

Abstract
Prostanoids are suggested to participate in diabetes pathology, but their roles are controversially discussed. The purpose of the current study was to examine the role of cyclooxygenase (prostaglandin synthase [PTGS]) enzymes and prostaglandin (PG) E2 signaling pathways in streptozotocin (STZ)-induced type 1 diabetes. Blood glucose, insulin, and survival rate were studied in mice with targeted disruption of the genes for PTGS and PGE receptors (PTGERs). PGE2 was found as the main prostanoid formed by the pancreas. Contrarily to PTGS-1, deficiency of PTGS-2 activity significantly amplified STZ effect, causing dramatic loss of insulin production and rise in blood glucose and death rate. STZ metabolism was unaffected by PTGS deficiency. Diabetogenicity of STZ in PTGER1−/−, PTGER2−/−, PTGER3−/−, and PTGER4−/− mice was comparable to control mice. In striking contrast, combined knockout of PTGER2 and PTGER4 by blocking PTGER4 in PTGER2−/− mice strongly enhanced STZ pathology. Treatment of PTGS-2−/− and wild-type mice with PTGER2/PTGER4 agonists partially protected against STZ-induced diabetes and restored β-cell function. Our data uncover a previously unrecognized protective role of PTGS-2–derived PGE2 in STZ-induced diabetes mediated by the receptor types PTGER2 and PTGER4. These findings offer the possibility to intervene in early progression of type 1 diabetes by using PTGER-selective agonists.
Type 1 diabetes is an autoimmune disease characterized by islet destruction followed by loss of insulin synthesis and hyperglycemia. Interactions between environmental factors, the immune system, and host genes are suggested to lead to type 1 diabetes, but still, initial events in pathogenesis that cause the death of β-cells are unresolved. Toxins, such as N-nitroso compounds (1), and viral infections (2) have been investigated for their potential role in triggering β-cell death, which is regarded as an early event leading to T-cell activation and loss of tolerance (3). Prostanoids, metabolites derived from the cyclooxygenase (prostaglandin synthase [PTGS]) pathway of arachidonic acid, are known to be pleiotropic mediators, modulating immune response and cell death (4). Although, these mediators have been shown to contribute to β-cell function (5), their possible role in the pathogenesis of type 1 diabetes is unclear.
Two types of PTGS are known: PTGS-1, constitutively expressed in different cell types, and PTGS-2, mostly expressed at low levels but inducible by inflammatory and stress mediators (6). Most likely, both enzymes are expressed in pancreatic tissue, and there is some evidence that PTGS-2 rather than PTGS-1 is predominantly expressed in islets of Langerhans (7). The roles played by PTGS enzymes in β-cells have not been fully established, but there is good evidence that at least PTGS-2 is involved in cytokine-mediated damage of β-cells (8). In NOD mice, PTGS-2 expression disappeared concomitantly from β-cells with progression of diabetes (9). However, it is unknown whether PTGS-2 is directly linked to β-cell damage. PTGS enzymes metabolize arachidonic acid to prostaglandin (PG) endoperoxide, which in turn is converted to various prostanoids (PGE2, PGD2, PGF2α, PGI2, and thromboxane [TX] A2) by specific prostanoid-forming enzymes (10). We have found PGE2 as the major PTGS product in pancreatic tissue. PGE2 exerts its actions on target cells by interacting with one or more of its four G-protein–coupled receptor subtypes, PTGER1, PTGER2, PTGER3, and PTGER4 (also named EP1–EP4). These receptors are linked to different signaling pathways that involve phosphoinoside hydrolysis and Ca2+ increase (PTGER1), inhibition of adenylate cyclase (PTGER3), and activation of adenylate cyclase (PTGER2 and PTGER4) (11). Recently, mRNA expression of all four receptor types of PTGER has been reported for human islets (12), but still, expression and function of the prostanoid system in pancreatic tissue or in islets is a matter of debate. Regarding its role in pancreas physiology, PGE2 has been shown to inhibit glucose-stimulated insulin secretion (13,14), probably by interaction with the PTGER3 (15), but a recent study questioned this function of PGE2 (16). In addition, studies using human islets have found stimulation of insulin secretion in response to arachidonic acid, but not metabolites of arachidonic acid (12). The role of PGE2 in early diabetes pathology is unknown.
