effects on the nitric oxide synthase pathway.
gp120
The HIV-1 protein, gp120 also promotes ROS production. The envelope glycoprotein gp120 is expressed on the surface of HIV-1 virions and facilitates the receptor binding and subsequent membrane fusion required for HIV-1 infection [101]. In addition, soluble gp120, estimated to exist between 12–92 ng/mL in the serum of HIV-1 patients [102], can be shed from virus particles or infected cells into the circulation. As a result, gp120 can cause extensive cellular damage by stimulating oxidative stress pathways [103, 104], inflammatory cytokine release [105], apoptosis [106, 107], and tight junction injury [108, 109].
Several studies indicate that gp120 stimulate ROS release in numerous cell types. For example, exposure to gp120 for 24 hours causes an almost 6-fold elevation in MDA levels in astroglial cell homogenates. This effect was significantly antagonized by pretreatment with the antioxidant, NAC [110]. gp120 exposure also induces marked increases in human retinal epithelial cell MDA and NO production, as well as inducible nitric oxide synthase expression over a 72-hour interval when compared to untreated controls [111]. Additionally, gp120 induces marked staining for HNE, an indicator of lipid peroxidation, in cells expressing the endothelial cell marker, CD31. These increases in ROS production were associated with elevations in MMP-9, and gene delivery of the antioxidant enzymes GPx and SOD1 returned MMP-9 to control levels [112]. Research also demonstrates that low concentrations of gp120 promote ROS release. Recombinant gp120 at a concentration of 340 nM increases ROS production in human monocyte-derived macrophages [113]. Also, gp120 concentration of 40 nM was also shown to increase intracellular H2O2 in lymphoid cells [114]. Picomolar concentrations of gp120 induce ROS release in U937 cells, whereas co-administration of catalase and SOD decreased the gp120-induced oxidative damage by 81% [115]. Also, in vivo studies also established that injecting 500 ng of gp120 significantly increases MDA levels.
In addition to the increases in ROS, gp120 has also been shown to alter antioxidant regulation. Seventy two hours of gp120 exposure significantly decreases the mRNA expression of the Nrf2 transcription factor in the L2 epithelial cell line [116]. The decrease in Nrf2 mRNA expression, however, did not produce a significant reduction in Nrf2 protein expression. Moreover, HIV-1 transgene expression in rat alveolar epithelial cells produced a 30% attenuation in Nrf2 mRNA expression [116]. Overall, these data suggest a role of gp120 in ROS release. In addition, the effects of gp120 on Nrf2 expression may act as a possible mechanism underlying HIV-induced antioxidant depletion.
Vpr
HIV-1 Viral Protein R, or Vpr is a 14 kDa HIV-1 accessory protein that is highly conserved in HIV-1 and simian immunodeficiency virus [117]. In addition to the HIV-1 proteins Tat and gp120, Vpr is linked to several pathways associated with HIV-1 infection and replication as well as cellular function. Studies show that Vpr transactivates long terminal repeat (LTR), promotes cell cycle arrest, induces DNA damage and apoptosis, and regulates nuclear factor-kappaB (NF-kB) activity [118]. Moreover, when exogenously administered, Vpr induces viral reproduction in latently infected cells [119]. Research also indicates that Vpr is present in the plasma of infected patients at nanomolar concentrations [120, 121].
Recent studies conducted by Deshmane showed that infection with a Vpr-expressing adenovirus induces a 4-fold elevation in H2O2 generation and an approximate 15-fold increase in ROS production in the mitochondrial compartments of infected microglial cells. These effects are consistent with results showing that infection with the JR-FL strain of HIV-1 generates very similar increases in microglia H2O2 release following 24-hours of infection [122]. Studies performed using human monocyte-derived macrophages also demonstrate that recombinant Vpr administration induces mitochondrial dysfunction, release of the pro-inflammatory cytokine IL-6, and oxidized phosphatidylcholine generation. These effects were significantly inhibited by the administration of the antioxidant, NAC [123].
Collectively, these data confirm that HIV-1 proteins significantly contribute to HIV-induced ROS generation and reduce antioxidant activity. While it is unclear whether the concentrations of HIV-1 proteins tested in vitro are physiologically relevant or appropriately effective to induce these changes, it is clear that specific HIV-1 proteins significantly impact ROS levels. Research indicates that HIV-infected monocytes and macrophages serve as reservoirs for HIV. Whether these reservoirs have the ability to continuously secrete HIV-1 proteins into the bloodstream remains controversial and more studies are needed to determine the effect of HIV-1 HAART on circulating HIV-1 proteins.
Increased ROS - Contribution of Antiretroviral Therapies
Numerous studies implicate antiretrovirals (ARVs) as a major contributor to the increased ROS levels documented in HIV-infected patients. ARVs are divided into 5 major classes: the nucleoside reverse transcriptase inhibitors (NRTIs), non-nucleoside reverse transcriptase inhibitors (NNRTIs), protease inhibitors (PIs), integrase inhibitors, and entry inhibitors [124]. In order to better understand the effects of ARVs on ROS, numerous investigators have used both in vitro and in vivo models to examine individual and combined HIV-1 therapies. Similar to clinical studies, the effects of ARVs on ROS were dependent on the cell studied, drugs examined, and duration of treatment. The following section summarizes the effects of ARVs such as NRTI and/or PI on ROS generation and how these agents may induce these alterations.
Nucleoside Reverse Transcriptase Inhibitors (NRTIs)
NRTIs were the first drugs approved for the treatment of HIV-1 (in 1987) and constitute the majority of prescribed ARVs [124]. NRTIs block HIV-1 replication by inhibiting the action of reverse transcriptase, the enzyme necessary for production of the viral double-stranded DNA, which is subsequently integrated into the genetic material of infected cells. The antiviral activity of the nucleoside analogues requires phosphorylation to their respective tri-phosphorylated moieties by a variety of nucleoside- and nucleotide-specific host cell kinases. NRTIs are well-documented to have adverse clinical toxicities which include lactic acidosis and hyperlactatemia, peripheral neuropathy, skeletal myopathy, cardiomyopathy, pancreatitis, and lipodystrophy [125]. Toxic effects of NRTIs are commonly ascribed to competitive inhibition with endogenous nucleotides for cellular mitochondrial DNA polymerase-γ (pol-γ), the sole enzyme responsible for mitochondrial DNA (mtDNA) replication and repair [128]. However, mounting evidence indicates that the effects of NRTIs are not exclusively due to actions on pol-γ but may rely on ARV-induced ROS release and antioxidant dysregulation. This evidence is discussed below.
In a study assessing the effect of NRTIs on oxidative stress, HIV-1 patients receiving ARV therapies demonstrated significantly greater plasma F2 isoprostane concentrations when compared to untreated HIV-infected subjects [126]. In a more recent study, 6 months of ARV treatment with stavudine (d4T), lamivudine (3TC), and the NNRTI, nevirapine (NEV) in HIV seropositive patients reduced serum GSH and increased MDA levels compared to non-treated HIV-infected subjects [127]. HIV-1 HAART-treated subjects also show significant elevations in serum GPx as well as a reduction in glutathione reductase (GR) following 12 months of HAART treatment compared to baseline levels [128]. Furthermore, although glutathione disulfide (GSSG) concentrations were similar among baseline, 6-, and 12-month time points, GSH levels in the epithelial lining fluid of asymptomatic HIV-1 patients are dramatically decreased following 6 months of ARV, though CD4+ lymphocyte counts are only moderately altered [129].
In addition, long-term exposure to NRTIs promotes endothelial ROS production. Bovine aortic endothelial cells (BAEC) exposed to 1 μM azidothymidine (AZT) for 14 days produce 30% greater amounts of superoxide than untreated controls [130]. In 2009, our group showed that 3- and 5-weeks of AZT exposure concomitantly decrease human aortic endothelial cell (HAEC) GSH levels and increase both total intracellular and mitochondrial superoxide production. Interestingly, d4T had no significant effect on HAEC levels of GSH or superoxide, emphasizing that different NRTIs may have divergent effects on ROS levels [131].
Animal studies investigating the effects of NRTIs on oxidative stress make similar conclusions. Mice administered 100mg/kg of AZT for 5 days display 50–60% reductions in aortic aconitase activity, a marker for ROS production due to its high susceptibility to inactivation by ROS [132]. Aortas from mice treated with AZT for 35 days show marked increases in superoxide staining when compared to vehicle-treated controls [130]. AZT administration for 8 months markedly reduces plasma and heart homogenate antioxidant levels, increases gp91phox expression, and stimulates membrane-associated p47phox levels in Wistar-Kyoto rats. These AZT-induced effects were attenuated by treatment with vitamin C, further supporting a ROS contribution [133].
Numerous studies demonstrate mitochondrial-specific ROS generation in response to NRTIs. Since mitochondria serve as the primary intracellular site of oxygen reduction, they have the greatest potential for ROS formation and are highly susceptible to ROS toxicity. Extensive research demonstrates that exposure to the NRTI AZT induces significant mitochondrial dysfunction and ROS production in a variety of cell types including endothelial cells, cardiomyocytes, and lymphoid cells [134, 135]. AZT concentrations up to 10 μmol mg−1 induce an approximate 3-fold increase in H2O2 production in mitochondria from rat liver. However, AZT co-administration with mildronate, a drug shown to protect mitochondrial membranes from damage by free fatty acids and prevent NF-κB activation in cardiac tissues, reduces H2O2 production from rat liver mitochondria by 55% [136]. Treatment with AZT has also been shown to significantly elevate mitochondrial ROS generation in human primary cardiomyocytes. These studies show that increases in AZT-induced mitochondrial ROS stimulate caspase-3 and -7 activation as well as PARP-1-mediated cell death pathways [137]. Studies in cultured fibroblasts also demonstrate that the NRTIs, d4T, and AZT induce mitochondrial dysfunction and elevate ROS generation. AZT and d4T increase fibroblast mitochondrial mass by 3- to 4-fold when assessed by Mitotracker Red fluorescence. AZT and d4T administration also significantly increases fibroblast ROS production when compared to untreated cells. Exposure to the NRTIS abacavir, lamivudine, didanosine, or tenofovir produced no change in fibroblast mitochondrial mass or ROS production [138]. Furthermore, 40 nM concentrations of the NRTI, 2′, 3′-dideoxycytidine (ddC) induces significant increases in isolated mitochondria protein oxidation levels after 6 hours, but similar concentrations of ddC fail to affect mitochondrial lipid peroxidation levels [139]. In an in vivo study of AZT-induced oxidative stress, Kohler demonstrated that daily AZT administration induces significant increases in heart mitochondrial H2O2 and reduced cardiac aconitase activity in SOD2 knockout mice, however, transgenic mice over-expressing SOD2 or mitochondrially-targeted catalase demonstrate no alterations in heart mitochondrial H2O2 release following AZT administration [140]. This suggests that mitochondria-specific ROS generation and antioxidant depletion both play an important role in AZT-induced oxidative stress.
At least In the case of AZT, data suggest that the pol-γ effect is not the sole mechanism for ROS generation. Data indicate that AZT and AZT-MP directly interact with Complex I of the mitochondrial electron transport chain and prevent its cAMP-dependent phosphorylation independently of pol-γ inhibition [141]. In addition, researchers have found that AZT and its azido-containing derivatives (AZT-MP, AZT-TP, and glucoronidated-AZT) have direct pro-oxidant activities in an in vitro cell-free chemical system, while non-azido-containing derivatives do not [142]. Furthermore, AZT and AZT-MP induce lipid peroxidation in membrane preparations devoid of cellular constituents and presumably most secondary targets [143]. These findings suggest that a secondary target likely contributes to the pro-oxidative properties of NRTIs.
Non-nucleoside Reverse Transcriptase Inhibitors (NNRTIs)
NNRTIs (Efavirenz, Nevirapine and Delavirdine) are non-competitive inhibitors of reverse transcriptase, and, in contrast to NRTIs, do not require phosphorylation to achieve their anti-viral effects. The first drug of this class was approved by the FDA in 1996. Data indicate that NNRTIs likely cause any significant effect on ROS generation alone. However, since the efficacy of NNRTI drugs is impaired by rapid emergence of drug-resistance mutations, NNRTIs are often prescribed with agents of other ARV classes. The high-level resistance to NNRTIs arises due to the development of point mutations within the allosteric binding site of the reverse transcriptase [144]. Since NNRTIs share similar binding sites, mutations commonly cause cross-class resistance [145]. Although second generation NNRTIs, such as etravirine and rilpivirine, hold great promise for their resistance to mutations and possible use in ARV naïve patients, further investigation is needed to examine their effects on ROS and antioxidant levels.
Protease Inhibitors (PI)
PIs such as, saquinavir, ritonavir, indinavir, lopinavir, nelfinavir, amprenavir, and darunavir were first approved for HIV-1 therapy in 1995. These agents prevent HIV-1 replication by inhibiting the cleavage of viral polypeptides into mature and functional proteins [124]. In vitro studies examining PIs demonstrate significant increases in oxidative stress in endothelial cells [146, 147], adipocytes [148, 149], and macrophages [150]. For example, PIs, either alone or as part of a HAART regimen, increase the generation of ROS in human aortic endothelial cells [151]. The PIs ritonavir and/or lopinavir markedly increase ROS production in human coronary artery endothelial cells after 30 days of exposure. However, NAC or MnTBAP administration during the final 24 hours of the 30-day exposure dramatically reduced PI-induced ROS [152]. Studies also report very similar effects in other in vitro models. The PI nelfinavir, but not saquinavir or atazanavir, increases ROS production in the rat pancreatic insulinoma cell line, INS-1, after 24 hours of exposure. Nelfinavir also decreases GSH content, as well as SOD1 expression and activity [153]. Additionally, macrophage-derived foam cells exposed to RTV produce significantly greater amounts of superoxide than untreated controls [154].
In ex vivo studies, endothelial cells from coronary vessels also demonstrate decreased eNOS mRNA levels following RTV, APV, and saquinavir (SQV) exposure. PIs are shown to produce similar effects in porcine coronary arteries as exposure to ritonavir (RTV) and amprenavir (APV) increases superoxide levels in vessel endothelial layers by 47% and 52%, respectively, when compared to untreated controls. Vessel treatment with RTV and ATV also reduces nitrite release by approximately one-third when compared to controls. These alterations likely contribute to the altered endothelial-dependent relaxation in vessel rings treated with 15 μM concentrations of RTV, ATV, and SQV [155]. Interestingly, in later studies, treatment with curcumin [156] and the ginsenosides, Rb1, Rc, and Re [157] reversed the ritonavir-induced effects on superoxide generation, eNOS expression, nitrite release, and endothelial-dependent relaxation.
In addition to the independent effects of NRTI and PI on ROS levels, several studies have examined the combined effect of NRTIs and PIs on cellular ROS regulation. Exposure to the HAART drug combination of azidothymidine (AZT), a NRTI, and indinavir (IDV), a PI, for 48-hours dose dependently increases ROS production in human brain microvascular endothelial cells (hBMEC). Moreover, 72-hours of AZT and IDV exposure dose-dependently reduce endothelial intracellular GSH levels and increase the lipid peroxidation metabolite, MDA [158].
Other ARVs
Integrase inhibitors (i.e. raltegravir), fusion inhibitors, and entry inhibitors (i.e. enfuvirtide, maraviroc) are relatively new to HIV-1 therapy and are typically given to HAART-experienced patients demonstrating therapeutic failure. The effects of these newer agents on ROS are currently unknown. Future studies may provide information regarding the effect of these agents on ROS release and/or antioxidant activity.
Overall, these data demonstrate how ARVs increase ROS and ROS-mediated effects. More importantly, however, the data demonstrates how the effects of HIV-1 and ARV on ROS production may have combined effects on the development and progression of HIV-induced vascular disorders.
Reactive Oxygen Species – Sources and Regulation
Research demonstrates that HIV-1 and HIV-1 ART severely alters ROS sources and regulation pathways, creating an environment of oxidative stress. These data underscore the possible role of oxidative stress in HIV-1 pathogenesis and the development of HIV-associated vascular pathologies. ROS, such as superoxide, hydrogen peroxide (H2O2), and hydroxyl radical (HO·) as well as the reactive nitrogen species nitric oxide (NO) and peroxynitrite (ONOO−) are biologically active species known to play important roles in vascular biology via redox signaling pathways [159–161]. These oxidants are produced by numerous sources such as the NADPH oxidases (Noxes), xanthine oxidase, cytochrome P450, uncoupled endothelial nitric oxide synthase (eNOS) and as byproducts of the mitochondrial respiratory chain [162]. To balance ROS levels and combat their toxic effects, cells employ several antioxidant enzymes including the superoxide dismutases (SOD), catalase, glutathione peroxidase (GPx), thioredoxins (Trx), and peroxiredoxins (Prx). Non-enzymatic antioxidant mechanisms also exist, including the vitamins E and C as well as glutathione, which acts as a reducing substrate for glutathione peroxidase [163]. These antioxidant systems are localized throughout the cell and function either in an independent or complementary manner to scavenge ROS (Figure 1). Overall, the balance between ROS generation and antioxidants is essential for normal cell function. However, research demonstrates marked alterations in ROS and antioxidant levels in HIV-1 patients. These data suggest a potential correlation between HIV-1 and oxidative stress, and raise the question of whether the oxidant/antioxidant imbalance found in HIV-1 patients contributes to the characteristic pathologies associated with this population.
