

Abstract
The cellular recognition of viruses evokes the secretion of type-I interferons (IFNs) that induce an antiviral protective state. By live-cell imaging, we show that key steps of virus-induced signal transduction, IFN-β expression, and induction of IFN-stimulated genes (ISGs) are stochastic events in individual cells. The heterogeneity in IFN production is of cellular—and not viral—origin, and temporal unpredictability of IFN-β expression is largely due to cell-intrinsic noise generated both upstream and downstream of the activation of nuclear factor-κB and IFN regulatory factor transcription factors. Subsequent ISG induction occurs as a stochastic all-or-nothing switch, where the responding cells are protected against virus replication. Mathematical modelling and experimental validation show that reliable antiviral protection in the face of multi-layered cellular stochasticity is achieved by paracrine response amplification. Achieving coherent responses through intercellular communication is likely to be a more widely used strategy by mammalian cells to cope with pervasive stochasticity in signalling and gene expression.
Keywords: bacterial artificial chromosomes, gene expression, interferon regulatory factor-7, live-cell microscopy, multi-scale modelling
Introduction
The type-I interferon (IFN) system provides a powerful defence against viral infections (Kunzi and Pitha, 2003; Sadler and Williams, 2008; Takeuchi and Akira, 2009). Pathogen recognition receptors, such as RIG-I, stimulate an intracellular signalling cascade that leads to the activation of nuclear factor κB (NF-κB) and IFN regulatory factors (IRFs) 3 and 7 (Brennan and Bowie, 2010). Subsequent induction of the IFN-β gene and other type-I IFNs is a hallmark of the early response to infection (Theofilopoulos et al, 2005). Upon secretion and binding of type-I IFNs to their specific receptor, the Jak/STAT signalling pathway is activated to reprogram gene expression (Brierley and Fish, 2005). By activation of a wide set of these genes, IFNs act directly by establishing an antiviral state and indirectly through recruiting innate and adaptive immune cells.
On the one hand, IFN production must be tightly regulated to avoid harmful inflammation and autoimmune disease (Trinchieri, 2010). On the other hand, pathogenic viruses inhibit the expression of IFNs or IFN-stimulated genes (ISGs) by diverse mechanisms, indicating that maintaining the efficiency of the IFN system in the face of these viral challenges might be an overriding objective of its evolution (Versteeg and Garcia-Sastre, 2010). Therefore, the finding that only a fraction of cells in a virus-infected cell population expresses IFNs has been surprising (Zawatzky et al, 1985; Hu et al, 2007). Biochemical studies have suggested that stochastic IFN induction results from host cell-intrinsic causes, such as a mechanism of IFN gene induction (Hu et al, 2007; Apostolou and Thanos, 2008) or cellular heterogeneity in expression of the viral sensor RIG-I (Hu et al, 2011). Alternatively, heterocellularity of IFN expression could be caused by the infecting virus (Chen et al, 2010; Killip et al, 2011). It is conceivable that several factors might shape the dynamics of IFN induction, depending on host cell type and virus. Live imaging provides a suitable tool to study dynamics and variability at single-cell resolution (Spiller et al, 2010), but, such an analysis has not yet been carried out for the IFN system.
To understand the functional consequences of cell-to-cell variability in IFN induction, the cellular response to IFNs, particularly the expression of ISGs, must also be defined. Two recent quantitative analyses of IFN-stimulated signalling have modelled the dynamics at the cell-population level (Maiwald et al, 2010) and considered effects of cell-to-cell variability (Levin et al, 2011). However, the expression of ISGs and the resulting cell fate have not yet been characterized at single-cell level.
Here, we study in living cells and at single-cell resolution both type-I IFN induction and the cellular response to secreted IFN. Using fluorescent reporters based on bacterial artificial chromosomes (BACs) and chimeric transcription factors (Supplementary Figure S1), we imaged successive key steps of IFN induction and response in a prototypical model system, the infection of murine cells in culture with a single-stranded RNA virus. To link our single-cell data to antiviral protection at the cell-population level, we developed a mathematical model based on these data and tested its predictions experimentally.
Our results show that cell-to-cell heterogeneity is a pervasive feature of the IFN system. This heterogeneity manifests itself not only in the virus-induced expression of IFN but also in the IFN-induced protective response. It is to a large part due to cell-intrinsic stochasticity occurring in three key steps: virus-induced signalling, IFN gene induction, and expression of ISGs. In contrast to gene-expression noise described in bacteria and yeast that causes protein levels to fluctuate around a mean value in individual cells (Raj and van Oudenaarden, 2008), the stochasticity found here in the IFN system is an all-or-nothing phenomenon: cells switch on signalling or gene expression or not, and the switching cells do so at widely variable time points. Nevertheless, we find that a reliable antiviral response is achieved through powerful paracrine propagation of the signal. Thus, our results show that the functional dynamics of the IFN response must be understood in terms of the collective spatio-temporal dynamics of stochastically reacting single cells.
Results
Cell-to-cell heterogeneity in IFN induction
In order to monitor authentic IFN-β expression in real time, we stably transfected murine fibroblasts with a BAC-encoded reporter expressing TurboGFP under the control of the Ifnb promoter (IFN-β–tGFP). A representative cell clone was selected showing stable expression of the reporter. These cells were infected with Newcastle Disease Virus (NDV), which replicates and induces IFN in the cells via the double-stranded RNA sensor RIG-I (Kato et al, 2005), without viral interference with this pathway (Childs et al, 2007). As the newly generated viral particles cannot re-infect the mouse cells (Rott, 1979), this system allows us to study in a controlled manner the IFN induction elicited by the primary infection. To quantitatively determine the kinetics and dose response of IFN-β–tGFP expression, reporter cells were infected with NDV and subjected to flow cytometry (Figure 1A). We observed a rise in IFN-β–tGFP-positive cells that faithfully reflected the accumulation of type-I IFNs in the supernatant (Figure 1B). The fraction of IFN producers increased nearly linearly over a broad range of NDV dose, whereas the average expression level already reached ∼70% of its maximal value at very low NDV dose (Figure 1C). The frequency of IFN-β expression was very similar for different clones, showing the highly reproducible behaviour of the BAC-based IFN-β–tGFP reporter (Supplementary Figure S2). Thus, the production of IFN-β in response to viral infection is controlled by the fraction of responding cells.
Figure 1.