A widely accepted animal model to study pathogenesis of type 1 diabetes uses streptozotocin (STZ) as a diabetogenic drug (17). The selective entry of STZ in β-cells is achieved by solute carrier family 2 member 2 (SLC2A2) (also named GLUT2), a glucose transporter selectively expressed in pancreatic islets that also transports STZ (18). When administered in a single high dose, STZ induces rapid onset of diabetes by generating DNA adducts and subsequent β-cell death by necrosis. The purpose of the current study was to examine the role of the PTGS system in β-cell destruction in type 1 diabetes. Therefore we studied expression and function of PTGS isoenzymes in the high-dose STZ diabetes model and present first evidence that inhibition of PTGS-2 activity strongly deteriorates the diabetogenic effect of STZ. Experiments using PTGER agonists disclosed a protective role of PTGS-2–derived PGE2, which is mediated by the receptor types PTGER2 and PTGER4.
RESEARCH DESIGN AND METHODS
Animals and treatment.
Animal care and experimental procedures were in accordance with institutional guidelines and approved by the Animal Welfare Committee of the State Agency (Darmstadt, Germany). C57/BL6J wild-type mice were obtained from Jackson Laboratories (Bar Harbor, ME). Breeder pairs of PTGS-1 and PTGS-2 knockout mice were provided by R. Langenbach (National Institute of Environmental Health Sciences, Research Triangle Park, NC). PTGER1-, PTGER2-, PTGER3-, and PTGER4-deficient mice were generated as previously described (19). All knockout mice were back-crossed to C57BL6J >10 times. Food and water were provided ad libitum. A single dose of 200 mg/kg STZ prepared in 0.05 mol/L citrate buffer, pH 4.5, immediately before injection, or citrate buffer alone (control) was injected intraperitoneally to weight-matched 8–10-week-old male mice. For the experiments using low-dose STZ, 40 mg/kg was injected intraperitoneally once a day for five consecutive days.
Measurement of blood glucose levels.
Glucose levels were monitored before and on day 4 after injection of STZ with a glucose meter (ACCU-CHEK Compact) in blood samples obtained from tail vein at 10:00 a.m. Diabetic hyperglycemia was defined as a nonfasting blood glucose concentration >250 mg/dL.
Measurement of plasma insulin concentration and plasma activity of liver enzymes.
Insulin concentrations were measured in plasma samples using an ELISA for mouse insulin (Shibayagi, Ishihara, Japan) according to the instructions of the manufacturer. Plasma activities of the liver enzymes alanine amino transferase (ALT) and aspartate amino transferase (AST) were determined by routine laboratory measurements (Laboklin, Kissingen, Germany).
mRNA detection by reverse transcriptase PCR.
Pancreata from mice were dissected and immediately quick frozen in RNAlater. Total RNA was isolated, reverse transcribed, and amplified as previously described (20). PCR primers used were as follows: for β-actin, 5′-GCTACAGCTTCACCACCACA-3′ and 5′-AAGGAAGGCTGGAAAAGAGC-3′; for PTGS-1, 5′-GTGGCTATTTCCTGCAGCTC-3′ and 5′-CAGTGCCTCAACCCCATAGT-3′; for PTGS-2, 5′-CCCCCACAGTCAAAGACACT-3′ and 5′-CTCATCACTCCACTCAGGAT-3′; for SLC2A2, 5′-GCCTGTGTATGCAACCATTG-3′ and 5′-TGGCCCAATCTCAAAGAAAC-3′; for proinsulin, 5′-GGCTTCTTCTACACACCCA-3′ and 5′-CAGTAGTTCTCCAGCTGGTA-3′; for PTGER1, 5′-GCTGTACGCCTCGCATCGTGG-3′ and 5′-CCTTGAGCCCAGCCCAGAATG-3′; for PTGER2, 5′-CCTGCCGCTGCTCAACTACG-3′ and 5′-GTCTCCTCTGCCATCGAAGTCCTC-3′; for PTGER3, 5′-GCTGGCTCTGGTGGTGACCTT-3′ and 5′-AAGCCAGGCGAACTGCAATTAGAA-3′; and for PTGER4, 5′-CTGAACAGCCCGGTGACCATTCC-3′ and 5′GCCGGCCAGCCGCTTGTCCAC-3′.
