

Abstract
Herein we present the outcome of a high throughput screening (HTS) campaign-based strategy for the rapid identification and optimization of selective and general chemotypes for both kappa (κ) opioid receptor (KOR) activation and inhibition. In this program, we have developed potent antagonists (IC50 < 120 nM) or agonists of high binding affinity (Ki < 3 nM). In contrast to many important KOR ligands, the compounds presented here are highly modular, readily synthesized, and, in most cases, achiral. The four new chemotypes hold promise for further development into chemical tools for studying the KOR or as potential therapeutic lead candidates.
Keywords: Kappa opioid receptor agonist, kappa opioid receptor antagonist, high-throughput screening
The kappa (κ) opioid receptor (KOR) regulates a wide range of physiological functions, including addiction,1,2 depression,3−5 pain relief, and the immune response. Its involvement in addiction pathways indicates that κ opioid agonist, and antagonist compounds are potential tools for the treatment of substance dependence. However, a complete understanding of how KOR-associated signaling pathways modulate behaviors is still emerging. Although dopamine regulation is generally accepted to play a major role, many unanswered questions remain about the mechanisms that influence the maintenance of dopaminergic tone. For instance, what are the chemical determinates that enable KOR ligands to selectively activate some or many of the diverse signaling cascades available to the receptor, and how does KOR homodimer or heterodimer formation affect the chemical space, hence pharmacological profile of the receptor?6 The extent to which these and other receptor mechanisms modulate KOR mediated processes such as antinociception7 through changes in dopamine sensitivity and dopamine dependent physiology are not fully understood. Furthermore, the chemical tools available to study these KOR mediated-processes are somewhat limited.
In this study, we report four new classes of KOR ligands for the study of the KOR in the etiology of drug addiction and relapse behaviors. They are representative of the successful outcome of a high throughput screening (HTS) campaign-based strategy for the rapid identification and optimization of selective and general chemotypes for both KOR activation and inhibition. In this program, we have developed potent antagonists (IC50 < 120 nM) or agonists of high binding affinity (Ki < 3 nM). In contrast to many important KOR binders, the compounds presented here are highly modular, readily synthesized, and, in most cases, achiral. The rapid identification and optimization of potent and selective new ligands for the KOR further validates the strategy of ligand discovery by HTS of an academic compound collection and its optimization through collaborative structure–activity relationship (SAR) studies.8
Results and Discussion
Screening Goals
Mounting evidence suggests that the seven transmembrane (7TM) class of receptors, which includes the KOR, possesses multiple binding sites capable of producing a range of physiological responses.9 The discovery of new chemotypes for the KOR, especially those with novel binding modes, should produce probes with selective downstream regulation or even therapeutically relevant compounds with different pharmacological, pharmacodynamic, or side-effect profiles.
With these ideas in mind, this project was initiated under the umbrella of the NIH Molecular Libraries Program (MLP) to develop new chemical probes,10 with the objective that these probes would also satisfy an unmet need in chemical biology or pharmacology.11−14We sought probes with the following characteristics: (1) a novel scaffold that would be chemically attractive in terms of synthetic accessibility and structural malleability, (2) potency of 1 μM or better, and (3) selectivity of 100-fold for KOR over the μ and δ opioid receptors (MOR and DOR, respectively). These requirements guided the logical selection and prioritization of chemotypes from the HTS campaign through to probe nomination. As per the guidelines for MLP supported projects, probe reports for agonist15 (ML138 and ML139) and antagonist compounds16 (ML140 and ML190) discovered in the present study are available online.
KOR Active Compounds
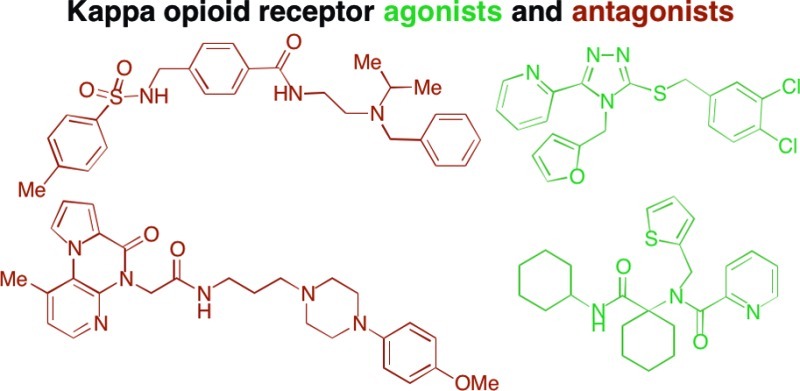
A few notable examples of the numerous molecules known to modulate the KOR are depicted in Figure 1. A survey of KOR ligands17 reveals that a majority of nonpeptide compounds share common structural elements and/or lineage with naturally occurring opioids. Many of these molecules, such as GNTI,18 derive directly from opioid alkaloids, whereas others contain structural elements seen in synthetic opioids such as meperidine (e.g., JDTic19 and ketazocine20). These established potent and selective KOR binders tend to be structurally complex, containing multiple stereocenters and requiring lengthy synthetic routes to construct modified analogues. For example, the natural product salvinorin A,21 a unique non-nitrogenous KOR ligand, has comparable structural complexity to many of the isolated opiates. Even the widely utilized, simplified agonist compound, U-50,48822 contains two stereogenic centers. In addition to their structural complexity, three of the most potent and selective of the KOR antagonists, NorBNI, GNTI, and JDTic, have long acting properties at the KOR that may be associated with JNK activation, which complicates SAR assessment in this series.23
Figure 1.

Structures of representative KOR ligands.
Assay Platforms
Two assay platforms24 were employed to evaluate KOR activity and selectivity: the KOR DiscoveRx β-arrestin PathHunter assay25 and an imaging based β-arrestin translocation assay26 for confirmatory and selectivity assays. The KOR PathHunter assay is a commercial platform for direct measurement of GPCR activation by detection of β-arrestin binding to the KOR using enzyme fragment complementation. In this system, β-arrestin is fused to an N-terminal deletion mutant of β-galactosidase (termed the enzyme acceptor or EA) and the GPCR of interest is fused to a smaller (42 amino acid), weakly complementing fragment (termed ProLink). In cells that stably express these fusion proteins, ligand stimulation results in the recruitment of β-arrestin by the GPCR. This encourages the complementation of the two β-galactosidase fragments results in a functional enzyme that converts substrate in the assay medium to a detectable signal. The imaging based β-arrestin translocation assays are based upon the redistribution of β-arrestin linked to green fluorescent protein (GFP) from the cytosolic compartment to the plasma membrane, to coated pits, and finally to endosomal vesicles.26
Initially, 290 000 compounds from the MLSMR compound collection were tested in the KOR DiscoveRx β-arrestin primary kappa opioid (KOR) agonist and antagonist screens (Pubchem AID 1777 and 1778, respectively27) at a single concentration point (10 μM). Compounds with activities of >50% compared to Dynorphin stimulation were retested with compound solutions resupplied from the MLSMR collection to confirm single concentration activity. The confirmed compounds were further tested in concentration response screens using the DiscoveRx β-arrestin primary KOR agonist and antagonist screens to obtain EC50 or IC50 values (Pubchem AID 2284 and 2285, respectively). These compounds were concurrently tested in a β-galactosidase counterscreen assay (Pubchem AID 1966) to assess the possibility that these compounds might inhibit the reporting enzyme. The activity of the validated compounds was confirmed in the KOR agonist and antagonist high content imaging β-arrestin translocation assays (Pubchem AID 2359 and 2348, respectively). The KOR:DOR:MOR selectivity of the compounds was determined using the analogous β-arrestin translocation assays for the DOR (Pubchem AID 2370 and 2357, for agonists and antagonists, respectively) and the MOR (Pubchem AID 2352 and 2420, for agonists and antagonists, respectively). The minimum threshold for the identification of an interesting new chemotype was set as an EC50 (agonist) or IC50 (antagonist) of less than 1 μM in the KOR PathHunter assay and greater than 100-fold selective for the KOR over either the MOR or DOR in the β-arrestin translocation assay (or within the detection limits of the assay platform). Following data analysis and attrition of compounds from the counterscreens, eight classes of compounds emerged from the combined agonist and antagonist screens possessing a strong potential for optimization and bearing no resemblance to known opioid ligands (Figure 2).
Figure 2.

Individual compounds illustrating validated KOR chemotypes from HTS.
