


Abstract
A flexible statistical framework is developed for the analysis of read counts from RNA-Seq gene expression studies. It provides the ability to analyse complex experiments involving multiple treatment conditions and blocking variables while still taking full account of biological variation. Biological variation between RNA samples is estimated separately from the technical variation associated with sequencing technologies. Novel empirical Bayes methods allow each gene to have its own specific variability, even when there are relatively few biological replicates from which to estimate such variability. The pipeline is implemented in the edgeR package of the Bioconductor project. A case study analysis of carcinoma data demonstrates the ability of generalized linear model methods (GLMs) to detect differential expression in a paired design, and even to detect tumour-specific expression changes. The case study demonstrates the need to allow for gene-specific variability, rather than assuming a common dispersion across genes or a fixed relationship between abundance and variability. Genewise dispersions de-prioritize genes with inconsistent results and allow the main analysis to focus on changes that are consistent between biological replicates. Parallel computational approaches are developed to make non-linear model fitting faster and more reliable, making the application of GLMs to genomic data more convenient and practical. Simulations demonstrate the ability of adjusted profile likelihood estimators to return accurate estimators of biological variability in complex situations. When variation is gene-specific, empirical Bayes estimators provide an advantageous compromise between the extremes of assuming common dispersion or separate genewise dispersion. The methods developed here can also be applied to count data arising from DNA-Seq applications, including ChIP-Seq for epigenetic marks and DNA methylation analyses.
INTRODUCTION
The cost of DNA sequencing continues to decrease at a staggering rate (1). As it does, sequencing technologies become more and more attractive as platforms for studying gene expression. Current ‘next-generation’ sequencing technologies measure gene expression by generating short reads or sequence tags, that is, sequences of 35–300 base pairs that correspond to fragments of the original RNA. There are a number of technologies and many different protocols. Popular approaches are either tag-based methods including Tag-Seq (2), deepSAGE (3), SAGE-Seq (4), which sequence from one or more anchored positions in each gene, or RNA-Seq (5–8), which sequences random fragments from the entire transcriptome. Both approaches have proven successful in investigating gene expression and regulation (9–11). In this article, we will use the term RNA-Seq generically to include any of the tag-based or RNA-Seq variants in which very high-throughput sequencing is applied to RNA fragments.
For the purposes of evaluating differential expression between conditions, read counts are summarized at the genomic level of interest, such as genes or exons. Although RNA-Seq can be used to search for novel exons or for splice-variants and isoform-specific expression (7,12–14), transcript assembly (15) or allele-specific expression (16,17), our focus in this article is on differential expression for pre-determined genomic features. Nevertheless, the methods developed here are relevant for inferring isoform-specific differential expression when applied to sub-gene features such as exons or exon-junctions.
Linear modelling methods have been highly successful for analysing microarray experiments with multiple explanatory factors (18,19). It is becoming common-place for RNA-Seq to be used for similar experiments, so there is a pressing need for statistical methods that can provide the same flexibility and rigor for complex RNA-Seq experiments (20).
One strategy for RNA-Seq data analysis is to standardize and transform the read counts to approximate normality, then analyse as for microarray data (3,6,21). This approach is however not fully tuned to the characteristics of read count data. One issue is that very small counts are far from normally distributed, even after transformation, although this issue is rapidly mitigated for larger counts. A more pervasive and important problem is that count data typically shows a strong mean–variance relationship which is not respected by existing normal-based analyses, leading to potentially inefficient statistical inferences. Transformations such as square-root (3) can reduce but do not remove the mean–variance dependence entirely. Calculating exact probabilities for the read counts using appropriate distributions therefore gives the possibility of more sensitive statistical procedures than simply transforming to normality (22–25). Simulations suggest that count models give more statistical power to detect differential expression than approximate normal models (26). Another advantage of explicit count models is that they give more refined possibilities for separating biological from technical variability (22,23).
Despite decreasing sequencing costs, RNA-Seq experiments remain expensive for many researchers, often limiting RNA-Seq studies to only a small number of libraries. There is often very little replication. Yet the basic scientific need to assess differential expression relative to biological variation remains undiminished (27). There is therefore a need to estimate biological variation as reliably as possible from a very small number of replicate libraries. The problem is further complicated by the fact that different genes or transcripts may have different degrees of biological variation. In microarray analysis, this problem has been solved by regularized t-tests (28) or more formally by empirical Bayes or related methods that share information between genes (18,19,29).
A DNA sample can be thought of as a population of cDNA fragments, and each genomic feature can be thought of as a species for which the population size is to be estimated. Sequencing a DNA sample can be thought of as random sampling of each of these species, with the aim of estimating the relative abundance of each species in the population. If each cDNA fragment has the same chance of being selected for sequencing, and the fragments are selected independently, then the number of read counts for a given genomic feature should follow a Poisson variation law across repeated sequence runs of the same cDNA sample. The Poisson model implies that the mean equals the variance, a relationship that has been validated in one of the early RNA-Seq studies using the same initial source of RNA distributed across multiple lanes of an Illumina GA sequencer (30).
The Poisson model does not take account of biological variability or any technical sources that might cause the relative abundance of different genes to vary between different RNA samples. When abundance is not constant between samples, read counts will be over-dispersed relative to Poisson, i.e. the variance must be higher than the mean. Over-dispersed binomial (31,32) or Poisson (32–36) models have been proposed for Serial Analysis of Gene Expression (SAGE) or RNA-Seq data. None of the these proposals have the ability to share information between genes, restricting them to experiments with large numbers of replicate libraries.
