

Abstract
In the aldohexopyranose idose, the unique presence of three axial ring hydroxyl groups causes considerable conformational flexibility, rendering it challenging to study experimentally and an excellent model for rationalising the relationship between puckering and anomeric configuration. Puckering in methyl α- and β-l-idopyranosides was predicted from kinetically rigorous 10 μs simulations using GLYCAM11 and three explicit water models (TIP3P, TIP4P and TIP4P-EW). In each case, computed pyranose ring three-bond (vicinal) 1H-1H spin-couplings (3JH,H) trended with NMR measurements. These values, calculated puckering exchange rates and free energies were independent of the water model. The α- and β-anomers were 1C4 chairs for 85% and >99% of their respective trajectories and underwent 1C4→4C1 exchange at rates of 20 μs-1 and 1 μs-1. Computed α-anomer 1C4↔4C1 puckering rates depended on the exocyclic C6 substituent, comparing hydroxymethyl with carboxyl from previous work. The slower kinetics and restricted pseudorotational profile of the β-anomer were caused by water occupying a cavity bounded by the anomeric 1-O-methyl and the C6 hydroxymethyl groups. This finding rationalises the different methyl α- and β-l-idopyranoside 3JH,H values. Identifying a relationship between idopyranose anomeric configuration, μs-puckering and water structure facilitates engineering of biologically and commercially important derivatives and underpins deciphering presently elusive structure-function relationships in the glycome.
Keywords: anomer, microsecond simulation, idopyranosides, pucker, water structure
Introduction
Understanding the conformational dynamics of carbohydrates in aqueous solution is central to rationalising their plethora of bioactivities. This goal promises to enable structure-based design of new medicines and materials, but its realisation remains challenging due to experimental and computational difficulties of probing carbohydrate motions, which occur over a broad range of timescales. Glycosidic linkages librate on nanosecond timescales while pyranose ring conformational exchange (or puckering) and anomerization are microsecond1 and millisecond timescale phenomena, respectively. The latter process is influenced by the anomeric effect, whose electronic origin remains controversial.2
The methyl l-idopyranosides 1 and 2 (Figure 1) are unique among the simple l-aldohexopyranosides because, in the 1C4 chair conformation, they exhibit three axial hydroxyl groups at C2, C3 and C4; all other aldohexopyranosides contain a mixture of axial and equatorial hydroxyl substituents at these sites. The idohexopyranosyl ring configuration is thus associated with considerable conformational heterogeneity in water that depends on anomeric configuration.3 Rationalising this association could help explain structure-function relationships in the wider glycome (starch and cellulose differ only in their linkage stereochemistry), but it would be difficult to explore in sugars with more stable puckering signatures (e.g., glucose).4 Idohexopyranoses are important precursors in much needed new approaches to heparan sulphate synthesis.5 Furthermore, methyl α-l-idopyranosiduronic acid (α-l-IdoA- 3, Figure 1), a constituent in heparan and dermatan sulfates and the widely used antithrombic drug heparin,6 was recently predicted1 to undergo microsecond puckering that depends on substituents and epimerization and is crucial to bioactivity (e.g., anticoagulation).7
Figure 1.
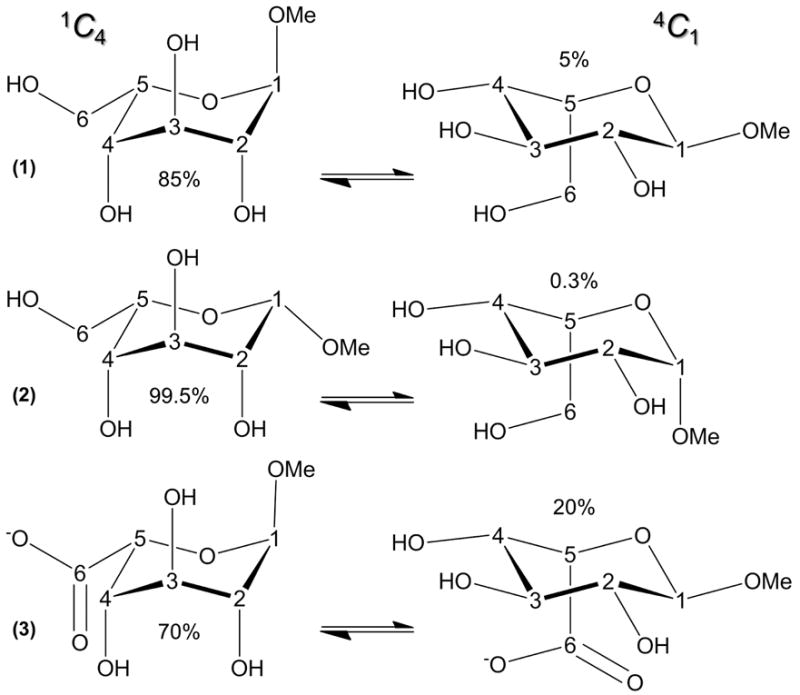
Chair puckers and calculated populations of methyl α- (1) and β- (2) l-idohexopyranosides studied using 10 μs unbiased explicit solvent molecular dynamics and NMR spectroscopy (d-isomers). Structures and computed populations of methyl α-l-idopyranuronate (3), studied previously,1 are illustrated for comparison.