Increased ROS in Vascular Disorders
Although cells utilize a variety of mechanisms to regulate ROS generation and inactivation, ROS are essential for normal vascular function and act as key second messengers in numerous signal transduction pathways [164–166]. Moreover, ROS levels regulate the activity of important transcription factors implicated in vascular function including NF-κB, activator protein-1 (AP-1), and HIF-1α [167–169]. Therefore, the excess generation of and/or the reduced ability to remove ROS can lead to detrimental effects such as dysregulated apoptotic or proliferative states, vascular smooth muscle cell migration and endothelial dysfunction. For example, low concentrations of H2O2 induce cellular proliferation, whereas high concentrations promote apoptosis and cell cycle arrest [170]. ROS also contribute to TNF-α-induced activation of endothelial apoptosis [171] and stimulate vascular smooth muscle cell migration by modulating the matrix metalloproteinases (MMP) -2 and -9 [172]. Furthermore, superoxide can cause endothelial dysfunction by combining with nitric oxide to produce the highly reactive radical, peroxynitrite (ONOO-) and decrease NO levels [173]. Peroxynitrite is then able to oxidize the essential eNOS cofactor, tetrahydrobiopterin (BH4), stimulating eNOS uncoupling and further contributing to endothelial dysfunction by increasing superoxide and reducing NO availability [174]. ROS-induced endothelial dysfunction is pivotal to vascular injury and the inflammatory response and endothelial dysfunction is known to be an early predictor of cardiovascular events in patients without [175] and with known vascular disease [176, 177]. The extensive effects of ROS on the vessel wall support a role for ROS in the development of numerous vascular disorders including PH and atherosclerosis. For example, increased superoxide production has been observed in experimental models of PH [178] and biomarkers of oxidative stress are elevated in PH patients [179]. PH patients exhibit low NO levels in their exhaled breath [180, 181], which suggests that scavenging by superoxide radicals may mediate the reductions in NO bioactivity [182]. Moreover, superoxide regulates characteristic PH pathologies such as modulating pulmonary vasoconstriction [183] and stimulating pulmonary smooth muscle cell proliferation [184]. Superoxide and other oxygen radicals also promote atherosclerosis by altering NO and activating redox-sensitive pathways that mediate vessel remodeling and plaque stability [185]. In addition, coronary arteries from CAD patients express greater levels of the NADPH oxidase subunits, p22phox, p67phox, and p47phox [186] and produce significantly larger amounts of superoxide [187].
In vivo studies demonstrate that apoE (-/-) mice deficient in NADPH oxidases have attenuated atherosclerosis due to decreased superoxide release via deficient NADPH oxidases [188]. Conversely, an increased production of ROS in vascular endothelial cells is implicated in the oxidation of LDL and the expression of ROS-sensitive inflammatory genes such as VCAM-1 [189]. For example, the overexpression of extracellular SOD suppresses endothelial cell-mediated LDL oxidation [190] and vitamin E treatment reduces endothelial VCAM-1 and ICAM-1 expression induced by oxidized LDL [191]. Furthermore, ROS increase monocyte chemotactic protein-1 [192, 193] and mediate endothelial monocyte adhesion and infiltration [194].
Oxidative stress also enhances vessel inflammation by stimulating the release of pro-inflammatory cytokines such as endothelin-1 (ET-1) and interleukin-6 (IL-6). Patients with atherosclerosis demonstrate elevations in plasma levels [195] and immunoreactive staining for ET-1 in the vasculature [196]. These increases in ET-1 and IL-6 [74, 197–199] are implicated in PH development. ROS mediate the secretion of ET-1 from endothelial cells [200, 201] and polymorphonuclear leukocytes [202]. Also, the Tat-induced release of IL-6 from macrophages is attenuated by NADPH oxidase inhibition [203]. Antioxidant administration, however, inhibits the IL-6 and ET-1 release in healthy volunteers [204], as well as, ET-1-induced release of IL-6 from vascular smooth muscle cells [205]. Altogether, ROS and redox-sensitive pathways greatly contribute to vessel injury and vascular disease pathogenesis which further support a role for ROS in HIV-1 pathogenesis and HIV-associated vascular disease development.
ROS, HIV-1 Infection and Replication
Elevated ROS can further potentiate the effects of HIV-1 infection because oxidative stress plays a crucial role in viral replication, inflammatory response, immune cell proliferation, loss of immune function, and increased sensitivity to drug toxicity [206–208]. While it remains largely unknown how HIV-1 promotes oxidative stress, research demonstrates that HIV-1 increases in the activity of ROS sources, and reductions in cellular antioxidant capacity lead to enhanced viral replication. For example, H2O2 stimulates activation of the HIV-1 LTR in patients with latent HIV-1 [209]. In vitro studies also show that lowering intracellular GSH levels decreases cell survival [210], increases HIV replication, and increases NF-κB activation [211, 212]. HIV replication is associated with an increase in MDA levels and nitrotyrosine formation, which are both recognized as indicators of lipid peroxidation from free radical overproduction. Therefore, the impact of HIV-induced ROS on replication can result in a further insult to the vascular wall. In addition, treatments that increase oxidative stress are also known to activate HIV-1 replication [213], which appears to be mediated by ROS-induced activation of NF-κB [214].
Conversely, antioxidants suppress HIV replication in chronically infected cells [215–217]. For example, antioxidant molecules such as GSH, glutathione ester, and NAC are able to suppress HIV expression in infected monocytic cells [218, 219]. Exogenous GSH administration also suppresses the release of virus particles and HIV-1 proteins from chronically infected cells [220]. Similarly, MnTBAP, a synthetic peroxynitrite decomposition catalyst, demonstrates antiviral activity in both acutely- and chronically-infected monocytes/macrophages [68]. The NAC pro-drug, I-152, and beta-mercaptoethylamine (MEA) increase intracellular GSH in human MDMs and significantly decrease viral replication in HIV-infected MDMs [221]. Moreover, infection of human primary monocytes is blocked or substantially reduced by GSH or NAC treatment. GSH and NAC administration reduce the amount of virus released by infected cells by 90% [222] as well as viral replication and disease progression in murine AIDS [223, 224]. In vivo studies demonstrate that the combination of GSH-loaded erythrocytes with AZT and DDI treatments significantly decreases proviral DNA content following 10 weeks of infection in bone marrow macrophages and brain samples [225].
Cellular redox state may also negatively affect HIV-1 gene expression and transcription by altering HIV-1 Tat internalization and function. Previous studies demonstrate that the oxidation state of the cysteine-rich region of Tat strongly influences its capacity to enter cells [226]. In addition, more recent studies indicate that the redox state of Tat alters protein uptake by macrophages and promotes protein aggregation thereby hindering hinders Tat biological activity [227]. Research also indicates that cellular redox state regulates Tat transactivation [228]. Normally, Tat stimulates transcriptional elongation from the viral LTR through a specific interaction with a 59-residue stem-loop RNA known as the transactivation-response element (TAR). Recent studies demonstrate that 1 hour of NAC exposure reduces Tat-induced HIV-1 LTR transactivation in MAGI cells by over 60% [85]. Selenium administration also inhibits Tat-dependent LTR activity in human MDM and in U937 cells [229].
Contributing and Alternative Mechanisms
While oxidative stress can directly impact vascular function, the effects of HIV-1 on the vasculature are likely multi-factorial and may include HIV-mediated chronic immune activation and inflammation. Chronic immune activation is a hallmark of HIV-1 and is associated with the depletion of CD4+ lymphocytes, elevations in lipopolysaccharide levels, proinflammatory cytokine release, deregulation of hematopoiesis pathways, CD8+ T cell activation and apoptosis, as well as accelerated immunosenescence [230]. Although these events provide a functional role in HIV-1 pathogenesis, they are also linked to the development of vascular complications. For example, HIV-1 infection increases the release of the toll-like receptor ligand, LPS. Whereas the injection of LPS alone is insufficient to cause atherosclerosis [231], it is able to exacerbate atherosclerosis-associated pathologies [232, 233]. Also, mice deficient in toll-like receptor 4 are resistant to chronic hypoxia-induced PH development [234]. Furthermore, elevations in plasma LPS levels stimulate the release of numerous pro-inflammatory cytokines such TNF-α, IL-6, and IL-1β from peripheral macrophages and dendritic cells. Abnormal levels of these cytokines have detrimental effects throughout the vasculature and are implicated in the development and progression of atherosclerosis [235–238] and PH [198, 199, 239–241]. Chronic immune activation in HIV-1 patients is also associated with a deregulation of hematopoiesis, leading to reduced numbers of progenitor cells and a decline in the generation of new cells [242–244]. This phenomenon may mediate PH development, as the direct injection of endothelial progenitor cells exhibits therapeutic effects in experimental models of PH [245, 246]. In addition, abnormalities in CD8+ T-cell function may also contribute to the increased susceptibility of HIV-1 patients to develop PH [247, 248] and atherosclerosis [249].
HIV-induced ROS may also alter the innate immune response of infected patients. Increased ROS release is shown to mediate both the effector and induction phases of the immune response. It is well established that immune cells generate ROS causing deleterious effects to nearby tissues and cells during immune activation. However, ROS may also regulate the immune response by altering key signaling pathways in antigen-presenting cells (APC). Research demonstrates that GSH depletion triggers a Th2 response pattern [250, 251]. These studies indicate that the redox state of APC not only regulate T-cell activation but may also suppress the T helper (Th1) immune response and induce the Th2 response. This Th2 response pattern is associated with significant increases in IL-4 and IL-10 production and the stimulation of various antibody responses which can impact vascular cell responses [252].
Additionally, the physiological declines seen in HIV-1 patients show incredible similarities to those noted in the uninfected elderly population, such as dementia, unintentional weight loss, and fatigue [253, 254]. These alterations suggest that HIV-1 infection may induce an accelerated aging phenotype in seropositive patients, potentially contributing to the hallmark immune deficiency of HIV-1 patients and increased occurrence of vascular disorders. Furthermore, other factors may mediate the oxidant/antioxidant alterations noted in HIV-1 patients. HIV-1 patients often suffer from co-morbidities such as alcohol and drug abuse, malnutrition, and nicotine addiction. In addition, ART induces hyperlipidemia, hyperglycemia, insulin resistance, and central obesity, which may also increase ROS levels [255]. Although these factors may prove difficult to account for in clinical and experimental studies, they may modulate the dysregulation of ROS production and antioxidant availability documented in HIV-1 patients.
Potential Therapies
Nutritional Antioxidants
HIV-1 reduces levels of plasma antioxidants [256], such as ascorbate, or vitamin C [257] and these decreases in antioxidant concentrations persist in HIV-1 patients although their dietary intake is sufficient for healthy individuals [258]. Also, low intakes of vitamin C [259] and low plasma concentrations of vitamin E [260] are associated with a greater risk of progression to AIDS in HIV-infected US subjects. These studies emphasize the importance of dietary antioxidant vitamins in HIV-1 seropositive individuals and suggest a potential therapeutic benefit for HIV-1 patients. However, research examining the effect of dietary supplementation in HIV-1 patients provides conflicting results. Multi-nutrient supplements containing vitamins C and E led to a lower risk of death due to HIV infection in Tanzanian women [261] and a small increase in CD4+ T lymphocyte counts in Kenyan women [262]. Vitamin C and E supplements were found to reduce oxidative damage and attenuate disease severity in HIV-positive Canadian adults [263]. Also, α-tocopherol, or vitamin E, (800 mg/day) administration decreased viral load in HIV-1 patients over a 60-day period [264]. In addition, research demonstrates that antioxidant molecules such as GSH, glutathione ester, and NAC are able to suppress HIV expression in infected monocytic cells [218, 219] as well as viral replication and disease progression in murine AIDS [223, 224]. These data argue that GSH administration may provide therapeutic benefit against HIV-1 replication in addition to its cardiovascular benefits.
Conversely, daily selenium administration has shown no significant effect on CD4+ cell counts or viral load in pregnant, HIV-infected women [265]. However, the results of a 9-month selenium supplementation study performed in 450 HIV-1 seropositive men and women may provide an explanation for the recent conflicting results and highlight the importance of treatment adherence. Study subjects with a selenium change less than or equal to 26.1 microgram/L, indicating poor subject compliance, were found to have an increase in HIV-1 viral load and a decrease in CD4+ lymphocyte counts after the 9-month treatment period. Conversely, subjects with an increase in serum selenium levels greater than 26.1 microgram/L demonstrate no change in viral load and increases in CD4+ cell counts [266]. The administration of alpha-lipoic acid, a glutathione-replenishing disulfide, three times daily increased total glutathione levels but failed to alter HIV RNA levels or improve CD4+ lymphocyte counts after 6 months [267]. These results of these intervention trials may not be completely attributable to the antioxidant actions of the supplements, but the data suggest that proper multi-nutrient and antioxidant supplementation may diminish the severity of HIV disease. Still, the varying outcomes of these studies underscore the need for further research in this area.
Nrf2 Activation
NF-E2 related factor 2 (Nrf2) is a ubiquitously expressed transcription factor that regulates antioxidant enzyme expression by binding to the antioxidant response element (ARE). Due to its function, the Nrf2 pathway is thought to play an essential role in cellular protection against ROS effects and oxidative stress [268]. Recent studies indicate that some vascular protective compounds act via the Nrf2 pathway. For example, resveratrol dose-dependently increases Nrf2 promoter activity and stimulates expression of Nrf2-regulated genes such as heme-oxygenase-1 in cultured primary human coronary arterial endothelial cells. Resveratrol also reduces mitochondrial and cellular ROS release following high glucose and TNF-α exposure in an Nrf2-dependent manner [269]. Additionally, Nrf2 is linked to the anti-atherogenic effects of the Ginkgo biloba extract (GBE). Studies indicate that GBE increases Nrf2 promoter activity and nuclear translocation in human aortic endothelial cells. Also, Nrf2 knockdown abolishes GBE-induced suppression of TNF-α-induced VCAM-1 expression in human aortic endothelial cells [270].
In addition, research indicates that Nrf2 activation protects vascular cells against ROS release and inflammation. For example, Nrf2 activation by sulforaphane reduces VCAM-1 signaling in human umbilical vein endothelial cells [271] and adenoviral Nrf2 overexpression prevents injury by ROS and inhibits monocyte adhesion in endothelial cells [272]. Nrf2 gene transfer via adenoviral transduction inhibits proliferation in human and rabbit smooth muscle cells. AdNrf2 also reduces inflammation and oxidized LDL accumulation following aortic balloon injury in rabbits [273]. Dh404 stimulates Nrf2 in primary cardiac myocytes and increases Nrf2 nuclear translocation and transcriptional activity in H9C2 cardiomyocytes. Although Nrf2-deficient mice display significant increases in liver MDA, Nrf2 knockdown in ApoE-deficient mice produced a decrease in aortic stiffness and a 61% reduction in plaque area after 20 weeks when compared to ApoE KO controls. Moreover, macrophages from ApoE/Nrf2 deficient mice demonstrate a reduced uptake of AcLDL, a commonly used indicator for OxLDL [274] and the Nrf2 activator, Dh404 inhibits basal and Angiotensin II-induced superoxide and peroxynitrite formation [275]. Also, treatment with the Nrf2 activator, CDDO-Imidazolide (CDDO-Im) prevents cigarette smoke-induced increases in RV pressures and alterations in RV diastolic and systolic functions and increases GSH levels and attenuates alveolar apoptosis and pulmonary oxidative damage [276]. Interestingly, numerous clinical trials are currently investigating the effect of Nrf2 activation and sulforaphane treatment in numerous diseases including cystic fibrosis, COPD, asthma, cancer, autism, and cardiovascular disease. As such, Nrf2 activation may serve as a therapeutic for HIV-associated vascular disorders.
Targeting Specific Antioxidant Pathways
Although extensive research implicates oxidative mechanisms in the pathogenesis of vascular diseases, studies using antioxidants as a major disease therapy produce controversial results regarding disease protection and reversal [277–280]. These outcomes, as well as recent advances in ROS detection, have led researchers to redefine the concept of oxidative stress and direct more attention to the balance of redox signaling, particularly the major thiol/disulfide couples such as glutathione (GSH)/glutathione disulfide (GSSG), reduced thioredoxin [Trx-(SH)2]/oxidized thioredoxin (Trx-SS), and cysteine (Cys)/cystine (CySS) [281]. These redox couples regulate numerous biological functions within the cell and evidence suggests that specific redox states may work together to perform distinct functions in various cellular locations [282].
Interestingly, the cellular localization of these redox couples and the resulting biological events may have considerable consequences on vascular function and hence, the development and progression of vascular diseases. For example, the expression and DNA-binding of redox-sensitive transcription factors, including NF-κB and AP-1, may be severely altered in response to modulations in nuclear redox states. Moreover, mitochondrial redox states may regulate mitochondrial permeability transition and thereby, trigger cellular necrosis or apoptosis. Differences in redox states, particularly those across the plasma membrane, are implicated in cell proliferation as well as monocyte adhesion to endothelial cells [283]. Variations in extracellular Cys/CySS redox states and those found in human plasma also enhance oxidant-induced apoptosis and mediate decreases in cell number [284]. Altogether, these data encourage researchers to obtain a better understanding of redox signaling to allow for more effective antioxidant treatments.
Peroxisome Proliferator-Activated Receptors (PPAR) Agonists
Agents that restore NO levels and reduce ROS generation would likely have a favorable impact on HIV-mediated vascular disease. Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors belonging to the nuclear hormone receptor superfamily [285]. PPARs regulate a variety of physiological processes ranging from lipogenesis to inflammation, and have been implicated in numerous disorders including diabetes and atherosclerosis. There are 3 PPAR isotypes: PPARα, PPAR β/δ, and PPARγ. These PPARs are expressed in multiple body tissues including the heart and vasculature. Of particular interest to vascular disorders is PPARγ, which regulates genes involved in characteristic vessel pathologies, such as cellular differentiation and growth, inflammation, ROS regulation, apoptosis, and angiogenesis.