A very simple method to share information between genes is to assume that all genes follow the same mean–variance relationship, so all genes with the same expected count have the same variance (23,25,37). This is almost certainly too simple, because it does not allow for the possibility that some genes may be more variable than others. Robinson and Smyth (22,24) developed a promising empirical Bayes approach using weighted likelihood to estimate biological variation in a genewise fashion, implemented in the Bioconductor package edgeR. Other more explicitly Bayesian methods have been proposed for SAGE (38,39) or RNA-Seq data (40), the latter implemented in the Bioconductor package baySeq. Comparisons, on both simulated and real data, show that edgeR and baySeq outperform alternative methods that do not allow for gene-specific variability or do not share information between genes (40). These approaches are however limited to comparisons between groups in a one-way layout.
Generalized linear models (GLMs) have been suggested for count data from SAGE or RNA-Seq experiments, with the counts treated as over-dispersed binomial (31,32,37), Poisson (21,41), over-dispersed Poisson (32,34) or Poisson with random effects (33). GLMs are non-linear models requiring iterative fitting, so an issue common to all these approaches is computational time and algorithmic failure for some genes for some datasets.
This article develops GLM algorithms for multifactor RNA-Seq experiments. Statistical methods are developed for estimating biological variation on a genewise basis and separating it from technical variation. Parallel computational approaches are developed to make GLM model fitting faster and more reliable. An empirical Bayes approach is developed for sharing information between genes, allowing for gene-specific variation even when only a few biological replicates are available. The methodology provides a pipeline for analysing arbitrarily complex RNA-Seq experiments provided that there is some degree of biological replication.
MATERIALS AND METHODS
Biological coefficient of variation
RNA-Seq profiles are formed from n RNA samples. Let πgi be the fraction of all cDNA fragments in the i-th sample that originate from gene g. Let G denote the total number of genes, so 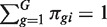 for each sample. Let
for each sample. Let 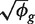 denote the coefficient of variation (CV) (standard deviation divided by mean) of πgi between the replicates i. We denote the total number of mapped reads in library i by Ni and the number that map to the g-th gene by ygi. Then
denote the coefficient of variation (CV) (standard deviation divided by mean) of πgi between the replicates i. We denote the total number of mapped reads in library i by Ni and the number that map to the g-th gene by ygi. Then
Assuming that the count ygi follows a Poisson distribution for repeated sequencing runs of the same RNA sample, a well known formula for the variance of a mixture distribution implies:
Dividing both sides by 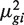 gives
gives
The first term 1/μgi is the squared CV for the Poisson distribution and the second is the squared CV of the unobserved expression values. The total CV2 therefore is the technical CV2 with which πgi is measured plus the biological CV2 of the true πgi. In this article, we call ϕg the dispersion and 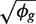 the biological CV although, strictly speaking, it captures all sources of the inter-library variation between replicates, including perhaps contributions from technical causes such as library preparation as well as true biological variation between samples.
the biological CV although, strictly speaking, it captures all sources of the inter-library variation between replicates, including perhaps contributions from technical causes such as library preparation as well as true biological variation between samples.
GLMs
GLMs are an extension of classical linear models to non-normally distributed response data (42,43). GLMs specify probability distributions according to their mean–variance relationship, for example the quadratic mean–variance relationship specified above for read counts. Assuming that an estimate is available for ϕg, so the variance can be evaluated for any value of μgi, GLM theory can be used to fit a log-linear model
for each gene (32,41). Here xi is a vector of covariates that specifies the treatment conditions applied to RNA sample i, and βg is a vector of regression coefficients by which the covariate effects are mediated for gene g. The quadratic variance function specifies the negative binomial GLM distributional family. The use of the negative binomial distribution is equivalent to treating the πgi as gamma distributed.
Fitting the GLMs
The derivative of the log-likelihood with respect to the coefficients βg is XTzg, where X is the design matrix with columns xi and zgi = (ygi − μgi)/(1 + ϕgμgi). The Fisher information matrix for the coefficients can be written as 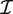 g = XTWgX, where Wg is the diagonal matrix of working weights from standard GLM theory (43). The Fisher scoring iteration to find the maximum likelihood estimate of βg is therefore
g = XTWgX, where Wg is the diagonal matrix of working weights from standard GLM theory (43). The Fisher scoring iteration to find the maximum likelihood estimate of βg is therefore 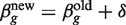 with δ = (XTWgX)−1
XTzg. This iteration usually produces an increase in the likelihood function, but the likelihood can also decrease representing divergence from the required solution. On the other hand, there always exists a stepsize modifier α with 0 < α < 1 such that
with δ = (XTWgX)−1
XTzg. This iteration usually produces an increase in the likelihood function, but the likelihood can also decrease representing divergence from the required solution. On the other hand, there always exists a stepsize modifier α with 0 < α < 1 such that 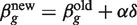 produces an increase in the likelihood. Choosing α so that this is so at each iteration is known as a line search strategy (44,45).
produces an increase in the likelihood. Choosing α so that this is so at each iteration is known as a line search strategy (44,45).
Fisher's scoring iteration can be viewed as an approximate Newton-Raphson algorithm, with the Fisher information matrix approximating the second derivative matrix. The line search strategy may be used with any approximation to the second derivative matrix that is positive definite. Our implemention uses a computationally convenient approximation. Without loss of generality, the linear model can be parametrized so that XTX = I. If this is done, and if the μgi also happen to be constant over i for a given gene g, then the information matrix simpifies considerably to μg/(1 + ϕgμg) times the identity matrix I. Taking this as the approximation to the information matrix, the Fisher scoring step with line search modification becomes simply δ = αXTzg, where the multiplier μg/(1 + ϕgμg) has been absorbed into the stepsize factor α. In this formulation, α is no longer constrained to be less than one. In our implementation, each gene has its own stepsize α that is increased or decreased as the iteration proceeds.