SAR Expansion and Optimization
The most potent and synthetically tractable chemotypes (Chemotypes I and III) were prioritized for optimization toward probe molecule nomination. In contrast, scaffolds bearing potentially unstable substructures (e.g., Chemotypes VI and VII from Figure 2) were not pursued further. Optimization on a second cluster of promising chemotypes (Chemotypes II, IV and V) was begun shortly behind the first batch. The synthesis of Chemotype V analogues proved more challenging than expected, and only seven analogues were prepared, none of which improved the potency to the probe threshold. The antagonist Chemotype VIII did not confirm as a KOR selective chemotype in the high-content imaging assays. Each successful chemotype represents a previously unknown class of general KOR agonists or antagonists. The modular synthetic routes to these four classes of compounds, summarized in Scheme 1, permitted the rapid exploration of structure–activity relationships (for detailed synthetic schemes and reaction details, see the Supporting Information). Each chemotype could be readily assembled from commercially available or easily accessed fragments, thus allowing the introduction of new functional groups or core modifications.
Scheme 1. General Retrosynthetic Pathways to KOR Chemotypes.

Agonists
The HTS and commercial purchasing phases of the project revealed a cluster of unnatural amino acid based compounds (Chemotype I) possessing promising potency and selectivity. The agonist functional activity (EC50 values, PathHunter assay) and KOR:DOR:MOR high content imaging assay selectivity for this initial subset of compounds already exceeded the criteria for a probe molecule (Table 1, entries 1–4, 7). The picolinic analogue 1{1} (entry 1) was singled out as the best candidate from this promising set and selected for nomination as a probe.23 Both furan- and thiophene-containing analogues were highly active, and the single case where a direct comparison was possible suggested the thiophene-containing side chain to be advantageous over the furan-containing side chain (entries 1 and 3). A clearer advantage was observed for the 2-pyridyl amides over the 2-pyrazinyl amides, as exemplified by comparison of the analogue pairs 1{1}/1{2} and 1{8}/1{9}. The glycine-based analogue 1{10} was synthesized and found to possess negligible KOR activity, confirming the requirement for substitution on the amino acid segment. In contrast, R1 on the secondary amide appears to have a less dramatic effect on potency, with the most potent analogue to date being R1 = cyclohexyl (entry 1). A striking observation was that a synergistic combination of substitution can have a profound effect, such as the loss of activity observed in 1{5}.
Table 1. SAR for Chemotype I κ-Opioid Receptor Agonist Compounds.

DiscoveRx β-arrestin PathHunter assay (n = 3, except for entry 10 where n = 2), compared to Dynorphin.
High content imaging based β-arrestin translocation assay (n = 2), compared to Dynorphin.
High content imaging based β-arrestin translocation assay (n = 1), compared to Dynorphin.
The numerous potent compounds derived from the disubstituted amino acid scaffolds reinforce the merits of this chemotype as a versatile template for probes in KOR research. Moreover, the synthetic route (Scheme 1 and Supporting Information), which utilizes Boc protection of the amine functional group followed by amine coupling of the carboxylic acid to avoid the inconvenience of zwitterionic intermediates, easily provides gram scale or larger quantities and is readily modified to provide analogues. A more comprehensive survey of substitution effects is currently underway and will be the subject of future reports.
The HTS screening campaign for the triazole-based Chemotype II originally uncovered four compounds with potencies around 2 μM and one example at 6.76 μM (Table 2, entries 1 and 3–6). While a number of Chemotype II analogues were commercially available, limited substitution on the phenyl ring and no available thiophene-containing analogues led us to adopt an entirely synthetic approach based on precedented chemistry. Beginning with the appropriate isothiocyanate and 2-picolynyl hydrazide, we synthesized the 1,2,4-triazole-3-thione scaffolds in two steps and excellent yields (77–82% overall yields) without chromatographic separations.28 The subsequent coupling with a wide range of benzyl halides proceeded smoothly in acetone facilitated by K2CO329 to readily furnish over 75 compounds, a selection of which is shown in Table 2.
Table 2. SAR Analysis for Chemotype II κ-Opioid Receptor Agonist Compounds.

DiscoveRx β-arrestin PathHunter assay (n = 3), compared to Dynorphin.
High content imaging based β-arrestin translocation assay (n = 2).
High content imaging based β-arrestin translocation assay (n = 1).
Styryl side chain in place of the substituted benzyl group.
Mindful of classical SAR substitution strategies,30,31 our approach toward the optimization of the Chemotype II compounds was inspired by the observation in the Chemotype I work that in some cases a switch from the furan to thiophene moiety afforded at least a 2-fold increase in potency (cf. Table 1, entries 1 and 3). In the present case, however, the thiophene and furan moieties afforded roughly equipotent compounds. A more productive approach was varying the substitution on the phenyl ring, which produced two submicromolar analogues (entries 8 and 9, Table 2). Interestingly, some compounds of this chemotype containing substituted phenyl rings displayed >100% Emax values at the highest concentration tested (as compared to dynorphin A (Emax = 100%)). Whether this is an artifact of the PathHunter assay platform or truly hyperactivation of the KOR is currently under investigation in conjunction with synthetic efforts exploring the replacement of the pyridyl and furan/thiophene side chains as well as more diverse substitution on the benzyl ring.
Antagonists
The HTS campaign for antagonists afforded a single Chemotype III compound already below the 1 μM threshold for a probe molecule (Table 3, entry 1) and one example with an IC50 of 6.72 μM (entry 2), as well as a number of analogues lacking potency (not shown). Although we had a compound in hand meeting the required potency, we wished to determine whether the potency could be enhanced by structural changes. The structure of the hit compound could easily be disconnected into three phenyl ring-containing fragments (Scheme 1). This modular route to Chemotype III analogues allowed the rapid synthesis of a number of analogues with structural modifications independently explored on all three fragments, a selection of which is shown in Table 3.
Table 3. SAR for Chemotype III κ-Opioid Receptor Antagonist Compounds.



DiscoveRx β-arrestin PathHunter assay (n = 4), compared to Dynorphin.
High content imaging based β-arrestin translocation assay (n = 2), compared to Dynorphin.
High content imaging based β-arrestin translocation assay (n = 1), compared to Dynorphin.
In place of the substituted phenyl ring.
Varying the substitution on either or both the far left- and right-hand aromatic rings afforded numerous analogues of close potency to the HTS hit from the DiscoveRx PathHunter KOR assay (Table 3, entries 5, 7, 9, 12, 17–20, 26, and 38). The HCS assay data for this subset of compounds contained more variability, most notable were a set of compounds with >5-fold increase in potency by this metric (entries 10, 16, 17, 19, 26, 38, and 39).
The subtle modifications above provided some intriguing selectivity patterns along with modest increases in potency. More substantial structural changes proved to be more fruitful for increasing analogue potency. The additional steric bulk of a tert-butyl group on the tertiary amine in 3{39} afforded a surprising >14-fold increase in potency over the HTS hit compound. In contrast, the incorporation of rings into the right-hand side of the structure did not improve potency. Interestingly, the constrained analogue 3{48} showed no significant (in fact showed negative) inhibition of the KOR at the higher test concentrations, although some DOR inhibition was observed. Based on this observation, several compounds (including 3{2} and 3{5}) were screened for KOR agonism, and 3{48} alone of these compounds confirmed as a moderately potent partial agonist (0.6 uM EC50, ∼20% Emax, Figure 3). Weak partial agonists may also prove of value in the study and potential treatment of polydrug addiction,32 so this class of compounds may warrant future investigation. An additional attribute of potential utility is the incorporation of a bromine atom in several analogues without detriment to the potency (entries 7, 20, and 21), providing a mass spectrum signature for tracking the localization of the compounds.
Figure 3.

Concentration-dependent inhibition (a) and activation (b) for sulfonamide 3{48}.
Overall, the Chemotype III antagonist scaffold possesses attributes that may be useful in KOR signaling investigations or in research on treatments for addiction and substance abuse. The potency of the HTS hit compound was quickly increased over 14-fold without significantly increasing the molecular weight of the compound. The limited SAR studies completed to date suggest that both the tether length as well as the substitution on the left-hand aromatic ring and right-hand nitrogen may be critical to maintain potency and selectivity. The versatile, modular route developed here enables the synthesis of additional analogues as well as compounds tailored toward investigator specific applications.
Along with the previous antagonist chemotype, the antagonist HTS campaign uncovered two Chemotype IV compounds with promising potency (entries 1 and 2, Table 4) as well as a number of analogues with IC50 values >10 uM. Both active compounds contained a p-methoxyphenyl-substituted piperazine attached to a pyrrolopyrazinone scaffold. We developed an amide-coupling route to this chemotype that permitted the synthesis of 15 analogues in a single round of SAR. The synthetic efforts were augmented with the purchase of 10 commercial analogues and a selection of the screening results is presented in Tables 4 and 5.
Table 4. SAR of Purchased Chemotype IV κ-Opioid Receptor Antagonist Compounds.