Research shows that PPARγ agonists and PPARγ overexpression increase endothelial NO release [286]. PPARγ agonists also decrease the expression of Nox-2 and -4 and significantly stimulate Cu/Zn SOD expression and activity in human umbilical vein endothelial cells [287]. Reduced endothelial PPARγ expression attenuates NO production and produces significant increases in serum d-ROMs, derivatives of reactive oxygen metabolites [288]. Overexpression of PPARα and PPARγ reduces HIV-induced dysregulation of tight junction proteins in brain endothelial cells, effects mediated by alterations in matrix metalloproteinase [289]. Moreover, PPARγ activation via rosiglitazone administration in brain microvascular endothelial cells inhibits adhesion and transendothelial migration of HIV-1 infected monocytes [290]. The PPARγ agonist, rosiglitazone, also attenuates LPS-induced inflammation in vascular smooth muscle cells [291]. Additionally, PPARs decrease endothelial-leukocyte interactions in atherosclerosis models [292] and PPAR agonists demonstrate antiviral activity. PPAR agonists, rosiglitazone, PgJ2, ciglitazone, troglitazone and fenofibrate, inhibit HIV replication in HIV-1 infected peripheral blood mononuclear cells (PBMCs). These data indicate that decreases in PPAR activity may mediate endothelial dysfunction, ROS regulation, vascular injury, and HIV-1 replication. Although several have been unsuccessful due to patient safety concerns, PPAR agonists serve as an encouraging therapeutic for preventing HIV-associated vascular disorders.
Conclusion
In this review, we summarize how HIV- and ART-induced increases in ROS can impact vascular cell apoptosis, proliferation, migration and cytokine release signaling pathways. These data delineate how HIV-1 induced ROS may contribute to the cardiovascular events, such as atherosclerosis and PH that are observed in people living with HIV-1/AIDS. Due to the increased ROS generation and antioxidant dysregulation in HIV-1 patients, the idea of antioxidant supplementation offers promise. However, conflicting outcomes from these studies emphasize a need for an improved understanding of ROS and redox-sensitive pathways. Recent efforts have provided a fresh perspective of oxidative stress and a new focus on redox-responsive signaling and ROS compartmentalization [282].
Although data indicate that ROS play a prominent role in HIV-vascular disorders, the pathogenesis of HIV-associated vascular disorders is likely multi-factorial. An increase in ROS may serve as a “priming” mechanism preparing vascular cells for a “second hit”. HIV-1 patients often also suffer from other co-morbidities, such as cancers, as well as smoking and drug and alcohol abuse. These conditions combined with elevated ROS may play a role in the increased succeptibility of HIV-1 patients to develop vascular. It is obvious that HIV-1 infection models, either in vitro or in vivo, cannot replicate the events that occur in seropositive individuals. Having said this, further research is needed for a better understanding of these disorders which may then lead to improved therapies and preventative measures for HIV-1 mediated cardiovascular disease.
Highlights.
Vascular diseases are one of the most recognized non-AIDS events in HIV-1 patients
HIV-1 proteins and antiretrovirals impact antioxidants and superoxide production
Increased reactive oxygen species (ROS) likely promote vascular disease
ROS also potentiate HIV-1 effects by altering viral replication and immune function
Targeting ROS sources or restoring antioxidants may reduce HIV vascular disease
Acknowledgments
We would like to express our great thanks to Drs. C. Michael Hart, Erik Kline and Manu Platt for their assistance in the critique and revision of this review. We sincerely appreciate your time and effort. In addition, we thank Mr. Donn Johnson, MSMI of the Atlanta VA Medical Center Medical Media department for his amazing skill and extreme patience during the development of the review image. This work was facilitated by support from the NIH (HL070892) and the Center for AIDS Research at Emory University (P30 AI050409).
Abbreviations
- AIDS
Acquired Immunodeficiency Syndrome
- AP-1
Activator Protein-1
- APC
Antigen-presenting Cells
- APV
Amprenavir
- ARV
Antiretrovirals
- AZT
Azidothymidine
- BH4
Tetrahydrobiopterin
- Cys
Cysteine
- Cyss
Cystine
- d4T
Stavudine
- ddC
2′, 3′-dideoxycytidine
- d-ROM
Derivatives of Reactive Oxygen Metabolites
- DPI
Diphenyleneiodonium
- eNOS
endothelial Nitric Oxide Synthase
- ET-1
Endothelin-1
- FMD
Flow-mediated Dilation
- GPx
Glutathione Peroxidase
- GR
Glutathione Reductase
- GSH
Glutathione
- GSSG
Glutathione Disulfide
- H2O2
Hydrogen Peroxide
- HAART
Highly Active Antiretroviral Therapies
- HAEC
Human Aortic Endothelial Cells
- HPAEC
Human Pulmonary Artery Endothelial Cells
- HIV
Human Immunodeficiency Virus type 1
- ICAM
Intracellular Adhesion Molecule
- IL-1β
Interleukin-1beta
- IL-6
Interleukin-6
- IDV
Indinavir
- iNOS
Inducible Nitric Oxide Synthase
- LPS
Lipopolysaccharide
- MDA
Malondialdehyde
- MDM
Monocyte-derived Macrophages
- MMP
Matrix Metalloproteinase
- NAC
N-acetylcysteine
- NF-kB
Nuclear factor-kappaB
- NO
Nitric Oxide
- Nox
NADPH Oxidase
- NRTI
Nucleoside Reverse Transcriptase Inhibitors
- NNRTI
Non-Nucleoside Reverse Transcriptase Inhibitors
- ONOO−
Peroxynitrite
- PBMCs
Peripheral blood mononuclear cells
- PH
Pulmonary Hypertension
- PI
Protease Inhibitors
- PPAR
Peroxisome Proliferator-activated Receptors
- ROS
Reactive Oxygen Species
- RTV
Ritonavir
- RV
Right Ventricle
- SOD
Superoxide Dismutase
- SQV
Saquinavir
- Tg
Transgenic
- TNF-α
Tumor Necrosis Factor-alpha
- Trx
Thioredoxin
- VCAM-1
Vascular Cell Adhesion Molecule-1
Footnotes
Reviewers:
Rakesh Patel, Ph.D.: rakeshp@uab.edu; Phone: 205-975-9225
Johnny (Changyi) Chen, M.D., Ph.D.: jchen@bcm.edu; Phone: 713-798-4401
Michal Toborek, M.D.: michal.toborek@uky.edu; Phone: (859) 323-4094
Mariana Gerschenson, Ph.D.: gerschen@hawaii.edu; Phone: 808.441.1573
Sonia Flores, Ph.D.: Sonia.Flores@ucdenver.edu; Phone: (303) 315-0055
Lance Terada, M.D.: lance.terada@utsouthwestern.edu; Phone: 214-645-8300
Authors Have No Conflicts of Interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Palella FJJ, Baker RK, Moorman AC, Chmiel JS, Wood KC, Brooks JT, Holmberg SD Investigators, HOS. Mortality in the Highly Active Antiretroviral Therapy Era: Changing Causes of Death and Disease in the HIV Outpatient Study. JAIDS Journal of Acquired Immune Deficiency Syndromes. 2006;43:27–34. doi: 10.1097/1001.qai.0000233310.0000290484.0000233316. [DOI] [PubMed] [Google Scholar]
- 2.Corey DM, Kim HW, Salazar R, Illescas R, Villena J, Gutierrez L, Sanchez J, Tabet SR. Brief Report: Effectiveness of Combination Antiretroviral Therapy on Survival and Opportunistic Infections in a Developing World Setting: An Observational Cohort Study. JAIDS Journal of Acquired Immune Deficiency Syndromes. 2007;44:451–4554. doi: 10.1097/QAI.1090b1013e31802f38512. [DOI] [PubMed] [Google Scholar]
- 3.Neuhaus J, Angus B, Kowalska JD, Rosa AL, Sampson J, Wentworth D, Mocroft A. SMART, f. t. I., groups, E. s. Risk of all-cause mortality associated with nonfatal AIDS and serious non-AIDS events among adults infected with HIV. AIDS. 2010;24:697–7066. doi: 10.1097/QAD.1090b1013e3283365356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barbaro G. Cardiovascular Manifestations of HIV Infection. Circulation. 2002;106:1420–1425. doi: 10.1161/01.cir.0000031704.78200.59. [DOI] [PubMed] [Google Scholar]
- 5.Krishnaswamy G, Chi DS, Kelley JL, Sarubbi F, Smith JK, Peiris A. The cardiovascular and metabolic complications of HIV infection. Cardiol Rev. 2000;8:260–268. doi: 10.1097/00045415-200008050-00005. [DOI] [PubMed] [Google Scholar]
- 6.Hsue PY, Lo JC, Franklin A, Bolger AF, Martin JN, Deeks SG, Waters DD. Progression of Atherosclerosis as Assessed by Carotid Intima-Media Thickness in Patients With HIV Infection. Circulation. 2004;109:1603–1608. doi: 10.1161/01.CIR.0000124480.32233.8A. [DOI] [PubMed] [Google Scholar]
- 7.Hsue PY, Bolger AF, Martin JN. Pulmonary Hypertension in HIV-Infected Individuals. Clinical Infectious Diseases. 2011;53:96. doi: 10.1093/cid/cir277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crum NF, Riffenburgh RH, Wegner S, Agan BK, Tasker SA, Spooner KM, Armstrong AW, Fraser S, Wallace MR Consortium, o. B. o. t. T. A. C. Comparisons of Causes of Death and Mortality Rates Among HIV-Infected Persons: Analysis of the Pre-, Early, and Late HAART (Highly Active Antiretroviral Therapy) Eras. JAIDS Journal of Acquired Immune Deficiency Syndromes. 2006;41:194–200. doi: 10.1097/01.qai.0000179459.31562.16. 110.1097/1001.qai.0000179459.0000131562.0000179416. [DOI] [PubMed] [Google Scholar]
- 9.Seaberg EC, Benning L, Sharrett AR, Lazar JM, Hodis HN, Mack WJ, Siedner MJ, Phair JP, Kingsley LA, Kaplan RC. Association Between Human Immunodeficiency Virus Infection and Stiffness of the Common Carotid Artery. Stroke. 2010;41:2163–2170. doi: 10.1161/STROKEAHA.110.583856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guaraldi G, Zona S, Orlando G, Carli F, Ligabue G, Fiocchi F, Menozzi M, Rossi R, Modena MG, Raggi P. Human immunodeficiency virus infection is associated with accelerated atherosclerosis. Journal of Antimicrobial Chemotherapy. 2011;66:1857–1860. doi: 10.1093/jac/dkr206. [DOI] [PubMed] [Google Scholar]
- 11.Weir EK. The United States experience with the acute and chronic treatment of primary pulmonary hypertension. European Heart Journal. 1988;9:33–38. doi: 10.1093/eurheartj/9.suppl_j.33. [DOI] [PubMed] [Google Scholar]
- 12.Nunes H, Humbert M, Sitbon O, Morse JH, Deng Z, Knowles JA, Le Gall C, Parent F, Garcia G, Herve P, Barst RJ, Simonneau G. Prognostic Factors for Survival in Human Immunodeficiency Virus-associated Pulmonary Arterial Hypertension. Am J Respir Crit Care Med. 2003;167:1433–1439. doi: 10.1164/rccm.200204-330OC. [DOI] [PubMed] [Google Scholar]
- 13.Petrosillo N, Chinello P, Cicalini S. Pulmonary hypertension in individuals with HIV infection. AIDS. 2006;20:2128–2129. doi: 10.1097/01.aids.0000247569.03504.8b. 2110.1097/2101.aids.0000247569.0000203504.0000247568b. [DOI] [PubMed] [Google Scholar]
- 14.Opravil MPM, Speich R, Joller-Jemelka HI, Jenni R, Russi EW, Hirschel B, Luthy R. HIV-associated primary pulmonary hypertension. A case control study. Swiss HIV Cohort Study. Am J Respir Crit Care Med. 1997;155:990–995. doi: 10.1164/ajrccm.155.3.9117037. [DOI] [PubMed] [Google Scholar]
- 15.Yuan JX-J, Rubin LJ. Pathogenesis of Pulmonary Arterial Hypertension. Circulation. 2005;111:534–538. doi: 10.1161/01.CIR.0000156326.48823.55. [DOI] [PubMed] [Google Scholar]
- 16.Degano BGM, Savale L, Montani D, Jais X, Yaici A, Le Pavec J, Humbert, Simmonneau G, Sitbon O. HIV-associated pulmonary arterial hypertension: survival and prognostic factors in the modern therapeutic era. AIDS. 2010;24:67–75. doi: 10.1097/QAD.0b013e328331c65e. [DOI] [PubMed] [Google Scholar]
- 17.Fontas E, van Leth F, Sabin CA, Friis-Møller N, Rickenbach M, d’Arminio Monforte A, Kirk O, Dupon M, Morfeldt L, Mateu S, Petoumenos K, El-Sadr W, de Wit S, Lundgren JD, Pradier C, Reiss P Group, f. t. DA. DS. Lipid Profiles in HIV-Infected Patients Receiving Combination Antiretroviral Therapy: Are Different Antiretroviral Drugs Associated with Different Lipid Profiles? Journal of Infectious Diseases. 2004;189:1056–1074. doi: 10.1086/381783. [DOI] [PubMed] [Google Scholar]
- 18.Spieker LE, Karadag B, Binggeli C, Corti R. Rapid progression of atherosclerotic coronary artery disease in patients with human immunodeficiency virus infection. Heart and Vessels. 2005;20:171–174. doi: 10.1007/s00380-004-0790-8. [DOI] [PubMed] [Google Scholar]
- 19.Maggi P, Lillo A, Perilli F, Maserati R, Chirianni A Group, o. b. o. t. P. Colour-Doppler ultrasonography of carotid vessels in patients treated with antiretroviral therapy: a comparative study. AIDS. 2004;18:1023–1028. doi: 10.1097/00002030-200404300-00010. [DOI] [PubMed] [Google Scholar]
- 20.Meng Q, Lima JAC, Lai H, Vlahov D, Celentano DD, Strathdee SA, Nelson KE, Wu KC, Chen S, Tong W, Lai S. Coronary artery calcification, atherogenic lipid changes, and increased erythrocyte volume in black injection drug users infected with human immunodeficiency virus-1 treated with protease inhibitors. American Heart Journal. 2002;144:642–648. doi: 10.1067/mhj.2002.125009. [DOI] [PubMed] [Google Scholar]
- 21.Chironi G, Escaut L, Gariepy J, Cogny A, Monsuez J-J, Levenson J, Simon A, Vittecoq D. Carotid Intima-Media Thickness in Heavily Pretreated HIV-Infected Patients. JAIDS Journal of Acquired Immune Deficiency Syndromes. 2003;32:490–493. doi: 10.1097/00126334-200304150-00004. [DOI] [PubMed] [Google Scholar]
- 22.van Vonderen MGA, Hassink EAM, van Agtmael MA, Stehouwer CDA, Danner SA, Reiss P, Smulders Y. Increase in Carotid Artery Intima-Media Thickness and Arterial Stiffness but Improvement in Several Markers of Endothelial Function after Initiation of Antiretroviral Therapy. Journal of Infectious Diseases. 2009;199:1186–1194. doi: 10.1086/597475. [DOI] [PubMed] [Google Scholar]
- 23.McComsey GA, O’Riordan M, Hazen SL, El-Bejjani D, Bhatt S, Brennan M-L, Storer N, Adell J, Nakamoto DA, Dogra V. Increased carotid intima media thickness and cardiac biomarkers in HIV infected children. AIDS. 2007;21:921–927. 910. doi: 10.1097/QAD.0b013e328133f29c. 1097/QAD.1090b1013e328133f328129c. [DOI] [PubMed] [Google Scholar]
- 24.Mercié P, Thiébaut R, Aurillac-Lavignolle V, Pellegrin JL, Yvorra-Vives MC, Cipriano C, Neau D, Morlat P, Ragnaud JM, Dupon M, Bonnet F, Lawson-Ayayi S, Malvy D, Roudaut R, Dabis F on behalf of the Groupe d’Epidemiologie Clinique du Sida en, A. Carotid intima–media thickness is slightly increased over time in HIV-1-infected patients*. HIV Medicine. 2005;6:380–387. doi: 10.1111/j.1468-1293.2005.00324.x. [DOI] [PubMed] [Google Scholar]
- 25.Currier JS, Kendall MA, Zackin R, Henry WK, Alston-Smith B, Torriani FJ, Schouten J, Mickelberg K, Li Y, Hodis HN Team, f. t. A. S. Carotid artery intima-media thickness and HIV infection: traditional risk factors overshadow impact of protease inhibitor exposure. AIDS. 2005;19:927–933. doi: 10.1097/01.aids.0000171406.53737.f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnsen S, Dolan SE, Fitch KV, Kanter JR, Hemphill LC, Connelly JM, Lees RS, Lee H, Grinspoon S. Carotid Intimal Medial Thickness in Human Immunodeficiency Virus-Infected Women: Effects of Protease Inhibitor Use, Cardiac Risk Factors, and the Metabolic Syndrome. Journal of Clinical Endocrinology & Metabolism. 2006;91:4916–4924. doi: 10.1210/jc.2006-1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masiá M, Bernal E, Padilla S, García N, Escribano JC, Martínez E, Gutiérrez Fl. A pilot randomized trial comparing an intensive versus a standard intervention in stable HIV-infected patients with moderate-high cardiovascular risk. Journal of Antimicrobial Chemotherapy. 2009;64:589–598. doi: 10.1093/jac/dkp250. [DOI] [PubMed] [Google Scholar]