Cox–Reid adjusted profile likelihood
The adjusted profile likelihood (APL) for ϕg is the penalized log-likelihood
where yg is the vector of counts for gene g, 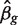 is the estimated coefficient vector, ℓ() is the log-likelihood function and
is the estimated coefficient vector, ℓ() is the log-likelihood function and 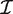 g is the Fisher information matrix. The Cholesky decomposition (46) provides a numerically stable and efficient algorithm for computing the determinant of the information matrix. Specifically, logdet
g is the Fisher information matrix. The Cholesky decomposition (46) provides a numerically stable and efficient algorithm for computing the determinant of the information matrix. Specifically, logdet 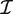 g is the sum of the logarithms of the diagonal elements of the Cholesky factor R, where
g is the sum of the logarithms of the diagonal elements of the Cholesky factor R, where 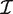 g = RT
R and R is upper triangular. The matrix R can be obtained as a by product of the QR-decomposition used in standard linear model fitting. In our implementation, the Cholesky calculations are carried out in a vectorized fashion, computed for all genes in parallel.
g = RT
R and R is upper triangular. The matrix R can be obtained as a by product of the QR-decomposition used in standard linear model fitting. In our implementation, the Cholesky calculations are carried out in a vectorized fashion, computed for all genes in parallel.
Simulations
Artificial data sets were generated with negative binomial distributed counts for a fixed total number of 10 000 genes. The expected count size varied between genes according to a gamma distribution with shape parameter 0.5, an ad hoc choice that happened to mimic the size distribution of the carcinoma data. The average dispersion was set to 0.16 (BCV = 0.4). In one simulation, all genes had the same true dispersion. In the other simulation, true dispersions were randomly generated around 0.16 according to an inverse chisquare distribution with 20 degrees of freedom.
RESULTS
Technical and biological variation
The starting point for an RNA-Seq experiment is a set of n RNA samples, typically associated with a variety of treatment conditions. Each sample is sequenced, short reads are mapped to the appropriate genome, and the number of reads mapped to each genomic feature of interest is recorded. For simplicity of terminology, we will assume in this article that counts are summarized at the gene level, although in practice the genomic features might just as well be transcripts, exons, SAGE-tags, exon-junctions or non-coding RNAs. The number of reads from sample i mapped to gene g will be denoted ygi. The set of genewise counts for sample i makes up the expression profile or library for that sample. The expected size of each count is the product of the library size and the relative abundance of that gene in that sample.
Two levels of variation can be distinguished in any RNA-Seq experiment. First, the relative abundance of each gene will vary between RNA samples, due mainly to biological causes. Second, there is measurement error, the uncertainty with which the abundance of each gene in each sample is estimated by the sequencing technology. If aliquots of the same RNA sample are sequenced, then the read counts for a particular gene should vary according to a Poisson law (30). If sequencing variation is Poisson, then it can be shown (‘Materials and Methods’ section) that the squared coefficient of variation (CV) of each count between biological replicate libraries is the sum of the squared CVs for technical and biological variation respectively,
Biological CV (BCV) is the coefficient of variation with which the (unknown) true abundance of the gene varies between replicate RNA samples. It represents the CV that would remain between biological replicates if sequencing depth could be increased indefinitely. The technical CV decreases as the size of the counts increases. BCV on the other hand does not. BCV is therefore likely to be the dominant source of uncertainty for high-count genes, so reliable estimation of BCV is crucial for realistic assessment of differential expression in RNA-Seq experiments. If the abundance of each gene varies between replicate RNA samples in such a way that the genewise standard deviations are proportional to the genewise means, a commonly occurring property of measurements on physical quantities, then it is reasonable to suppose that BCV is approximately constant across genes. We allow however for the possibility that BCV might vary between genes and might also show a systematic trend with respect to gene expression or expected count.
The magnitude of BCV is more important than the exact probabilistic law followed by the true gene abundances. For mathematical convenience, we assume that the true gene abundances follow a gamma distributional law between replicate RNA samples. This implies that the read counts follow a negative binomial probability law.
Linear models for multifactor experiments
The use of linear models to describe multifactor microarray experiments is well established (18,19). While linear models are associated with normally distributed data, negative binomial count data can be analysed using GLMs in a way that is closely analogous to normal linear models in all important respects. We assume a log-linear model for the expected read counts in terms of explanatory covariates that capture the treatment conditions applied to each RNA sample (‘Materials and Methods’ section). The total library size Ni serves as an offset in the linear model predictor, capturing the dependence of counts on sequencing depth. The library size may be defined as the total number of mapped reads, or it may be estimated from the data to effect some relative normalization between the different libraries (26).
GLMs are non-linear models for which the parameters must be estimated iteratively for each individual gene. An intuitive iterative computational algorithm was proposed to fit GLMs when they were first formulated (42), and almost all available GLM software uses this algorithm. Each iteration can be thought of as a least squares regression in which each count is weighted inversely to the total CV2 defined above (43,45). The model fitting process must be repeated until convergence is achieved. Previous applications of GLMs to RNA-Seq data have made genewise calls to standard univariate GLM software. Although the usual GLM algorithm is fairly reliable for univariate data, there is no guarantee that it will converge successfully, especially for very small or poorly fitting data sets. In the RNA-Seq context, the usual GLM algorithm frequently fails and is not sufficiently reliable for our purposes. We solve this problem by embellishing the usual algorithm with a line search modification (45). This modification checks for convergence at each iteration, reducing the step size to avoid divergence. The step size is repeatedly halved until an increase in the log-likelihood is achieved. This ensures convergence of the algorithm, unless floating point errors intervene. The line search algorithm is in practice extremely reliable.