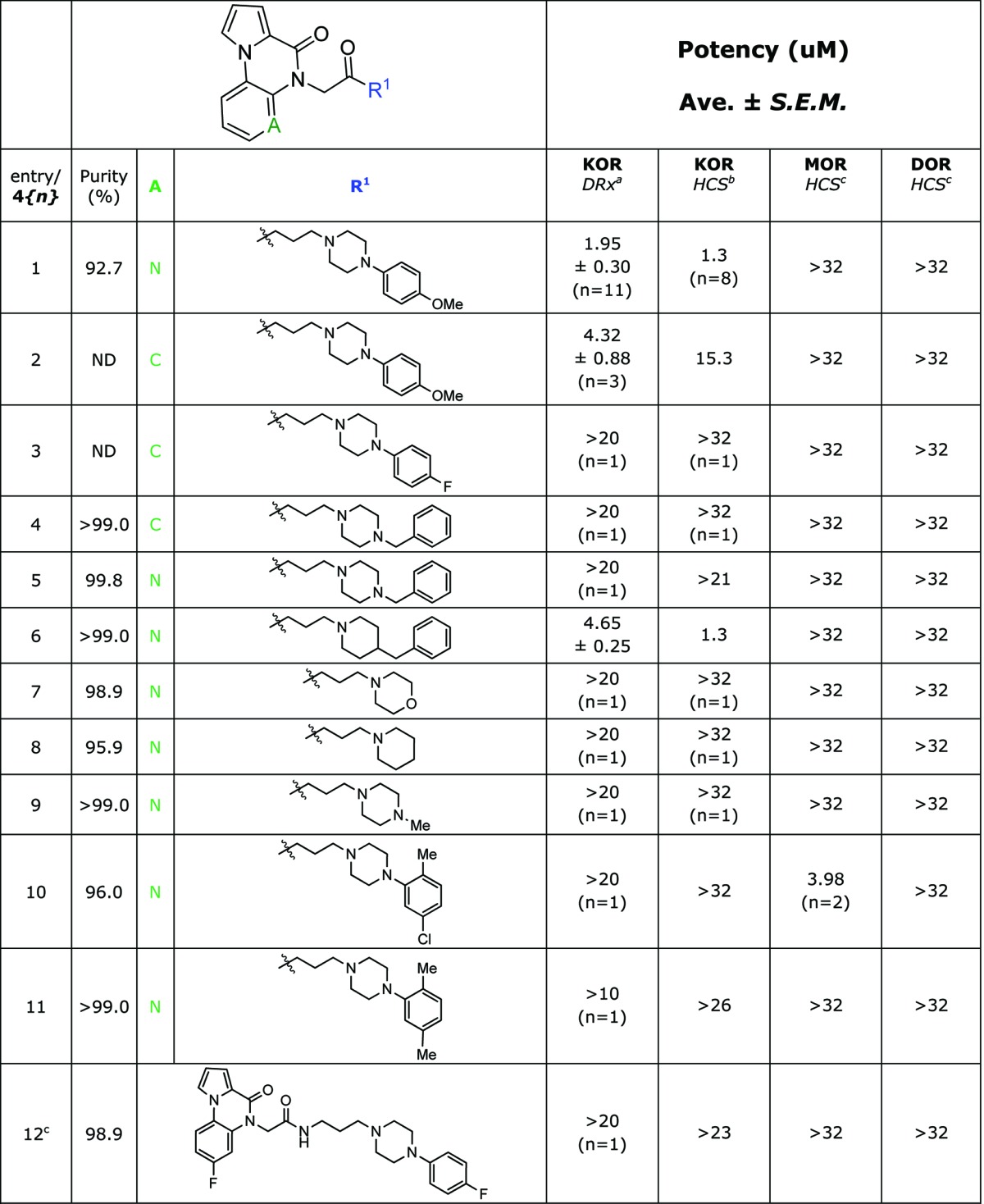
DiscoveRx β-arrestin PathHunter assay (n = 4), compared to Dynorphin.
High content imaging based β-arrestin translocation assay (n = 2), compared to Dynorphin.
High content imaging based ßβ-arrestin translocation assay (n = 1), compared to Dynorphin.
For entry 12 the full structure of the compound is given.
Table 5. SAR of Synthetic Chemotype IV κ-Opioid Receptor Antagonist Compounds.
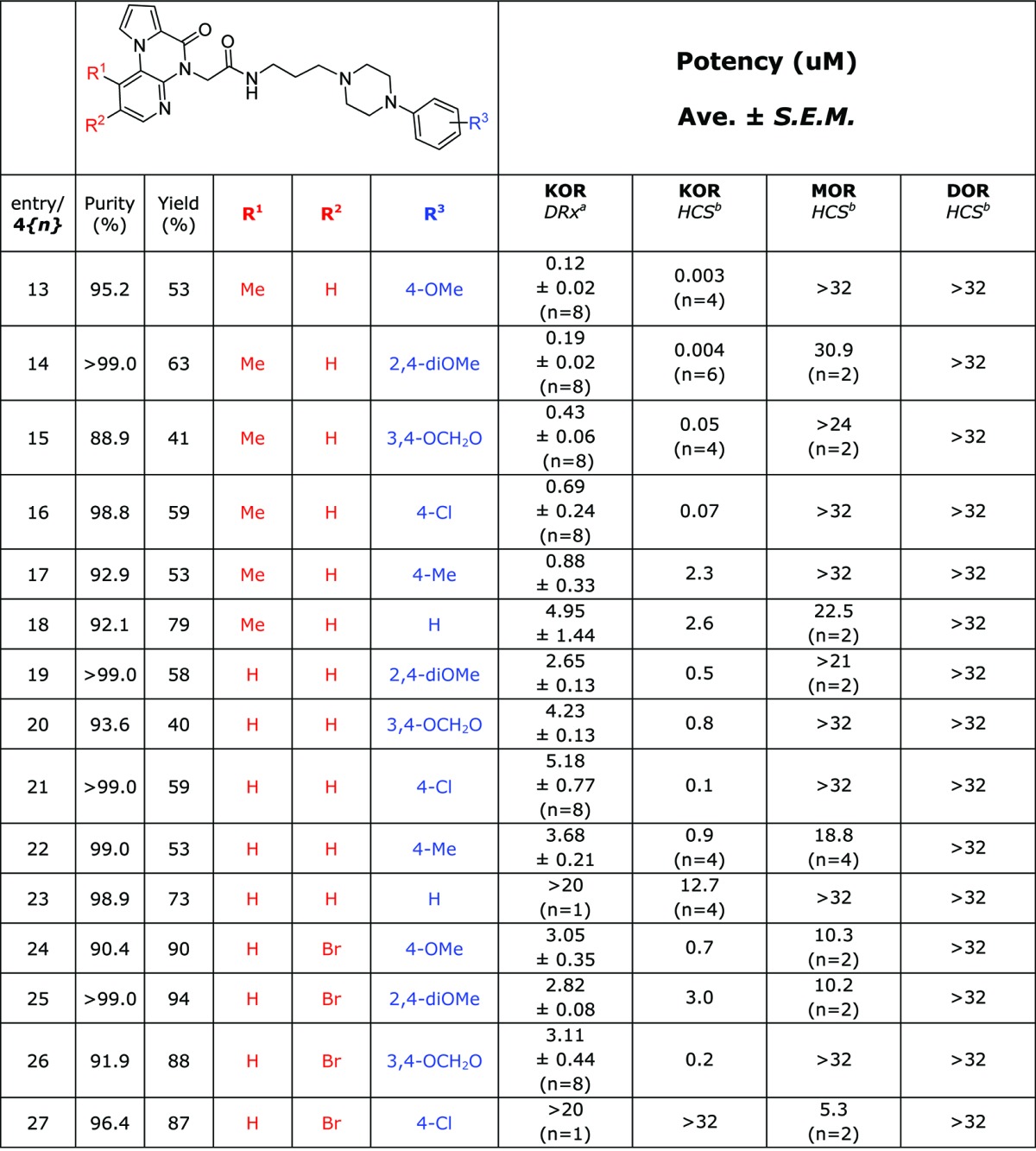
DiscoveRx β-arrestin PathHunter assay (n = 4), compared to Dynorphin.
High content imaging based β-arrestin translocation assay (n = 2), compared to Dynorphin.
High content imaging based β-arrestin translocation assay (n = 1), compared to Dynorphin.
Interestingly, the incorporation of a single methyl group on the heterocyclic core increased the potency of the p-methoxyphenyl substituted piperazine analogue by over 10-fold in the β-arrestin assay and afforded the KOR-selective compound 4{13} with a high content imaging assay IC50 of 3 nM. While less potent than JDTic (IC50 = 0.02 nM), this compound is over 10 000-fold selective for the KOR over both the DOR and MOR, an improved selectivity compared to JDTic (202-fold selective for the KOR over the MOR). Based on these merits, compound 4{13} has been recently nominated as an MLPCN probe compound.33 Several other compounds sharing the additional methyl group also exceeded the probe criteria to a lesser extent (Table 5, entries 14 through 17). For several compounds, we observed a slight loss in selectivity against the MOR counterscreen (entries 14, 15, 18, 19, 22, 24, 25, and 27). This trend was amplified for compound 4{27}, which was a MOR selective antagonist of modest potency (5.32 μM). The scaffold is highly amenable to modification via the present synthetic route and the effects of varying the substitution of this chemotype are currently being explored.