Quantitative and temporal heterogeneity of IFN-β induction. A BAC-based reporter construct in which the IFN-β gene is replaced by TurboGFP was integrated into murine NIH3T3 fibroblasts. A cell clone with a stable integration of the BAC and representative response towards NDV infection was used (error bars represent ±s.d. of triplicates). (A) Induction of IFN-β–tGFP expression upon NDV infection. IFN-β reporter cells were infected with NDV for 1 h. Expression of tGFP was determined 24 h post-infection by flow cytometry. Representative dot plots are shown for 10, 20, und 40 HAU/ml NDV. (B) IFN-β reporter reflects endogenous IFN production. IFN-β–tGFP expression frequencies after infection with 40 HAU/ml NDV were detected at various time points by flow cytometry. Frequencies were plotted against time points post-infection (black circles) and compared with titres of type-I IFN in the supernatant (grey rhombs). (C) IFN-β expression frequency increases with viral titre. Reporter cells infected with increasing concentrations of NDV (HAU/ml) were subjected to flow cytometry 24 h post-infection. Frequency of IFN-β–tGFP expression (circles) following infection with 1, 2, 5, 10, 20, 40, 80, and 100 HAU/ml and the geometric mean of their fluorescence intensity (triangles) are presented. Source data is available for this figure in the Supplementary Information.

To examine whether IFN expression correlates with viral replication, we jointly measured the viral protein, hemagglutinin-neuraminidase (HN), and IFN-β–tGFP by flow cytometry. In agreement with previous findings (Kumagai et al, 2009; Rehwinkel et al, 2010), only cells with replicating virus expressed IFN-β–tGFP. However, a large proportion of cells with replicating virus did not activate the Ifnb promoter. Strikingly, we observed no correlation between the extent of replication and the fraction of IFN-β–tGFP-expressing cells (Figure 2A and B; Supplementary Figure S3). These observations suggest that the presence of replicating virus in a cell is necessary but not sufficient to induce IFN-β.
Figure 2.
Viral replication is necessary but not sufficient to induce IFN-β expression. (A) Fractional IFN-β expression among productively infected cells. Reporter cells were infected with 40 HAU/ml NDV for 1 h. IFN-β–tGFP reporter expression and intracellular NDV HN protein was measured by flow cytometry at indicated time post-infection. Dot plots show IFN-β–tGFP expression among productively infected (NDV HN+) cells at indicated time post-infection. (B) Separate kinetics of viral replication and IFN-β expression. Frequency of IFN-β–tGFP (black circles) and NDV HN expression (grey squares) over time. (C) Unresponsiveness is not caused by the absence of inducing viral RNA. NDV-infected (80 HAU/ml) IFN-β–tGFP reporter cells were separated into GFP+ and GFP− fractions. Total RNA was isolated and transfected into naive IFN-β–tGFP reporter cells (lower graphs). RNA from non-infected cells served as a control (upper graph). The frequency of IFN-β–tGFP-expressing cells 20 h after transfection is presented. Source data is available for this figure in the Supplementary Information.

As an alternative explanation of heterogeneous IFN expression, it has been suggested that defective viruses are primarily responsible for inducing IFN during parainfluenza virus type 5 (PIV5) infection while during normal replication of PIV5 effective pathogen-associated molecular patterns (PAMPs) are not produced or exposed (Killip et al, 2011). Therefore, we examined the intracellular RNA of non-responding virus-infected cells for its ability to induce IFN in naive cells (Figure 2C; Supplementary Figure S4). RNA from non-responding cells induced IFN to a comparable extent as RNA from responding cells. This finding shows that the viral RNA in IFN-producing and non-producing cells is equally capable of inducing IFN and thus the heterogeneity of IFN-β induction occurs despite the presence of IFN-inducing viral RNA.
Time-lapse microscopic data revealed that the onset of IFN-β expression after infection of cells with NDV varied strongly among the IFN-β producers, with cell-to-cell differences as large as 20 h (Figure 3A; Supplementary Figure S11; Supplementary Movie S1). While the increase in viral load results in earlier onset, the relative temporal variability of IFN-β–tGFP expression differs only slightly. To dissect whether this variability is due to variable time points of infection or reflects cell-intrinsic properties, we bypassed viral infection by liposome-transfecting the cells with the dsRNA analogue poly I:C (liposome-free delivery of poly I:C did not lead to IFN-β expression). Among the IFN producers and independent of the poly I:C concentration, the onset time of IFN-β expression varied strongly and was quantitatively comparable to the case of viral infection, as seen by the same order of magnitude of the coefficient of variation (CV) (Figure 3A). Furthermore, unchanged temporal variability was obtained when synchronizing viral entry by a temperature shift (Supplementary Figure S5). Our data show that the IFN-β gene is induced with widely varying time delays in the producing cells that are not due to variable infection times. In summary, we conclude that the cell-to-cell heterogeneity in IFN-β expression is predominantly of cellular origin.
Figure 3.
Temporal variability in cellular IFN-β induction. (A) IFN-β expression onset in single cells. Variability of response timing is virus-independent. IFN-β–tGFP reporter cells infected for 1 h with indicated concentrations of NDV or transfected with poly I:C at given concentrations were subjected to time-lapse microscopy (15 min picture intervals). Distribution of tGFP expression onset over time (scatter plot, n=456 (NDV), n=140 (poly I:C)) and CVs are shown. (B) Synchronous activation of NF-κB and IRF-7. NIH3T3 cell clone stably expressing the fusion proteins IRF-7–CFP and NF-κB/p65–YFP were infected with 80 HAU/ml NDV for 1 h and subjected to time-lapse microscopy. Fluorescence pictures for CFP and YFP were taken every 20 min. Subcellular localization of IRF-7–CFP (left column) and p65–YFP (right column) at indicated time after infection. The diagram shows relative nuclear fluorescence for IRF-7–CFP and p65–YFP from sister cells. (C) Synchronicity is independent of response time. IRF-7–CFP and p65–YFP initial nuclear translocation were determined in individual cells and plotted against each other (n=65). Coloured dots indicate the frequency of data points. (D) Expression delay of an individual cell. NIH3T3 cell clone stably expressing IRF-7–CFP together with IFN-β–tGFP were infected with 80 HAU/ml NDV. Fluorescence pictures for CFP and GFP were taken every 20 min. Subcellular localization of IRF-7–CFP (left column) and IFN-β–tGFP expression (right column) at indicated time after infection. Graphs show relative IRF-7–CFP nuclear fluorescence and tGFP intensity. Tsig: time interval between infection and IRF-7–CFP nuclear translocation. Tgen: time interval between IRF-7–CFP nuclear translocation and onset of IFN-β–tGFP gene expression. (E) Response variation at distinct stages of IFN induction. The starting times for IRF-7–CFP nuclear translocation were plotted against the times of IFN-β–tGFP expression for individual cells (n=315). Source data is available for this figure in the Supplementary Information.