Isolation of pancreatic islets.
Islets were isolated from pancreatic tissue as previously described (21) with slight modification. In brief, pancreata from mice were dissected, cut into small pieces, and immediately digested with collagenase P solution for 30 min at 37°C. Tissues were disaggregated by shaking, and islets were purified by Ficoll-Paque Plus (Amersham, Freiburg, Germany) density gradient centrifugation. The enriched islet fraction was washed and homogenized in RNA extraction solution.
Determination of STZ.
STZ (200 mg/kg) was injected intraperitoneally and plasma samples were taken at time points 30 min, 60 min, and 12 h. Determination of STZ was achieved by liquid chromatography-electrospray ionization/tandem mass spectrometry (LC-ESI-MS/MS). Plasma (5 μL) was mixed with 995 μL cold methanol and centrifuged for 5 min at 10,000 g for protein precipitation, and 300 μL of the upper layer was transferred to a glass vial. Finally, 10 μL was injected into the chromatographic system, equipped with a Synergi Polar RP column (150 × 2 mm, 4 μm, 80 Å) and a precolumn of the same material. The chromatographic separation was carried out at 40°C under gradient elution mode using water and acetonitrile, both containing 0.1% formic acid, as mobile phases. The samples were analyzed in MRM modus using a 5500 Qtrap (AB Sciex, Darmstadt, Germany) and a one-point calibration approach.
Determination of prostanoid profile and 5-hydroxyeicosatetraenoic acid.
Quick-frozen pancreas tissue was briefly thawed on ice, weighed, and homogenized in two volumes of phosphate buffer containing 1 mmol/L phenol and protease inhibitor cocktail (Calbiochem, Nottingham, U.K.) using polytron homogenizer and ultrasonification. The supernatant of a 3,000-g centrifugation step was used for protein determination and activity assay. Arachidonic acid (50 μmol/L) was added to 100 μg protein. After incubation for 10 min at 37°C, the reaction was stopped by acidification using 1% formic acid. Prostanoids were extracted and amounts were determined by liquid chromatography tandem mass spectrometry (LC-MS/MS) (22). As controls, we analyzed assay samples omitting protein, samples with denatured protein, and samples incubated in the presence of 10 μmol/L indomethacin. Under these conditions, only negligible amounts of prostanoids were detected (data not shown). 5-Hydroxyeicosatetraenoic acid (5-HETE) was determined as previously described (22,23).
Statistics.
Results were expressed as mean ± SEM. ANOVA with Bonferroni multiple comparison post hoc analysis was used to determine statistical differences between multiple groups and t test for two-group comparison. Statistical analysis was performed using PRISM 4.0 (GraphPad Software, Inc.) software. P values <0.05 were considered statistically significant.
RESULTS
STZ induces PTGS-2 and increases PGE2 formation.