The collection of compounds in Figure 4 highlight the most promising molecules generated from this brief project. The structural characteristics of this collection provide wide diversity both among the chemotypes depicted here and also as compared to the prior art of KOR active compounds. The successful discovery of four new, distinct KOR agonist or antagonist chemotypes through a HTS campaign of the MLPCN compound collection and subsequent optimization demonstrate the powerful combination of pairing biology and chemistry research institutions together toward a common goal. Each of these molecules has been shown to be of useful or promising potency and very high selectivity (for the KOR over the DOR or MOR).
Figure 4.

Summary of Chemotypes discovered as KOR agonists or antagonists.
Preliminary Pharmacology and ADMET Studies
Binding Profiling
We were interested in characterizing the broader selectivity of the more potent compounds against additional targets and assessing their basic pharmacological properties. The probe molecule candidates 1{1} (ML139), 2{8} (ML138), 3{1} (ML140), and 4{13} (ML190) and the post probe Chemotype III compound 3{39} were subjected to a binding assay panel of 44 GPCR and other molecular targets by the Psychoactive Drug Screening Program (PDSP) (Figure 5; for full results (Ki values) from these binding assays, see the Supporting Information). The compounds were initially screened in radioligand binding assays at a constant concentration (10 μM) to identify possible activity of the compound. Results showing significant activity in the initial screen were selected for Ki determinations.
Figure 5.

Selectivity binding studies for representative KOR agonist or antagonist compounds.
The agonist probe 1{1} (ML139) displayed an impressive binding affinity for the KOR (Ki = 0.6 nM) and an even more remarkable selectivity across the 44 assay panel, displaying affinity only for the opioid receptors at concentrations up to 10 000 nM. The compound was found to possess a KOR:DOR selectivity ratio of over 1:2000 and a KOR:MOR selectivity ratio of 1:940 in the secondary binding assays. These data further confirm that this chemotype exemplified by the present probe constitutes a highly potent new scaffold for exploring KOR biology.
As a representative of the Chemotype II series, the MLPCN probe molecule 2{8} (ML138) was characterized against the PDSP assay panel. Although modestly more potent analogues were subsequently found, we view this selectivity data as representative of this series. The test compound was found to possess weak binding affinity for 11 nonopioid targets with Ki values in the 1000–10 000 nM range. In contrast, the binding affinity for the KOR was at least 500-fold more potent (Ki = 2.4 nM). The compound was found to possess a KOR:DOR selectivity ratio of over 1:2000 and a KOR:MOR selectivity ratio of 1:792 in the secondary binding assays.
For the antagonist Chemotype III series, the MLPCN probe molecule 3{1} (ML140) along with the most potent member of this class, 3{39}, were profiled against the PDSP assay panel in addition to the probe compound. The KOR secondary binding affinity for these compounds 3{1} (ML140) and 3{39} were found to be of similar potency (50 and 68 nM, respectively). It is interesting that the KOR binding affinity values of these compounds are so similar given that the functional antagonist potency for compound 3{39} was over 10-fold greater than that of 3{1} (ML140). Along with the improved functional assay potency, 3{39} was found to possess favorable improvements in its selectivity profiles. The tert-butyl analogue 3{39} had only three non-KOR secondary assay binding values below 500 nM, compared to seven such values for the probe compound. Furthermore, 3{39} showed no affinity for the adrenergic and dopaminergic receptors and possessed a much improved selectivity profile across the serotonin and M1 receptors, though the selectivity against the other muscarinic receptors was only marginally improved (see the Supporting Information for numeric Ki values). The remarkable effect on selectivity arising from such subtle structural modification is encouraging for the prospect of creating a highly selective analogue with retention of potency, and work toward this goal is currently under investigation.
The Chemotype IV probe 4{13} (ML190) was found to possess KOR selective binding affinity (Ki = 129 nM) compared to the DOR and MOR binding affinity values (Ki = 1443 nM and 1585 nM, respectively). In general, 4{13} (ML190) displays a slightly cleaner binding profile than the Chemotype III antagonist compounds, although this probe does possess a rather high affinity for the D3 receptor (Ki = 250 nM).This affinity, while not optimal, may also encourage its development as a D3 receptor antagonist as ligands at this receptor have also shown promise in the treatment of addictive disorders.34
In Vitro PK/Toxicity Measurements
In addition to profiling the selectivity of representative molecules from these new classes of KOR ligands, we were interested in assessing their basic physical properties. Thus, each of the compounds nominated as MLPCN probe candidates was characterized by the screens shown in Table 6.
Table 6. Basic Pharmacological Properties for Representative Examples of Each Chemotype.
| plasma protein binding (% bound) |
plasma stabilityd |
hepatic microsome stabilitye |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| cmpd | aqueous solubility (μg/mL)a(@ pH) | PAMPA Pe (×10–6 cm/s)b(@ pH) | BBB-PAMPA Pe (×10–6 cm/s)c | human 1 μM/10 μM | mouse 1 μM/10 μM | human (PBS only) | mouse (PBS only) | human | mouse | hepatic toxicity, LC50 (μM)f |
| 1{1}ML139 | 5.7 (5.0) | 908 (5.0) | 90 | 99.59/99.07 | 94.05/90.70 | 100 | 99.06 | 0 | 0.63 | 6.3 (avg, n = 3) |
| 6.6 (6.2) | 971 (6.2) | |||||||||
| 6.5 (7.4) | 916 (7.4) | |||||||||
| 2{8}ML138 | <0.1 (5.0) | 1793 (5.0) | 242g | 99.75/99.80 | 99.80/99.64 | 100 | 100 | 0.03 | 0.03 | >50 |
| 0.29 (6.2) | 1921 (6.2) | |||||||||
| 0.14 (7.4) | 2089 (7.4) | |||||||||
| 3{1}ML140 | 0.47 (5.0) | >1710 (5.0) | 419 | 99.67/99.73 | 98.90/98.92 | 100 | 100 | 0.04 | 0.07 | >50 |
| 0.12 (6.2) | >1853 (6.2) | |||||||||
| 0.79 (7.4) | >1944 (7.4) | |||||||||
| 4{13}ML190 | 96.6 (5.0) | 27 (5.0) | 51 | 93.96/88.54 | 88.46/80.07 | 100 (29.8) | 100 (30.8, avg) | 22.13 | 7.34 | >50 |
| 90.6 (6.2) | 227 (6.2) | |||||||||
| 12.4 (7.4) | 757 (7.4) | |||||||||
| 12.9 (PBS) | ||||||||||
In aqueous buffer, pH’s 5.0/6.2/7.4.
In aqueous buffer; donor compartment pH’s 5.0/6.2/7.4; acceptor compartment pH 7.4.
In aqueous buffer; donor compartment pH’s 7.4; acceptor compartment pH 7.4.
Percent remaining at 3 h.
Percent remaining at 1 h.
Toward Fa2N-4 immortalized human hepatocytes.
Unable to detect any compound at 1 uM.
The solubility of the Chemotypes I to III was generally low, with the Chemotype I representative displaying marginally better solubility across the pH range tested. However, the Chemotype IV compound 4{13} (ML190) possessed a much better solubility profile across the pH range tested. In the PAMPA (Parallel Artificial Membrane Permeability Assay) screen,35 both agonist probes 1{1} (ML139) and 2{8} (ML138) have excellent permeability. 1{1} (ML139) had moderate brain barrier permeabilty while 2{8} (ML138) had almost 3-fold selectivity in the blood brain PAMPA assay. In this assay, the antagonist probe 3{1} (ML140) had very good Pe (>1710–1940 × 10–6 cm/s) and BBB-Pe (419 × 10–6 cm/s). By comparison, the probe 4{13} (ML190) had a moderate permeability (Pe) of 27 x10–6 cm/s at pH 5 that rapidly increased to 757 × 10–6 cm/s as the pH rose to 7.4, consistent with loss of protonation and positive charge, which would improve permeability. This probe exhibited moderate permeability in the blood brain barrier (BBB-Pe) PAMPA assay of 51 × 10–6 cm/s. Both agonist probes, 1{1} (ML139) and 2{8}(ML138) as well as the antagonist 3{1} (ML140) were determined to be highly bound (90–99%) to both human and mouse plasma, while the Chemotype IV antagonist 4{13} (ML190) was marginally more available (80–94%). All four compounds screened show excellent stability (>99%) in both human and mouse plasma. Moreover, all four test compounds are rapidly metabolized in either human or mouse microsomes and 4{13} (ML190) being the longest lasting with 22% and 7.3% (for human and mouse microsomes, respectively) remaining after 1 h.