The second issue with iterative model fitting is computational time. The usual GLM algorithm requires a matrix decomposition to be formed at each iteration for each gene, a substantial computational burden. To address this issue, we have implemented a novel, simplified pseudo-Newton algorithm that can be more readily parallelized across genes than other algorithms. In our pseudo-Newton algorithm, a fixed approximation is used for the second-derivative matrix of the model coefficients. The linear model parametrization is first transformed so that the columns of the design matrix are orthogonal. Then the second-derivative matrix is approximated by the expected information matrix that would arise if the fitted values for each gene were equal. This is conveniently just a multiple of the identity matrix, eliminating the computational overhead of matrix factorizations entirely. Although the pseudo-Newton algorithm requires slightly more iterations on average than true Newton-Raphson or the customary Fisher scoring algorithm for GLMs, the pseudo-Newton algorithm remains competitive in conjunction with our line-search strategy, and the computational gains that arise from the simplification are enormous. The algorithm is implemented in R in such a way that the iteration is progressed for all genes in parallel rather than for one gene at a time. Our pure R implementation fits GLMs to most RNA-Seq data sets in a few seconds, whereas genewise calls to the glm() function in R typically require minutes at least, and indeed may fail entirely due to iterative divergence for one or more genes.
Hypothesis tests
Our software allows users to test the significance of any coefficient in the linear model, or of any contrast or linear combination of the coefficients in the linear model. Genewise tests are conducted by computing likelihood-ratio statistics to compare the null hypothesis that the coefficient or contrast is equal to zero against the two-sided alternative that it is different from zero. The log-likelihood-ratio statistics are asymptotically chi square distributed under the null hypothesis that the coefficient or contrast is zero. Simulations show that the likelihood ratio tests hold their size relatively well and generally give a good approximation to the exact test (23) when the latter is available (data not shown). Any multiple testing adjustment method provided by the p.adjust function in R can be used. By default, P-values are adjusted to control the false discovery rate by the method of Benjamini and Hochberg (47).
Estimation of biological CV
The remaining issue is to obtain a reliable estimate of the BCV for each gene. An estimator that is approximately unbiased and performs well in small samples is required. Maximum likelihood estimation of the BCV would underestimate the BCV, because of the need to estimate the coefficients in the log-linear model from the same data. Our earlier work used exact conditional likelihood to estimate the BCV (22,23). This approach has excellent performance, but does not easily generalize to GLMs. Instead we use an approximate conditional likelihood approach known as APL (48). APL is a form of penalized likelihood. Again, we have implemented the APL computation in a vectorized and computationally efficient manner, rather than computing quantities gene by gene.
Estimating common dispersion
Estimating the BCV for each gene individually should not be considered unless a large number of biological replicates are available. When less replication is available, sharing information between genes is essential for reliable inference. Regardless of the amount of replication, appropriate information sharing methods should result in some benefits.
Let ϕg denote the squared BCV for gene g, which we call the dispersion of that gene. The dispersion is the coefficient of the quadratic term in the variance function. The simplest method of sharing information between genes is to assume that all genes share the same dispersion, so that ϕg = ϕ (23). The common dispersion may be estimated by maximizing the shared likelihood function
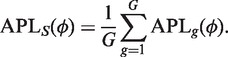 |
where APLg is the adjusted profile likelihood for gene g (‘Materials and Methods’ section). This maximization can be accomplished numerically in a number of ways, for example by a derivative-free approximate Newton algorithm (49).
Estimating trended dispersion
A generalization of the common dispersion is to model the dispersion ϕg as a smooth function of the average read count of each gene (25). Our software offers a number of methods to do this. A simple non-parametric method is to divide the genes into bins by average read count, estimate the common dispersion in each bin, then to fit a loess or spline curve through these bin-wise dispersions. A more sophisticated method is locally weighted APL. In this approach, each ϕg is estimated by making a local shared log-likelihood, which is a weighted average of the APLs for gene g and its neighbouring genes by average read count.
Estimating genewise dispersions
In real scientific applications, it is more likely that individual genes have individual BCVs depending on their genomic sequence, genomic length, expression level or biological function. We seek a compromise between entirely individual genewise dispersions ϕg and entirely shared values by extending the weighted likelihood empirical Bayes approach proposed by Robinson and Smyth (22). In this approach, ϕg is estimated by maximizing
where G0 is the weight given to the shared likelihood and APLSg(ϕg) is the local shared log-likelihood. This weighted likelihood approach can be interpreted in empirical Bayes terms, with the shared likelihood as the prior distribution for ϕg and the weighted likelihood as the posterior. The prior distribution can be thought of as arising from prior observations on a set of G0 genes. The number of prior genes G0 therefore represents the weight assigned to the prior relative to the actual observed data for gene g. The optimal choice for G0 depends on the variability of BCV between genes. Large values are best when the BCV is constant between genes. Smaller values are optimal when the BCVs vary considerably between genes. We have found that G0 = 20/df gives good results over a wide range of real data sets, where df is the residual degrees of freedom for estimating the BCV. For multigroup experiments, df is the number of libraries minus the number of distinct treatment groups. The default setting implies that the prior has the weight of 20 degrees of freedom for estimating the BCV, regardless of the actual number of libraries n or the complexity of the experimental design.
A desirable consequence of the empirical Bayes approach is that genewise dispersions are squeezed towards the prior value more or less strongly depending on how reliably the individual genewise dispersion can be estimated. Genes for which the counts are very low provide relatively little statistical information for estimating their own dispersion so, in these cases, the prior value dominates and the genewise dispersions are squeezed heavily towards the overall trend.
For computational convenience, the genewise and shared APL functions are evaluated on a grid of possible dispersion values. A cubic spline curve is used to interpolate the APL values on the grid for each gene, and the maximum of the spline curve is taken as the genewise dispersion estimate. Computing both the common and genewise dispersions for tens of thousands of genes takes around 20 s on a laptop computer.
Oral squamous cell carcinoma
A recent study investigated differential gene expression in oral squamous cell carcinomas (OSCC) (50). The study used the Applied Biosystems SOLiD System to construct RNA-Seq profiles of tumor and matched normal tissue from three patients with OSCC. The original analysis used an intuitive but ad hoc procedure to identify differentially expressed genes. Genes were first ranked by fold-change between tumour and normal for each patient. The top 300 up-regulated and top 300 down-regulated genes by median rank over the three patients were selected as differentially expressed (50). This simple analysis requires a gene to be highly ranked in two patients and then ignores the fold-change in the third patient. It was sufficient to obtain interesting biological results, but did not permit any assessment of statistical significance. It also treated all fold-changes as equally reliable regardless of the magnitude of the counts.