- 28.Hsue PY, Hunt PW, Schnell A, Kalapus SC, Hoh R, Ganz P, Martin JN, Deeks SG. Role of viral replication, antiretroviral therapy, and immunodeficiency in HIV-associated atherosclerosis. AIDS. 2009;23:1059–1067. doi: 10.1097/QAD.0b013e32832b514b. 1010.1097/QAD.1050b1013e32832b32514b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bonnet D, Aggoun Y, Szezepanski I, Bellal N, Blanche Sp. Arterial stiffness and endothelial dysfunction in HIV-infected children. AIDS. 2004;18:1037–1041. doi: 10.1097/00002030-200404300-00012. [DOI] [PubMed] [Google Scholar]
- 30.Sevastianova KSJ, Westerbacka J, Ristola M, Yki-Jarvinen H. Arterial stiffness in HIV-infected patients receiving highly active antiretroviral therapy. Antivir Ther. 2005;10:925–935. [PubMed] [Google Scholar]
- 31.Kristoffersen US, Kofoed K, Kronborg G, Giger AK, Kjaer A, Lebech AM. Reduction in circulating markers of endothelial dysfunction in HIV-infected patients during antiretroviral therapy. HIV Medicine. 2009;10:79–87. doi: 10.1111/j.1468-1293.2008.00661.x. [DOI] [PubMed] [Google Scholar]
- 32.Hsue PY, Hunt PW, Wu Y, Schnell A, Ho JE, Hatano H, Xie Y, Martin JN, Ganz P, Deeks SG. Association of abacavir and impaired endothelial function in treated and suppressed HIV-infected patients. AIDS. 2009;23:2021–2027. doi: 10.1097/QAD.0b013e32832e7140. 2010.1097/QAD.2020b2013e32832e37140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nolan D, Watts GF, Herrmann SE, French MA, John M, Mallal S. Endothelial function in HIV-infected patients receiving protease inhibitor therapy: does immune competence affect cardiovascular risk? QJM. 2003;96:825–832. doi: 10.1093/qjmed/hcg145. [DOI] [PubMed] [Google Scholar]
- 34.Blanco JJRo, García IsSr, Cerezo JGm, de Rivera JMaPaSn, Anaya PM, Raya PGa, García JGl, López JRnA, Hernández FJB, Rodríguez JJVz. Endothelial function in HIV-infected patients with low or mild cardiovascular risk. Journal of Antimicrobial Chemotherapy. 2006;58:133–139. doi: 10.1093/jac/dkl190. [DOI] [PubMed] [Google Scholar]
- 35.Joshi VV, Pawel B, Connor E, Sharer L, Oleske JM, Morrison S, Marin-Garcia J. Arteriopathy in children with acquired immune deficiency syndrome. Pediatr Pathol. 1987;7:261–275. doi: 10.1080/15513818709177129. [DOI] [PubMed] [Google Scholar]
- 36.Paton P, Tabib A, Loire R, Tete R. Coronary artery lesions and human immunodeficiency virus infection. Res Virol. 1993;144:225–231. doi: 10.1016/s0923-2516(06)80033-6. [DOI] [PubMed] [Google Scholar]
- 37.Mandell BF, Calabrese LH. Infections and systemic vasculitis. Curr Opin Rheumatol. 1998;10:51–57. doi: 10.1097/00002281-199801000-00008. [DOI] [PubMed] [Google Scholar]
- 38.Cebrian M, Miro O, Font C, Coll-Vinent B, Grau JM. HIV-related vasculitis. AIDS Patient Care STDS. 1997;11:245–258. doi: 10.1089/apc.1997.11.245. [DOI] [PubMed] [Google Scholar]
- 39.Maniker AH, Hunt CD. Cerebral aneurysm in the HIV patient: a report of six cases. Surg Neurol. 1996;46:49–54. doi: 10.1016/0090-3019(96)00030-4. [DOI] [PubMed] [Google Scholar]
- 40.Grunfeld C, Pang M, Doerrler W, Shigenaga JK, Jensen P, Feingold KR. Lipids, lipoproteins, triglyceride clearance, and cytokines in human immunodeficiency virus infection and the acquired immunodeficiency syndrome. J Clin Endocrinol Metab. 1992;74:1045–1052. doi: 10.1210/jcem.74.5.1373735. [DOI] [PubMed] [Google Scholar]
- 41.Schved JF, Gris JC, Arnaud A, Martinez P, Sanchez N, Wautier JL, Sarlat C. von Willebrand factor antigen, tissue-type plasminogen activator antigen, and risk of death in human immunodeficiency virus 1-related clinical disease: independent prognostic relevance of tissue-type plasminogen activator. J Lab Clin Med. 1992;120:411–419. [PubMed] [Google Scholar]
- 42.Lafeuillade A, Alessi MC, Poizot-Martin I, Boyer-Neumann C, Zandotti C, Quilichini R, Aubert L, Tamalet C, Juhan-Vague I, Gastaut JA. Endothelial cell dysfunction in HIV infection. J Acquir Immune Defic Syndr. 1992;5:127–131. [PubMed] [Google Scholar]
- 43.Wolf K, Tsakiris DA, Weber R, Erb P, Battegay M. Antiretroviral therapy reduces markers of endothelial and coagulation activation in patients infected with human immunodeficiency virus type 1. J Infect Dis. 2002;185:456–462. doi: 10.1086/338572. [DOI] [PubMed] [Google Scholar]
- 44.Greenwood AJ, Hughes J, Wallace G, Seed P, Stanford MR, Graham EM. Soluble intercellular adhesion molecule-1 (sICAM-1) and vascular cell adhesion molecule-1 (sVCAM-1) in patients with HIV/AIDS does not appear to correlate with cytomegalovirus retinitis. Int J STD AIDS. 1998;9:713–714. [PubMed] [Google Scholar]
- 45.Seigneur M, Constans J, Blann A, Renard M, Pellegrin JL, Amiral J, Boisseau M, Conri C. Soluble adhesion molecules and endothelial cell damage in HIV infected patients. Thromb Haemost. 1997;77:646–649. [PubMed] [Google Scholar]
- 46.Klein D, Hurley LB, Quesenberry CP, Jr, Sidney S. Do protease inhibitors increase the risk for coronary heart disease in patients with HIV-1 infection? J Acquir Immune Defic Syndr. 2002;30:471–477. doi: 10.1097/00126334-200208150-00002. [DOI] [PubMed] [Google Scholar]
- 47.Triant VA, Lee H, Hadigan C, Grinspoon SK. Increased acute myocardial infarction rates and cardiovascular risk factors among patients with human immunodeficiency virus disease. J Clin Endocrinol Metab. 2007;92:2506–2512. doi: 10.1210/jc.2006-2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.El-Sadr WM, Lundgren JD, Neaton JD, Gordin F, Abrams D, Arduino RC, Babiker A, Burman W, Clumeck N, Cohen CJ, Cohn D, Cooper D, Darbyshire J, Emery S, Fatkenheuer G, Gazzard B, Grund B, Hoy J, Klingman K, Losso M, Markowitz N, Neuhaus J, Phillips A, Rappoport C. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med. 2006;355:2283–2296. doi: 10.1056/NEJMoa062360. [DOI] [PubMed] [Google Scholar]
- 49.Blum A, Hadas V, Burke M, Yust I, Kessler A. Viral load of the human immunodeficiency virus could be an independent risk factor for endothelial dysfunction. Clin Cardiol. 2005;28:149–153. doi: 10.1002/clc.4960280311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Solages A, Vita JA, Thornton DJ, Murray J, Heeren T, Craven DE, Horsburgh CR., Jr Endothelial function in HIV-infected persons. Clin Infect Dis. 2006;42:1325–1332. doi: 10.1086/503261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bonnet D, Aggoun Y, Szezepanski I, Bellal N, Blanche S. Arterial stiffness and endothelial dysfunction in HIV-infected children. Aids. 2004;18:1037–1041. doi: 10.1097/00002030-200404300-00012. [DOI] [PubMed] [Google Scholar]
- 52.Oliviero U, Bonadies G, Apuzzi V, Foggia M, Bosso G, Nappa S, Valvano A, Leonardi E, Borgia G, Castello G, Napoli R, Saccà L. Human immunodeficiency virus per se exerts atherogenic effects. Atherosclerosis. 2009;204:586–589. doi: 10.1016/j.atherosclerosis.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 53.Lorenz MW, Stephan C, Harmjanz A, Staszewski S, Buehler A, Bickel M, von Kegler S, Ruhkamp D, Steinmetz H, Sitzer M. Both long-term HIV infection and highly active antiretroviral therapy are independent risk factors for early carotid atherosclerosis. Atherosclerosis. 2007 doi: 10.1016/j.atherosclerosis.2006.12.022. [DOI] [PubMed] [Google Scholar]
- 54.van Vonderen MG, Smulders YM, Stehouwer CD, Danner SA, Gundy CM, Vos F, Reiss P, Agtmael MA. Carotid intima-media thickness and arterial stiffness in HIV-infected patients: the role of HIV, antiretroviral therapy, and lipodystrophy. J Acquir Immune Defic Syndr. 2009;50:153–161. doi: 10.1097/QAI.0b013e31819367cd. [DOI] [PubMed] [Google Scholar]
- 55.Mandas A, Iorio EL, Congiu MG, Balestrieri C, Mereu A, Cau D, Dess, #236; S, Curreli N. Oxidative Imbalance in HIV-1 Infected Patients Treated with Antiretroviral Therapy. Journal of Biomedicine and Biotechnology. 2009 doi: 10.1155/2009/749575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suresh DRAV, Pratibha K, Prasad BV. Total antioxidant capacity--a novel early bio-chemical marker of oxidative stress in HIV infected individuals. J Biomed Sci. 2009;16:61. doi: 10.1186/1423-0127-16-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sonnerborg ACG, Akerlund B, Jarstrand C. Increased production of malondialdehyde in patients with HIV infection. Scand J Infect Dis. 1988;20:287–290. doi: 10.3109/00365548809032453. [DOI] [PubMed] [Google Scholar]
- 58.Revillard JPVC, Favier AE, Richard MJ, Zittoun M, Kazatchkine MD. Lipid peroxidation in human immunodeficiency virus infection. J Acquir Immune Defic Syndr. 1992;5:637–638. [PubMed] [Google Scholar]
- 59.Wang YWR. Potential therapeutics of vitamin E (tocopherol) in AIDS and HIV. Drugs. 1994;48:327–338. doi: 10.2165/00003495-199448030-00001. [DOI] [PubMed] [Google Scholar]
- 60.Lacey CJMM, Sanderson MJ, Monteiro EF, Vail A, Schorah CJ. Antioxidant-micronutrients and HIV infection. Int J STD AIDS. 1996;7:485–489. doi: 10.1258/0956462961918554. [DOI] [PubMed] [Google Scholar]
- 61.Buhl RJH, Holroyd KJ, Wells FB, Mastrangeli A, Saltini C, Cantin AM, Crystal RG. Systemic glutathione deficiency in symptom-free HIV-seropositive individuals. Lancet. 1989;2:1294–1298. doi: 10.1016/s0140-6736(89)91909-0. [DOI] [PubMed] [Google Scholar]
- 62.Eck HPGH, Hartmann M, Petzoldt D, Daniel V, Droge W. Low concentrations of acid-soluble thiol (cysteine) in the blood plasma of HIV-1 infected patients. Biol Chem Hoppe Seyler. 1989;370:101–108. doi: 10.1515/bchm3.1989.370.1.101. [DOI] [PubMed] [Google Scholar]
- 63.Staal FJT, Ela SW, Roederer M, Anderson MT, Herzenberg LA. Glutathione deficiency and human immunodeficiency virus infection. The Lancet. 1992;339:909–912. doi: 10.1016/0140-6736(92)90939-z. [DOI] [PubMed] [Google Scholar]
- 64.Staal FJRM, Israelski DM, Bubp J, Mole LA, McShane D, Deresinski SC, Ross W, Sussman H, Raju PA, et al. Intracellular glutathione levels in T cell subsets decrease in HIV-infected individuals. AIDS Res Hum Retroviruses. 1992;8:305–311. doi: 10.1089/aid.1992.8.305. [DOI] [PubMed] [Google Scholar]
- 65.Wanchu ASVR, Pallikkuth Suresh, Sachdeva Ravinder Kaur. Short Communication: Oxidative Stress in HIV-infected Individuals: A Cross-Sectional Study. 2009;25(12):1307. doi: 10.1089/aid.2009.0062. [DOI] [PubMed] [Google Scholar]
- 66.Nakamura H, De Rosa S, Roederer M, Anderson MT, Dubs JG, Yodoi J, Holmgren A, Herzenberg LA, Herzenberg LA. Elevation of plasma thioredoxin levels in HIV-infected individuals. International Immunology. 1996;8:603–611. doi: 10.1093/intimm/8.4.603. [DOI] [PubMed] [Google Scholar]
- 67.Watson WH, Yang X, Choi YE, Jones DP, Kehrer JP. Thioredoxin and Its Role in Toxicology. Toxicological Sciences. 2004;78:3–14. doi: 10.1093/toxsci/kfh050. [DOI] [PubMed] [Google Scholar]
- 68.Aquaro S, Muscoli C, Ranazzi A, Pollicita M, Granato T, Masuelli L, Modesti A, Perno C-F, Mollace V. The contribution of peroxynitrite generation in HIV replication in human primary macrophages. Retrovirology. 2007;4:76. doi: 10.1186/1742-4690-4-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Husain M, Meggs LG, Vashistha H, Simoes S, Griffiths KO, Kumar D, Mikulak J, Mathieson PW, Saleem MA, Del Valle L, Pina-Oviedo S, Wang JY, Seshan SV, Malhotra A, Reiss K, Singhal PC. Inhibition of p66ShcA Longevity Gene Rescues Podocytes from HIV-1-induced Oxidative Stress and Apoptosis. Journal of Biological Chemistry. 2009;284:16648–16658. doi: 10.1074/jbc.M109.008482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Choi J, Liu R-M, Kundu RK, Sangiorgi F, Wu W, Maxson R, Forman HJ. Molecular Mechanism of Decreased Glutathione Content in Human Immunodeficiency Virus Type 1 Tat-transgenic Mice. Journal of Biological Chemistry. 2000;275:3693–3698. doi: 10.1074/jbc.275.5.3693. [DOI] [PubMed] [Google Scholar]
- 71.Kline ER, Kleinhenz DJ, Liang B, Dikalov S, Guidot DM, Hart CM, Jones DP, Sutliff RL. Vascular oxidative stress and nitric oxide depletion in HIV-1 transgenic rats are reversed by glutathione restoration. American Journal of Physiology - Heart and Circulatory Physiology. 2008;294:H2792–H2804. doi: 10.1152/ajpheart.91447.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lassiter C, Fan X, Joshi P, Jacob B, Sutliff R, Jones D, Koval M, Guidot D. HIV-1 transgene expression in rats causes oxidant stress and alveolar epithelial barrier dysfunction. AIDS Research and Therapy. 2009;6:1. doi: 10.1186/1742-6405-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jacob BA, Porter KM, Elms SC, Cheng P-Y, Jones DP, Sutliff RL. HIV-1-induced pulmonary oxidative and nitrosative stress: exacerbated response to endotoxin administration in HIV-1 transgenic mouse model. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2006;291:L811–L819. doi: 10.1152/ajplung.00468.2005. [DOI] [PubMed] [Google Scholar]
- 74.Sadaie MR, Rappaport J, Benter T, Josephs SF, Willis R, Wong-Staal F. Missense mutations in an infectious human immunodeficiency viral genome: functional mapping of tat and identification of the rev splice acceptor. Proceedings of the National Academy of Sciences. 1988;85:9224–9228. doi: 10.1073/pnas.85.23.9224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Frankel AD, Young JAT. HIV-1: Fifteen Proteins and an RNA. Annual Review of Biochemistry. 1998;67:1–25. doi: 10.1146/annurev.biochem.67.1.1. [DOI] [PubMed] [Google Scholar]
- 76.Ensoli B, Buonaguro L, Barillari G, Fiorelli V, Gendelman R, Morgan RA, Wingfield P, Gallo RC. Release, uptake, and effects of extracellular human immunodeficiency virus type 1 Tat protein on cell growth and viral transactivation. J Virol. 1993;67:277–287. doi: 10.1128/jvi.67.1.277-287.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Westendorp M, Frank R, Ochsenbauer C, Stricker K, Dhein J, Walczak H, Debatin K, Krammer P. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature. 1995;375:497–500. doi: 10.1038/375497a0. [DOI] [PubMed] [Google Scholar]
- 78.Helland DE, Welles JL, Caputo A, Haseltine WA. Transcellular transactivation by the human immunodeficiency virus type 1 tat protein. J Virol. 1991;65:4547–4549. doi: 10.1128/jvi.65.8.4547-4549.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Marcuzzi A, Weinberger J, Weinberger OK. Transcellular activation of the human immunodeficiency virus type 1 long terminal repeat in cocultured lymphocytes. J Virol. 1992;66:4228–4232. doi: 10.1128/jvi.66.7.4228-4232.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Westendorp M, Shatrov VA, Schulze-Osthoff K, Frank R, Kraft M, Los M, Krammer PH, Droge W, Lehman V. HIV-1 Tat potentiates TNF-induced NF-kappa B activation and cytotoxicity by altering the cellular redox state. EMBO J. 1995;14:546–554. doi: 10.1002/j.1460-2075.1995.tb07030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xiao H, Neuveut C, Tiffany HL, Benkirane M, Rich EA, Murphy PM, Jeang K-T. Selective CXCR4 antagonism by Tat: Implications for in vivo expansion of coreceptor use by HIV-1. Proceedings of the National Academy of Sciences. 2000;97:11466–11471. doi: 10.1073/pnas.97.21.11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.András IE, Pu H, Deli MA, Nath A, Hennig B, Toborek M. HIV-1 Tat protein alters tight junction protein expression and distribution in cultured brain endothelial cells. Journal of Neuroscience Research. 2003;74:255–265. doi: 10.1002/jnr.10762. [DOI] [PubMed] [Google Scholar]