The agonist probe compound 1{1} (ML139) showed significant toxicity toward human hepatocytes, potentially limiting its in vivo utility. The other compounds tested showed no measurable toxicity in this screen. The thiophene moiety found in compound 1{1} (ML139) could be the likely source of the observed hepatic toxicity although the similar furan moiety in 2{8} (ML138) did not produce any observed toxicity.
Conclusions
These findings for the KOR demonstrate the utility of applying the synergistic efforts of complementary academic and institutional laboratories to problems beyond the scope of the individual participants. Under the umbrella of the MLP, our discovery and optimization studies have yielded four novel KOR chemotypes that hold promise for further development into ligands for basic research and clinical applications. The simplified structures of the scaffolds compared to those of currently utilized KOR agents and their modular synthetic routes make them attractive for further investigation. The potencies of these compounds are currently sufficient for their use in cells as probes to map out the contribution of different steps along the β-arrestin mediated signaling pathway, except for Chemotype II, are in a range sufficient for testing in small animals, and are approximating a threshold where their use as radioloabeled probes for studying receptor distribution and internalization is a distinct possibility. Though their utility as therapeutic leads will require some further SAR explorations to improve potency and address some pharmacokinetic liabilities, the developmental strategies that must be employed in these areas have been identified and the work is underway.
Methods
Representative Synthesis of Agonist Chemotype I Compounds
tert-Butyl (1-(cyclohexylcarbamoyl)cyclohexyl)carbamate
A microwave vial (2–5 mL rated capacity) was charged with 1-((tert-butoxycarbonyl)amino)cyclohexanecarboxylic acid36 (142 mg, 0.58 mmol), cyclohexylamine (75 mg, 0.76 mmol, 1.3 equiv), HOBt (89 mg, 0.58 mmol, 1.0 equiv), and diisopropylcarbodiimide (96 mg, 0.76 mmol, 1.3 equiv) in MeCN (3 mL). The reaction vial was heated in a microwave reactor at 100 °C for 10 min then cooled to rt. The reaction mixture was dissolved in MeOH (4 mL), adsorbed onto Celite and chromatographed to yield the product as a white solid (139 mg, 0.43 mmol, 73% yield). Rf = 0.70 (EtOAc/hexanes 1:1); mp 197–198 °C; 1H NMR (400 MHz, CDCl3) δ 1.13–1.19 (complex, 3 H), 1.28–1.45 (complex, 7 H), 1.45 (s, 9 H), 1.61–1.65 (complex, 4 H), 1.84–1.96 (complex, 6 H), 3.74 (m, 1 H), 4.72 (br s, 1 H), 6.72 (br s, 1 H); 13C NMR (101 MHz, CDCl3) δ d 28.2, 47.6; u 21.3, 24.5, 25.1, 25.5, 32.2, 32.8, 59.3, 154.8, 173.6; IR (neat) 3336, 3305, 2931, 2854, 1689, 1641 cm–1; HRMS (ESI) m/z calcd for C18H33N2O3([M+H]+) 325.2491, found 325.2498.
1-Amino-N-cyclohexylcyclohexanecarboxamide
tert-Butyl(1-(cyclohexylcarbamoyl)-cyclo-hexyl)carbamate (123 mg, 0.38 mmol) was dissolved in a mixture of trifluoroacetic acid (2 mL) and CH2Cl2(2 mL) and stirred at rt for 14 h. The solvents were removed in vacuo and the residue partitioned between saturated aqueous NaHCO3(5 mL) and CH2Cl2 (3 × 8 mL). The combined organics were dried with Na2SO3 and evaporated to yield the deprotected product as a white solid (84 mg, 0.37 mmol, 99% yield), which was used as obtained in the next step. Rf = 0.31 (EtOAc/hexanes 1:1); mp 98–101 °C; 1H NMR (400 MHz, CDCl3) δ 1.11–1.46 (complex, 10 H), 1.57–1.72 (complex, 6 H), 1.86 (dd, J = 3.6, 12.8 Hz, 2 H), 1.99 (dt, J = 4.0, 13.2 Hz, 2 H), 3.69 (m, 1 H), 7.71 (br s, 1 H); 13C NMR (101 MHz, CDCl3) δ d 47.4; u 21.2 (×2), 24.7 (×2), 25.1 (×2), 25.5 (×2), 33.0 (×2), 34.6 (×2), 56.9, 176.8; IR (neat) 3383, 3320, 2929, 2852, 1613 cm–1; HRMS (ESI) m/z calcd for C13H25N2O ([M+H]+) 225.1967, found 225.1977.
N-Cyclohexyl-1-((thiophen-2-ylmethyl)amino)cyclohexanecarboxamide
To a solution of 1-amino-N-cyclohexylcyclohexanecarboxamide (84 mg, 0.374 mmol) and thiophene-2-carboxaldehyde (84 mg, 0.749 mmol, 2.0 equiv) in dichloroethane (5 mL) was added sodium triacetoxyborohydride (238 mg, 1.12 mmol, 3.0 equiv). The reaction was stirred at rt for 16 h and partitioned between aqueous NaOH (1 N, 10 mL) and CH2Cl2 (3 × 10 mL). The combined organics were dried with Na2SO3, adsorbed onto Celite, and chromatographed to afford the reductive amination product as a light orange oil (101 mg, 0.314 mmol, 84% yield). Rf = 0.72 (EtOAc/hexanes 1:1); 1H NMR (400 MHz, CDCl3) δ 1.11–1.22 (complex, 3 H), 1.32–1.42 (complex, 7 H), 1.57–1.71 (complex, 5 H), 3.68–3.75 (m, 1 H), 3.76 (s, 2 H), 6.94 (dd, J = 0.8, 3.2 Hz, 1 H), 6.98 (dd, J = 3.2, 5.2 Hz, 1 H), 7.25 (dd, J = 1.2, 4.8 Hz, 1 H), 7.54 (br s, 1 H); 13C NMR (101 MHz, CDCl3) δ d 47.5, 124.3, 124.4, 126.7; u 21.5 (×2), 24.7 (×2), 25.1, 25.6, 31.8 (×2), 33.1 (×2), 42.2, 61.0, 144.2, 175.2; IR (neat) 3345, 2928, 2853, 1644 cm–1; HRMS (ESI) m/z calcd for C18H29N2OS ([M+H]+) 321.2001, found 321.2007.
N-(1-(Cyclohexylcarbamoyl)cyclohexyl)-N-(thiophen-2-ylmethyl)picolinamide 1{1} (ML139)
To a solution of N-cyclohexyl-1-((thiophen-2-ylmethyl)amino)cyclohexanecarboxamide (93 mg, 0.290 mmol) and triethylamine (117 mg, 1.16 mmol, 4.0 equiv) in CH2Cl2 (5 mL) was added picolinoyl chloride hydrochloride (103 mg, 0.58 mmol, 2.0 equiv). The reaction was stirred at rt for 18 h and partitioned between saturated aqueous NaHCO3 and CH2Cl2 (3 × 8 mL). The combined organics were dried with Na2SO3, adsorbed onto Celite and chromatographed to afford the bisamide product as an off-white solid (106 mg, 0.249 mmol, 86% yield). Rf = 0.36 (EtOAchexanes 1:1); mp 102–110 °C; 1H NMR (400 MHz, CDCl3) δ 1.06–1.22 (m, 3 H), 1.28–1.38 (m, 2 H), 1.42–1.83 (complex, 11 H), 2.14–2.26 (m, 4 H), 3.66 (m, 1 H), 4.98 (s, 2 H), 6.78 (d, J = 2.8 Hz, 1 H), 6.83 (t, J = 4.4 Hz, 1 H), 6.95 (br s, 1 H), 7.14 (d, J = 5.2 Hz, 1 H), 7.33 (dd, J = 0.8, 4.8 Hz, 1 H), 7.59 (d, J = 8.0 Hz, 1 H), 7.75 (dt, J = 1.6, 7.6 Hz, 1 H), 8.57 (d, J = 4.8 Hz, 1 H); 13C (100 MHz, CDCl3) δ d (CH, CH3) 47.9, 124.2, 124.7, 125.2, 126.4, 126.5, 137.0, 147.9; u (C, CH2) 22.4 (×2), 24.6 (×2), 25.5, 25.6, 32.7 (×2), 33.0 (×2), 44.8, 66.8, 141.8, 155.0, 171.6, 172.7; IR (neat) 2928, 2854, 1655 cm–1; HRMS (ESI) m/z calcd for C24H32N3O2S ([M+H]+),= 426.2215, found 426.2207.