Prior work has shown that three-bond (vicinal) 1H-1H spin-couplings (3JH,H) differ in the α- and β-d-idopyranoses,3 indicating different conformational equilibria. The distribution of puckers in the α-pyranose may include skew-boats in aqueous solution,3 however, the kinetics of 1C4↔4C1 exchange remain unknown. Unfortunately, experimental 3JH,H values provide no kinetic information, and puckering rates derived from previous non-equilibrium picosecond8 ab initio simulations and nanosecond9 enhanced sampling molecular dynamics (MD) must be treated with caution since perturbations applied to traverse high (≈8-10 kcal mol-1) free energy barriers associated with chair-chair interconversion distort the kinetics.10 In order to better understand puckering dynamics and the effect of C6 substitution in idohexopyranosyl rings and to facilitate exploitation of puckering in structure-based design, we sought to reproduce the experimentally observed dependence of the methyl idopyranoside puckering equilibrium on anomeric configuration using kinetically rigorous microsecond simulations, in which conformational sampling is not artificially enhanced (e.g. by elevated temperature).
Water plays a central role in defining oligosaccharide conformation by influencing glycosidic linkage structure.11 The effect of water and different water models on pyranose ring puckering has not been studied previously using microsecond simulations. The present explicit solvent MD (six 10 μs trajectories) converge aqueous puckering in the methyl l-idopyranosides 1 and 2 for the first time. Comparison of these new data with those from a previous 10 μs simulation of 3 shows that idopyranosyl ring 1C4↔4C1 exchange kinetics depend on the C6 substituent structure. Predicted 3JH,H values in 1 and 2 trended with NMR measurements made on the d isomers of 1 and 2 at 750 MHz. These computed properties, calculated chair-chair exchange rates, and relative free energies were unaffected by the type of water potential used in the simulations, comparing three models often used for biomolecular systems12: TIP3P,13 TIP4P14 and TIP4P-EW.15 The experimentally observed puckering behaviours of 1 and 2 were explained by different water structure around each anomer and not by intramolecular steric interactions.
Methods
Molecular dynamics
Periodic molecular dynamics simulations of charge-neutral methyl α- and β-l-idopyranosides 1 and 2 were extended to 10 μs using NVIDIA graphics processing units and a development version of ACEMD16 software (2108). Both monosaccharides were modelled using the GLYCAM11 force field (Supporting Information), each was solvated in an explicit water box of equal side length (28 Å) and three solvent potentials were compared: TIP3P,13 TIP4P14 and TIP4P-EW.15 Following initial conjugate-gradient energy minimisation (1000 steps), each system was heated from 0 to 298K and equilibrated in the NPT ensemble (for 20 ns) prior to 10.25 μs NVT production dynamics, the first 250 ns were discarded and data were recorded at 10 ps intervals. The velocity-Verlet integration algorithm and a hydrogen mass repartitioning scheme allowed a 4 fs time-step without affecting the equilibrium distribution.17 Hydrogen atoms were constrained using the M-SHAKE algorithm18 and electrostatic interactions were calculated via the PME method with a grid spacing of 0.9 Å (in the X, Y and Z dimensions). Electrostatic and van der Waals interactions were truncated at 9 Å and a scaling factor of 1.0 was employed for carbohydrate 1-4 interactions,19 the importance of this term has been addressed recently.20 Gas phase simulations were performed identically; however a cubic box of side length 20 Å and a PME grid spacing of 1.0 Å were used.
Calculation of molecular properties
All molecular properties were computed using the complete 10 μs trajectories (1,000,000 data points). Puckering was quantified using the Cremer-Pople21 parameters θ, ϕ and Q, which were derived using the GROMACS22 analysis tool g_puckering. Relative free energies (ΔG) of binned puckers were calculated using the standard relationship ΔG=kTln(p1/p2), where k is the Boltzmann constant, T is temperature (289K) and p is probability. Puckers were placed in bins of equal area on the Cremer-Pople sphere (unequal spacing in the azimuthal angle θ and equal spacing in the meridian angle ϕ). To achieve this, linear binned values were divided by sin θ to normalize for the area on the sphere (the circumference at azimuthal angle θ is 2πrsinθ on a sphere of radius r). Rotamer populations were computed as described previously23 and pyranose ring 1H-1H vicinal spin-couplings were calculated using the substituent-adjusted Karplus equations of Altona and Haasnoot24 (see Supporting Information). Radial distribution functions (RDFs) and water densities were derived using the AMBER25 tool ptraj (keywords “radial” and “grid”, respectively). The RDF bin spacing was set to 0.05 Å.
Synthesis of methyl α- and β-d-idopyranosides
d-Idose was prepared by cyanohydrin reduction using d-xylose and KCN as substrates.26 The C2-epimeric products, d-idose and d-gulose, were separated by chromatography on a column (3 cm × 100 cm) containing Dowex 50 × 8 (200-400 mesh) ion-exchange resin in the Ca+2 form;27 idoses eluted first, followed by gulose. d-Idose was converted into the methyl d-idopyranosides by Fischer glycosidation as described previously.28 After the reaction was complete (∼2 h), the resin catalyst was removed by vacuum filtration, and the methanolic solution was concentrated at 30° C in vacuo to give a syrup. 13C NMR of the syrup in 2H2O showed that it contained idofuranosides and idopyranosides, and the 1,6-anhydro derivative (the glycosidation reaction time was kept to a minimum to reduce the amount of the latter). This syrup was dissolved in a minimal amount of distilled water and the solution was applied to a column (2.5 cm × 100 cm) containing Dowex 1 × 8 (200-400 mesh) ion-exchange resin in the OH form.29 The column was eluted with distilled, decarbonated water, and the effluent was assayed with phenol-sulfuric acid30 to locate the eluting pyranosides. Careful pooling of fractions gave >95% pure samples of methyl α-d-idopyranoside and methyl β-d-idopyranoside as determined by 1H NMR; the anomers were assigned from their anomeric 1H NMR signal multiplicities by analogy to those reported for α- and β-d-idopyranoses.3
NMR Spectroscopy
High-resolution 1H NMR spectra of the methyl α- and β-d-idopyranosides in 2H2O were obtained at 750 MHz and 25° C. Spectra were collected with 2000-3000 Hz sweep widths and 32K points, and FIDs were zero-filled once before processing with resolution enhancement to improve spectral resolution. Since 1H spectra of 1 and 2 were not first-order at 750 MHz, the data were simulated using the MacNUTs program (MacNUTs Pro; Acorn NMR Inc.: Livermore, CA) to extract accurate chemical shifts and 3JH,H values. Reported 1H chemical shifts (Supporting Information) are accurate to ±0.002 ppm and reported 3JH,H values (Table 1) are accurate to ±0.2 Hz. The 1H chemical shifts were referenced to the internal residual HOD signal at 4.8 ppm. 13C[1H] NMR spectra of 1 and 2 were obtained at 150 MHz in 2H2O and 21° C. Spectra were collected with 8500 Hz sweep widths and 128K points, and FIDs were zero-filled once before processing with resolution enhancement to improve spectral resolution. 13C chemical shifts (Supporting Information) were referenced externally to the C1 signal of α-d-[1-13C]mannose (95.5 ppm).31
Table 1.