Both virus-induced signal transduction and Ifnb gene expression are sources of heterogeneity
To analyse mechanistically how cell-to-cell heterogeneity in IFN induction arises, we monitored the activation of the key transcription factors NF-κB and IRF-7 in dual reporter cells, representing the IKKα/β/γ–NF-κB and IKKε/TBK-1–IRF-3/IRF-7 pathways downstream of the dsRNA sensor RIG-I. Fluorescent protein-tagged NF-κB and IRF-7 (p65–YFP and IRF-7–CFP) were localized predominantly in the cytoplasm in uninfected cells and accumulated in the nucleus after NDV infection (Figure 3B). Nuclear translocation of the two transcription factors occurred at the same time in a given cell, but the joint translocation time varied widely between cells (Figure 3C). This finding was corroborated by antibody staining of endogenous IRF-3 and NF-κB (p65), showing that the activation of both factors upon NDV infection or poly I:C stimulation correlated in individual cells (Supplementary Figure S6). Thus, strong cell-to-cell variability arises in the shared upstream activation pathway of NF-κB and IRF-7.
To relate IFN induction to the activation of the latent transcription factors we used dual reporter cells expressing the IRF-7–CFP fusion protein and the transcriptional reporter IFN-β–tGFP (Figure 3D; Supplementary Figure S7). We found that the majority of cells translocating IRF-7 to the nucleus also activated the Ifnb promoter (91% at 80 HAU/ml NDV) (Figure 3E). Only few cells (9%) did not express IFN-β–tGFP upon IRF-7 nuclear accumulation, showing the same distribution of IRF-7 translocation times as the IFN-β-expressing cells (Figure 3E, bottom part). No IFN-β–tGFP induction was observed without preceding IRF-7 nuclear accumulation. Thus, IRF-7 nuclear translocation is strictly required for IFN-β expression. The tight coupling of the two events also shows the validity of the reporters used. In agreement with the results in Figure 3C, there was strong temporal heterogeneity in IRF-7 translocation between the cells (signalling delay from viral infection to IRF-7 translocation Tsig=11.7±4.0 h). The interval between IRF-7 translocation and the onset of IFN-β–tGFP gene expression also varied considerably (gene expression delay, Tgen=3.4±1.5 h; note that the CVs are similar, σsig/Tsig=0.34, σgen/Tgen=0.44). These quantitative data show that both cytoplasmic signalling from viral entry to the activation of latent transcription factors and induction of IFN-β expression cause strong heterogeneity in IFN-β production.
Sister-cell analysis reveals cell-intrinsic stochasticity
Cell-to-cell variability can arise from the intrinsic noise of biochemical reactions and from extrinsic factors, such as differences in cell-cycle stage or cellular environment (Elowitz et al, 2002; Maheshri and O’Shea, 2007; Paixão et al, 2007; Raj and van Oudenaarden, 2008; Snijder and Pelkmans, 2011). To minimize extrinsic cell-to-cell differences, we analysed sister cells after division (Spencer et al, 2009). Cells that divided after the 1 h period of infection were followed. IRF-7 activation in sister cells occurred mainly asynchronously, differing by >2 h in ∼50% of cell pairs (Figure 4A). The time between IRF-7–CFP activation and IFN-β–tGFP expression correlated even less in sister cells (Figure 4B), consistent with the previously described stochastic transcription of the Ifnb gene (Apostolou and Thanos, 2008). The coefficient of determination r2 was 0.6 for Tsig and 0.34 for Tgen, indicating that 40% of the variability in signalling and 66% of the variability in Ifnb gene expression are uncorrelated between sister cells and thus provide an estimate for the cell-intrinsic stochasticity. To examine whether different viral replication kinetics in sister-cell pairs are a source of variability, we subjected cells to poly I:C stimulation. Also, under these conditions cells displayed largely uncorrelated IRF-7 signalling and IFN-β gene expression (Figure 4E and F), with similar coefficients of determination (r2=0.54 for Tsig and 0.11 for Tgen) as with viral infection. Taken together, these findings show that cell-intrinsic stochasticity is a strong source of cell-to-cell heterogeneity in IFN-β expression.
Figure 4.
Temporal variability of signalling events in sister cells reveals stochasticity. NIH3T3 cell clone stably expressing IRF-7–CFP or IRF-7–TagRFP together with IFN-β–tGFP were infected with 80 HAU/ml NDV for 1 h (A–D, n=38 sister-cell pairs) or transfected with poly I:C (5 μg/ml) (E–H, n=36 sister-cell pairs) and subjected to time-lapse microscopy (20 min interval). Sister-pair analysis was carried out for IRF-7 nuclear translocation and IFN-β expression onset. Coloured dots indicate the frequency of data points. (A, E) Time of IRF-7 nuclear translocation onset (Tsig) in sister-cell pairs. (B, F) Time intervals between IRF-7 nuclear translocation and IFN-β–tGFP expression onset (Tgen) among sister cells. (C, G) Time elapsed from cell division to IRF-7 nuclear translocation (Tsig−Tdiv) of sister cells. (D, H) Time elapsed from cell division to IFN-β–tGFP expression (Tsig+Tgen−Tdiv). Source data is available for this figure in the Supplementary Information.

We checked whether the observed differences between sister cells relate to their time of division. For this purpose, we plotted the time differences between sister cells (ΔTsig and ΔTgen) versus the time elapsed since cell division. The very weak correlations argue against strong control of heterogeneity by the cell cycle (Figure 4C and D for NDV infection and Figure 4G and H for poly I:C stimulation).
Taken together, the sister-cell analysis indicates a role for both ‘extrinsic’ variability between cells and cell-intrinsic stochasticity. The cell-intrinsic component is strong, accounting for approximately half of the variability in the kinetics of antiviral signalling and IFN-β induction in individual cells. This intrinsic stochasticity provides a rationale for the lack of correlation of IFN-β expression with the extent of viral replication (cf. Figure 2A).
Antiviral protection is an IFN concentration-dependent switch in individual cells
The intrinsic stochasticity indicates that the responsiveness of IFN-β induction towards virus is not maximized (i.e., many cells with replicating virus do not express IFN-β or do so only very late after infection). Therefore, we asked how IFN-β production translates into antiviral protection. We chose IRF-7 as a prototypical ISG (Honda et al, 2005) and measured its expression upon IFN-β stimulation in cells stably transfected with a BAC-encoded IRF-7–mCherry fusion gene (Supplementary Movie S2). Flow-cytometric analysis showed a digital pattern of IRF-7–mCherry levels, with distinct expressing and non-expressing subpopulations, where the expressing subpopulation increased with extracellular IFN-β concentration (Figure 5A). The binary IRF-7 response was consistently found for several IRF-7–mCherry clones (Supplementary Figure S8); it was not caused by competition of cells for IFN-β, as IFN-β was still detectable in the supernatant for >30 h (Supplementary Figure S9).