Representative Synthesis of Agonist Chemotype II Compounds
N-(Thiophen-2-ylmethyl)-2-picolinoylhydrazinecarbothioamide
2-Picolynyl hydrazide (883 mg, 6.44 mmol) and thiophene isothiocyanate (1000 mg, 6.44 mmol) in MeCN (20 mL) were stirred for 16 h at rt. The reaction mixture was filtered, and the precipitate washed with additional MeCN (3 × 10 mL) and dried under vacuum to afford the thioamide as an off-white solid (1,642 mg, 5.62 mmol, 87% yield), which was used without further purification. Mp 175–178 °C; 1H NMR (DMSO-d6) δ 4.84 (d, J = 6.0 Hz, 2 H), 6.93 (m, 1 H), 7.00 (m, 1 H), 7.36 (dd, J = 1.2, 4.8 Hz, 1 H), 7.63 (m, 1 H), 8.03 (m, 2 H), 8.56 (br s, 1 H), 8.66 (d, J = 4.8 Hz, 1 H), 9.50 (br s, 1 H), 10.60 (s, 1 H); 13C NMR (DMSO-d6) δ d 122.5, 124.9, 125.8, 126.2, 126.9, 137.6, 148.4; u 42.1, 141.9, 149.3, 181.4, 198.3; IR (neat) 3141, 1672, 1527, 1499, 1466 cm–1; HRMS (ESI) m/z calcd for C12H13N4OS2([M+H]+) 293.0531, found 293.0516.
4-(Thiophene-2-ylmethyl)-3-(pyridin-2-yl)-1H-1,2,4-triazole-5(4H)-thione
To a slurry of the above thioamide (602 mg, 2.06 mmol) in water (25 mL) was added NaOH (4.00 g, 100 mmol). The reaction was heated at reflux for 2 h; the starting thioamide dissolving promptly upon warming. The reaction was cooled to rt, diluted with aqueous HCl (1 N, 20 mL) and acidified to pH = 6 with concentrated HCl. The solid precipitate was filtered, washed with water (2 × 15 mL) and dried under vacuum to afford the thiophene thione as a white solid (530 mg, 1.93 mmol, 94% yield), which was used without further purification. Mp 229–231 °C; 1H NMR (DMSO-d6) δ 6.02 (s, 2 H), 6.87 (dd, J = 3.2, 4.8 Hz, 1 H), 7.09 (d, J = 2.4 Hz, 1 H), 7.34 (dd, J = 0.8, 5.2 Hz, 1 H), 7.59 (q, J = 4.4 Hz, 1 H), 7.99 (d, J = 4.4 Hz, 2 H), 8.80 (d, J = 4.8 Hz, 1 H), 14.2 (br s, 1 H), 10.60 (s, 1 H); 13C NMR (DMSO-d6) δ d 122.8, 125.5, 126.3, 126.5, 128.1, 138.0, 149.0; u 42.3, 137.8, 145.5, 147.8, 168.3; IR (neat) 3019, 2896, 1584, 1549, 1501, 1462 cm–1; HRMS (ESI) m/z calcd for C12H11N4S2 ([M+H]+) 275.0475, found 275.0412.
2-(5-((3,4-Dichlorobenzyl)thio)-4-(thiophen-2-ylmethyl)-4H-1,2,4-triazol-3-yl)pyridine 2{9}
The thiophene thione (65 mg, 0.24 mmol), K2CO3 (66 mg, 0.48 mmol) and 2,4-dichlorobenzyl chloride (56 mg, 0.28 mmol) were combined in acetone (3 mL) and stirred in a sealed vial for 15 h. The solvent was removed and the residue washed with CH2Cl2 (2 × 3 mL) then filtered. The combined filtrates were concentrated and purified by silica chromatography to afford the triazole product as an off-white solid (92 mg, 0.21 mmol, 90% yield). Mp 162–163 °C; Rf = 0.24 (1:1 hexanes/ EtOAc); 1H NMR (CDCl3) δ 4.42 (s, 2 H), 5.93 (s, 2 H), 6.85 (dd, J = 3.6, 4.8 Hz, 1 H), 6.98 (d, J = 2.4 Hz, 1 H), 7.15 (dd, J = 0.8, 4.8 Hz, 1 H), 7.23 (dd, J = 1.6, 8.0 Hz, 1 H), 7.33 (m, 2 H), 7.48 (d, J = 2.0 Hz, 1 H), 7.80 (dt, J = 1.6, 8.0 Hz, 1 H), 8.29 (d, J = 8.0 Hz, 1 H), 8.66 (d, J = 3.6 Hz, 1 H); 13C NMR (CDCl3) δ d 123.2, 124.3, 126.4, 126.5, 127.7, 128.6, 130.6, 131.0, 137.1, 148.6; u 36.6, 43.8, 131.9, 132.6, 137.7, 147.6 (×2), 152.0, 152.5; IR (neat) 3052, 1589, 1568, 1463 cm–1; HRMS (ESI) m/z calcd for C19H15Cl2N4S2 ([M+H]+) 433.0115, found 433.0108.
Representative Synthesis of Antagonist Chemotype III Compounds
N-(2-(Benzyl(isopropyl)amino)ethyl)-4-((4-methylphenylsulfonamido)methyl)benzamide 3{1} (ML140)
4-((4-Methylphenylsulfonamido)methyl)benzoyl chloride (120 mg, 0.37 mmol), triethylamine (94 mg, 0.93 mmol), and N1-benzyl-N1-isopropylethane-1,2-diamine (71 mg, 0.37 mmol) were combined in CH2Cl2 (3 mL) and stirred at rt 15 h. The reaction was diluted with saturated aqueous NaHCO3 (2 mL), and all solvents evaporated in vacuo. The residue was extracted with MeOH (3 × 5 mL) and the combined methanol extracts evaporated in vacuo. The crude product was purified by preparative reverse phase HPLC purification to afford the pure sulfonamide amide product as a white solid (92 mg, 0.19 mmol, 52% yield). Rf = 0.21 (EtOAc/hexanes 1:1); mp 131–133 °C; 1H NMR (400 MHz, CDCl3) δ 1.07 (d, J = 6.8 Hz, 6 H), 2.42 (s, 3 H), 2.67 (t, J = 6.0 Hz, 2 H), 3.00 (sept, J = 6.8 Hz, 1 H), 3.34 (q, J = 5.2 Hz, 2 H), 3.55 (s, 2 H), 4.14 (s, 2 H), 5.34 (br s, 1 H), 6.56 (br s, 1 H), 7.21–7.31 (complex, 9 H), 7.45 (d, J = 8.0 Hz, 2 H), 7.77 (d, J = 8.4 Hz, 2 H); 13C (100 MHz, CDCl3) δ d (CH, CH3) 18.0, 21.5, 49.7, 126.9, 127.0, 127.1, 127.7, 128.4, 128.5, 129.7; u (C, CH2) 37.5, 46.8, 47.8, 53.6, 134.0, 136.9, 139.8, 140.7, 143.5, 166.6; IR (neat) 2964, 1639, 1156 cm–1; HRMS (ESI) m/z calcd for C27H33N3O3S ([M+H]+) 480.2315, found 480.2316.
Representative Synthesis of Antagonist Chemotype IV Compounds
2-Fluoro-4-methyl-3-(1H-pyrrol-1-yl)pyridine
The protocol of Melander and co-workers was utilized.37 Thus, 2-fluoro-4-methylpyridin-3-amine (1.00 g, 7.93 mmol) and 2,5-dimethoxytetrahydrofuran (1.08 mL, 1.05 equiv) were suspended in acetic acid (3 mL) and refluxed for 2 h, and then the reaction was cooled down to rt. The solvents were removed in vacuo, and the residue was purified by silca gel chromatography to afford the product as a light colored oil (1.00 g, 5.68 mmol, 72% yield). Rf = 0.30 (EtOAc/hexanes, 1:8); 1H NMR (400 MHz, CDCl3) δ 2.26 (s, 3H), 6.40 (t, J = 2.1 Hz, 2H), 6.74 (td, J = 2.1, 0.9 Hz, 2H), 7.17 (d, J = 5.1 Hz, 1H), 8.10 (dd, J = 0.8, 5.1 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 17.2, 17.3, 109.8, 122.1, 123.7, 123.8, 145.4, 145.5, 149.5, 149.6, 157.8, 160.2; HRMS (ESI) m/z calcd for C10H10FN2 ([M+H]+) 177.0828, found 177.0827.