Calculated (TIP3P, TIP4P, TIP4P-EW) and experimental (EXPT) 3JH,H values (Hz) for 1 and 2.
| Anomer | 3JH,H | TIP3P | TIP4P | TIP4P-EW | EXPT |
|---|---|---|---|---|---|
| α | J1,2 | 2.4 | 2.6 | 2.6 | 4.3 |
| α | J2,3 | 2.8 | 3.0 | 2.9 | 6.7 |
| α | J3,4 | 3.1 | 3.2 | 3.2 | 6.1 |
| α | J4,5 | 1.3 | 1.6 | 2.8 | 3.6 |
| β | J1,2 | 1.6 | 1.7 | 1.7 | 1.7 |
| β | J2,3 | 3.0 | 2.9 | 2.9 | 4.1 |
| β | J3,4 | 3.0 | 2.9 | 2.9 | 4.6 |
| β | J4,5 | 1.1 | 1.4 | 2.8 | 2.5 |
Calculated 3JH,H values were derived from 10 μs trajectories using the substituent-adjusted Karplus equations of Altona and Haasnoot.24 Computed and experimental values correspond to l- and d-forms of the sugars, respectively. Calculated values not trending with the experimental data (where the β-anomer 3JH,H value was equal to or greater than the α-anomer 3JH,H value) are bold.
Results and Discussion
Molecular 3D-properties and comparison with experiment
Based on our previous metric for conformational convergence (determined by the average value of cos θ, where θ is the Cremer-Pople21 polar angle),1 the dynamic 3D-ensemble of ring puckers in 1 and 2 approached the simulation average pucker after approximately 5 μs of explicit solvent MD, irrespective of the water model used (see Supporting Information). Puckering convergence took longer than found previously for net charge-neutral explicit solvent simulations of 3, methyl 2-O-sulfo-IdoA (dianion) and methyl β-d-glucuronate (anion),1 methyl β-d-glucopyranoside, β-d-galactopyranoside, N-acetyl-β-d-galactosaminide, α-d-mannopyranoside and α-l-fucopyranoside,4 and five methyl and free α-anomer d-hexosamines23 (the appropriate number of Na+ counterions were used to neutralise the ensembles). Both 1 and 2 underwent 1C4↔4C1 transitions with all three water models and their average puckers were independent of the water potential (see Supporting Information). In 1, the average Cremer-Pople21 pucker parameters θ, ϕ and Q (see Methods) for 1C4 chairs were 169° (±6°), 135° (±70°) and 0.53 (±0.04); in 2, these values were 171 ° (±6°), 195° (±80°) and 0.57 (±0.04), respectively. These results show a larger average value of ϕ in the β-anomer, indicating a shift from puckers in the 2SO-region (ϕ≈150°) to those in the 1S3-region (ϕ ≈210°). The increased Q value in the β-anomer indicates a more chair-like (less flat) pucker relative to the α-anomer. For the 4C1 chairs of 1, the average θ, ϕ and Q values were 12° (±6°), ≈158° (±100°) and 0.54 (±0.04); in 2 these values were 14° (±7°), ≈151° (±55°) and 0.53 (±0.04), respectively. The 4C1 chair thus appears to be less sensitive to anomeric configuration than the 1C4 form.
In the 1C4 pucker of 1 and 2, all ring vicinal proton-proton pairs are gauche (axial-equatorial or equatorial-equatorial), and therefore relatively small 3JH,H values are expected (≈1-4 Hz). In the 4C1 chair, 3J1,2, 3J2,3 and 3J3,4 in the α-anomer, and 3J2,3 and 3J3,4 in the β-anomer, correspond to trans (diaxial) coupled protons, and thus large couplings are expected (≈10 Hz). Computed 3JH,H values were in general agreement with these expectations and with prior3 and new experimental data (Table 1) in that couplings for the α-anomer were larger and thus indicative of a greater proportion of 4C1 form in aqueous solution than found for the β-anomer. Three minor exceptions were noted (bold in Table 1): J2,3 (TIP3P and TIP4P-EW) and J4,5 (TIP4P-EW), in which the computed 3JH,H values were either identical in both anomers or at most 0.2 Hz greater in the α-anomer (i.e., within the estimated experimental error of ±0.2 Hz). The computed 3JH,H values for the α-anomer were all smaller than the corresponding experimental observations (≈2-4Hz). In the β-anomer, the maximum difference between calculated and experimental 3JH,H values was ±2 Hz. All computed 3JH,H values were effectively independent of the water model (within 0.2 Hz of each other), except for 3J4,5, which differed at most by 1.7 Hz. Although the absolute computed and experimental 3JH,H values for 1 differed by several Hz, the comparisons in Table 1 provide confidence that the force fields reproduced qualitatively the relative differences between the α- and β-l-idopyranoside equilibrium puckering distributions. The NMR data predicts a more balanced 1C4:4C1 equilibria than does the MD data (see Supporting Information, two-site models). Improvement of the computed 3JH,H values for the very finely balanced conformational equilibria of 1 and 2 can be achieved via small corrections to force field parameters.