Figure 5.

Bimodal antiviral response towards IFN. A BAC-based reporter construct containing mCherry fused to the C-terminus of the chromosomal IRF-7 gene was integrated into NIH3T3 cells. Experiments were performed with a cell clone exhibiting a stable integration of the BAC and a representative response towards IFN. (A) Binary dose- and time-dependent IRF-7–mCherry expression. IRF-7–mCherry reporter cells were stimulated with the indicated concentrations of IFN-β, mCherry expression was determined by flow cytometry. (B, C) Bimodality of IRF-7 expression is reflected in ISG transcription and antiviral protection. (B) Reporter cells were treated with 500 U/ml IFN-β for 16 h and subjected to FACS. IRF-7–mCherry positive (2) and negative (1) populations indicated by the shaded areas were separated. RNA was prepared from both populations and analysed by qRT–PCR for expression of the indicated ISGs and Rps9 as a control. The results were normalized to β-actin mRNA and are shown as fold increase of untreated reporter cells. (C) Reporter cells treated with 500 U/ml IFN-β for 8 and 24 h were infected with 80 HAU/ml NDV. In all, 20 h after infection, cells positive (blue) or negative (red) for NDV HN (left column, intracellular antibody staining) were analysed for IFN-stimulated IRF-7–mCherry expression (right column) by flow cytometry.
To correlate IRF-7–mCherry with the expression of endogenous ISGs, we separated IRF-7–mCherry expressing and non-expressing cells by fluorescence-activated cell sorting (FACS) and subjected isolated RNA from these cells to qPCR analysis. All tested ISGs showed enhanced mRNA levels in IRF-7–mCherry expressing cells (Figure 5B). These findings suggest that a distinct subpopulation of cells that is identified by IRF-7–mCherry expression coordinately expresses an antiviral gene programme. To test this, we stimulated IRF-7–mCherry reporter cells with IFN-β for 8 or 24 h to allow induction of ISGs and then infected the cells with NDV. Virus replicated in IRF-7–mCherry non-expressing cells but not in the expressing cells (Figure 5C). This result shows that, as predicted, only the IRF-7–mCherry positive subpopulation of cells has been protected by IFN. Taken together, these data demonstrate that antiviral protection is a stochastic, IFN-β concentration-dependent, switch in individual cells.
Mathematical modelling predicts paracrine propagation of the IFN response
Our data show a remarkable heterogeneity of type-I IFN expression and subsequent induction of an antiviral state. To understand the resulting dynamics of the IFN system in an infected cell population, we developed a multi-scale mathematical model that describes viral replication and the expression of IFN and ISGs in individual cells together with intercellular communication through secreted IFN (Figure 6A; Supplementary Text S1). According to the sister-cell data, signalling and gene expression contribute strong cell-intrinsic stochasticity (cf. Figure 4). For this reason, we attribute cell-to-cell variability in the model to intrinsic stochasticity in both kinds of processes.
Figure 6.
A multi-scale mathematical model of IFN induction and response. (A) Scheme of the state transitions of an individual cell. The model describes a population of individual cells with virus replication, induction of IFN genes through RIG-I signalling to NF-κB and IRFs, induction of ISGs (including IRF-7) by IFN, and cell communication via secreted IFN. The possible state transitions of an individual cell with the propensity functions w are shown. The colour code indicates nuclear translocation of IRFs/NF-κB (cyan nucleus), induction of IFN (green cytoplasm), and induction of IRF-7 (red cytoplasm), as imaged experimentally. Induction of IFN-β (dashed arrow wI+) is explicitly modelled as a multi-step process to match the available experimental data (see (C) below); all other transitions are modelled as single steps (solid arrows). (B) Model simulation of NF-κB/IRF nuclear translocation times versus experimental data. The distribution computed with the model (black line) matches the data (histogram) obtained for high infection dose (80 HAU/ml). (C) Model simulation of IFN-β–tGFP onset times versus experimental data. The distribution computed with the model (black line) matches the data (histogram) obtained for high infection dose (80 HAU/ml). (D) Comparison between model and experimental data for high-dose NDV infection. The model reproduces the observed dynamics of viral load (as measured by HN expression), IFN-β–tGFP induction, extracellular IFN titre and IRF-7–mCherry expression (solid lines, model; dots, experimental data; infection dose 40 HAU/ml). Note that the smooth model curves in (B, C) are mean values obtained by simulating 104 cells. Source data is available for this figure in the Supplementary Information.

Two sources of heterogeneous IFN induction are the (i) activation of the latent NF-κB and IRF transcription factors downstream of viral sensing by RIG-I and (ii) induction of the Ifnb gene by nuclear NF-κB and IRFs. Importantly, nuclear transcription factor concentration and IFN-β protein increase quantitatively very rapidly once nuclear translocation and gene induction, respectively, have been triggered. By contrast, the waiting times for these events in an individual cell are much longer, so that IFN-β induction is switch-like in time (cf. sample trajectories in Figure 3B and D). Therefore, we describe the dynamics of these events as stochastic transitions 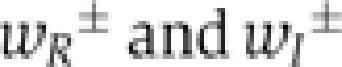 between discrete cell states (virus-infected, NF-κB/IRF-activated, IFN-β expressing along the horizontal axis in Figure 6A) (Mariani et al, 2010). The intracellular replication of virus particles is modelled as a birth–death process. Secreted IFN diffuses in the extracellular space and induces ISGs in an autocrine or paracrine manner. Given the high diffusion coefficient of IFN (Kreuz and Levy, 1965), diffusion on the relevant length scale of several cell diameters takes place within minutes, compared with the hour-timescale of IFN and ISG expression, so that we assume uniform distribution of secreted IFN. To account for the switch-like induction of ISGs, such as IRF-7 (cf. Figure 5), we modelled the IFN-dependent activation rate of STAT1/2 nuclear translocation,
between discrete cell states (virus-infected, NF-κB/IRF-activated, IFN-β expressing along the horizontal axis in Figure 6A) (Mariani et al, 2010). The intracellular replication of virus particles is modelled as a birth–death process. Secreted IFN diffuses in the extracellular space and induces ISGs in an autocrine or paracrine manner. Given the high diffusion coefficient of IFN (Kreuz and Levy, 1965), diffusion on the relevant length scale of several cell diameters takes place within minutes, compared with the hour-timescale of IFN and ISG expression, so that we assume uniform distribution of secreted IFN. To account for the switch-like induction of ISGs, such as IRF-7 (cf. Figure 5), we modelled the IFN-dependent activation rate of STAT1/2 nuclear translocation, 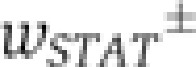 (Lillemeier et al, 2001; Vinkemeier, 2004), followed by the rate of ISG induction,
(Lillemeier et al, 2001; Vinkemeier, 2004), followed by the rate of ISG induction, 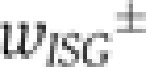 , resulting in a bimodal distribution of ISG-expressing and non-expressing cells. Virus ceases to replicate in cells expressing ISGs.