Here, we describe a more formal analysis that assesses statistical significance relative to biological variation. First we downloaded the read count data from Supplementary Table S1 of Tuch et al. (50). The table gives read counts summarized by RefSeq transcript, and filtered to include only those transcripts with at least 50 aligned reads for at least one tissue (tumour or normal) in all three patients. The full sequence data from the study is available from the GEO database (www.ncbi.nlm.nih.gov/geo, accession number GSE20116), but working from the summarized counts ensures that our analysis is based on the identical counts used for the original analysis. We mapped the RefSeq identifiers to the latest official gene symbols using the Bioconductor annotation package org.Hs.eg.db (version 2.5.0), discarding any RefSeq identifiers no longer in the database. The RefSeq transcript with the greatest number of exons was chosen to represent each unique gene, and redundant RefSeq transcripts were removed. This left 10 464 transcripts each representing a unique gene. Effective library sizes were then estimated using the weighted trimmed mean of M-values scale-normalization method (26).
The overall (common) BCV between the three normal tissue profiles is estimated as 40% (Figure 1). The BCV between the three tumour tissue profiles is distinctly higher at 52%, showing that the tumours are more heterogeneous than the normal tissues. It is therefore of interest to detect at least two classes of differentially expressed genes: first, those that are consistently different in all the tumours versus matched normal tissue, and second, those that show expression changes specific to one or two out of the three tumours.
Figure 1.

Multidimensional scaling plot of the squamous cell carcinoma profiles in which distances correspond to BCV between pairs of samples. Pairwise BCVs were computed from the 500 most heterogeneous genes. Samples are labelled with patient number and either ‘T’ for tumour or ‘N’ or normal. The first plot dimension roughly corresponds to tissue source (normal or tumour) and the second to patient differences. The tumour samples are more heterogeneous than the normals.
The study design has two explanatory factors, one being patient ID, with three levels, and the other being the tissue type, with two levels (normal or tumour). The data is analysed by fitting three successive log-linear models to the read counts for each gene (Table 1). The first model represents baseline expression differences between the three patients. The second, an additive model, allows for consistent relative expression changes in tumour versus normal tissue. The third, an interaction model, allows for patient-specific tumour effects.
Table 1.
Log-linear models fitted to the oral carcinoma data
| Model | Interpretation | Genes detected |
|---|---|---|
| Patient | Baseline patient differences | |
| Patient + tissue | Consistent tumour differences | 1276 |
| Patient × tissue | Patient-specific tumour differences | 202 |
Differentially expressed genes are detected by likelihood ratio tests between successive models (FDR<0.05).
Our first analysis looks for genes that are consistently differentially expressed in cancer as compared to normal tissue. For this analysis, dispersion estimation is based on the additive model, which has two residual degrees of freedom. A common BCV across all genes was found to be too simple, with 39 genes showing strong evidence of greater variability than implied by the common BCV (Figure 2) at a family-wise error rate of 0.05 (51). Allowing an abundance trend on the BCV did not reduce the number of outlier genes for which the BCV is rejected (Figure 2). On the other hand, permitting genewise BCV, with empirical Bayes moderation with prior G0 = 10, shows no remaining lack of fit (Figure 2). This provides a statistical justification for the use of genewise BCVs in the following analysis. There is also a biological justification, which is that genes that have inconsistent tumour versus normal differences in the three patients will receive higher BCV estimates, and hence be demoted in the list of differentially expressed genes. The use of genewise BCV therefore allows us to focus on genes that have consistent tumour versus normal differences.
Figure 2.

QQ-plots of goodness of fit statistics using common, trended or empirical Bayes genewise (tagwise) dispersions. Genewise deviance statistics were transformed to normality, and plotted against theoretical normal quantiles. Points in blue are those genes with a significantly poor fit (Holm-adjusted P-value < 0.05). When using genewise dispersions, no genes show a significantly poor fit.
Using the genewise BCV values, we test for differential expression between tumour and normal tissue by comparing the additive with the baseline model. This analysis adjusts for baseline differences between the patients, in a way that is analogous to computing a paired t-test for each gene, but adapted to count data. It yielded 1276 genes at false discovery rate (FDR) < 0.05 (Table 1, Supplementary Table S1). Included prominently among these genes are those previously identified as differentially expressed between tumour and normal tissues in head and neck squamous cell carcinoma studies. Of 25 genes reported by Yu et al. (52), 18 were included in our list at FDR < 0.05 (Supplementary Table S2). Another two (TNC and FN1) show fold-changes greater than two-fold and FDR around 0.4. The remaining five genes show small fold-changes and no evidence of differential expression (Supplementary Table S3). Tuch et al. (50) discussed nine genes of particular biological interest. Six of the genes (CASQ1, INHBA, MMP1, HMGA2, SHANK2 and WIF1) are confirmed to be strongly differentially expressed in our analysis with FDR < 0.001 (Supplementary Tables S4 and S5). This includes one gene (HMGA2) validated by RT-qPCR. Note that the original study (50) validated 16 genes by PCR, but only HMGA2 was identified by name.
To demonstrate further the biological relevance of the detected genes, we tested for enrichment of curated gene sets from the MSigDB database (53) using the mean-rank gene-set enrichment test (54). At FDR < 0.05 this yielded 417 gene sets enriched in the up-regulated genes and 268 gene sets enriched in the down-regulated genes. Significantly enriched sets were overwhelmingly cancer related and concordant, suggesting an enhanced WNT1 pathway in the tumours, and an expression signature similar to other cancers such as basal-like breast cancer (Supplementary Tables S7 and S8). Gene ontology analysis (55) found 146 GO terms enriched for up-regulated genes and 264 terms enriched for down-regulated genes. The GO terms for up-regulated genes tend to be associated with cell development, proliferation and differentiation and associated processes concordant with tumour development (Supplementary Tables S9 and S10).