The computed exocyclic hydroxymethyl rotamer populations in 1 and 2 were similar and unaffected by the water models used (Supporting Information). Using the TIP3P water model, the tg:gt:gg states for the α- and β-l-idopyranoside were populated (±1%) at 17:14:69 and 18:8:75, respectively. The experimental populations for the d isomers, in which the gt and gg states are reversed with respect to the l isomer, were (±5%) 24:0:76 (α-anomer) and 29:4:67 (β-anomer), indicating a slight over-estimation of gt in the simulations and faithful representation of the experimentally observed trend for gg >> tg > gt.
Puckering thermodynamics and kinetics
In the simulations of 1 and 2 using three water models, the 1C4 and 4C1 conformers were the two most populated (lowest energy) ring puckers (Figure 2 and Supporting Information). Ratios of 1C4:4C1 chairs in the a-anomer simulations were 85:5 (TIP3P) and 84:7 (TIP4P) and 84:8 (TIP4P-EW), resulting in computed free energy differences (ΔG) between the two chairs of 1.7 (TIP3P) and 1.4 kcal mol-1 (TIP4P and TIP4P-EW). For the β-anomer, the 1C4:4C1 ratios were 99.5:0.3 (TIP3P), 99.4:0.4 (TIP4P) and 99.8:0.1 (TIP4P-EW) and the computed ΔG values were, respectively, 3.5, 3.3 ad 3.9 kcal mol-1. In addition to having a higher energy 4C1 pucker, pivotal 3D-intermediates in the β-anomer simulation were predicted to have higher free energies when compared to the corresponding α-anomer puckers (Figure 2 and Supporting Information). For example, the lowest energy non-chair puckers in the α- and β-anomer simulations (for all water models) were B1,4 and 5S1, respectively, which were ≈2 and ≈4 kcal mol-1 higher in free energy cf. 1C4. In the α-anomer, 5S1, 3S1, 2SO and 1S3 puckers were ≤ 3 kcal mol-1 above the 1C4 energy. For the β-anomer, the next lowest energy puckers (B1,4, 2,5B, 1,4B and 2SO) were predicted to be > 5 kcal mol-1 above the 1C4 form.
Figure 2.
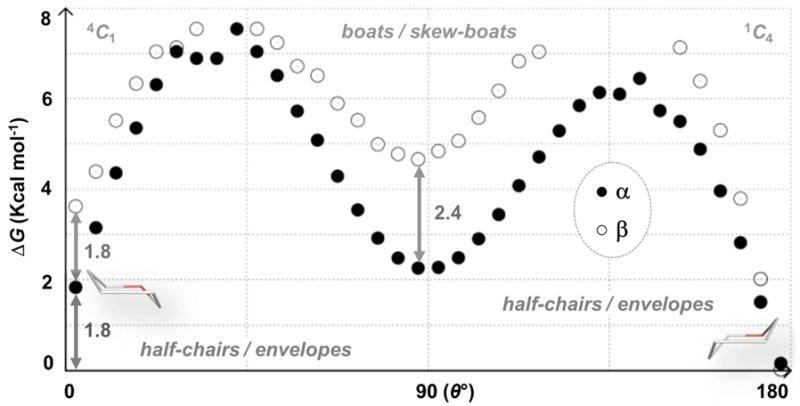
One-dimensional puckering free energy (G) landscapes for 1 and 2 computed from 10 μs explicit solvent (TIP3P) simulations. Free energy differences of 4C1 and equatorial puckers are noted.
Figure 2 footnote: This simplified representation facilitates comparison with other sugars. Computed free energies for all sampled canonical puckers are reported in the Supporting Information.
Computed rates of 1C4↔4C1 exchange were also independent of the water potential employed (Table 2). Rates for the forward (1C4→4C1) transition were 20 μs-1 and 1 μs-1 in the α- and β-anomers, respectively; the backward (4C1→1C4) rates were in the range 200-300 μs-1 in 1 and 150-250 μs-1 in 2. Our previous simulation of 3 predicted forward and backward rates of 4 μs-1 and 19 μs-1, respectively.1 This comparison suggests that the C6 carboxyl substituent of 3 slows the forward and backward puckering exchange rates by approximately five- and ten-fold, respectively, in idopyranosyl rings.
Table 2.