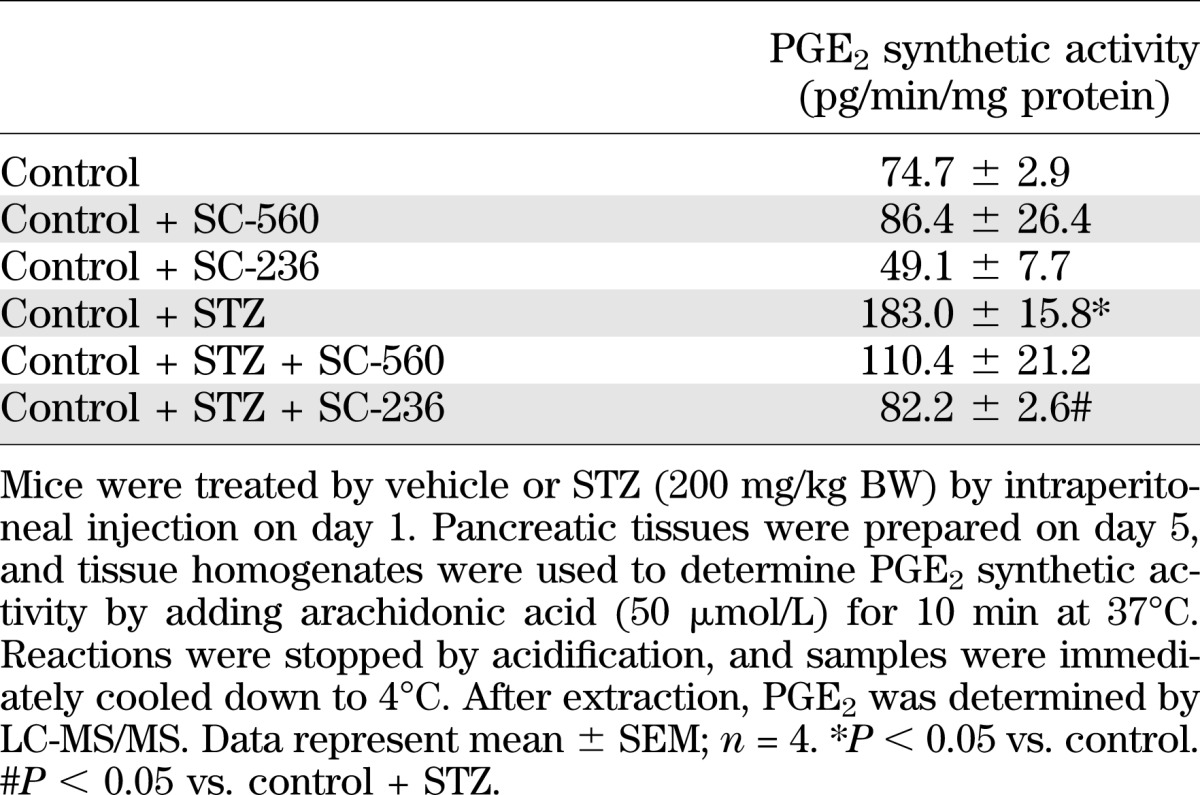
Pancreatic tissue is able to form various prostanoids. After addition of arachidonic acid to tissue homogenates, we observed PGE2 as the main prostanoid (Table 1). Less amounts of PGD2, PGF2α, 6-keto-PGF1α, and thromboxane B2 (TXB2) were formed. In tissue homogenates prepared from STZ-treated mice, prostanoid-synthesizing activity was enhanced, but PGE2 still represented the main product (Table 1). Expression of mRNA for PTGS-1 and PTGS-2 was found in pancreatic tissue, and also in isolated islet cells (Fig. 1A). The observed strong mRNA signals for SLC2A2 and proinsulin indicate the enrichment of islet cells in our preparation. STZ application increased PTGS-2, but not PTGS-1, expression in mouse pancreas (Fig. 1B), indicating that PTGS-2 is responsible for enhanced prostanoid-forming activity. In line with this observation, we noticed that PTGS-2–specific inhibitor SC-236, but not PTGS-1–selective inhibitor SC-560, was able to block STZ-induced PGE2 synthesis (Table 2).
TABLE 1.