Next, we looked for genes with heterogeneous tumour versus normal differences. Ideally this analysis should be conducted relative to BCV between independent tissue extracts from the same patients. However, the interaction model fully fits the available data, leaving no residual degrees of freedom, hence cannot be used to estimate the BCV. Instead we conduct this analysis using genewise BCVs estimated from differences between the three normal patients. These BCVs represent inter-patient rather than intra-patient differences, and so should over-estimate somewhat the desired BCV. Hence our analysis will be conservative to some extent in terms of P-values and FDRs. The BCVs between the normal patients are generally similar in size to the BCVs from the additive model, so the conservatism may be relatively minor. Using these conservative BCVs, a comparison of the interaction and additive models yields 202 differentially expressed genes at FDR < 0.05. The top-ranked gene in this analysis is CDKN2B, which was identified by Tuch et al. (50) as of biological interest based on correlation of expression level with copy number variation in Patient 8. The other two genes (CCND1, CTTN) similarly identified by Tuch et al. (50) have FDR around 0.1 in our interaction analysis (Supplementary Table S6).
Simulation study
Simulations were used to investigate the performance of our dispersion estimators. The first scenario simulated was the simplest design with a single group of three replicate libraries, with a constant true BCV of 0.4 for all genes, chosen to match the carcinoma data. In this simple scenario, conditional likelihood provides the least biased and most accurate estimation of dispersion, with Cox–Reid closely comparable (Figure 3a). Other estimators commonly used for generalized linear models based on Pearson or deviance residuals do not perform as well. These results agree with previous simulations (23).
Figure 3.

Boxplots of common BCV estimates from 100 simulated data sets. The left panel shows results for the one group case, with three replicate samples in the group. The right panel shows results for a paired-design with two groups and three blocks. The horizontal lines indicate the true common BCV of 0.4, chosen to match with the carcinoma case study. Conditional maximum likelihood (qCML) is the most accurate in the former case. For generalized linear models, Cox–Reid APL is the best performer.
The second scenario matches the carcinoma case study, with six libraries and with a 2 × 3 additive log-linear model fitted to each gene. In this case, conditional likelihood is not applicable, and Cox–Reid APL is the best performer of the remaining possibilities (Figure 3b).
Next we generated random true dispersions (BCV2) according to an inverse-chi square distribution, using the same 2 × 3 design as previously. In this case, empirical Bayes provides the most precise estimators of the genewise dispersions. The genewise dispersions were estimated most accurately when the prior weight G0 was in the range 10–12, corresponding to 20–24 prior degrees of freedom (Figure 4). Neither separate genewise estimation (G0 = 0) nor common dispersion (G0 = ∞) perform as well.
Figure 4.

Mean-square error with which empirical Bayes genewise dispersions estimate the true dispersion (BCV2), when true dispersions are randomly generated. In this case, the optimal prior weight is 10–12 prior genes, equivalent to 20–24 prior degrees of freedom. The common BCV estimator is equivalent to using infinite weight for the prior. Boxplots show results for 10 simulations.
DISCUSSION
The methods described in this article are implemented in the software package edgeR (24), available as part of the Bioconductor project for open-source genomic software (56). The methods provide a flexible and powerful approach to analyse read counts from gene expression experiments using RNA-Seq technologies. Models based on the negative binomial distribution facilitate an intuitively interpretable separation of biological from technical variation. Generalized linear models allow for arbitrarily complex experiments. Empirical Bayes methods allow for gene-specific variability, in a way that remains useful even when relatively few biological replicates are available.
The case study analysis of carcinoma data demonstrated the ability of generalized linear model methods to detect differential expression in a paired design, and even to detect tumour-specific expression changes. The results were more detailed and richer than provided by more ad hoc methods. The case study also demonstrated the need to allow for gene-specific variability, rather than assuming constant BCV or a constant abundance–variability relationship. Apart from other advantages, estimation of genewise BCV allows the main analysis to focus on changes that are consistent between biological replicates, by de-prioritizing genes with inconsistent results.
The estimation of biological variation is crucially important. Statistical methods based on Poisson models, for example, would drastically underestimate the amount of variability in data from biological replicates and potentially result in large numbers of false discoveries. The pipeline developed here provides a defensible means to incorporate biological variation into the analysis, even for the smallest possible experiments when only one condition is replicated. This is not to downplay the importance of obtaining a scientifically appropriate number of replicates for the experiment at hand (27,34). Rather the methods provided allow data analysts to make the best use of whatever data is available.
The BCV of 40% observed in the carcinoma case study is typical of what we have observed in other RNA-Seq or deepSAGE studies with human subjects. On the other hand, experiments with genetically identical model organisms tend in our experience to yield smaller variability between replicates, typically around 10% BCV (data not shown).
Our numerical implementations solve many common problems associated with fitting non-linear models to genomic data. Our models fits are very fast and have reliable convergence. For a typical data set, fitting genewise generalized linear models, hypothesis testing and fold-change estimation takes only a second on a laptop computer. Empirical Bayes estimation of genewise dispersions, a process requiring many model fits, takes around 20 s on a laptop computer. Simulations demonstrate the ability of Cox–Reid adjusted profile likelihood and empirical Bayes estimators to return accurate estimators of BCV in complex situations. When biological variation is gene-specific, empirical Bayes estimation can provide an advantageous compromise, superior to either of the extremes of common dispersion or separate genewise dispersion.