, resulting in a bimodal distribution of ISG-expressing and non-expressing cells. Virus ceases to replicate in cells expressing ISGs.
To determine the model parameters from the experimental data, we focused on our extensive data for high-dose infections, 40 and 80 HAU/ml NDV (Figure 6; Supplementary Text S1; Supplementary Figure S10). We first computed the distributions for the NF-κB/IRF-7 nuclear translocation times and the IFN-β–tGFP switching-on times. Parameter optimization using as objective function the squared sum of residuals between measured and computed (binned) distributions yielded a good fit (Figure 6B and C and green trace in Figure 6D). This fit required that (i) induction of RIG-I-mediated signalling by virus is cooperative (Supplementary Figure S10B) and (ii) Ifnb gene induction is modelled as a multi-step process (Supplementary Figure S10C). Both implications of the quantitative modelling of the data—cooperative RIG-I signalling and multi-step assembly of the IFN-β enhanceosome—are consistent with experimental findings (Ford and Thanos, 2010; Onoguchi et al, 2010).
Next, we parameterized IRF-7/ISG induction by fitting the dose response of IRF-7-expressing cells versus external IFN-β (Supplementary Figure S10D) and determining the IFN-β secretion rate per cell to match the observed time course of extracellular IFN-β (Figure 6D, blue trace) to the fraction of IFN-β producers (Figure 6D, green trace). With these parameters (Supplementary Table S1), the simulated kinetics of IRF-7-expressing cells agreed with the data (Figure 6D, red trace; note that IRF-7 induction takes place during prolonged elevation of extracellular IFN-β). Importantly, the stochastic switching in individual cells translates into predictable dynamics of IFN-secreting and protected cell fractions at the population level.
The comparison between model and data in Figure 6D suggests that the IFN-β produced by a single cell can induce ISGs in several cells. To examine this further, we simulated the model for smaller infection doses where IFN-β-producing cells are sparser. Measuring ISG expression, we found good agreement between model and experiment over a range of small doses of infection (Figure 7A). Importantly, the model predicted strong paracrine amplification of antiviral protection through IFN, with up to 40 times as many ISG-expressing cells as IFN-secreting cells. This prediction proved remarkably accurate when tested experimentally (Figure 7B and C). Thus, at the level of the cell population, paracrine amplification transforms stochastic single-cell responses into efficient and predictable antiviral protection. Few IFN-producing cells suffice as sentinels of viral infection to protect a large number of cells surrounding the infection site.
Figure 7.
Paracrine amplification of the IFN response is predicted by the model and verified experimentally. (A) Comparison between model and experiment for low infection doses. For low viral doses yielding sparse infection (1, 2, and 5 HAU/ml), the model predicts IFN response kinetics of the correct magnitude (solid lines, model; dots, experimental data). (B) Paracrine propagation of the signal. The rarity of IFN-β-producing cells at low infection doses reveals strong paracrine propagation of the IFN response because up to ∼40 times as many cells respond with IRF-7–mCherry expression (IFN responders). The agreement between model (squares) and experimental data (black dots) is remarkable, given that the model was calibrated only for a single condition (40 HAU/ml, marked with blue square) while the red squares represent predictions that were subsequently tested. (C) Co-culture of IFN-β–tGFP (green) and IRF-7–mCherry (red) cells illustrates paracrine communication. IFN-β–tGFP reporter cells were infected with 40 HAU/ml NDV for 1 h. Then IRF-7–mCherry cells were added at same density and cells were subjected to time-lapse microscopy. Merged fluorescent pictures for IFN-β–tGFP and IRF-7–mCherry at selected time points post-infection are shown. Source data is available for this figure in the Supplementary Information.

Discussion
In this paper, we have dissected the dynamics of the type-I IFN system in an experimental setting that allowed reliable quantitation in living cells. In addition to heterogeneous IFN induction, we describe for the first time bimodal, all-or-nothing expression of ISGs that governs resistance to viral infection. Thus, heterogeneity of single-cell decisions is a pervasive feature of the IFN system. Imaging signal transduction and gene-expression dynamics, we show that these processes contribute quantitatively to heterogeneous expression of the Ifnb gene, and that cell-intrinsic stochasticity prevails in both antiviral signalling and IFN-β expression. Importantly, however, we find that paracrine propagation of the IFN signal can mediate reliable antiviral protection at the cell-population level.
The origin of heterogeneous IFN induction has been controversial; both cell-intrinsic stochastic events in gene expression (Hu et al, 2007; Apostolou and Thanos, 2008) and virus-related mechanisms (Chen et al, 2010; Killip et al, 2011) have been implicated. Our live-cell imaging data obtained with both virus and viral-surrogate (poly I:C) stimuli demonstrate that cell-intrinsic mechanisms generate large-scale heterogeneity in the decision to express IFN-β and in the timing of expression. This establishes intrinsic host cell variability as a decisive factor shaping the antiviral response at the single-cell level while not excluding additional virus effects in other systems (e.g., interference with IFN-β induction, which is a hallmark of many pathogenic viruses).
Cell-intrinsic stochasticity occurs at multiple levels of regulation to generate heterogeneity in IFN-β expression. Quantifying the relative contributions of gene expression and signal transduction activating IRFs and NF-κB, we found that both processes have strong stochastic components. The decision whether to express the Ifnb gene was made primarily at the level of signal transduction, indicating a limitation in RIG-I-induced signalling. Stochasticity in gene expression contributed to temporal variability. These data show that heterogeneous IFN-β expression cannot be attributed solely to stochastic gene regulation, which has previously been the main suspect for IFN-β and other cytokines (Hu-Li et al, 2001; Guo et al, 2004; Apostolou and Thanos, 2008; Raj and van Oudenaarden, 2008; Mariani et al, 2010). Rather, both virus-induced signalling and gene regulation could have different control on cell-to-cell variability in different cell types, depending, for example, on the expression levels of pathway components.