- 83.Toborek MLY, Pu H, Malecki A, Flora G, Garrido R, Hennig B, Bauer HC, Nath A. HIV-Tat protein induced oxidative and inflammatory pathways in brain endothelium. J Neurochem. 2003;84:169–179. doi: 10.1046/j.1471-4159.2003.01543.x. [DOI] [PubMed] [Google Scholar]
- 84.Price TOEN, Nakaoke R, Banks WA. HIV-1 viral proteins gp120 and Tat induce oxidative stress in brain endothelial cells. Brain Res. 2005;1045:57–63. doi: 10.1016/j.brainres.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 85.Zhang H-S, Li H-Y, Zhou Y, Wu M-R, Zhou H-S. Nrf2 is involved in inhibiting Tat-induced HIV-1 long terminal repeat transactivation. Free Radical Biology and Medicine. 2009;47:261–268. doi: 10.1016/j.freeradbiomed.2009.04.028. [DOI] [PubMed] [Google Scholar]
- 86.Opalenik SR, Ding Q, Mallery SR, Thompson JA. Glutathione Depletion Associated with the HIV-1 TAT Protein Mediates the Extracellular Appearance of Acidic Fibroblast Growth Factor. Archives of Biochemistry and Biophysics. 1998;351:17–26. doi: 10.1006/abbi.1997.0566. [DOI] [PubMed] [Google Scholar]
- 87.Banerjee A, Zhang X, Manda KR, Banks WA, Ercal N. HIV proteins (gp120 and Tat) and methamphetamine in oxidative stress-induced damage in the brain: Potential role of the thiol antioxidant N-acetylcysteine amide. Free Radical Biology and Medicine. 2010;48:1388–1398. doi: 10.1016/j.freeradbiomed.2010.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Richard M-J, Guiraud P, Didier C, Seve M, Flores SC, Favier A. Human Immunodeficiency Virus Type 1 Tat Protein Impairs Selenoglutathione Peroxidase Expression and Activity by a Mechanism Independent of Cellular Selenium Uptake: Consequences on Cellular Resistance to UV-A Radiation. Archives of Biochemistry and Biophysics. 2001;386:213–220. doi: 10.1006/abbi.2000.2197. [DOI] [PubMed] [Google Scholar]
- 89.Flores SC, Marecki JC, Harper KP, Bose SK, Nelson SK, McCord JM. Tat protein of human immunodeficiency virus type 1 represses expression of manganese superoxide dismutase in HeLa cells. Proceedings of the National Academy of Sciences. 1993;90:7632–7636. doi: 10.1073/pnas.90.16.7632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Marecki JCC-GA, Vaitaitis GM, Honda JR, Porntadivity S, St Clair DK, Flores SC. HIV-1 Tat regulates the SOD2 basal promoter by altering Sp1/Sp3 binding activity. Free Radic Biol Med. 2004;37:869–890. doi: 10.1016/j.freeradbiomed.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 91.Gu Y, Wu RF, Xu YC, Flores SC, Terada LS. HIV Tat Activates c-Jun Amino-terminal Kinase through an Oxidant-Dependent Mechanism. Virology. 2001;286:62–71. doi: 10.1006/viro.2001.0998. [DOI] [PubMed] [Google Scholar]
- 92.Wu R-F, Ma Z, Liu Z, Terada LS. Nox4-Derived H2O2 Mediates Endoplasmic Reticulum Signaling through Local Ras Activation. Mol Cell Biol. 2010;30:3553–3568. doi: 10.1128/MCB.01445-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Geyer MFO, Peterlin BM. Structure-function relationships in HIV-1 Nef. EMBO Rep. 2001;2:580–585. doi: 10.1093/embo-reports/kve141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Harris M. HIV: a new role for Nef in the spread of HIV. Current Biology. 1999;9:R463–R463. doi: 10.1016/s0960-9822(99)80282-6. [DOI] [PubMed] [Google Scholar]
- 95.Villhardt FPO, Sawada M, Suzuki K, Wiznerowicz M, Kiyokawa E, Trono D, Krause KH. The HIV-1 Nef protein and phagocyte NADPH oxidase activation. J Biol Chem. 2002;277:42136–42143. doi: 10.1074/jbc.M200862200. [DOI] [PubMed] [Google Scholar]
- 96.Salmen S, Colmenares M, Peterson DL, Reyes E, Rosales JD, Berrueta L. HIV-1 Nef associates with p22-phox, a component of the NADPH oxidase protein complex. Cellular Immunology. 2010;263:166–171. doi: 10.1016/j.cellimm.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 97.Duffy P, Wang X, Lin PH, Yao Q, Chen C. HIV Nef Protein Causes Endothelial Dysfunction in Porcine Pulmonary Arteries and Human Pulmonary Artery Endothelial Cells. Journal of Surgical Research. 2009;156:257–264. doi: 10.1016/j.jss.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Marecki JC, Cool CD, Parr JE, Beckey VE, Luciw PA, Tarantal AF, Carville A, Shannon RP, Cota-Gomez A, Tuder RM, Voelkel NF, Flores SC. HIV-1 Nef Is Associated with Complex Pulmonary Vascular Lesions in SHIV-nef-infected Macaques. Am J Respir Crit Care Med. 2006;174:437–445. doi: 10.1164/rccm.200601-005OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sehgal PB, Mukhopadhyay S, Patel K, Xu F, Almodóvar S, Tuder RM, Flores SC. Golgi dysfunction is a common feature in idiopathic human pulmonary hypertension and vascular lesions in SHIV-nef-infected macaques. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2009;297:L729–L737. doi: 10.1152/ajplung.00087.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Almodovar S, Hsue PY, Morelli J, Huang L, Flores SC on behalf of the Lung HIV Study. Pathogenesis of HIV-Associated Pulmonary Hypertension: Potential Role of HIV-1 nef. Proc Am Thorac Soc. 2011;8:308–312. doi: 10.1513/pats.201006-046WR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Freed EO, Martin MA. The Role of Human Immunodeficiency Virus Type 1 Envelope Glycoproteins in Virus Infection. Journal of Biological Chemistry. 1995;270:23883–23886. doi: 10.1074/jbc.270.41.23883. [DOI] [PubMed] [Google Scholar]
- 102.Oh SKCW, Raina J, Blanchard GC, Adler WH, Walker J, Kornfeld H. Identification of HIV-1 envelope glycoprotein in the serum of AIDS and ARC patients. J Acquir Immune Defic Syndr. 1992;5:251–256. [PubMed] [Google Scholar]
- 103.Holguin A, O’Connor KA, Biedenkapp J, Campisi J, Wieseler-Frank J, Milligan ED, Hansen MK, Spataro L, Maksimova E, Bravmann C, Martin D, Fleshner M, Maier SF, Watkins LR. HIV-1 gp120 Stimulates proinflammatory cytokine-mediated pain facilitation via activation of nitric oxide synthase-I (nNOS) Pain. 2004;110:517–530. doi: 10.1016/j.pain.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 104.Bagetta GPS, Del Duca C, Morrone LA, Rombola L, Nappu G, De Alba J, Knowles RG, Corasaniti MT. Inducible nitric oxide synthase is involved in the mechanisms of cocaine enhanced neuronal apoptosis induced by HIV-1 gp120 in the neocortex of rat. Neuroscience Letters. 2004;235:183–186. doi: 10.1016/j.neulet.2003.11.065. [DOI] [PubMed] [Google Scholar]
- 105.Gendelman H, Genis P, Jett M, Zhai QH, Nottet HS. An experimental model system for HIV-1-induced brain injury. Adv Neuroimmunol. 1994;4:189–193. doi: 10.1016/s0960-5428(06)80256-1. [DOI] [PubMed] [Google Scholar]
- 106.Singhal PC, Reddy K, Franki N, Ding G. HIV-1 gp120 envelope protein modulates proliferation of human glomerular epithelial cells. Journal of Cellular Biochemistry. 2000;76:61–70. doi: 10.1002/(sici)1097-4644(20000101)76:1<61::aid-jcb7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 107.Huang M-B, Khan M, Garcia-Barrio M, Powell M, Bond VC. Apoptotic Effects in Primary Human Umbilical Vein Endothelial Cell Cultures Caused by Exposure to Virion-Associated and Cell Membrane-Associated HIV-1 gp120. JAIDS Journal of Acquired Immune Deficiency Syndromes. 2001;27:213–221. doi: 10.1097/00126334-200107010-00001. [DOI] [PubMed] [Google Scholar]
- 108.Annunziata P, Cioni C, Toneatto S, Paccagnini E. HIV-1 gp120 increases the permeability of rat brain endothelium cultures by a mechanism involving substance P. AIDS. 1998;12:2377–2385. doi: 10.1097/00002030-199818000-00006. [DOI] [PubMed] [Google Scholar]
- 109.Kanmogne GD, Primeaux C, Grammas P. HIV-1 gp120 Proteins Alter Tight Junction Protein Expression and Brain Endothelial Cell Permeability: Implications for the Pathogenesis of HIV-Associated Dementia. Journal of Neuropathology & Experimental Neurology. 2005;64:498–505. doi: 10.1093/jnen/64.6.498. [DOI] [PubMed] [Google Scholar]
- 110.Visalli V, Muscoli C, Sacco I, Sculco F, Palma E, Costa N, Colica C, Rotiroti D, Mollace V. N-acetylcysteine prevents HIV gp 120-related damage of human cultured astrocytes: correlation with glutamine synthase dysfunction. BMC Neuroscience. 2007;8:106. doi: 10.1186/1471-2202-8-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yu QRZZ, Zhang H, Lin HT, Li XM, Bai L, Cai WB. Inducible nitric oxide synthase is involved in the oxidation stress induced by HIV-1 gp120 in human retina pigment epithelial cells. Chin Med J (Engl) 2008;121:2578–2583. [PubMed] [Google Scholar]
- 112.Louboutin J-P, Agrawal L, Reyes BAS, Van Bockstaele EJ, Strayer DS. HIV-1 gp120-Induced Injury to the Blood-Brain Barrier: Role of Metalloproteinases 2 and 9 and Relationship to Oxidative Stress. Journal of Neuropathology & Experimental Neurology. 2010;69:801–816. doi: 10.1097/NEN.0b013e3181e8c96f. 810.1097/NEN.1090b1013e3181e1098c1096f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pietraforte D, Tritarelli E, Testa U, Minetti M. gp120 HIV envelope glycoprotein increases the production of nitric oxide in human monocyte-derived macrophages. Journal of Leukocyte Biology. 1994;55:175–182. doi: 10.1002/jlb.55.2.175. [DOI] [PubMed] [Google Scholar]
- 114.Shatrov V, Ratter F, Gruber A, Droge W, Lehmann V. HIV type 1 glycoprotein amplifies tumor necrosis factor-induced NF-kappa B activation in Jurkat cells. AIDS Res Hum Retroviruses. 1996;12:1209–1216. doi: 10.1089/aid.1996.12.1209. [DOI] [PubMed] [Google Scholar]
- 115.Foga IONA, Hasinoff BB, Geiger JD. Antioxidants and dipyridamole inhibit HIV-1 gp120-induced free radical-based oxidative damage to human monocytoid cells. J Acquir Immune Defic Syndr Hum Retrovirol. 1997;16:223–229. doi: 10.1097/00042560-199712010-00001. [DOI] [PubMed] [Google Scholar]
- 116.Fan X, Joshi PC, Koval M, Guidot DM. Chronic Alcohol Ingestion Exacerbates Lung Epithelial Barrier Dysfunction in HIV-1 Transgenic Rats. Alcoholism: Clinical and Experimental Research. 2011:no–no. doi: 10.1111/j.1530-0277.2011.01531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cohen EA, Dehni G, Sodroski JG, Haseltine WA. Human immunodeficiency virus vpr product is a virion-associated regulatory protein. J Virol. 1990;64:3097–3099. doi: 10.1128/jvi.64.6.3097-3099.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kogan M, Rappaport J. HIV-1 Accessory Protein Vpr: Relevance in the pathogenesis of HIV and potential for therapeutic intervention. Retrovirology. 2011;8:25. doi: 10.1186/1742-4690-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Levy D, Refaeli Y, Weiner D. Extracellular Vpr protein increases cellular permissiveness to human immunodeficiency virus replication and reactivates virus from latency. J Virol. 1995;69:1243–1252. doi: 10.1128/jvi.69.2.1243-1252.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Levy DN, Refaeli Y, MacGregor RR, Weiner DB. Serum Vpr regulates productive infection and latency of human immunodeficiency virus type 1. Proceedings of the National Academy of Sciences. 1994;91:10873–10877. doi: 10.1073/pnas.91.23.10873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hoshino S, Sun B, Konishi M, Shimura M, Segawa T, Hagiwara Y, Koyanagi Y, Iwamoto A, Mimaya J-I, Terunuma H, Kano S, Ishizaka Y. Vpr in Plasma of HIV Type 1-Positive Patients Is Correlated with The HIV Type 1 RNA Titers. AIDS Research and Human Retroviruses. 2007;23:391–397. doi: 10.1089/aid.2006.0124. [DOI] [PubMed] [Google Scholar]
- 122.Deshmane SL, Mukerjee R, Fan S, Del Valle L, Michiels C, Sweet T, Rom I, Khalili K, Rappaport J, Amini S, Sawaya BE. Activation of the Oxidative Stress Pathway by HIV-1 Vpr Leads to Induction of Hypoxia-inducible Factor 1α Expression. Journal of Biological Chemistry. 2009;284:11364–11373. doi: 10.1074/jbc.M809266200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hoshino S, Konishi M, Mori M, Shimura M, Nishitani C, Kuroki Y, Koyanagi Y, Kano S, Itabe H, Ishizaka Y. HIV-1 Vpr induces TLR4/MyD88-mediated IL-6 production and reactivates viral production from latency. Journal of Leukocyte Biology. 2010;87:1133–1143. doi: 10.1189/jlb.0809547. [DOI] [PubMed] [Google Scholar]
- 124.Kline ER, Sutliff RL. The Roles of HIV-1 Proteins and Antiretroviral Drug Therapy in HIV-1-Associated Endothelial Dysfunction. Journal of Investigative Medicine. 2008;56:752–769. doi: 10.1097/JIM.0b013e3181788d15. 710.1097/JIM.1090b1013e3181788d3181715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pinti M, Salomoni P, Cossarizza A. Anti-HIV drugs and the mitochondria. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2006;1757:700–707. doi: 10.1016/j.bbabio.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 126.Hulgan T, Morrow J, D’Aquila RT, Raffanti S, Morgan M, Rebeiro P, Haas DW. Oxidant Stress Is Increased during Treatment of Human Immunodeficiency Virus Infection. Clinical Infectious Diseases. 2003;37:1711–1717. doi: 10.1086/379776. [DOI] [PubMed] [Google Scholar]
- 127.Gil L, Tarinas A, Hernández D, Riverón BV, Pérez D, Tápanes R, Capo V, Pérez J. Altered oxidative stress indexes related to disease progression marker in human immunodeficiency virus infected patients with antiretroviral therapy. Biomedicine & Pharmacotherapy. 2010 doi: 10.1016/j.biopha.2010.09.009. In Press, Corrected Proof. [DOI] [PubMed] [Google Scholar]
- 128.Sundaram M, Saghayam S, Priya B, Venkatesh KK, Balakrishnan P, Shankar EM, Murugavel KG, Solomon S, Kumarasamy N. Changes in antioxidant profile among HIV-infected individuals on generic highly active antiretroviral therapy in southern India. International Journal of Infectious Diseases. 2008;12:e61–e66. doi: 10.1016/j.ijid.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 129.Pacht ER, Diaz P, Clanton T, Hart J, Gadek JE. Alveolar Fluid Glutathione Decreases in Asymptomatic HIV-Seropositive Subjects Over Time. Chest. 1997;112:785–788. doi: 10.1378/chest.112.3.785. [DOI] [PubMed] [Google Scholar]
- 130.Sutliff RL, Dikalov S, Weiss D, Parker J, Raidel S, Racine AK, Russ R, Haase CP, Taylor WR, Lewis W. Nucleoside reverse transcriptase inhibitors impair endothelium-dependent relaxation by increasing superoxide. American Journal of Physiology - Heart and Circulatory Physiology. 2002;283:H2363–H2370. doi: 10.1152/ajpheart.00151.2002. [DOI] [PubMed] [Google Scholar]
- 131.Kline E, Bassit L, Hernandez-Santiago B, Detorio M, Liang B, Kleinhenz D, Walp E, Dikalov S, Jones D, Schinazi R, Sutliff R. Long-Term Exposure to AZT, but not d4T, Increases Endothelial Cell Oxidative Stress and Mitochondrial Dysfunction. Cardiovascular Toxicology. 2009;9:1–12. doi: 10.1007/s12012-008-9029-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jiang B, Khandelwal AR, Rogers LK, Hebert VY, Kleinedler JJ, Zavecz JH, Shi W, Orr AW, Dugas TR. Antiretrovirals Induce Endothelial Dysfunction via an Oxidant-Dependent Pathway and Promote Neointimal Hyperplasia. Toxicological Sciences. 2010;117:524–536. doi: 10.1093/toxsci/kfq213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Papparella I, Ceolotto G, Berto L, Cavalli M, Bova S, Cargnelli G, Ruga E, Milanesi O, Franco L, Mazzoni M, Petrelli L, Nussdorfer GG, Semplicini A. Vitamin C prevents zidovudine-induced NAD(P)H oxidase activation and hypertension in the rat. Cardiovascular Research. 2007;73:432–438. doi: 10.1016/j.cardiores.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 134.Yamaguchi T, Katoh I, Kurata S-i. Azidothymidine causes functional and structural destruction of mitochondria, glutathione deficiency and HIV-1 promoter sensitization. European Journal of Biochemistry. 2002;269:2782–2788. doi: 10.1046/j.1432-1033.2002.02954.x. [DOI] [PubMed] [Google Scholar]