4-Methyl-3-(1H-pyrrol-1-yl)pyridin-2-amine
2-Fluoro-4-methyl-3-(1H-pyrrol-1-yl)pyridine (3.90 g, 22.1 mmol) was dissolved in an ammonia solution (80 mL, 7 N in MeOH) in a 250 mL pressure vessel. The mixture was heated at 150 °C for 2 days protected by a blast shield. The mixture was cooled to rt, then cooled in an ice bath for 30 min, and the mixture was filtered. The filtrate was evaporated to dryness and purified by flash chromatography to give the product as a white solid (2.90 g, 16.7 mmol, 76% yield). Rf = 0.50 (EtOAc/hexanes, 1:1); 1H NMR (400 MHz, CDCl3) δ 2.02 (s, 3H), 4.52 (s, 2H), 6.41 (t, J = 2.1 Hz, 2H), 6.60 (d, J = 5.2 Hz, 1H), 6.67 (t, J = 2.1 Hz, 2H), 7.96 (d, J = 5.1 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 16.7, 110.0, 116.0, 121.2, 121.5, 145.9, 147.2, 156.1; HRMS (m/z) calcd for C10H12N3 ([M+H]+) 174.1031, found 174.1026.
Methyl 2-(1-methyl-6-oxopyrido[2,3-e]pyrrolo[1,2-a]pyrazin-5(6H)-yl)acetate
4-Methyl-3-(1H-pyrrol-1-yl)pyridin-2-amine (1.00 g, 5.80 mmol) and triphosgene (2.60 g, 8.70 mmol) were dissolved in toluene (100 mL). The mixture was refluxed for 3 h, then cooled to rt. The red solid was collected by filtration and washed with CH3CN to afford the crude product (0.50 g, 2.51 mmol, 43% yield). The material was used directly for next step reaction without purification. For 1-methylpyrido[2,3-e]pyrrolo[1,2-a]pyrazin-6(5H)-one: 1H NMR (400 MHz, DMSO) δ 2.81 (s, 3H), 6.75 (dd, J = 2.9, 3.9 Hz, 1H), 7.20–7.08 (m, 2H), 8.13 (d, J = 4.9 Hz, 1H), 8.15 (dd, J = 1.4, 2.9 Hz, 1H), 11.64 (s, 1H).
To a solution of 1-methylpyrido[2,3-e]pyrrolo[1,2-a]pyrazin-6(5H)-one (50 mg, 0.25 mmol) in DMF (2 mL) was added NaH (60%, 11 mg, 0.28 mmol). The mixture was stirred at rt for 1 h, then methyl bromoacetate (0.027 mL, 0.28 mmol) was added. The mixture was stirred for 16 h, the solvents removed under vacuum, and the residue was purified by silca gel flash chromatography to afford the product as a light yellow solid (37 mg, 0.14 mmol, 54% yield). Rf = 0.5 (DCM/MeOH, 1:10); 1H NMR (400 MHz, CDCl3) δ 2.83 (s, 3H), 3.77 (s, 3H), 5.25 (s, 2H), 6.71 (dd, J = 2.9, 4.0 Hz, 1H), 7.00 (d, J = 4.9 Hz, 1H), 7.36 (dd, J = 1.5, 4.0 Hz, 1H), 7.95 (dd, J = 1.5, 2.9 Hz, 1H), 8.15 (d, J = 4.9 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 22.9, 41.9, 52.3, 113.4, 113.5, 120.4, 122.6, 122.7, 124.2, 134.8, 142.1, 143.1, 155.5, 169.3; HRMS (ESI) m/z calcd for C14H14N3O3 ([M+H]+) 272.1035, found 272.1042.
2-(1-Methyl-6-oxopyrido[2,3-e]pyrrolo[1,2-a]pyrazin-5(6H)-yl)acetic acid
Methyl 2-(1-methyl-6-oxopyrido[2,3-e]pyrrolo[1,2-a]pyrazin-5(6H)-yl)acetate (517 mg, 1.91 mmol) was dissolved in a mixture of MeOH/H2O/THF (20 mL, 1:1:4 ratio). LiOH (68.5 mg, 2.86 mmol) was added. The mixture was stirred at room temperature for 16 h. The solvents were removed in vacuo and residue was dissolved in water, washed with ether, and then neutralized with 2 N HCl to pH = 3. The precipitate was filtered and dried under vacuum to afford the product as a white solid (356 mg, 1.38 mmol, 73% yield). 1H NMR (400 MHz, DMSO) δ 2.85 (s, 3H), 5.02 (s, 2H), 6.80 (dd, J = 2.9, 3.9 Hz, 1H), 7.29–7.19 (m, 2H), 8.27–8.17 (m, 2H), 12.90 (s, 1H); 13C NMR (101 MHz, DMSO) δ 22.2, 41.6, 112.7, 113.3, 119.5, 122.8, 123.3, 123.9, 135.9, 141.4, 143.1, 154.5, 169.8; HRMS (ESI) m/z calcd for C13H12N3O3 ([M+H]+) 258.0879, found 258.0894.
N-(3-(4-(4-Methoxyphenyl)piperazin-1-yl)propyl)-2-(1-methyl-6-oxopyrido[2,3-e]pyrrolo[1,2-a]pyrazin-5(6H)-yl)acetamide 4{13} (ML190)
2-(1-Methyl-6-oxopyrido[2,3-e]pyrrolo[1,2-a]pyrazin-5(6H)-yl)acetic acid (30 mg, 0.12 mmol), 3-(4-(4-methoxyphenyl)piperazin-1-yl)propan-1-amine (43.6 mg, 0.17 mmol), and DMAP (1.4 mg, 0.012 mmol) were dissolved in DCM (1 mL). Diisopropylcarbodiimide (0.09 mL, 0.58 mmol) was added, and the mixture stirred at rt for 16 h. The solvents were removed in vacuo, and the residue was purified by silica gel flash chromatography to afford the product as a white solid (30 mg, 0.061 mmol, 53% yield). Rf = 0.5 (DCM/MeOH = 10:1); 1H NMR (400 MHz, CDCl3) δ 1.75–1.69 (m, 2H), 2.49 (t, J = 6.4 Hz, 2H), 2.63–2.54 (m, 4H), 2.74 (s, 3H), 3.02–2.91 (m, 4H), 3.41 (dd, J = 5.8, 12.0 Hz, 2H), 3.78 (s, 3H), 5.11 (s, 2H), 6.68 (dd, J = 2.9, 4.0 Hz, 1H), 6.84 (s, 4H), 6.99 (d, J = 5.0 Hz, 1H), 7.12 (s, 1H), 7.33 (dd, J = 1.4, 4.0 Hz, 1H), 7.87 (dd, J = 1.4, 2.9 Hz, 1H), 8.17 (d, J = 4.9 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 22.8, 25.2, 39.4, 44.2, 50.4, 53.4, 55.6, 57.3, 113.4, 113.5, 114.4, 118.0, 120.4, 122.6, 122.8, 124.1, 134.8, 142.3, 143.2, 145.4, 153.8, 155.8, 167.9; HRMS (ESI) m/z calcd for C27H33N6O3 ([M+H]+) 489.2609, found 489.2600.
Acknowledgments
We thank Bryan Roth, Jon Evans, and Vincent Setola of the Psychoactive Drug Screening Program at the University of North Carolina, Chapel Hill for conducting receptor profiling and Ki determinations, Benjamin Nuenswander for performing HPLC purification of samples and obtaining high resolution mass spectra data, Patrick Porubsky for compound management services, and Thomas O. Painter and Steven A. Rogers for reviewing the experimental sections.
Supporting Information Available
Experimental details and characterization for all new compounds, Ki values for the receptor binding assays in Figure 5, Pubchem compound ID (CID) numbers of all final compounds, a summary of assays and corresponding Pubchem assay ID (AID) numbers and assay protocols. This material is available free of charge via the Internet at http://pubs.acs.org.