Prostanoid profile by mouse pancreatic tissue
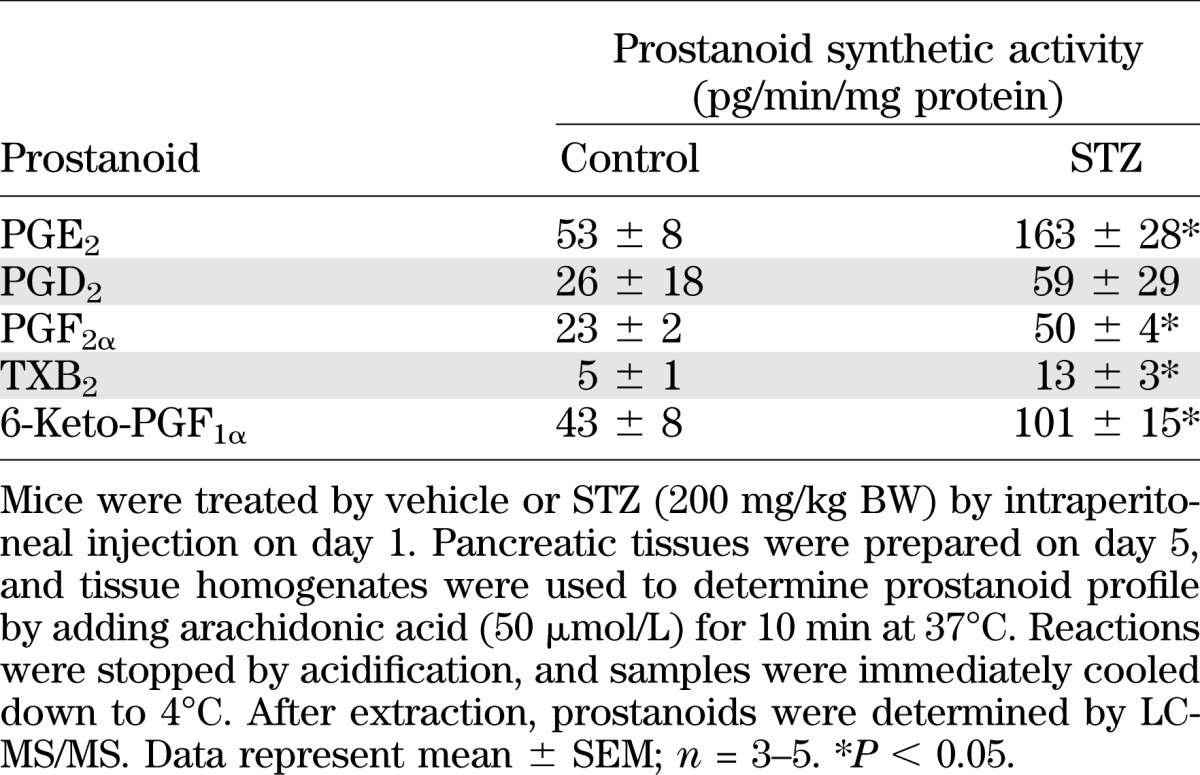
FIG. 1.

Expression of PTGS isoforms in pancreas. A: Expression of PTGS mRNA in pancreas and isolated islets from mouse. RNA was isolated from pancreas and from isolated islets, and mRNA expression for PTGS-1 and PTGS-2 was analyzed by PCR in comparison with SLC2A2 and proinsulin mRNA expression. B: Effect of STZ on pancreatic expression of PTGS-1 and PTGS-2. STZ or vehicle was injected on day 1. On day 4, pancreata were prepared, and mRNA expression for PTGS-1 and PTGS-2 was analyzed by PCR in comparison with β-actin mRNA expression. CTRL, wild-type mice. Data are shown as mean ± SEM; n = 3. *P < 0.05.
TABLE 2.
PGE2 synthesis by mouse pancreatic tissue
PTGS-2 deficiency increases STZ-induced hyperglycemia.
In order to elucidate the role of PTGS-1 and PTGS-2 in our diabetes model, we studied the effect of STZ in PTGS-deficient mice. The basal glucose levels in PTGS-1−/− and PTGS-2−/− mice were not different from control mice. In PTGS-1−/−, STZ application caused an increase in blood glucose concentration from 159.4 ± 4.4 to 448.5 ± 40.7 mg/dL (P < 0.05), quite indistinguishable from control mice. However, STZ injection in PTGS-2−/− caused a significantly stronger induction of hyperglycemia (from 140.5 ± 6.5 to 855.2 ± 114.9 mg/dL, P < 0.001; compared with STZ-treated control mice, P = 0.0136) (Fig. 2A). To exclude bias in STZ effect by genetic background variations in the course of gene knockout, we studied the effect of STZ in wild-type mice treated with the selective PTGS-2 inhibitor SC-236 (10 mg/day/kg body weight [BW]). At the used dose, SC-236 blocks >90% of PTGS-2 activity (20). After SC-236 application, STZ raised blood glucose to a similar level as in PTGS-2−/− mice (Fig. 2A). Application of multiple low doses of STZ (40 mg/kg) causes type 1 diabetes that is more dependent on immune modulatory mechanisms than the β-cell–toxic effect of STZ (24). In this type of model, we observed a significantly stronger hyperglycemic effect of STZ in PTGS-2−/− mice only in the early phase of diabetes development (day 5) (Fig. 2B). At later time points, blood glucose levels were similar in wild-type, PTGS-1−/−, and PTGS-2−/− mice (day 23) (Fig. 2B).