Our model prediction that viral induction of signalling exhibits cooperativity might be linked to the clustering of the adaptor IPS-1 downstream of RIG-I (Onoguchi et al, 2010). Indeed, the formation of large macromolecular complexes in signalling transduction as well as gene expression (Ford and Thanos, 2010) are likely to be slow stochastic processes (Luijsterburg et al, 2010) that could determine both frequency and strong temporal heterogeneity of IFN-β expression. Distributed expression levels of signalling molecules, transcription factors etc. may further add to this cell-intrinsic stochasticity in creating cell-to-cell heterogeneity (Feinerman et al, 2008). A recent study has correlated variability of both viral and cellular factors with stochastic IFN-β expression (Zhao et al, 2012).
Despite the large heterogeneity of single-cell decisions to express IFN-β and ISGs, the cell-population response is remarkably predictable. Our data-driven model indicates that this is a robust feature of the system that requires simply an appropriate tuning of IFN-β production per cell and IFN responsiveness of ISG induction, allowing paracrine propagation of the IFN signal. In this way, an individual IFN-secreting cell can bring a defined territory surrounding it into an antiviral state.
Thus, heterogeneous and stochastic IFN induction could primarily reflect mechanistic limitations of the host cells (as has been speculated for other cytokines; Mariani et al, 2010) that are readily overcome through paracrine signal propagation. Alternatively, heterogeneity may have functional benefits. In particular, the widely distributed onset times of producers could sustain signalling if the IFN production period per cell is limited. Moreover, we expect that different interference strategies of pathogenic viruses, inhibiting IFN versus ISG induction (Versteeg and Garcia-Sastre, 2010), will have differential effects on the spatio-temporal dynamics of the IFN system which, in turn, might control the viral ‘choice’ between persistent versus acute lytic infection.
Our demonstration of paracrine cell-to-cell communication as an efficient mechanism for quenching single-cell stochasticity will likely be relevant also for other systems. It underscores the importance of studying single cell behaviour within cell-population context for rationalizing cell physiology.
Materials and methods
Plasmids and BAC constructs
Cloning of BACs was performed by homologous recombination (Zhang et al, 2000) in Escherichia coli strain GS1783 (Tischer et al, 2010). BACs of the RPCI-24 library were obtained from Children’s Hospital Oakland Research Institute. IFN-β–tGFP is based on clone RP24-325J11, where the Ifnb ORF was replaced by TurboGFP ORF (Evrogen) containing an N-terminal triple FLAG epitope and a FRT-site flanked neomycine phosphotransferase gene under control of the PGK promoter as described earlier (Pulverer et al, 2010). IRF-7–mCherry is based on clone RP24-266F8, in which the Irf7 stop codon and 3′ adjacent ∼300 bp were replaced by the mCherry (Shaner et al, 2004) coding sequence and the above-mentioned neomycine selection cassette. Cassettes containing TurboGFP or mCherry together with the selection marker were amplified by PCR introducing 50 bp target sequence homologies (Ifnb–tGFP forward: 5′-GCCTTTGCCTCATCTTGCAGGTAGCAGCCGACACCAGCCTGGCTTCCATCATGGACTACAAAGACCACGACGGAG-3′; Ifnb–tGFP reverse: 5′-GATGCCACAGTCATTTCCCTAGATGCTTTAGATTTCCCATTATCTCTTACCCTTACAATTTACGCCCTTAAGATCC-3′; Irf7–mCherry forward: 5′-CAACAGTCTCTACGAAGACATCGAACACTTCCTCATGGACCTGGGTCAGTGGCCTTCAGGTGGAGGCGGTATGGTGAGCAAGGGCGAGGAGGATA-3′; Irf7–mCherry reverse: 5′-CCCCCAGTCCCTACACACCAGGCCTCCTTTGCAGCACTCACTTTCATAATCAAGACTCAAGGGCATCGGTCGACGGATCC-3′). PCR products were transferred into E. coli by electroporation. Correctness of recombination and integrity of the BAC molecules was checked by PCR, restriction analysis and sequencing of the integration site.
p65–YFP was created by inserting amplified murine p65 cDNA into pMBC-1 (Dirks et al, 1994) containing linker and YFP encoding sequences. pIRF-7–CFP and pIRF7–tagRFP were created by introducing Irf7 cDNA (C57BL/6) (amplified by forward: 5′-CGAATTCCACCATGGCTGAAGTGAGG-3′; reverse: 5′-CGCCGGCCCGCCTCCACCTGAAGGCCACTGACCCAGGTCC-3′) into pMBC-1 vector containing linker and sequences for CFP and tagRFP (Evrogen), respectively, via EcoRI and NaeI restriction sites.
Cell culture and DNA transfection
Murine fibroblastoid NIH 3T3 cells were grown under standard tissue culture conditions in DMEM medium (Sigma) supplemented with 10% fetal calf serum, glutamine, penicillin and streptomycin. Transfections of BAC or plasmid DNA were performed with Metafectene (Biontex) according to the manufacturer’s instructions. G418- or puromycin-selected representative clonal cells showing stable expression of the reporter construct and strong signal to background ratio of the fluorescent marker were used. Transfections of BAC or plasmid DNA were performed with Metafectene (Biontex).
IFN stimulation
Infections with Newcastle Disease Virus LaSota (Lohmann Tierzucht, Cuxhaven, Germany) were performed after three times washing with serum-free medium. After 1 h, residual virus was removed by washing three times with serum-containing medium. Low molecular weight poly I:C (InvivoGen) was transfected via Lipofectamine2000 (Invitrogen) according to the manufacturer’s instructions. Total RNA of fractionated cell populations was isolated using RNeasy (QIAGEN) according to the manufacturer’s instruction and transfected using Lipofectamine2000.
IFN source and detection
Murine IFN-β was recombinantly produced in stably expressing BHK-21 cells and obtained from supernatant as described before (Pulverer et al, 2010). IFN activity was measured with a bioassay based on Mx2-Luc reporter gene expression (Kugel et al, 2011).
Time-lapse microscopy
Time-lapse microscopy used an Olympus IX81 fluorescence microscope, with a PRIOR motorized stage and climate chamber. The Olympus CellˆM autofocus function was used to compensate Z-drift. Cells were seeded into ibidi eight-well μ-slides coated with collagen IV. Image acquisition was performed at intervals between 15 and 30 min.
Image and data analysis
Microscopical picture series were analysed with ImageJ (NIH, Bethesda, MD) and built-in plugins as well as MTrackJ (E Meijering). Time points of p65 or IRF-7 activation were plotted upon significant increase of nuclear signal that eventually led to maximal accumulation. Time of cell division was counted when sister cells were clearly separated. Figures and statistical analysis were done with MATLAB, Microsoft Excel 2007 and GraphPad Prism 5.