Calculated rates of exchange (μs-1) between chair puckers in the six α- and β-l-idopyranose 10 μs simulations.
| Anomer | Transition | TIP3P (μs-1) | TIP4P (μs-1) | TIP4P-EW (μs-1) |
|---|---|---|---|---|
| α | 1C4 → 4C1 | 20.1 | 19.8 | 20.4 |
| β | 1C4 → 4C1 | 0.9 | 0.9 | 0.7 |
| α | 4C1 → 1C4 | 292.0 | 200.0 | 193.0 |
| β | 4C1 → 1C4 | 167.0 | 149.0 | 250.0 |
Puckering itineraries
Currently, it is not possible to quantify μs-timescale puckering itineraries using experiments alone. Kinetically rigorous simulations are the best way to improve our understanding of puckering and to complement the burgeoning field of functional glycomics. Previous density functional theory studies of 1 predicted 1C4↔4C1 exchange and that the most likely 3D-itinerary connects the 4C1 chair and the boat B3,O through the transition-state envelope pucker E3.32 A recent metadynamics study found the skew-boat and boat puckers 1S3, OS2, 1,4B and B2,5 to be the lowest energy non-chair 3D-intermediates in α- and β-d-idopyranose.9 Our computed puckering free energy landscapes for 1 and 2 were very similar for the TIP3P, TIP4P and TIP4P-EW water models (see Supporting Information). All 38 canonical puckers (chairs, half-chairs, boats, skew-boats and envelope puckers) were populated in the α-anomer simulations. In the β-anomer simulations, all 1C4-hemisphere puckers were populated, however, specific half-chair and envelope puckers were not explored, including 4C1-hemisphere 4H3, 4E and 4H5 (θ≈30-70° and ϕ≈210-270°) and 1C4-hemisphere 1HO and 1E (θ≈100-140° and ϕ≈30-90°) (Figures 3 and 4). Three equatorial puckers, 3,OB, OS2 and B2,5 (θ≈70-110° and ϕ≈0/360, 330 and 300°), were also precluded in the β-anomer. By inspection of these 3D-intermediates, the limited pseudorotational profile and reduced number of inter-hemisphere transitions in 2 could not be attributed to intramolecular steric hindrance (e.g., between the bulky anomeric 1-O-methyl and C6 hydroxymethyl groups). This observation implicates water-pyranose interactions as the cause of the experimentally observed different puckering 3D-ensembles of 1 and 2, with which our calculated 3JH,H values are consistent.
Figure 3.
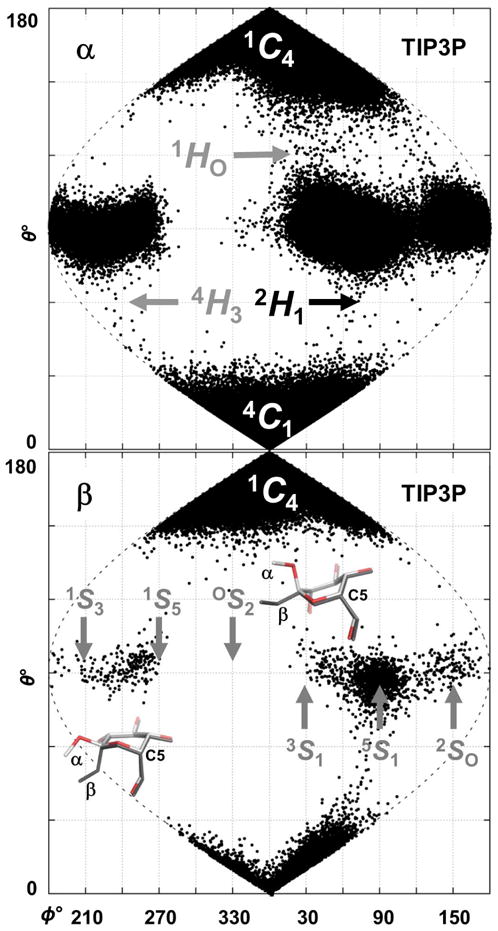
Sinusoidal projection of computed puckering in 1 (top) and 2 (bottom).
Figure 3 footnote: Data are from 10 μs simulations using GLYCAM11 and the TIP3P solvent model. All sampled puckers are shown. Predicted 3D-intermediates linking chair and equatorial puckers in the α-anomer (1HO and 4H3) which were precluded in the β-anomer simulation, are indicated in grey (top). Models of these puckers (α- and β-anomers) are shown at their respective positions in puckering space on the β-anomer plot (bottom), in which equatorial skew-boat (S) puckers are labelled above/below their respective positions (grey). By inspection of these overlaid 3D-structures, it is apparent that no intermolecular steric hindrance results from anomerisation. Analogous plots for TIP4P and TIP4P-EW water models are reported in the Supporting Information.
Figure 4.
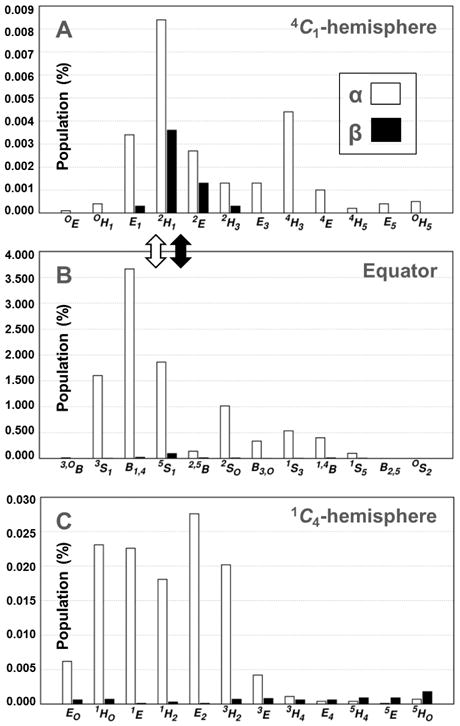
Histograms showing the occupancy of pyranose puckers for (1) and (2) in the 4C1-hemisphere (A), equatorial region (B) and 1C4-hemisphere (C) during 10 μs simulations (TIP3P water model).
Figure 4 footnote: The most likely transitions (based on pucker populations) between the equator and the 4C1-hemisphere are denoted by arrows. Multiple conformational itineraries between the equator and the 1C4-hemisphere were predicted for (1), e.g., B1,4 ↔ 1E.