- 135.Lewis W. Defective mitochondrial DNA replication and NRTIs: pathophysiological implications in AIDS cardiomyopathy. Am J Physiol Heart Circ Physiol. 2003;284:H1–9. doi: 10.1152/ajpheart.00814.2002. [DOI] [PubMed] [Google Scholar]
- 136.Pupure J, Fernandes MAS, Santos MS, Moreno AJM, Kalvinsh I, Klusa V, Oliveira CR. Mitochondria as the target for mildronate’s protective effects in azidothymidine (AZT)-induced toxicity of isolated rat liver mitochondria. Cell Biochemistry and Function. 2008;26:620–631. doi: 10.1002/cbf.1486. [DOI] [PubMed] [Google Scholar]
- 137.Gao RYMP, Mohanraj R, Wang H, Horvath B, Yin S, Pacher P. Resveratrol attenuates azidothymidine-induced cardiotoxicity by decreasing mitochondrial reactive oxygen species generation in human cardiomyocytes. Mol Med Report. 2011;4:151–155. doi: 10.3892/mmr.2010.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Caron MAM, Vissian A, Vigouroux C, Capeau J. Contribution of mitochondrial dysfunction and oxidative stress to cellular premature senescence induced by antiretroviral thymidine analogues. Antivir Ther. 2008;13:27–38. [PubMed] [Google Scholar]
- 139.Opii WO, Nukala VN, Sultana R, Pandya JD, Day KM, Merchant ML, Klein JB, Sullivan PG, Butterfield DA. Proteomic Identification of Oxidized Mitochondrial Proteins following Experimental Traumatic Brain Injury. Journal of Neurotrauma. 2007;24:772–789. doi: 10.1089/neu.2006.0229. [DOI] [PubMed] [Google Scholar]
- 140.Kohler JJ, Cucoranu I, Fields E, Green E, He S, Hoying A, Russ R, Abuin A, Johnson D, Hosseini SH, Raper CM, Lewis W. Transgenic mitochondrial superoxide dismutase and mitochondrially targeted catalase prevent antiretroviral-induced oxidative stress and cardiomyopathy. Lab Invest. 2009;89:782–790. doi: 10.1038/labinvest.2009.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lund KC, Wallace KB. Adenosine 3′,5′-cyclic monophosphate (cAMP)-dependent phosphoregulation of mitochondrial complex I is inhibited by nucleoside reverse transcriptase inhibitors. Toxicology and Applied Pharmacology. 2008;226:94–106. doi: 10.1016/j.taap.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Komarov AM, Hall JM, Weglicki WB. Azidothymidine promotes free radical generation by activated macrophages and hydrogen peroxide-iron-mediated oxidation in a cell-free system. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2004;1688:257–264. doi: 10.1016/j.bbadis.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 143.Mak ITNL, Weglicki WB. Pro-oxidant properties cytotoxicity of AZT-monophosphate and AZT. Cardiovasc Toxicol. 2004;4:109–115. doi: 10.1385/ct:4:2:109. [DOI] [PubMed] [Google Scholar]
- 144.Erik DC. The role of non-nucleoside reverse transcriptase inhibitors (NNRTIs) in the therapy of HIV-1 infection. Antiviral Research. 1998;38:153–179. doi: 10.1016/s0166-3542(98)00025-4. [DOI] [PubMed] [Google Scholar]
- 145.Adams J, Patel N, Mankaryous N, Tadros M, Miller CD. Nonnucleoside Reverse Transcriptase Inhibitor Resistance and the Role of the Second-Generation Agents. The Annals of Pharmacotherapy. 2010;44:157–165. doi: 10.1345/aph.1M359. [DOI] [PubMed] [Google Scholar]
- 146.Shankar SS, Dubé MP, Gorski JC, Klaunig JE, Steinberg HO. Indinavir impairs endothelial function in healthy HIV-negative men. American Heart Journal. 2005;150:933.e931–933.e937. doi: 10.1016/j.ahj.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 147.Chen C, Lu X-H, Yan S, Chai H, Yao Q. HIV protease inhibitor ritonavir increases endothelial monolayer permeability. Biochemical and Biophysical Research Communications. 2005;335:874–882. doi: 10.1016/j.bbrc.2005.07.155. [DOI] [PubMed] [Google Scholar]
- 148.Ben-Romano RRA, Etzion S, Potashnik R, Kagan E, Greenbaum U, Bashan N. Nelfinavir induces adipocyte insulin resistance through the induction of oxidative stress: differential protective effect of antioxidant agents. Antivir Ther. 2006;11:1051–1060. [PubMed] [Google Scholar]
- 149.Vincent Sp, Tourniaire F, El Yazidi CM, Compe E, Manches O, Plannels R, Roche Rg. Nelfinavir Induces Necrosis of 3T3F44-2A Adipocytes by Oxidative Stress. JAIDS Journal of Acquired Immune Deficiency Syndromes. 2004;37:1556–1562. doi: 10.1097/00126334-200412150-00003. [DOI] [PubMed] [Google Scholar]
- 150.Lagathu CEB, Prot M, Frantz D, Gu Y, Bastard JP, Maachi M, Azoulav S, Briggs M, Caron M, Capeau J. Some HIV antiretrovirals increase oxidative stress and alter chemokine, cytokine or adiponectin production in human adipocytes and macrophages. Antivir Ther. 2007;12:489–500. [PubMed] [Google Scholar]
- 151.Mondal DPL, Ali M, Agrawal KC. HAART drugs induce oxidative stress in human endothelial cells and increase endothelial recruitment of mononuclear cells: exacerbation by inflammatory cytokines and amelioration by antioxidants. Cardiovasc Toxicol. 2004;4:287–302. doi: 10.1385/ct:4:3:287. [DOI] [PubMed] [Google Scholar]
- 152.Lefèvre C, Auclair M, Boccara F, Bastard J-P, Capeau J, Vigouroux C, Caron-Debarle M. Premature Senescence of Vascular Cells Is Induced by HIV Protease Inhibitors. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30:2611–2620. doi: 10.1161/ATVBAHA.110.213603. [DOI] [PubMed] [Google Scholar]
- 153.Chandra S, Mondal D, Agrawal KC. HIV-1 Protease Inhibitor Induced Oxidative Stress Suppresses Glucose Stimulated Insulin Release: Protection with Thymoquinone. Exp Biol Med. 2009;234:442–453. doi: 10.3181/0811-RM-317. [DOI] [PubMed] [Google Scholar]
- 154.Wang X, Mu H, Chai H, Liao D, Yao Q, Chen C. Human Immunodeficiency Virus Protease Inhibitor Ritonavir Inhibits Cholesterol Efflux from Human Macrophage-Derived Foam Cells. The American Journal of Pathology. 2007;171:304–314. doi: 10.2353/ajpath.2007.060965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Chai H, Yang H, Yan S, Li M, Lin PH, Lumsden AB, Yao Q, Chen C. Effects of 5 HIV Protease Inhibitors on Vasomotor Function and Superoxide Anion Production in Porcine Coronary Arteries. JAIDS Journal of Acquired Immune Deficiency Syndromes. 2005;40:12–19. doi: 10.1097/01.qai.0000172368.05327.7b. [DOI] [PubMed] [Google Scholar]
- 156.Chai H, Yan S, Lin P, Lumsden AB, Yao Q, Chen C. Curcumin Blocks HIV Protease Inhibitor Ritonavir-Induced Vascular Dysfunction in Porcine Coronary Arteries. Journal of the American College of Surgeons. 2005;200:820–830. doi: 10.1016/j.jamcollsurg.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 157.Chai H, Zhou W, Lin P, Lumsden A, Yao Q, Chen C. Ginsenosides block HIV protease inhibitor ritonavir-induced vascular dysfunction of porcine coronary arteries. American Journal of Physiology - Heart and Circulatory Physiology. 2005;288:H2965–H2971. doi: 10.1152/ajpheart.01271.2004. [DOI] [PubMed] [Google Scholar]
- 158.Manda KR, Banerjee A, Banks WA, Ercal N. Highly active antiretroviral therapy drug combination induces oxidative stress and mitochondrial dysfunction in immortalized human blood-brain barrier endothelial cells. Free Radical Biology and Medicine. 2011;50:801–810. doi: 10.1016/j.freeradbiomed.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. The Journal of Clinical Investigation. 2005;115:500–508. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kondo T, Hirose M, Kageyama K. Roles of Oxidative Stress and Redox Regulation in Atherosclerosis. Journal of Atherosclerosis and Thrombosis. 2009;16:532–538. doi: 10.5551/jat.1255. [DOI] [PubMed] [Google Scholar]
- 161.Go Y-M, Jones DP. Cysteine/cystine redox signaling in cardiovascular disease. Free Radical Biology and Medicine. 2011;50:495–509. doi: 10.1016/j.freeradbiomed.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Papaharalambus CA, Griendling KK. Basic Mechanisms of Oxidative Stress and Reactive Oxygen Species in Cardiovascular Injury. Trends in Cardiovascular Medicine. 2007;17:48–54. doi: 10.1016/j.tcm.2006.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nordberg J, Arnér ESJ. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radical Biology and Medicine. 2001;31:1287–1312. doi: 10.1016/s0891-5849(01)00724-9. [DOI] [PubMed] [Google Scholar]
- 164.Irani K. Oxidant Signaling in Vascular Cell Growth, Death, and Survival: A Review of the Roles of Reactive Oxygen Species in Smooth Muscle and Endothelial Cell Mitogenic and Apoptotic Signaling. Circulation Research. 2000;87:179–183. doi: 10.1161/01.res.87.3.179. [DOI] [PubMed] [Google Scholar]
- 165.Griendling KK, Sorescu D, Lassègue B, Ushio-Fukai M. Modulation of Protein Kinase Activity and Gene Expression by Reactive Oxygen Species and Their Role in Vascular Physiology and Pathophysiology. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20:2175–2183. doi: 10.1161/01.atv.20.10.2175. [DOI] [PubMed] [Google Scholar]
- 166.Ushio-Fukai M. Compartmentalization of Redox Signaling Through NADPH Oxidase-Derived ROS. Antioxidants & Redox Signaling. 2009;11:1289–1299. doi: 10.1089/ars.2008.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Canty TG, Boyle EM, Farr A, Morgan EN, Verrier ED, Pohlman TH. Oxidative Stress Induces NF-κB Nuclear Translocation Without Degradation of IκBα. Circulation. 1999;100:II-361–II-364. doi: 10.1161/01.cir.100.suppl_2.ii-361. [DOI] [PubMed] [Google Scholar]
- 168.Wung BS, Cheng JJ, Hsieh HJ, Shyy YJ, Wang DL. Cyclic Strain–Induced Monocyte Chemotactic Protein-1 Gene Expression in Endothelial Cells Involves Reactive Oxygen Species Activation of Activator Protein 1. Circulation Research. 1997;81:1–7. doi: 10.1161/01.res.81.1.1. [DOI] [PubMed] [Google Scholar]
- 169.Wellman TL, Jenkins J, Penar PL, Tranmer B, Zahr R, Lounsbury KM. Nitric oxide and reactive oxygen species exert opposing effects on the stability of hypoxia inducible factor-1α (HIF-1α) in explants of human pial arteries. The FASEB Journal. 2003 doi: 10.1096/fj.03-0143fje. [DOI] [PubMed] [Google Scholar]
- 170.Baas AS, Berk BC. Differential Activation of Mitogen-Activated Protein Kinases by H2O2 and O2- in Vascular Smooth Muscle Cells. Circulation Research. 1995;77:29–36. doi: 10.1161/01.res.77.1.29. [DOI] [PubMed] [Google Scholar]
- 171.Xia Z, Liu M, Wu Y, Sharma V, Luo T, Ouyang J, McNeill JH. N-acetylcysteine attenuates TNF-[alpha]-induced human vascular endothelial cell apoptosis and restores eNOS expression. European Journal of Pharmacology. 2006;550:134–142. doi: 10.1016/j.ejphar.2006.08.044. [DOI] [PubMed] [Google Scholar]
- 172.Luchtefeld M, Grote K, Grothusen C, Bley S, Bandlow N, Selle T, Strüber M, Haverich A, Bavendiek U, Drexler H, Schieffer B. Angiotensin II induces MMP-2 in a p47phox-dependent manner. Biochemical and Biophysical Research Communications. 2005;328:183–188. doi: 10.1016/j.bbrc.2004.12.152. [DOI] [PubMed] [Google Scholar]
- 173.Bauersachs J, Popp R, Hecker M, Sauer E, Fleming I, Busse R. Nitric Oxide Attenuates the Release of Endothelium-Derived Hyperpolarizing Factor. Circulation. 1996;94:3341–3347. doi: 10.1161/01.cir.94.12.3341. [DOI] [PubMed] [Google Scholar]
- 174.Kuzkaya N, Weissmann N, Harrison DG, Dikalov S. Interactions of Peroxynitrite, Tetrahydrobiopterin, Ascorbic Acid, and Thiols. Journal of Biological Chemistry. 2003;278:22546–22554. doi: 10.1074/jbc.M302227200. [DOI] [PubMed] [Google Scholar]
- 175.Suwaidi JA, Hamasaki S, Higano ST, Nishimura RA, Holmes DR, Lerman A. Long-Term Follow-Up of Patients With Mild Coronary Artery Disease and Endothelial Dysfunction. Circulation. 2000;101:948–954. doi: 10.1161/01.cir.101.9.948. [DOI] [PubMed] [Google Scholar]
- 176.Schachinger VZA. Atherosclerosis-associated endothelial dysfunction. Z Kardiol. 2000;89(Suppl 9):70–74. doi: 10.1007/s003920070033. [DOI] [PubMed] [Google Scholar]
- 177.Gokce N, Keaney JF, Hunter LM, Watkins MT, Nedeljkovic ZS, Menzoian JO, Vita JA. Predictive value of noninvasivelydetermined endothelial dysfunction for long-term cardiovascular events inpatients with peripheral vascular disease. Journal of the American College of Cardiology. 2003;41:1769–1775. doi: 10.1016/s0735-1097(03)00333-4. [DOI] [PubMed] [Google Scholar]
- 178.Brennan LA, Steinhorn RH, Wedgwood S, Mata-Greenwood E, Roark EA, Russell JA, Black SM. Increased Superoxide Generation Is Associated With Pulmonary Hypertension in Fetal Lambs. Circulation Research. 2003;92:683–691. doi: 10.1161/01.RES.0000063424.28903.BB. [DOI] [PubMed] [Google Scholar]
- 179.Bowers RCC, Murphy RC, Tuder RM, Hopken MW, Flores SC, Voelkel NF. Oxidative stress in severe pulmonary hypertension. Am J Respir Crit Care Med. 2004;169:764–769. doi: 10.1164/rccm.200301-147OC. [DOI] [PubMed] [Google Scholar]
- 180.KANEKO FT, ARROLIGA ALEJANDROC, DWEIK RAEDA, COMHAIR SUZYA, LASKOWSKI D, OPPEDISANO R, THOMASSEN MARYJ, ERZURUM SERPILC. Biochemical Reaction Products of Nitric Oxide as Quantitative Markers of Primary Pulmonary Hypertension. Am J Respir Crit Care Med. 1998;158:917–923. doi: 10.1164/ajrccm.158.3.9802066. [DOI] [PubMed] [Google Scholar]
- 181.Machado RFLNM, Dweik RA, Hammel J, Janocha A, Pyle J, Laskowski D, Jennings C, Arroloiga AC, Erzurum SC. Nitric oxide and pulmonary arterial pressures in pulmonary hypertension. Free Radical Biology & Medicine. 2004;37:1010–1017. doi: 10.1016/j.freeradbiomed.2004.06.039. [DOI] [PubMed] [Google Scholar]
- 182.Cai H, Harrison DG. Endothelial Dysfunction in Cardiovascular Diseases: The Role of Oxidant Stress. Circulation Research. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 183.Liu JQ, Zelko IN, Folz RJ. Reoxygenation-induced Constriction in Murine Coronary Arteries. Journal of Biological Chemistry. 2004;279:24493–24497. doi: 10.1074/jbc.M402920200. [DOI] [PubMed] [Google Scholar]
- 184.Wedgwood S, Black SM. Induction of apoptosis in fetal pulmonary arterial smooth muscle cells by a combined superoxide dismutase/catalase mimetic. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2003;285:L305–L312. doi: 10.1152/ajplung.00382.2002. [DOI] [PubMed] [Google Scholar]
- 185.Szöcs K, Lassègue B, Sorescu D, Hilenski LL, Valppu L, Couse TL, Wilcox JN, Quinn MT, Lambeth JD, Griendling KK. Upregulation of Nox-Based NAD(P)H Oxidases in Restenosis After Carotid Injury. Arteriosclerosis, Thrombosis, and Vascular Biology. 2002;22:21–27. doi: 10.1161/hq0102.102189. [DOI] [PubMed] [Google Scholar]
- 186.Guzik TJ, Sadowski J, Guzik B, Jopek A, Kapelak B, Przybylowski P, Wierzbicki K, Korbut R, Harrison DG, Channon KM. Coronary Artery Superoxide Production and Nox Isoform Expression in Human Coronary Artery Disease. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26:333–339. doi: 10.1161/01.ATV.0000196651.64776.51. [DOI] [PubMed] [Google Scholar]
- 187.Sorescu D, Weiss D, Lassègue B, Clempus RE, Szöcs K, Sorescu GP, Valppu L, Quinn MT, Lambeth JD, Vega JD, Taylor WR, Griendling KK. Superoxide Production and Expression of Nox Family Proteins in Human Atherosclerosis. Circulation. 2002;105:1429–1435. doi: 10.1161/01.cir.0000012917.74432.66. [DOI] [PubMed] [Google Scholar]
- 188.Vendrov AE, Hakim ZS, Madamanchi NR, Rojas M, Madamanchi C, Runge MS. Atherosclerosis Is Attenuated by Limiting Superoxide Generation in Both Macrophages and Vessel Wall Cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27:2714–2721. doi: 10.1161/ATVBAHA.107.152629. [DOI] [PubMed] [Google Scholar]