We have focused on genewise analyses in this article, but the software may just as well be used to perform exon-level analyses, or indeed analyses of read-counts for other genomic features. This article has focused on RNA-Seq and gene expression, but the methodology and software is applicable to differential count analyses for other types of genomic data. Such applications include the search for differentially methylated promoters using methylated DNA immunoprecipitation sequencing (MeDIP-Seq) (57,58), ChIP-Seq for finding differentially enriched regions for either transcriptional factor binding or for epigenetic histone marks, differential analysis of spectral counts in tandem mass spectrometry (59) or the analysis of species counts in metagenomics studies (60). All these technologies produce genome-scale count data on which the methods described here could fruitfully be brought to bear.
The edgeR software contains many features and options in addition to those described in this article, and opens up flexible possibilities for RNA-Seq data analysis. For example, the use of offsets in the log-linear models can easily accommodate non-linear normalization procedures, including those based on quantile normalization or GC-sequence content (61). The use of generalized linear models also provides the potential to incorporate quantitative weights, and hence to integrate quality weights for RNA samples into a differential expression analysis as has been done for microarrays (62).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–10.
FUNDING
National Health and Medical Research Council (Program Grant 490036 and Research Fellowship to G.K.S.); Autralian Government (Australian Postgraduate Research Award to Y.C.). Funding for open access charge: National Health and Medical Research Council Program Grant 490036.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
Thanks to Mark Robinson for many helpful discussions and to Brian Tuch for advice on the squamous cell carcinoma study data.
REFERENCES
- 1. National Human Genome Research Institute (2011). DNA sequencing costs. http://www.genome.gov/sequencingcosts/ [Google Scholar]
- 2.Morrissy AS, Morin RD, Delaney A, Zeng T, McDonald H, Jones S, Zhao Y, Hirst M, Marra MA. Next-generation tag sequencing for cancer gene expression profiling. Genome Res. 2009;19:1825–1835. doi: 10.1101/gr.094482.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.'t Hoen PAC, Ariyurek Y, Thygesen HH, Vreugdenhil E, Vossen RHAM, Menezes RXD, Boer JM, Ommen GJBV, Dunnen JTD. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res. 2008;36:e141. doi: 10.1093/nar/gkn705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu ZJ, Meyer CA, Choudhury S, Shipitsin M, Maruyama R, Bessarabova M, Nikolskaya T, Sukumar S, Schwartzman A, Liu JS, et al. Gene expression profiling of human breast tissue samples using SAGE-Seq. Genome Res. 2010;20:1730–1739. doi: 10.1101/gr.108217.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mortazavi A, Williams BA, Mccue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Meth. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 6.Cloonan N, Forrest ARR, Kolle G, Gardiner BBA, Faulkner GJ, Brown MK, Taylor DF, Steptoe AL, Wani S, Bethel G, et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Meth. 2008;5:613–619. doi: 10.1038/nmeth.1223. [DOI] [PubMed] [Google Scholar]
- 7.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nat. Rev. Genet. 2011;12:87–98. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li B, Ruotti V, Stewart RM, Thomson JA, Dewey CN. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics. 2010;26:493–500. doi: 10.1093/bioinformatics/btp692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Meth. 2009;6:377–82. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- 11.Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, Seifert M, Borodina T, Soldatov A, Parkhomchuk D, et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321:956–960. doi: 10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- 12.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 13.Denoeud F, Aury JM, Silva CD, Noel B, Rogier O, Delledonne M, Morgante M, Valle G, Wincker P, Scarpelli C, et al. Annotating genomes with massive-scale RNA sequencing. Genome Biol. 2008;9:R175. doi: 10.1186/gb-2008-9-12-r175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li J, Jiang H, Wong WH. Modeling non-uniformity in short-read rates in RNA-Seq data. Genome Biol. 2010;11:R50. doi: 10.1186/gb-2010-11-5-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–5. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Degner JF, Marioni JC, Pai AA, Pickrell JK, Nkadori E, Gilad Y, Pritchard JK. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics. 2009;25:3207–3212. doi: 10.1093/bioinformatics/btp579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Montgomery SB, Sammeth M, Gutierrez-Arcelus M, Lach RP, Ingle C, Nisbett J, Guigo R, Dermitzakis ET. Transcriptome genetics using second generation sequencing in a caucasian population. Nature. 2010;464:773–777. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wright G, Simon R. A random variance model for detection of differential gene expression in small microarray experiments. Bioinformatics. 2003;19:2448–2455. doi: 10.1093/bioinformatics/btg345. [DOI] [PubMed] [Google Scholar]
- 19.Smyth G. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Molec. Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article 3. [DOI] [PubMed] [Google Scholar]
- 20.Oshlack A, Robinson MD, Young MD. From RNA-seq reads to differential expression results. Genome Biol. 2010;11:220. doi: 10.1186/gb-2010-11-12-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langmead B, Hansen KD, Leek JT. Cloud-scale RNA-sequencing differential expression analysis with Myrna. Genome Biol. 2010;11:R83. doi: 10.1186/gb-2010-11-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson MD, Smyth GK. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics. 2007;23:2881–2887. doi: 10.1093/bioinformatics/btm453. [DOI] [PubMed] [Google Scholar]
- 23.Robinson MD, Smyth GK. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics. 2008;9:321–332. doi: 10.1093/biostatistics/kxm030. [DOI] [PubMed] [Google Scholar]