Immunocytochemistry and flow cytometry
NDV-infected cells were stained in suspension following fixation with 4% paraformaldehyde (10 min) and Triton X-100 (0.1%) permeabilization. Primary mouse anti-HN14f (Santa Cruz) was used at 1:500. After washing with PBS supplemented with saponin (0.1%), secondary Cy3- or Cy5-labelled antibody (goat anti-mouse IgG+IgM, Dianova) was used. Analysis of living and fixed cells in flow cytometry was performed with BD FACscalibur and LSR II (analytical) or FACSaria (sorting), using appropriate laser and filter settings for TurboGFP, mCherry, and chemical fluorophores. Results were quantified with FlowJo 7.6 software.
Real-time PCR
mRNA was isolated with RNeasy (QIAGEN), cDNA hybrid strands were generated with first-strand cDNA synthesis kit (GE Healthcare). Reactions were performed with QIAGEN QuantiTect SYBR green kit in a Roche LightCycler 480 using specific intron-spanning primers (Mx2: forward 5′-TCACCAGAGTGCAAGTGAGG-3′, reverse 5′-CATTCTCCCTCTGCCACATT-3′; Rsad2: forward 5′ GTCCTGTTTGGTGCCTGAAT-3′, reverse 5′-GCCACGCTTCAGAAACATCT-3′; Isg20: forward 5′-TAAGCGCCTGCTACACAAGA-3′, reverse 5′-GCAGCTTCTAACCCTGGATG-3′; Usp18: forward 5′-AAGGACCAGATCACGGACAC-3′, reverse 5′-CATCCTCCAGGGTTTTCAGA-3′; Irf9: forward 5′-ACCACGGAACCAGAAATCAC-3′, reverse 5′-GTTGCAGTTGCTGTTGCTGT-3′; Irf7: forward 5′-GAAGACCCTGATCCTGGTGA-3′, reverse 5′-CCAGGTCCATGAGGAAGTGT-3′; Irf1: forward 5′-CTCACCAGGAACCAGAGGAA-3′, reverse 5′-TGAGTGGTGTAACTGCTGTGG-3′; Actb: forward 5′-TGGAATCCTGTGGCATCCATGAAA-3′, reverse 5′-TAAAACGCAGCTCAGTAACAGTCCG-3′).
Mathematical modelling
We simulated in 104 cells the stochastic intracellular dynamics for virus replication, IRF and NF-κB activation by virus, induction of IFN, activation of STAT1/2 by IFN, and expression of IRF-7 as a representative ISG, using the Gillespie’s algorithm. Extracellular IFN was considered as rapidly diffusing in cell culture and iterated in parallel with a deterministic Euler method. Initially, signal transduction and gene expression were inactive in all cells, and the cells were infected with virus. Parameters were chosen according to experimental measurements as explained in Supplementary Information. MATLAB codes for all model simulations are available as Supplementary Information.
Supplementary Material
Supplementary Figures S1-11, Supplementary Table S1
Acknowledgments
We acknowledge M Freund for excellent technical assistance, N Hänsgen for qPCR, and L Gröbe for cell sorting. We thank R Tsien for providing mCherry and E Meijering for MTrackJ image analysis plugin. UR and JS were supported by a Georg-Christoph-Lichtenberg stipend by the federal state of Lower Saxony and the Helmholtz International Research School for Infection Biology. This work was supported in part by grants from the German Research Foundation (SFB 900), the BMBF MedSys and ForSys initiative (ViroQuant), and the Initiative and Networking Fund of the Helmholtz Association within the Helmholtz Alliance on Systems Biology/SBCancer.
Author contributions: MK, HH, UR, and TH conceived the study. UR, MK, JS, GN, MN, and AK carried out the experiments. MR, MF, KK, and TH performed the modelling. UR, MR, MK, HH, MF, and TH analysed the data. UR, MK, HH, MR, and TH wrote the paper.
Footnotes
The authors declare that they have no conflict of interest.
References
- Apostolou E, Thanos D (2008) Virus infection induces NF-κB-dependent interchromosomal associations mediating monoallelic IFN-β gene expression. Cell 134: 85–96 [DOI] [PubMed] [Google Scholar]
- Brennan K, Bowie AG (2010) Activation of host pattern recognition receptors by viruses. Curr Opin Microbiol 13: 503–507 [DOI] [PubMed] [Google Scholar]
- Brierley MM, Fish EN (2005) Stats: Multifaceted regulators of transcription. J Interferon Cytokine Res 25: 733–744 [DOI] [PubMed] [Google Scholar]
- Chen S, Short JAL, Young DF, Killip MJ, Schneider M, Goodbourn S, Randall RE (2010) Heterocellular induction of interferon by negative-sense RNA viruses. Virology 407: 247–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs K, Stock N, Ross C, Andrejeva J, Hilton L, Skinner M, Randall R, Goodbourn S (2007) mda-5, but not RIG-I, is a common target for paramyxovirus V proteins. Virology 359: 190–200 [DOI] [PubMed] [Google Scholar]
- Dirks W, Schaper F, Kirchhoff S, Morelle C, Hauser H (1994) A multifunctional vector family for gene expression in mammalian cells. Gene 149: 387–388 [DOI] [PubMed] [Google Scholar]
- Elowitz MB, Levine AJ, Siggia ED, Swain PS (2002) Stochastic gene expression in a single cell. Science 297: 1183–1186 [DOI] [PubMed] [Google Scholar]
- Feinerman O, Veiga J, Dorfman JR, Germain RN, Altan-Bonnet G (2008) Variability and robustness in T cell activation from regulated heterogeneity in protein levels. Science 321: 1081–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford E, Thanos D (2010) The transcriptional code of human IFN-beta gene expression. Biochim Biophys Acta 1799: 328–336 [DOI] [PubMed] [Google Scholar]
- Guo L, Hu-Li J, Paul WE (2004) Probabilistic regulation of IL-4 production in Th2 cells: accessibility at the Il4 locus. Immunity 20: 193–203 [DOI] [PubMed] [Google Scholar]
- Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T (2005) IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434: 772–777 [DOI] [PubMed] [Google Scholar]
- Hu-Li J, Pannetier C, Guo L, Löhning M, Gu H, Watson C, Assenmacher M, Radbruch A, Paul WE (2001) Regulation of expression of IL-4 alleles: analysis using a chimeric GFP/IL-4 gene. Immunity 14: 1–11 [DOI] [PubMed] [Google Scholar]
- Hu J, Nudelman G, Shimoni Y, Kumar M, Ding Y, López C, Hayot F, Wetmur JG, Sealfon SC (2011) Role of cell-to-cell variability in activating a positive feedback antiviral response in human dendritic cells. PLoS One 6: e16614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Sealfon SC, Hayot F, Jayaprakash C, Kumar M, Pendleton AC, Ganee A, Fernandez-Sesma A, Moran TM, Wetmur JG (2007) Chromosome-specific and noisy IFNB1 transcription in individual virus-infected human primary dendritic cells. Nucleic Acids Res 35: 5232–5241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S (2005) Cell type-specific involvement of RIG-I in antiviral response. Immunity 23: 19–28 [DOI] [PubMed] [Google Scholar]