A role for water mediating idopyranose μs-puckering
Two computed metrics derived from the explicit solvent MD support the hypothesis that the water structure is different around 1 and 2. Firstly, calculated radial distribution functions (RDFs) for hydrogen-bonding interactions between hydroxyl hydrogen and water oxygen atoms differed in the α- and β-l-idopyranose simulations at the O2 and O4 positions. The relatively decreased peak height in the β-l-idopyranose RDFs at 1.8 Å indicates comparatively fewer solute-solvent hydrogen bonding interactions (cf. α-l-idopyranose) in the first hydration shell at both positions (Figure 5). The inequalities observed in the second solvation shell trended similarly and also indicate a different water structure at O2 and O4. Integration of the RDFs in the first shell (1.0 to 2.5 Å) showed the presence of 0.55 and 0.48 water molecules in the α- and β-anomers, respectively. In the second shell (3.0 to 5.0 Å), the respective water occupancy was computed to be and 1.14 and 1.05. No differences were observed in RDFs computed for interactions between water oxygen atoms and hydroxyl hydrogen atoms at the pyranose O3 and O6 positions.
Figure 5.
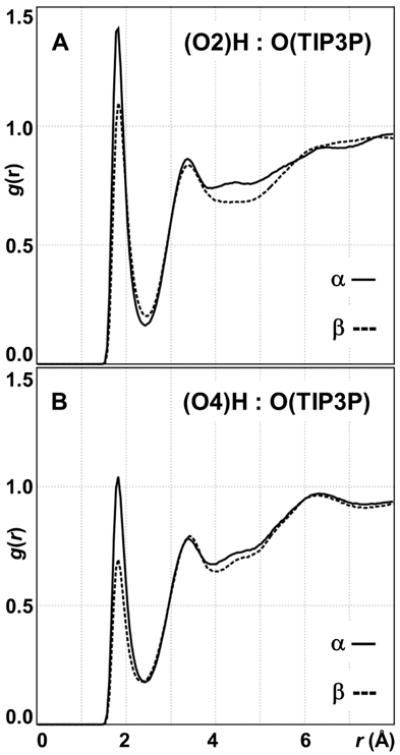
Radial distribution functions for the through-space interaction of TIP3P water oxygen atoms and hydroxyl hydrogen atoms at the α- (1) and β (2) -l-trajectories using the substituentidohexopyranosyl ring positions O2 (A) and O4 (B).
Secondly, the computed water density in close proximity to the solute, over the lifetime of the 10 μs trajectories, was different in the simulations of 1 and 2. Inequalities were observed in the computed water densities using the TIP3P solvent model, with the most significant difference being the greater propensity of water to cluster in the cavity bounded by the anomeric 1-O-methyl substituent and the C6 hydroxymethyl group in the β-anomer, compared to the α-anomer (Figure 6). The explicit solvent simulations therefore suggest that water molecules present within this cavity in the β-l-idopyranoside slow the μs-kinetics of pyranose puckering.
Figure 6.
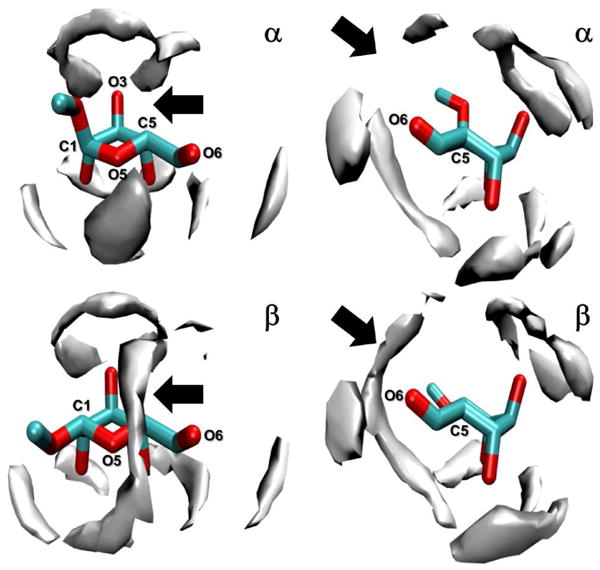
Isosurface representations of computed water densities surrounding 1 and 2 during 10 μs explicit solvent (TIP3P) simulations.
Figure 6 footnote: Two views of each anomer, in the 1C4 pucker, are shown. The cavity bounded by the anomeric 1-O-methyl substituent and the C6-hydroxymethyl group is indicated by an arrow.
This hypothesis was further supported by analysis of 10 μs periodic gas phase MD data for 1 and 2. The principal difference from the explicit solvent MD was a three-fold increase in the computed forward 1C4→4C1 transition rate in the β-anomer, computed to be 1 μs-1 in the aqueous MD and 3 μs-1 in the gas phase (Supporting Information). Calculated α-anomer 1C4→4C1 puckering rates were identical in the water and gas phase MD. This finding is consistent with the proposal that water structure causes the slower puckering kinetics of the β-anomer, compared to the α-anomer. Together, these predictions rationalise the smaller measured and calculated 3JH,H values in the β-anomer (cf. the α-anomer). Mechanistically, the explicit solvent MD findings suggest that water molecules limit the capacity of the β-anomer to adopt the 3D-intermediates 1HO and 4H3 highlighted in Figure 3.
Conclusions
Pyranose ring puckering is crucial to carbohydrate-protein interactions and concomitant bioactivity, exemplified by the heparin-antithrombin III interaction,7 which initiates anticoagulation. Previous work has shown that pyranose substituents and chiral configurations (epimers) impact μs-puckering kinetics.1, 4, 23 To further understand the effects of stereochemical changes on puckering, here we used flexible idopyranosides as a model to explore the dependence of pyranose μs-exchange on anomeric configuration, which is also a key regulator of bioactivity (e.g., glycan-lectin33 binding).