- 24.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hansen KD, Wu Z, Irizarry RA, Leek JT. Sequencing technology does not eliminate biological variability. Nat Biotechnol. 2011;29:572–573. doi: 10.1038/nbt.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl Acad. Sci. USA. 2001;98:5116. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baldi P, Long A. A Bayesian framework for the analysis of microarray expression data: regularized t-test and statistical inferences of gene changes. Bioinformatics. 2001;17:509. doi: 10.1093/bioinformatics/17.6.509. [DOI] [PubMed] [Google Scholar]
- 30.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baggerly K, Deng L, Morris J, Aldaz C. Overdispersed logistic regression for SAGE: modelling multiple groups and covariates. BMC Bioinformatics. 2004;5:144. doi: 10.1186/1471-2105-5-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu J, Tomfohr J, Kepler T. Identifying differential expression in multiple SAGE libraries: an overdispersed log-linear model approach. BMC Bioinformatics. 2005;6:165. doi: 10.1186/1471-2105-6-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blekhman R, Marioni JC, Zumbo P, Stephens M, Gilad Y. Sex-specific and lineage-specific alternative splicing in primates. Genome Res. 2010;20:180–189. doi: 10.1101/gr.099226.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Auer PL, Doerge RW. Statistical design and analysis of RNA sequencing data. Genetics. 2010;185:405–416. doi: 10.1534/genetics.110.114983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Srivastava S, Chen L. A two-parameter generalized Poisson model to improve the analysis of RNA-seq data. Nucleic Acids Res. 2010;38:e170. doi: 10.1093/nar/gkq670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Auer PL, Doerge RW. A two-stage Poisson model for testing RNA-Seq data. Stat. Appl. Genet. Molec. Biol. 2011;10:1–28. [Google Scholar]
- 37.Zhou YH, Xia K, Wright FA. A powerful and flexible approach to the analysis of RNA sequence count data. Bioinformatics. 2011;27:2672–2678. doi: 10.1093/bioinformatics/btr449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vencio RZ, Brentani H, Pereira CA. Using credibility intervals instead of hypothesis tests in SAGE analysis. Bioinformatics. 2003;19:2461–2464. doi: 10.1093/bioinformatics/btg357. [DOI] [PubMed] [Google Scholar]
- 39.Vêncio RZN, Brentani H, Patrão DFC, Pereira CAB. Bayesian model accounting for within-class biological variability in Serial Analysis of Gene Expression (SAGE) BMC Bioinformatics. 2004;5:119. doi: 10.1186/1471-2105-5-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hardcastle TJ, Kelly KA. baySeq: empirical Bayesian methods for identifying differential expression in sequence count data. BMC Bioinformatics. 2010;11:422. doi: 10.1186/1471-2105-11-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bullard J, Purdom E, Hansen K, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics. 2010;18:11–94. doi: 10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nelder JA, Wedderburn RWM. Generalized linear models. J. Roy. Stat. Soc. A. 1972;135:370–384. [Google Scholar]
- 43.McCullagh P, Nelder JA. Generalized Linear Models. 2nd edn. Boca Raton, Florida: Chapman & Hall/CRC; 1989. [Google Scholar]
- 44.Osborne MR. Fisher's method of scoring. International Statistical Review. 1992;60:99–117. [Google Scholar]
- 45.Smyth GK. Optimization and nonlinear equations. In: Armitage P, Colton T, editors. Encyclopedia of Biostatistics. London: Wiley; 1998. pp. 3174–3180. [Google Scholar]
- 46.Stewart G. Computer Science and Applied Mathematics. NY: Academic Press; 1973. Introduction to Matrix Computations. [Google Scholar]
- 47.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- 48.Cox DR, Reid N. Parameter orthogonality and approximate conditional inference. J. Roy. Stat. Soc. B. 1987;49:1–39. [Google Scholar]
- 49.Brent R. Algorithms for Minimization without Derivatives. Englewood Cliffs N.J.: Prentice-Hall; 1973. [Google Scholar]
- 50.Tuch BB, Laborde RR, Xu X, Gu J, Chung CB, Monighetti CK, Stanley SJ, Olsen KD, Kasperbauer JL, Moore EJ, et al. Tumor transcriptome sequencing reveals allelic expression imbalances associated with copy number alterations. PLoS ONE. 2010;5:e9317. doi: 10.1371/journal.pone.0009317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Holm S. A simple sequentially rejective multiple test procedure. Scand. J. Stat. 1979;6:65–70. [Google Scholar]
- 52.Yu YH, Kuo HK, Chang KW, Rutherford S. The evolving transcriptome of head and neck squamous cell carcinoma: a systematic review. PLoS ONE. 2008;3:e3215. doi: 10.1371/journal.pone.0003215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Michaud J, Simpson KM, Escher R, Buchet-Poyau K, Beissbarth T, Carmichael C, Ritchie ME, Schütz F, Cannon P, Liu M, et al. Integrative analysis of RUNX1 downstream pathways and target genes. BMC Genomics. 2008;9:363. doi: 10.1186/1471-2164-9-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11:R14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bock C, Tomazou EM, Brinkman A, Müller F, Simmer F, Gu H, Jäger N, Gnirke A, Stunnenberg HG, Meissner A. Genome-wide mapping of DNA methylation: a quantitative technology comparison. Nature Biotechnol. 2010;28:1106. doi: 10.1038/nbt.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robinson MD, Stirzaker C, Statham AL, Coolen MW, Song JZ, Nair SS, Strbenac D, Speed TP, Clark SJ. Evaluation of affinity-based genome-wide dna methylation data: effects of CpG density, amplification bias, and copy number variation. Genome Res. 2010;20:1719–1729. doi: 10.1101/gr.110601.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carvalho P, Hewel J, Barbosa V, Yates JR., 3rd Identifying differences in protein expression levels by spectral counting and feature selection. Genetics and Mol. Res. 2008;7:342–356. doi: 10.4238/vol7-2gmr426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.White JR, Nagarajan N, Pop M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput Biol. 2009;5:e1000352. doi: 10.1371/journal.pcbi.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hansen KD, Irizarry RA, Wu Z. Removing technical variability in RNA-seq data using conditional quantile normalization. Biostatistics. 2012;13:204–216. doi: 10.1093/biostatistics/kxr054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ritchie M, Diyagama D, Neilson J, Laar RV, Dobrovic A, Holloway A, Smyth G. Empirical array quality weights in the analysis of microarray data. BMC Bioinformatics. 2006;7:261. doi: 10.1186/1471-2105-7-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.