- Killip MJ, Young DF, Ross CS, Chen S, Goodbourn S, Randall RE (2011) Failure to activate the IFN-beta promoter by a paramyxovirus lacking an interferon antagonist. Virology 415: 39–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuz LE, Levy AH (1965) Physical properties of chick interferon. J Bacteriol 89: 462–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugel D, Pulverer JE, Köster M, Hauser H, Staeheli P (2011) Novel nonviral bioassays for mouse type I and type III interferon. J Interferon Cytokine Res 31: 345–349 [DOI] [PubMed] [Google Scholar]
- Kumagai Y, Kumar H, Koyama S, Kawai T, Takeuchi O, Akira S (2009) Cutting edge: TLR-dependent viral recognition along with type I IFN positive feedback signaling masks the requirement of viral replication for IFN-α production in plasmacytoid dendritic cells. J Immunol 182: 3960–3964 [DOI] [PubMed] [Google Scholar]
- Kunzi MS, Pitha PM (2003) Interferon targeted genes in host defense. Autoimmunity 36: 457–461 [DOI] [PubMed] [Google Scholar]
- Levin D, Harari D, Schreiber G (2011) Stochastic receptor expression determines cell fate upon interferon treatment. Mol Cell Biol 31: 3252–3266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillemeier BF, Köster M, Kerr IM (2001) STAT1 from the cell membrane to the DNA. EMBO J 20: 2508–2517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijsterburg MS, von Bornstaedt G, Gourdin AM, Politi AZ, Mone MJ, Warmerdam DO, Goedhart J, Vermeulen W, van Driel R, Höfer T (2010) Stochastic and reversible assembly of a multiprotein DNA repair complex ensures accurate target site recognition and efficient repair. J Cell Biol 189: 445–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maheshri N, O'Shea EK (2007) Living with noisy genes: how cells function reliably with inherent variability in gene expression. Annu Rev Biophys Biomol Struct 36: 413–434 [DOI] [PubMed] [Google Scholar]
- Maiwald T, Schneider A, Busch H, Sahle S, Gretz N, Weiss TS, Kummer U, Klingmuller U (2010) Combining theoretical analysis and experimental data generation reveals IRF9 as a crucial factor for accelerating interferon alpha-induced early antiviral signalling. FEBS J 277: 4741–4754 [DOI] [PubMed] [Google Scholar]
- Mariani L, Schulz EG, Lexberg MH, Helmstetter C, Radbruch A, Löhning M, Höfer T (2010) Short-term memory in gene induction reveals the regulatory principle behind stochastic IL-4 expression. Mol Syst Biol 6: 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onoguchi K, Onomoto K, Takamatsu S, Jogi M, Takemura A, Morimoto S, Julkunen I, Namiki H, Yoneyama M, Fujita T (2010) Virus-infection or 5'ppp-RNA activates antiviral signal through redistribution of IPS-1 mediated by MFN1. PLoS Pathog 6: e1001012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paixão T, Carvalho TP, Calado DP, Carneiro J (2007) Quantitative insights into stochastic monoallelic expression of cytokine genes. Immunol Cell Biol 85: 315–322 [DOI] [PubMed] [Google Scholar]
- Pulverer JE, Rand U, Lienenklaus S, Kugel D, Zietara N, Kochs G, Naumann R, Weiss S, Staeheli P, Hauser H, Köster M (2010) Temporal and spatial resolution of type I and III interferon responses in vivo. J Virol 84: 8626–8638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, van Oudenaarden A (2008) Nature, nurture, or chance: stochastic gene expression and its consequences. Cell 135: 216–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehwinkel J, Tan CP, Goubau D, Schulz O, Pichlmair A, Bier K, Robb N, Vreede F, Barclay W, Fodor E, Reis e Sousa C (2010) RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 140: 397–408 [DOI] [PubMed] [Google Scholar]
- Rott R (1979) Molecular basis of infectivity and pathogenicity of myxovirus. Brief review. Arch Virol 59: 285–298 [DOI] [PubMed] [Google Scholar]
- Sadler AJ, Williams BRG (2008) Interferon-inducible antiviral effectors. Nat Rev Immunol 8: 559–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, Tsien RY (2004) Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 22: 1567–1572 [DOI] [PubMed] [Google Scholar]
- Snijder B, Pelkmans L (2011) Origins of regulated cell-to-cell variability. Nat Rev Mol Cell Biol 12: 119–125 [DOI] [PubMed] [Google Scholar]
- Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK (2009) Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 459: 428–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiller DG, Wood CD, Rand DA, White MR (2010) Measurement of single-cell dynamics. Nature 465: 736–745 [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Akira S (2009) Innate immunity to virus infection. Immunol Rev 227: 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theofilopoulos AN, Baccala R, Beutler B, Kono DH (2005) Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol 23: 307–336 [DOI] [PubMed] [Google Scholar]
- Tischer BK, Smith GA, Osterrieder N (2010) En passant mutagenesis: a two step markerless red recombination system. Methods Mol Biol 634: 421–430 [DOI] [PubMed] [Google Scholar]
- Trinchieri G (2010) Type I interferon: friend or foe? J Exp Med 207: 2053–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versteeg GA, Garcia-Sastre A (2010) Viral tricks to grid-lock the type I interferon system. Curr Opin Microbiol 13: 508–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinkemeier U (2004) Getting the message across, STAT Design principles of a molecular signaling circuit. J Cell Biol 167: 197–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawatzky R, De Maeyer E, De Maeyer-Guignard J (1985) Identification of individual interferon-producing cells by in situ hybridization. Proc Natl Acad Sci USA 82: 1136–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Muyrers JPP, Testa G, Stewart AF (2000) DNA cloning by homologous recombination in Escherichia coli. Nat Biotechnol 18: 1314–1317 [DOI] [PubMed] [Google Scholar]
- Zhao M, Zhang J, Phatnani H, Scheu S, Maniatis T (2012) Stochastic expression of the interferon-beta gene. PLoS Biol 10: e1001249. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures S1-11, Supplementary Table S1