- 189.Marui NOM, Swerlick R, Kunsch C, Rosen CA, Ahmad M, Alexander RW, Medford RM. Vascular cell adhesion molecule-1 (VCAM-1) gene transcription and expression are regulated through an antioxidant-sensitive mechanism in human vascular endothelial cells. J Clin Invest. 1993;92:1866–1874. doi: 10.1172/JCI116778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Takatsu H, Tasaki H, Kim H-N, Ueda S, Tsutsui M, Yamashita K, Toyokawa T, Morimoto Y, Nakashima Y, Adachi T. Overexpression of EC-SOD Suppresses Endothelial-Cell-Mediated LDL Oxidation. Biochemical and Biophysical Research Communications. 2001;285:84–91. doi: 10.1006/bbrc.2001.5114. [DOI] [PubMed] [Google Scholar]
- 191.Cominacini L, Garbin U, Pasini AF, Davoli A, Campagnola M, Contessi GB, Pastorino AM, Lo Cascio V. Antioxidants Inhibit the Expression of Intercellular Cell Adhesion Molecule-1 and Vascular Cell Adhesion Molecule-1 Induced by Oxidized LDL on Human Umbilical Vein Endothelial Cells. Free Radical Biology and Medicine. 1997;22:117–127. doi: 10.1016/s0891-5849(96)00271-7. [DOI] [PubMed] [Google Scholar]
- 192.Wung BS, Cheng JJ, Shyue S-K, Wang DL. NO Modulates Monocyte Chemotactic Protein-1 Expression in Endothelial Cells Under Cyclic Strain. Arteriosclerosis, Thrombosis, and Vascular Biology. 2001;21:1941–1947. doi: 10.1161/hq1201.099428. [DOI] [PubMed] [Google Scholar]
- 193.Chen X-L, Zhang Q, Zhao R, Medford RM. Superoxide, H2O2, and iron are required for TNF-α-induced MCP-1 gene expression in endothelial cells: role of Rac1 and NADPH oxidase. American Journal of Physiology - Heart and Circulatory Physiology. 2004;286:H1001–H1007. doi: 10.1152/ajpheart.00716.2003. [DOI] [PubMed] [Google Scholar]
- 194.Devaraj S, Li D, Jialal I. The effects of alpha tocopherol supplementation on monocyte function. Decreased lipid oxidation, interleukin 1 beta secretion, and monocyte adhesion to endothelium. The Journal of Clinical Investigation. 1996;98:756–763. doi: 10.1172/JCI118848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Lerman A, Edwards BS, Hallett JW, Heublein DM, Sandberg SM, Burnett JC. Circulating and Tissue Endothelin Immunoreactivity in Advanced Atherosclerosis. New England Journal of Medicine. 1991;325:997–1001. doi: 10.1056/NEJM199110033251404. [DOI] [PubMed] [Google Scholar]
- 196.Zeiher AM, Goebel H, Schächinger V, Ihling C. Tissue Endothelin-1 Immunoreactivity in the Active Coronary Atherosclerotic Plaque: A Clue to the Mechanism of Increased Vasoreactivity of the Culprit Lesion in Unstable Angina. Circulation. 1995;91:941–947. doi: 10.1161/01.cir.91.4.941. [DOI] [PubMed] [Google Scholar]
- 197.Savale L, Tu L, Rideau D, Izziki M, Maitre B, Adnot S, Eddahibi S. Impact of interleukin-6 on hypoxia-induced pulmonary hypertension and lung inflammation in mice. Respiratory Research. 2009;10(6) doi: 10.1186/1465-9921-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 Overexpression Induces Pulmonary Hypertension. Circulation Research. 2009;104:236–244. doi: 10.1161/CIRCRESAHA.108.182014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Golembeski SM, West J, Tada Y, Fagan KA. Interleukin-6 Causes Mild Pulmonary Hypertension and Augments Hypoxia-Induced Pulmonary Hypertension in Mice*. Chest. 2005;128:572S–573S. doi: 10.1378/chest.128.6_suppl.572S-a. [DOI] [PubMed] [Google Scholar]
- 200.Michael JR, Markewitz BA, Kohan DE. Oxidant stress regulates basal endothelin-1 production by cultured rat pulmonary endothelial cells. American Journal of Physiology - Lung Cellular and Molecular Physiology. 1997;273:L768–L774. doi: 10.1152/ajplung.1997.273.4.L768. [DOI] [PubMed] [Google Scholar]
- 201.Sethi AS, Lees DM, Douthwaite JA, Dawnay AB, Corder R. Homocysteine-Induced Endothelin-1 Release Is Dependent on Hyperglycaemia and Reactive Oxygen Species Production in Bovine Aortic Endothelial Cells. Journal of Vascular Research. 2006;43:175–183. doi: 10.1159/000090947. [DOI] [PubMed] [Google Scholar]
- 202.Syeda F, Tullis E, Slutsky AS, Zhang H. Human neutrophil peptides upregulate expression of COX-2 and endothelin-1 by inducing oxidative stress. American Journal of Physiology - Heart and Circulatory Physiology. 2008;294:H2769–H2774. doi: 10.1152/ajpheart.00211.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Turchan-Cholewo J, Dimayuga VM, Gupta S, Gorospe RMC, Keller JN, Bruce-Keller AJ. NADPH Oxidase Drives Cytokine and Neurotoxin Release from Microglia and Macrophages in Response to HIV-Tat. Antioxidants & Redox Signaling. 2008;11:193–204. doi: 10.1089/ars.2008.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Böhm F, Settergren M, Pernow J. Vitamin C blocks vascular dysfunction and release of interleukin-6 induced by endothelin-1 in humans in vivo. Atherosclerosis. 2007;190:408–415. doi: 10.1016/j.atherosclerosis.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 205.Browatzki M, Schmidt J, Kübler W, Kranzhöfer R. Endothelin-1 induces interleukin-6 release via acctivation of the transcription factor NF-κB in human vascular smooth muscle cells. Basic Research in Cardiology. 2000;95:98–105. doi: 10.1007/s003950050170. [DOI] [PubMed] [Google Scholar]
- 206.Pitrak DL, Mullane KM, Bilek ML, Stevens P, Allen RC. Impaired phagocyte oxidative capacity in patients with human immunodeficiency virus infection. Journal of Laboratory and Clinical Medicine. 1998;132:284–293. doi: 10.1016/s0022-2143(98)90041-5. [DOI] [PubMed] [Google Scholar]
- 207.Koziel H, Li X, Armstrong MYK, Richards FF, Rose RM. Alveolar Macrophages from Human Immunodeficiency Virus-Infected Persons Demonstrate Impaired Oxidative Burst Response to Pneumocystis carinii In Vitro. Am J Respir Cell Mol Biol. 2000;23:452–459. doi: 10.1165/ajrcmb.23.4.4084. [DOI] [PubMed] [Google Scholar]
- 208.Pace GW, Leaf CD. The role of oxidative stress in HIV disease. Free Radic Biol Med. 1995;19:523–528. doi: 10.1016/0891-5849(95)00047-2. [DOI] [PubMed] [Google Scholar]
- 209.Kurata S-i. Sensitization of the HIV-1-LTR upon Long Term Low Dose Oxidative Stress. Journal of Biological Chemistry. 1996;271:21798–21802. doi: 10.1074/jbc.271.36.21798. [DOI] [PubMed] [Google Scholar]
- 210.Adamson GMBR. Tumor necrosis factor induced oxidative stress in isolated mouse hepatocytes. Arch Biochem Biophys. 1992;294:223–229. doi: 10.1016/0003-9861(92)90161-o. [DOI] [PubMed] [Google Scholar]
- 211.Staal FJ, Roederer M, Herzenberg LA. Intracellular thiols regulate activation of nuclear factor kappa B and transcription of human immunodeficiency virus. Proceedings of the National Academy of Sciences. 1990;87:9943–9947. doi: 10.1073/pnas.87.24.9943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Mihm SEJ, Pessara U, Kurth R, Droge W. Inhibition of HIV-1 replication and NK-kappa B activity by cysteine and cysteine derivatives. AIDS. 1991;5:497–503. doi: 10.1097/00002030-199105000-00004. [DOI] [PubMed] [Google Scholar]
- 213.Vicenzi E, Poli G. Regulation of HIV expression by viral genes and cytokines. Journal of Leukocyte Biology. 1994;56:328–334. doi: 10.1002/jlb.56.3.328. [DOI] [PubMed] [Google Scholar]
- 214.Schreck RRP, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991;10:2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Roederer M, Raju PA, Staal FJ, Herzenberg LA, Herzenberg LA. N-acetylcysteine inhibits HIV expression in chronically infected cells. AIDS Res Hum Retroviruses. 1991;7:563–567. doi: 10.1089/aid.1991.7.563. [DOI] [PubMed] [Google Scholar]
- 216.Staal FJ, Anderson MT, Herzenberg LA. Redox regulation of NF-kappa B transcription factor complex: effects of N-acetylcysteine. Methods Enzymol. 1995;252:168–174. doi: 10.1016/0076-6879(95)52019-8. [DOI] [PubMed] [Google Scholar]
- 217.Staal FJ, Roederer M, Raju PA, Anderson MT, Ela SW, Herzenberg LA. Antioxidants inhibit stimulation of HIV transcription. AIDS Res Hum Retroviruses. 1993;9:299–306. doi: 10.1089/aid.1993.9.299. [DOI] [PubMed] [Google Scholar]
- 218.Kalebic T, Kinter A, Poli G, Anderson ME, Meister A, Fauci AS. Suppression of human immunodeficiency virus expression in chronically infected monocytic cells by glutathione, glutathione ester, and N-acetylcysteine. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:986–990. doi: 10.1073/pnas.88.3.986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Garaci E, Palamara AT, Ciriolo MR, D’Agostini C, Abdel-Latif MS, Aquaro S, Lafavia E, Rotilio G. Intracellular GSH content and HIV replication in human macrophages. J Leukoc Biol. 1997;62:54–59. doi: 10.1002/jlb.62.1.54. [DOI] [PubMed] [Google Scholar]
- 220.Palamara AT, Perno CF, Aquaro S, Bue MC, Dini L, Garaci E. Glutathione inhibits HIV replication by acting at late stages of the virus life cycle. AIDS Res Hum Retroviruses. 1996;12:1537–1541. doi: 10.1089/aid.1996.12.1537. [DOI] [PubMed] [Google Scholar]
- 221.Oiry J, Mialocq P, Puy JY, Fretier P, Dereuddre-Bosquet N, Dormont D, Imbach JL, Clayette P. Synthesis and biological evaluation in human monocyte-derived macrophages of N-(N-acetyl-L-cysteinyl)-S-acetylcysteamine analogues with potent antioxidant and anti-HIV activities. Journal of medicinal chemistry. 2004;47:1789–1795. doi: 10.1021/jm030374d. [DOI] [PubMed] [Google Scholar]
- 222.Ho W, Douglas SD. Glutathione and N-acetylcysteine suppression of human immunodeficiency virus replication in human monocytes/macrophages in vitro. AIDS Res Hum Retroviruses. 1992;8:1249–1253. doi: 10.1089/aid.1992.8.1249. [DOI] [PubMed] [Google Scholar]
- 223.Magnani M, Fraternale A, Casabianca A, Schiavano GF, Chiarantini L, Palamara AT, Ciriolo MR, Rotilio G, Garaci E. Antiretroviral effect of combined zidovudine and reduced glutathione therapy in murine AIDS. AIDS Res Hum Retroviruses. 1997;13:1093–1099. doi: 10.1089/aid.1997.13.1093. [DOI] [PubMed] [Google Scholar]
- 224.Palamara AT, Garaci E, Rotilio G, Ciriolo MR, Casabianca A, Fraternale A, Rossi L, Schiavano GF, Chiarantini L, Magnani M. Inhibition of murine AIDS by reduced glutathione. AIDS Res Hum Retroviruses. 1996;12:1373–1381. doi: 10.1089/aid.1996.12.1373. [DOI] [PubMed] [Google Scholar]
- 225.Fraternale A, Casabianca A, Orlandi C, Cerasi A, Chiarantini L, Brandi G, Magnani M. Macrophage protection by addition of glutathione (GSH)-loaded erythrocytes to AZT and DDI in a murine AIDS model. Antiviral Res. 2002;56:263–272. doi: 10.1016/s0166-3542(02)00128-6. [DOI] [PubMed] [Google Scholar]
- 226.Siddappa NB, Venkatramanan M, Venkatesh P, Janki MV, Jayasuryan N, Desai A, Ravi V, Ranga U. Transactivation and signaling functions of Tat are not correlated: biological and immunological characterization of HIV-1 subtype-C Tat protein. Retrovirology. 2006;3:53. doi: 10.1186/1742-4690-3-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Pierleoni R, Menotta M, Antonelli A, Sfara C, Serafini G, Dominici S, Laguardia ME, Salis A, Damonte G, Banci L, Porcu M, Monini P, Ensoli B, Magnani M. Effect of the redox state on HIV-1 tat protein multimerization and cell internalization and trafficking. Molecular and cellular biochemistry. 2010;345:105–118. doi: 10.1007/s11010-010-0564-9. [DOI] [PubMed] [Google Scholar]
- 228.Fanales-Belasio E, Moretti S, Nappi F, Barillari G, Micheletti F, Cafaro A, Ensoli B. Native HIV-1 Tat protein targets monocyte-derived dendritic cells and enhances their maturation, function, and antigen-specific T cell responses. J Immunol. 2002;168:197–206. doi: 10.4049/jimmunol.168.1.197. [DOI] [PubMed] [Google Scholar]
- 229.Kalantari P, Narayan V, Natarajan SK, Muralidhar K, Gandhi UH, Vunta H, Henderson AJ, Prabhu KS. Thioredoxin reductase-1 negatively regulates HIV-1 transactivating protein Tat-dependent transcription in human macrophages. J Biol Chem. 2008;283:33183–33190. doi: 10.1074/jbc.M807403200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Appay V, Sauce D. Immune activation and inflammation in HIV-1 infection: causes and consequences. The Journal of Pathology. 2008;214:231–241. doi: 10.1002/path.2276. [DOI] [PubMed] [Google Scholar]
- 231.Kerttula YVM, Pyhala L, Sariola H, Kostiainen E, Huttunen JK. Effect of bacterial lipopolysaccharide on serum lipids and on the development of aortic atherosclerosis in rabbits. Atherosclerosis. 1986;59:307–312. doi: 10.1016/0021-9150(86)90126-7. [DOI] [PubMed] [Google Scholar]
- 232.Ostos MA, Recalde D, Zakin MM, Scott-Algara D. Implication of natural killer T cells in atherosclerosis development during a LPS-induced chronic inflammation. FEBS Letters. 2002;519:23–29. doi: 10.1016/s0014-5793(02)02692-3. [DOI] [PubMed] [Google Scholar]
- 233.den Dekker WK, Cheng C, Pasterkamp G, Duckers HJ. Toll like receptor 4 in atherosclerosis and plaque destabilization. Atherosclerosis. 2010;209:314–320. doi: 10.1016/j.atherosclerosis.2009.09.075. [DOI] [PubMed] [Google Scholar]
- 234.Young KC, Hussein SMA, Dadiz R, deMello D, Devia C, Hehre D, Suguihara C. Toll-like receptor 4– deficient mice are resistant to chronic hypoxia-induced pulmonary hypertension. Experimental Lung Research. 2010;36:111–119. doi: 10.3109/01902140903171610. [DOI] [PubMed] [Google Scholar]
- 235.Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y, Asano M, Moriwaki H, Seishima M. Lack of Interleukin-1β Decreases the Severity of Atherosclerosis in ApoE-Deficient Mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23:656–660. doi: 10.1161/01.ATV.0000064374.15232.C3. [DOI] [PubMed] [Google Scholar]
- 236.Lim JHUH, Park JW, Lee IK, Kwon TK. Interleukin-1beta promotes the expression of monocyte chemoattractant protein-1 in human aorta smooth muscle cells via multiple signaling pathways. Exp Mol Pathol. 2009;41:757–764. doi: 10.3858/emm.2009.41.10.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Takeda N, Manabe I, Shindo T, Iwata H, Iimuro S, Kagechika H, Shudo K, Nagai R. Synthetic Retinoid Am80 Reduces Scavenger Receptor Expression and Atherosclerosis in Mice by Inhibiting IL-6. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26:1177–1183. doi: 10.1161/01.ATV.0000214296.94849.1c. [DOI] [PubMed] [Google Scholar]
- 238.Saremi A, Anderson RJ, Luo P, Moritz TE, Schwenke DC, Allison M, Reaven PD. Association between IL-6 and the extent of coronary atherosclerosis in the veterans affairs diabetes trial (VADT) Atherosclerosis. 2009;203:610–614. doi: 10.1016/j.atherosclerosis.2008.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Humbert MMG, Brenot F, Sitbon O, Portier A, Grandeot-Keros L, Duroux P, Galanaud P, Simonneau G, Emilie D. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med. 1995;151:1628–1631. doi: 10.1164/ajrccm.151.5.7735624. [DOI] [PubMed] [Google Scholar]
- 240.Bhargava AKA, Yuan N, Gewitz MH, Mathew R. Monocrotaline induced interleukin-6 mRNA expression in rat lungs. Heart Dis. 1999;1:126–132. [PubMed] [Google Scholar]
- 241.Hagen M, Fagan K, Steudel W, Carr M, Lane K, Rodman DM, West J. Interaction of interleukin-6 and the BMP pathway in pulmonary smooth muscle. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2007;292:L1473–L1479. doi: 10.1152/ajplung.00197.2006. [DOI] [PubMed] [Google Scholar]