The aqueous simulations described herein explain the different experimentally observed puckering ensembles in 1 and 2 and reveal intermediate puckers that are less favourable in the β-anomer compared to the α-anomer, in water. Comparison with a previous1 explicit solvent 10 μs simulation of methyl α-l-idopyranuronate 3 intimated that conversion of the C6 carboxyl group to a hydroxymethyl group in an α-l-idopyranosyl ring significantly increases the forward (1C4→4C1) and backward (4C1→1C4) puckering exchange rates by five- and ten-fold, respectively. Based on previous work concerning the effect of 1-O-methylation on aqueous monosaccharide μs-puckering,23 we anticipate that the free reducing idopyranoses will exhibit different puckering 3D-ensembles from those seen in the simulations of 1 and 2. The effect of C6 substitution in the free reducing idopyranoses also remains to be studied.
The present work reaffirms the role of water as a major contributor to carbohydrate 3D-structure,11 suggesting that water determines pucker populations and chair-chair exchange kinetics, and also provides evidence that three- and four-site water models perform similarly with respect to computed μs-puckering 3D-structural, kinetic and thermodynamic properties. It will likewise be informative to test the performance of simpler (e.g., implicit) and more complex (e.g., five-site) water models when such simulations become practical on the timescales required to converge pyranose puckering (≈2-5 μs).1, 4, 23 The observation that water structure plays a role in pyranose μs-puckering kinetics can be exploited to help engineer exchange rates and bioactivity. For example, substituents that stabilise potential biologically important puckers in 1, 2, 3 and structurally-related molecules can be studied in silico on μs-timescales and in water, prior to synthesis and biological testing. In this regard, biologically relevant, kinetically rigorous and experimentally validated microsecond simulations, such as those reported here, are an important step towards deciphering glycomic structure-function relationships and rational design of new carbohydrate-based medicines and materials.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge M. J. Harvey and G. De Fabritiis (acellera.com) for enabling use of a development version of ACEMD software. R. J. Woods wishes to thank the National Institutes of Health (P41RR005351, P41GM103390 and GM094919 (EUREKA) as well as the Science Foundation of Ireland (08/IN.1/B2070) and the European Research Development Fund for support.
Footnotes
Supporting Information Available: Detailed methods, analyses and molecular properties (computational and experimental) are provided. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Sattelle BM, Hansen SU, Gardiner J, Almond A. Free energy landscapes of iduronic acid and related monosaccharides. J Am Chem Soc. 2010;132(38):13132–4. doi: 10.1021/ja1054143. [DOI] [PubMed] [Google Scholar]
- 2.(a) Wang C, Ying F, Wu W, Mo Y. Sensing or No Sensing: Can the Anomeric Effect Be Probed by a Sensing Molecule? J Am Chem Soc. 2011;133(34):13731–13736. doi: 10.1021/ja205613x. [DOI] [PubMed] [Google Scholar]; (b) Mo Y. Computational evidence that hyperconjugative interactions are not responsible for the anomeric effect. Nat Chem. 2010;2(8):666–71. doi: 10.1038/nchem.721. [DOI] [PubMed] [Google Scholar]
- 3.Snyder JR, Serianni AS. D-Idose - a One-Dimensional and Two-Dimensional NMR Investigation of Solution Composition and Conformation. J Org Chem. 1986;51(14):2694–2702. [Google Scholar]
- 4.Sattelle BM, Almond A. Assigning kinetic 3D-signatures to glycocodes. Phys Chem Chem Phys. 2012;14(16):5843–5848. doi: 10.1039/c2cp40071e. [DOI] [PubMed] [Google Scholar]
- 5.Reichardt NC, Czechura P, Guedes N, Kopitzki S, Vazquez N, Martin-Lomas M. A new linker for solid-phase synthesis of heparan sulfate precursors by sequential assembly of monosaccharide building blocks. Chem Comm. 2011;47(8):2390–2392. doi: 10.1039/c0cc04686h. [DOI] [PubMed] [Google Scholar]
- 6.Liu H, Zhang Z, Linhardt RJ. Lessons learned from the contamination of heparin. Nat Prod Rep. 2009;26(3):313–21. doi: 10.1039/b819896a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hricovini M, Guerrini M, Bisio A, Torri G, Petitou M, Casu B. Conformation of heparin pentasaccharide bound to antithrombin III. Biochem J. 2001;359(Pt 2):265–72. doi: 10.1042/0264-6021:3590265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schnupf U, Willett JL, Momany F. DFTMD studies of glucose and epimers: anomeric ratios, rotamer populations, and hydration energies. Carbohydr Res. 2010;345(4):503–11. doi: 10.1016/j.carres.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 9.Autieri E, Sega M, Pederiva F, Guella G. Puckering free energy of pyranoses: A NMR and metadynamics-umbrella sampling investigation. J Chem Phys. 2010;133(9) doi: 10.1063/1.3476466. [DOI] [PubMed] [Google Scholar]