We gratefully acknowledge financial support from grants 5U54HG005033 (John Reed, PI) and 5U54HG005031 (J.A.) from the Molecular Libraries Initiative, and 1X01DA026208 (L.S.B.) and 5U01DA022950-03 (M.G.C.) from the National Institutes of Drug Abuse. Psychoactive Drug Screening Program at the University of North Carolina, Chapel Hill: National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271-2008-00025-C (NIMH PDSP).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Prisinzano T. E.; Tidgewell K.; Harding W. W. (2005) κ Opioids as potential treatments for stimulant dependence. AAPS J. 7, E592–E599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick S. D.; Maisonneuve I. M.; Raucci J.; Archer S. (1995) Kappa-opioid inhibition of morphine and cocaine self-administration. Brain Res. 681, 147–152. [DOI] [PubMed] [Google Scholar]
- Reindl J. D.; Rowan K.; Carey A. N.; Peng X.; Neumeyer J. L.; McLaughlin J. P. (2008) Antidepressant-like effects of the novel kappa opioid antagonist MCL-144B in the forced-swim test. Pharamcology 81, 229–235. [DOI] [PubMed] [Google Scholar]
- Knoll A. T.; Meloni E. G.; Thomas J. B.; Carroll F. I.; Carlezon W. A. Jr. (2007) Anxiolytic-like effects of kappa-opioid receptor antagonists in models of unlearned and learned fear in rats. J. Pharmacol. Exp. Ther. 323, 838–845. [DOI] [PubMed] [Google Scholar]
- Mague S. D.; Pliakas A. M.; Todtenkopf M. S.; Thomasiewicz H. C.; Zhang Y.; Stevens W. C. J.; Jones R. M.; Portoghese P. S.; Carlezon W. A. Jr. (2003) Antidepressant-like effects of κ-opioid receptor antagonists in the forced swim test in rats. J. Pharmacol. Exp. Ther. 305, 323–330. [DOI] [PubMed] [Google Scholar]
- Tang Y.; Yang J.; Lunzer M. M.; Powers M. D.; Portoghese P. S. (2011) A κ opioid pharmacophore becomes a spinally selective κ-δ agonist when modified with a basic extender arm. ACS Med. Chem. Lett. 2, 7–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin J. P.; Myers L. C.; Zarek P. E.; Caron M. G.; Lefkowitz R. J.; Czyzyk T. A.; Pintar J. E.; Chavkin C. (2004) Prolonged kappa opioid receptor phosphorylation mediated by G-protein receptor kinase underlies sustained analgesic tolerance. J. Biol. Chem. 279, 1810–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachovchin D. A.; Mohr J. T.; Speers A. E.; Wang C.; Berlin J. M.; Spicer T. P.; Fernandez-Vega V.; Chase P.; Hodder P. S.; Schürer S. C.; Nomura D. K.; Rosen H.; Fu G. C.; Cravatt B. F. (2011) Organic Synthesis Toward Small-Molecule Probes and Drugs Special Feature: Academic cross-fertilization by public screening yields a remarkable class of protein phosphatase methylesterase-1 inhibitors. Proc. Natl. Acad. Sci. U.S.A. 108, 6811–6816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanakin T.; Miller L. J. (2010) Seven transmemberane receptors as shapeshifting proteins: The impact of allosteric modulation and functional selectivity on new drug discovery. Pharm. Rev. 62, 265–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy A. (2010) The NIH Molecular Libraries Program: identifying chemical probes for new medicines. Chem. Biol. 549–550. [DOI] [PubMed] [Google Scholar]
- Frye S. V. (2010) The art of the chemical probe. Nat. Chem. Biol. 6, 159–161. [DOI] [PubMed] [Google Scholar]
- Li Q.; Cheng T.; Wang Y.; Bryant S. H. (2010) PubChem as a public resource for drug discovery. Drug Discovery Today 15, 1052–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Serna R.; Mestres J. (2011) Chemical probes for biological systems. Drug Discovery Today 16, 99–106. [DOI] [PubMed] [Google Scholar]
- Workman P.; Collins I. (2010) Probing the probes: fitness factors for small molecule tools. Chem. Biol. 17, 561–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick M. P.et al. Selective KOP receptor agonists: probe 1 and probe 2. http://www.ncbi.nlm.nih.gov/books/NBK47352/; PMID: 21433386. [PubMed]
- Hedrick M. P.et al. Selective KOP receptor antagonists: probe 1. http://www.ncbi.nlm.nih.gov/books/NBK47352/; PMID: 21433381. [PubMed]
- McCurdy C. R., and Prisinzano T. E. (2010) Opioid receptor ligands. In Burger’s Medicinal Chemistry, Drug Discovery and Development (Abraham D. J., Rotella D. P., Eds.), 7th ed., John Wiley & Sons: New York. [Google Scholar]
- Jones R. M.; Hjorth S. A.; Schwartz T. W.; Portoghese P. S. (1998) Mutational evidence for a common κ antagonist binding pocket in the wild-type κ and mutant μ[K303E] opioid receptors. J. Med. Chem. 41, 4911. [DOI] [PubMed] [Google Scholar]
- Thomas J. B.; Atkinson R. N.; Rothman R. B.; Fix S. E.; Mascarella S. W.; Vinson N. A.; Xu H.; Dersch C. M.; Lu Y.; Cantrell B. E.; Zimmerman D. M.; Carroll F. I. (2001) Identification of the first trans-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine derivative to possess highly potent and selective opioid kappa receptor antagonist activity. J. Med. Chem. 44, 2687–2690. [DOI] [PubMed] [Google Scholar]
- Merz H.; Stockhaus K. (1979) N-[(Tetrahydrofuryl)alkyl] and N-(alkoxyalkyl) derivatives of (−)-normetazocine, compounds with differentiated opioid action profiles. J. Med. Chem. 22, 1475–1483. [DOI] [PubMed] [Google Scholar]
- Roth B. L.; Baner K.; Westkaemper R.; Siebert D.; Rice K. C.; Steinberg S.; Ernsberger P.; Rothman R. B. (2002) Salvinorin A: a potent naturally occurring nonnitrogenous kappa opioid selective agonist. Proc. Natl. Acad. Sci. U.S.A. 99, 11934–11939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VonVoigtlander P. F.; Szmuszkovicz J. (1982) Benzeneacetamide amines: structurally novel non-mu opioids. J. Med. Chem. 25, 1125–1126. [DOI] [PubMed] [Google Scholar]
- Bruchas M. R.; Yang T.; Schreiber S.; DeFino M.; Kwan S. C.; Li S.; Chavkin C. (2007) Long-acting κ opioid antagonists disrupt receptor signaling and produce noncompetitive effects by activating C-Jun N-terminal kinase. J. Biol. Chem. 282, 29803–29811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn L. M.; McDonald P. H. (2010) Seeking ligand bias: assessing GPCR coupling to ß-arrestins for drug discovery. Drug Discovery Today: Technol. 7, e37–e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X.; Jones A.; Olson K. R.; Peng K.; Wehrman T.; Park A.; Mallari R.; Nebalasca D.; Young S. W.; Xiao S.-H. (2008) A homogeneous enzyme fragment complementation-based ß-arrestin translocation assay for high-throughput screening of G-protein-coupled receptors. J. Biomol. Screening 13, 737–747. [DOI] [PubMed] [Google Scholar]
- Barak L. S.; Ferguson S. S. G.; Zhang J.; Caron M. G. (1997) A ß-arrestin/green fluorescent protein biosensor for detecting G protein-coupled receptor activation. J. Biol. Chem. 272, 27497–27500. [DOI] [PubMed] [Google Scholar]
- Pubchem is a publicly accessible database that contains data resulting from MLPCN projects: http://Pubchem.ncbi.nlm.nih.gov/.
- Burbuliene M. M.; Sakociute V.; Vainilavicius P. (2009) Synthesis and characterization of new pyrimidine-based 1,3,4-oxa(thia)diazoles, 1,2,4-triazoles and 4-thiazolidinones. ARKIVOC 281–289. [Google Scholar]
- Dilanyan E. R.; Hovsepyan T. R.; Melik-Ohanjanyan R. G. (2008) Synthesis of some substituted 1,2,4-triazole and 1,3,4-thiadiazole derivatives. Chem. Heterocycl. Compd. 44, 1395–1397. [Google Scholar]
- Topliss J. G. (1977) A manual method for applying the Hansch approach to drug design. J. Med. Chem. 20, 463–469. [DOI] [PubMed] [Google Scholar]
- Hajduk P. J.; Sauer D. R. (2008) Statistical analysis of the effects of common chemical substituents on ligand potency. J. Med. Chem. 51, 553–564. [DOI] [PubMed] [Google Scholar]
- Carlezon W. A. Jr.; Béguin C.; Knoll A. T.; Cohen B. M. (2009) Kappa-opioid ligands in the study and treatment of mood disorders. Pharmacol. Ther. 123, 334–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick M. P.et al. Selective KOP receptor antagonists: probe 2, in press. [PubMed]
- Heidbreder C. A.; Newman A. H. (2010) Current perspectives on selective dopamine D(3) receptor antagonists as pharmacotherapeutics for addictions and related disorders. Ann. N.Y. Acad. Sci. 1187, 4–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kansy M.; Senner F.; Gubernator K. (1998) Physiochemical high throughput screening: parallel artificial membrane permeation assay in the description of passive absorption processes. J. Med. Chem. 41, 1007–1010. [DOI] [PubMed] [Google Scholar]
- Costanzo M. J.; Almond H. R. Jr.; Hecker L. R.; Schott M. R.; Yabut S. C.; Zhang H.-C.; Andrade-Gordon P.; Corcoran T. W.; Giardino E. C.; Kauffman J. A.; Lewis J. M.; de Garavilla L.; Haertlein B. J.; Maryanoff B. E. (2005) In-depth study of tripeptide-based α-ketoheterocycles as inhibitors of Thrombin. Effective utilization of the S1′ subsite and its implications to structure-based drug design. J. Med. Chem. 48, 1984–2008. [DOI] [PubMed] [Google Scholar]
- Richards J. J.; Reed C. S.; Melander C. (2008) Effects of N-pyrrole substitution on the anti-biofilm activities of oroidin derivatives against Acinetobacter baumannii. Bioorg. Med. Chem. Lett. 18, 4325–4327. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.