- 242.Marandin A, Katz A, Oksenhendler E, Tulliez M, Picard F, Vainchenker W, Louache F. Loss of primitive hematopoietic progenitors in patients with human immunodeficiency virus infection. Blood. 1996;88:4568–4578. [PubMed] [Google Scholar]
- 243.Jenkins M, Hanley MB, Moreno MB, Wieder E, McCune JM. Human Immunodeficiency Virus-1 Infection Interrupts Thymopoiesis and Multilineage Hematopoiesis In Vivo. Blood. 1998;91:2672–2678. [PubMed] [Google Scholar]
- 244.Moses A, Nelson J, Bagby GC. The Influence of Human Immunodeficiency Virus-1 on Hematopoiesis. Blood. 1998;91:1479–1495. [PubMed] [Google Scholar]
- 245.Zhao YD, Courtman DW, Deng Y, Kugathasan L, Zhang Q, Stewart DJ. Rescue of Monocrotaline-Induced Pulmonary Arterial Hypertension Using Bone Marrow-Derived Endothelial-Like Progenitor Cells. Circulation Research. 2005;96:442–450. doi: 10.1161/01.RES.0000157672.70560.7b. [DOI] [PubMed] [Google Scholar]
- 246.Ormiston ML, Deng Y, Stewart DJ, Courtman DW. Innate Immunity in the Therapeutic Actions of Endothelial Progenitor Cells in Pulmonary Hypertension. Am J Respir Cell Mol Biol. 2010;43:546–554. doi: 10.1165/rcmb.2009-0152OC. [DOI] [PubMed] [Google Scholar]
- 247.Taraseviciene-Stewart L, Nicolls MR, Kraskauskas D, Scerbavicius R, Burns N, Cool C, Wood K, Parr JE, Boackle SA, Voelkel NF. Absence of T Cells Confers Increased Pulmonary Arterial Hypertension and Vascular Remodeling. Am J Respir Crit Care Med. 2007;175:1280–1289. doi: 10.1164/rccm.200608-1189OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Austin ED, Rock MT, Mosse CA, Vnencak-Jones CL, Yoder SM, Robbins IM, Loyd JE, Meyrick BO. T lymphocyte subset abnormalities in the blood and lung in pulmonary arterial hypertension. Respiratory Medicine. 2010;104:454–462. doi: 10.1016/j.rmed.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Kolbus D, Ramos O, Berg K, Persson J, Wigren M, Bjorkbacka H, Fredrikson G, Nilsson J. CD8+ T cell activation predominate early immune responses to hypercholesterolemia in Apoe−/− mice. BMC Immunology. 2010;11:58. doi: 10.1186/1471-2172-11-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Kamide Y, Utsugi M, Dobashi K, Ono A, Ishizuka T, Hisada T, Koga Y, Uno K, Hamuro J, Mori M. Intracellular glutathione redox status in human dendritic cells regulates IL-27 production and T-cell polarization. Allergy. 2011;66:1183–1192. doi: 10.1111/j.1398-9995.2011.02611.x. [DOI] [PubMed] [Google Scholar]
- 251.Peterson JD, Herzenberg LA, Vasquez K, Waltenbaugh C. Glutathione levels in antigen-presenting cells modulate Th1 versus Th2 response patterns. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:3071–3076. doi: 10.1073/pnas.95.6.3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Romagnani S. Lymphokine production by human T cells in disease states. Annual review of immunology. 1994;12:227–257. doi: 10.1146/annurev.iy.12.040194.001303. [DOI] [PubMed] [Google Scholar]
- 253.McArthur JCHD, Bacellat H, Miller EN, Cohen BA, Becker JT, Graham NM, McArthur JH, Selnes OA, Jacobson LP, et al. Dementia in AIDS patients: incidence and risk factors. Multicenter AIDS Cohort Study. Neurology. 1993;43:2245–2253. doi: 10.1212/wnl.43.11.2245. [DOI] [PubMed] [Google Scholar]
- 254.Desquilbet LJL, Fried LP, Phair JP, Jamieson BD, Holloway M, Margolick JB Multicenter AIDS Cohort Study. HIV-1 infection is associated with an earlier occurrence of a phenotype related to frailty. J Gerontol A Biol Med Sci. 2007;62:1279–1286. doi: 10.1093/gerona/62.11.1279. [DOI] [PubMed] [Google Scholar]
- 255.Hurwitz BEKN, Llabre MM, Maher KJ, Skyler JS, Bilsker MS, McPherson-Baker S, Lawrence PJ, Laperriere AR, Greeson JM, Klaus JR, Lawrence R, Schneiderman N. HIV, metabolic syndrome X, inflammation, oxidative stress, and coronary heart disease risk: role of protease inhibitor exposure. Cardiovasc Toxicol. 2004;4:303–316. doi: 10.1385/ct:4:3:303. [DOI] [PubMed] [Google Scholar]
- 256.Halliwell B. Antioxidant defence mechanisms: from beginning to the end (of the beginning) Free Radic Res. 1999;31:261–272. doi: 10.1080/10715769900300841. [DOI] [PubMed] [Google Scholar]
- 257.Stephenson CBMG, Jacob RA, Kruzich LA, Douglas SD, Wilson CM. Vitamins C and E in adolescents and young adults with HIV infection. Am J Clin Nutr. 2006;83:870–879. doi: 10.1093/ajcn/83.4.870. [DOI] [PubMed] [Google Scholar]
- 258.Kruzich LA, Marquis GS, Carriquiry AL, Wilson CM, Stephensen CB. US youths in the early stages of HIV disease have low intakes of some micronutrients important for optimal immune function. Journal of the American Dietetic Association. 2004;104:1095–1101. doi: 10.1016/j.jada.2004.04.021. [DOI] [PubMed] [Google Scholar]
- 259.Tang AMGN, Kirby AJ, McCall LD, Willet WC, Saah AJ. Dietary micronutrient intake risk of progression to acquired immunodeficiency syndrome (AIDS) in human immunodeficiency virus type 1 (HIV-1)-infected homosexual men. Am J Epidemiol. 1993;138:937–951. doi: 10.1093/oxfordjournals.aje.a116814. [DOI] [PubMed] [Google Scholar]
- 260.Tang AM, Graham NMH, Semba RD, Saah AJ. Association between serum vitamin A and E levels and HIV-1 disease progression. AIDS. 1997;11:613–620. doi: 10.1097/00002030-199705000-00009. [DOI] [PubMed] [Google Scholar]
- 261.Fawzi WW, Msamanga GI, Spiegelman D, Wei R, Kapiga S, Villamor E, Mwakagile D, Mugusi F, Hertzmark E, Essex M, Hunter DJ. A Randomized Trial of Multivitamin Supplements and HIV Disease Progression and Mortality. New England Journal of Medicine. 2004;351:23–32. doi: 10.1056/NEJMoa040541. [DOI] [PubMed] [Google Scholar]
- 262.McClelland RS, Baeten JM, Overbaugh J, Richardson BA, Mandaliya K, Emery S, Lavreys L, Ndinya-Achola JO, Bankson DD, Bwayo JJ, Kreiss JK. Micronutrient Supplementation Increases Genital Tract Shedding of HIV-1 in Women: Results of a Randomized Trial. JAIDS Journal of Acquired Immune Deficiency Syndromes. 2004;37:1657–1663. doi: 10.1097/00126334-200412150-00021. [DOI] [PubMed] [Google Scholar]
- 263.Allard JP, Aghdassi E, Chau J, Tam C, Kovacs CM, Salit IE, Walmsley SL. Effects of vitamin E and C supplementation on oxidative stress and viral load in HIV-infected subjects. AIDS. 1998;12:1653–1659. doi: 10.1097/00002030-199813000-00013. [DOI] [PubMed] [Google Scholar]
- 264.Spada C, Treitinger Ac, Reis M, Masokawa IY, Verdi JlC, Luiz MC, Silveira MVS, Oliveira OV, Michelon CM, Avila-Junior Sl, Gil IDO, Ostrowsky S. An Evaluation of Antiretroviral Therapy Associated with α-Tocopherol Supplementation in HIV-infected Patients. Clinical Chemistry and Laboratory Medicine. 2002;40:456–459. doi: 10.1515/CCLM.2002.078. [DOI] [PubMed] [Google Scholar]
- 265.Kupka R, Mugusi F, Aboud S, Msamanga GI, Finkelstein JL, Spiegelman D, Fawzi WW. Randomized, double-blind, placebo-controlled trial of selenium supplements among HIV-infected pregnant women in Tanzania: effects on maternal and child outcomes. The American Journal of Clinical Nutrition. 2008;87:1802–1808. doi: 10.1093/ajcn/87.6.1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Hurwitz BE, Klaus JR, Llabre MM, Gonzalez A, Lawrence PJ, Maher KJ, Greeson JM, Baum MK, Shor-Posner G, Skyler JS, Schneiderman N. Suppression of Human Immunodeficiency Virus Type 1 Viral Load With Selenium Supplementation: A Randomized Controlled Trial. Arch Intern Med. 2007;167:148–154. doi: 10.1001/archinte.167.2.148. [DOI] [PubMed] [Google Scholar]
- 267.Jariwalla RJ, Lalezari J, Cenko D, Mansour SE, Kumar A, Gangapurkar B, Nakamura D. Restoration of Blood Total Glutathione Status and Lymphocyte Function Following α-Lipoic Acid Supplementation in Patients with HIV Infection. The Journal of Alternative and Complementary Medicine. 2008;14:139–146. doi: 10.1089/acm.2006.6397. [DOI] [PubMed] [Google Scholar]
- 268.Kaspar JW, Niture SK, Jaiswal AK. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol Med. 2009;47:1304–1309. doi: 10.1016/j.freeradbiomed.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Ungvari Z, Bagi Z, Feher A, Recchia FA, Sonntag WE, Pearson K, de Cabo R, Csiszar A. Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am J Physiol Heart Circ Physiol. 2010;299:H18–24. doi: 10.1152/ajpheart.00260.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Chen JS, Huang PH, Wang CH, Lin FY, Tsai HY, Wu TC, Lin SJ, Chen JW. Nrf-2 mediated heme oxygenase-1 expression, an antioxidant-independent mechanism, contributes to anti-atherogenesis and vascular protective effects of Ginkgo biloba extract. Atherosclerosis. 2011;214:301–309. doi: 10.1016/j.atherosclerosis.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 271.Zakkar M, Van der Heiden K, Luong le A, Chaudhury H, Cuhlmann S, Hamdulay SS, Krams R, Edirisinghe I, Rahman I, Carlsen H, Haskard DO, Mason JC, Evans PC. Activation of Nrf2 in endothelial cells protects arteries from exhibiting a proinflammatory state. Arterioscler Thromb Vasc Biol. 2009;29:1851–1857. doi: 10.1161/ATVBAHA.109.193375. [DOI] [PubMed] [Google Scholar]
- 272.Chen XL, Dodd G, Thomas S, Zhang X, Wasserman MA, Rovin BH, Kunsch C. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am J Physiol Heart Circ Physiol. 2006;290:H1862–1870. doi: 10.1152/ajpheart.00651.2005. [DOI] [PubMed] [Google Scholar]
- 273.Levonen AL, Inkala M, Heikura T, Jauhiainen S, Jyrkkanen HK, Kansanen E, Maatta K, Romppanen E, Turunen P, Rutanen J, Yla-Herttuala S. Nrf2 gene transfer induces antioxidant enzymes and suppresses smooth muscle cell growth in vitro and reduces oxidative stress in rabbit aorta in vivo. Arterioscler Thromb Vasc Biol. 2007;27:741–747. doi: 10.1161/01.ATV.0000258868.80079.4d. [DOI] [PubMed] [Google Scholar]
- 274.Sussan TE, Jun J, Thimmulappa R, Bedja D, Antero M, Gabrielson KL, Polotsky VY, Biswal S. Disruption of Nrf2, a key inducer of antioxidant defenses, attenuates ApoE-mediated atherosclerosis in mice. PloS one. 2008;3:e3791. doi: 10.1371/journal.pone.0003791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Ichikawa T, Li J, Meyer CJ, Janicki JS, Hannink M, Cui T. Dihydro-CDDO-trifluoroethyl amide (dh404), a novel Nrf2 activator, suppresses oxidative stress in cardiomyocytes. PloS one. 2009;4:e8391. doi: 10.1371/journal.pone.0008391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Sussan TE, Rangasamy T, Blake DJ, Malhotra D, El-Haddad H, Bedja D, Yates MS, Kombairaju P, Yamamoto M, Liby KT, Sporn MB, Gabrielson KL, Champion HC, Tuder RM, Kensler TW, Biswal S. Targeting Nrf2 with the triterpenoid CDDO-imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:250–255. doi: 10.1073/pnas.0804333106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Redout EM, van der Toorn A, Zuidwijk MJ, van de Kolk CWA, van Echteld CJA, Musters RJP, van Hardeveld C, Paulus WJ, Simonides WS. Antioxidant treatment attenuates pulmonary arterial hypertension-induced heart failure. American Journal of Physiology - Heart and Circulatory Physiology. 2010;298:H1038–H1047. doi: 10.1152/ajpheart.00097.2009. [DOI] [PubMed] [Google Scholar]
- 278.Lachmanová V, Hnilicková O, Povýsilová V, Hampl V, Herget J. N-acetylcysteine inhibits hypoxic pulmonary hypertension most effectively in the initial phase of chronic hypoxia. Life Sciences. 2005;77:175–182. doi: 10.1016/j.lfs.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 279.Antoniades C, Tousoulis D, Tentolouris C, Toutouzas P, Stefanadis C. Oxidative Stress, Antioxidant Vitamins, and Atherosclerosis. Herz. 2003;28:628–638. doi: 10.1007/s00059-003-2417-8. [DOI] [PubMed] [Google Scholar]
- 280.Violi FMF, Juliano L. Antioxidants and atherosclerosis. European Heart Journal Supplements. 2004;4(Supplement B):B17–B21. [Google Scholar]
- 281.Jones DP. Redefining Oxidative Stress. Antioxidants & Redox Signaling. 2006;8:1865–1879. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- 282.Hansen JM, Go Y-M, Jones DP. NUCLEAR AND MITOCHONDRIAL COMPARTMENTATION OF OXIDATIVE STRESS AND REDOX SIGNALING. Annual Review of Pharmacology and Toxicology. 2006;46:215–234. doi: 10.1146/annurev.pharmtox.46.120604.141122. [DOI] [PubMed] [Google Scholar]
- 283.Go Y-M, Jones DP. Intracellular Proatherogenic Events and Cell Adhesion Modulated by Extracellular Thiol/Disulfide Redox State. Circulation. 2005;111:2973–2980. doi: 10.1161/CIRCULATIONAHA.104.515155. [DOI] [PubMed] [Google Scholar]
- 284.Jiang S, Moriarty-Craige SE, Orr M, Cai J, Sternberg P, Jones DP. Oxidant-Induced Apoptosis in Human Retinal Pigment Epithelial Cells: Dependence on Extracellular Redox State. Investigative Ophthalmology & Visual Science. 2005;46:1054–1061. doi: 10.1167/iovs.04-0949. [DOI] [PubMed] [Google Scholar]
- 285.Isseman IGS. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 286.Calnek DS, Mazzella L, Roser S, Roman J, Hart CM. Peroxisome Proliferator-Activated Receptor γ Ligands Increase Release of Nitric Oxide From Endothelial Cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23:52–57. doi: 10.1161/01.atv.0000044461.01844.c9. [DOI] [PubMed] [Google Scholar]
- 287.Hwang J, Kleinhenz DJ, Lassègue B, Griendling KK, Dikalov S, Hart CM. Peroxisome proliferator-activated receptor-γ ligands regulate endothelial membrane superoxide production. American Journal of Physiology - Cell Physiology. 2005;288:C899–C905. doi: 10.1152/ajpcell.00474.2004. [DOI] [PubMed] [Google Scholar]
- 288.Kleinhenz JM, Kleinhenz DJ, You S, Ritzenthaler JD, Hansen JM, Archer DR, Sutliff RL, Hart CM. Disruption of endothelial peroxisome proliferator-activated receptor-γ reduces vascular nitric oxide production. American Journal of Physiology - Heart and Circulatory Physiology. 2009;297:H1647–H1654. doi: 10.1152/ajpheart.00148.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Huang W, Eum SY, András IE, Hennig B, Toborek M. PPARα and PPARγ attenuate HIV-induced dysregulation of tight junction proteins by modulations of matrix metalloproteinase and proteasome activities. The FASEB Journal. 2009;23:1596–1606. doi: 10.1096/fj.08-121624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Ramirez SH, Heilman D, Morsey B, Potula R, Haorah J, Persidsky Y. Activation of Peroxisome Proliferator-Activated Receptor γ (PPARγ) Suppresses Rho GTPases in Human Brain Microvascular Endothelial Cells and Inhibits Adhesion and Transendothelial Migration of HIV-1 Infected Monocytes. The Journal of Immunology. 2008;180:1854–1865. doi: 10.4049/jimmunol.180.3.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Ji Y, Liu J, Wang Z, Li Z. PPAR[gamma] agonist rosiglitazone ameliorates LPS-induced inflammation in vascular smooth muscle cells via the TLR4/TRIF/IRF3/IP-10 signaling pathway. Cytokine. 2011 doi: 10.1016/j.cyto.2011.05.020. In Press, Corrected Proof. [DOI] [PubMed] [Google Scholar]
- 292.Kurebayashi S, Xu X, Ishii S, Shiraishi M, Kouhara H, Kasayama S. A novel thiazolidinedione MCC-555 down-regulates tumor necrosis factor-[alpha]-induced expression of vascular cell adhesion molecule-1 in vascular endothelial cells. Atherosclerosis. 2005;182:71–77. doi: 10.1016/j.atherosclerosis.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 293.Prakash O, Teng S, Ali M, Zhu X, Coleman R, Dabdoub RA, Chambers R, Aw TY, Flores SC, Joshi BH. The Human Immunodeficiency Virus Type 1 Tat Protein Potentiates Zidovudine-Induced Cellular Toxicity In Transgenic Mice. Archives of Biochemistry and Biophysics. 1997;343:173–180. doi: 10.1006/abbi.1997.0168. [DOI] [PubMed] [Google Scholar]