- 10.Schlick T. Molecular dynamics-based approaches for enhanced sampling of long-time, large-scale conformational changes in biomolecules. F1000 Biol Rep. 2009;1:51. doi: 10.3410/B1-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Kirschner KN, Woods RJ. Solvent interactions determine carbohydrate conformation. Proc Natl Acad Sci U S A. 2001;98(19):10541–10545. doi: 10.1073/pnas.191362798. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Corzana F, Motawia MS, Du Penhoat CH, Perez S, Tschampel SM, Woods RJ, Engelsen SB. A hydration study of (1->4) and (1->6) linked alpha-glucans by comparative 10 ns molecular dynamics simulations and 500-MHz NMR. J Comput Chem. 2004;25(4):573–86. doi: 10.1002/jcc.10405. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Corzana F, Motawia MS, Herve du Penhoat C, van den Berg F, Blennow A, Perez S, Engelsen SB. Hydration of the amylopectin branch point. Evidence of restricted conformational diversity of the alpha-(1->6) linkage. J Am Chem Soc. 2004;126(40):13144–55. doi: 10.1021/ja048622y. [DOI] [PubMed] [Google Scholar]
- 12.Jorgensen WL, Tirado-Rives J. Potential energy functions for atomic-level simulations of water and organic and biomolecular systems. Proc Natl Acad Sci U S A. 2005;102(19):6665–70. doi: 10.1073/pnas.0408037102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926–935. [Google Scholar]
- 14.Jorgensen WL, Madura JD. Temperature and Size Dependence for Monte-Carlo Simulations of Tip4p Water. Mol Phys. 1985;56(6):1381–1392. [Google Scholar]
- 15.Horn HW, Swope WC, Pitera JW, Madura JD, Dick TJ, Hura GL, Head-Gordon T. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J Chem Phys. 2004;120(20):9665–78. doi: 10.1063/1.1683075. [DOI] [PubMed] [Google Scholar]
- 16.Harvey MJ, Giupponi G, De Fabritiis G. ACEMD: Accelerating biomolecular dynamics in the microsecond time scale. J Chem Theory Comput. 2009;5(6):1632–1639. doi: 10.1021/ct9000685. [DOI] [PubMed] [Google Scholar]
- 17.Buch I, Harvey MJ, Giorgino T, Anderson DP, De Fabritiis G. High-throughput all-atom molecular dynamics simulations using distributed computing. J Chem Inf Model. 2010;50(3):397–403. doi: 10.1021/ci900455r. [DOI] [PubMed] [Google Scholar]
- 18.Krautler V, Van Gunsteren WF, Hunenberger PH. A fast SHAKE: Algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J Comput Chem. 2001;22(5):501–508. [Google Scholar]
- 19.Kirschner KN, Yongye AB, Tschampel SM, Gonzalez-Outeirino J, Daniels CR, Foley BL, Woods RJ. GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J Comput Chem. 2008;29(4):622–55. doi: 10.1002/jcc.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spiwok V, Kralova B, Tvaroska I. Modelling of β-D-glucopyranose ring distortion in different force fields: a metadynamics study. Carbohydr Res. 2010;345(4):530–7. doi: 10.1016/j.carres.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 21.Cremer D, Pople JA. General definition of ring puckering coordinates. J Am Chem Soc. 1975;97(6):1354–1358. [Google Scholar]
- 22.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. JCTC. 2008;4(3):435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 23.Sattelle BM, Almond A. Is N-acetyl-D-glucosamine a rigid 4C1-chair? Glycobiology. 2011;21(12):1651–1662. doi: 10.1093/glycob/cwr101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haasnoot CAG, Deleeuw FAAM, Altona C. The Relationship between Proton-Proton Nmr Coupling-Constants and Substituent Electronegativities .1. An Empirical Generalization of the Karplus Equation. Tetrahedron. 1980;36(19):2783–2792. [Google Scholar]
- 25.Case DA, Cheatham TE, 3rd, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem. 2005;26(16):1668–88. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.(a) Serianni AS, Nunez HA, Barker R. Carbon-13-Enriched Carbohydrates - Preparation of Aldononitriles and Their Reduction with a Palladium Catalyst. Carbohyd Res. 1979;72(Jul):71–78. [Google Scholar]; (b) Serianni AS, Vuorinen T, Bondo PB. Stable Isotopically-Enriched D-Glucose - Strategies to Introduce Carbon, Hydrogen and Oxygen Isotopes at Various Sites. J Carbohyd Chem. 1990;9(5):513–541. [Google Scholar]
- 27.Angyal SJ, Bethell GS, Beveridge RJ. Complexes of Carbohydrates with Metal Cation .10. Separation of Sugars and of Polyols on Cation-Exchange Resins in the Calcium Form. Carbohyd Res. 1979;73(Aug):9–18. [Google Scholar]
- 28.Podlasek CA, Wu J, Stripe WA, Bondo PB, Serianni AS. [13C]-Enriched Methyl Aldopyranosides - Structural Interpretations of 13C-1H Spin-Coupling Constants and 1H Chemical-Shifts. J Am Chem Soc. 1995;117(33):8635–8644. [Google Scholar]
- 29.Austin PW, Baddiley J, Hardy FE, Buchanan JG. Separation of Isomeric Glycosides on Basic Ion-Exchange Resins. J Chem Soc. 1963;(Nov):5350–5353. [Google Scholar]
- 30.Hodge JE, Hofreiter BT. Determination of reducing sugar and carbohydrate. Methods Carbohydr Chem. 1962;1:380–394. [Google Scholar]
- 31.King-Morris MJ, Serianni AS. 13C NMR-Studies of [1-13C]Aldoses - Empirical Rules Correlating Pyranose Ring Configuration and Conformation with 13C Chemical-Shifts and 13C/13C Spin Couplings. J Am Chem Soc. 1987;109(12):3501–3508. [Google Scholar]
- 32.Kurihara Y, Ueda K. An investigation of the pyranose ring interconversion path of alpha-L-idose calculated using density functional theory. Carbohydr Res. 2006;341(15):2565–2574. doi: 10.1016/j.carres.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 33.Gabius HJ. Glycans: bioactive signals decoded by lectins. Biochem Soc Trans. 2008;36(Pt 6):1491–6. doi: 10.1042/BST0361491. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.