FIG. 2.

Hyperglycemic effect of STZ in PTGS-deficient mice. A: Hyperglycemic effect of high dose of STZ. STZ (200 mg/kg BW) or vehicle was given intraperitoneally to wild-type mice (CTRL) and PTGS-1−/−, PTGS-2−/−, and wild-type mice treated with PTGS-2 inhibitor SC-236. On experimental day 4, blood glucose concentrations were measured by glucose meter. Data are shown as mean ± SEM; n = 6. *P < 0.05; #P < 0.05 vs. STZ-treated CTRL. B: Hyperglycemic effect of multiple low doses of STZ. STZ (40 mg/kg BW) was given intraperitoneally once daily for 5 days to CTRL, PTGS-1−/−, and PTGS-2−/− mice. Blood glucose concentrations were measured before STZ application (day 0) and on days 5 and 23. Data are shown as mean ± SEM; n = 6. *P < 0.05 vs. day 0.
Toxic effect of STZ on β-cells is enhanced by PTGS-2 deficiency.
The effects of high-dose STZ on blood glucose were reflected by significant and comparable decreases in plasma insulin concentrations in control and PTGS-1−/− mice (Fig. 3A). However, we observed a much stronger decline in insulin concentration in PTGS-2−/− mice and wild-type mice pretreated with PTGS-2 inhibitor SC-236. In both groups, the fall in plasma insulin concentration was significantly stronger than in STZ-treated control mice. To study the effect of STZ on insulin-producing β-cells, we immunostained paraformaldehyde-fixed pancreas sections of control and PTGS-2−/− using anti-insulin antibodies. Compared with vehicle-treated control, immunosignals for insulin declined significantly, by ∼50%, after application of STZ, indicating reduced viability of β-cells (Fig. 3B and C). In PTGS-2−/− mice, STZ nearly totally ablated anti-insulin signal.
FIG. 3.

Insulin concentration and expression in islets. A: Effect of STZ on plasma insulin concentration in PTGS-deficient mice. Wild-type (CTRL), PTGS-1−/−, and PTGS-2−/− mice and mice receiving SC-236 were treated by a single injection of STZ (200 mg/kg BW), and on day 4, insulin concentrations were measured in plasma samples by enzyme immunosorbent assay. B: Effect of STZ on insulin expression in islets. Pancreata were isolated and cryopreserved and sections were immunostained using insulin-specific antibodies. Representative stainings are shown (n = 3; original magnification ×200). C: Insulin-immunostained islet area. After immunostaining, sections were captured digitally, and areas stained by anti-insulin antibody were determined. Data are shown as mean ± SEM; n = 5. *P < 0.05 vs. control; #P < 0.05 vs. STZ-treated CTRL.
PTGS-2 deficiency increases lethality of STZ.
Long-term observation, up to 10 days after a single injection of high-dose STZ, disclosed high lethality in PTGS-2−/− mice. Kaplan-Meier analysis revealed a survival rate of >90% for control and PTGS-1−/− mice. In striking contrast, STZ-treated PTGS-2−/− mice (P < 0.0001) as well as control mice pretreated with PTGS-2 inhibitor SC-236 (P < 0.0001) all died within 6 days (Fig. 4A). To test whether knockout of specific PTGS isoforms may cause redistribution of arachidonic acid to the lipoxygenase pathway and thereby modulate STZ effects, we measured 5-HETE in pancreas homogenates after addition of arachidonic acid in STZ-treated control (15.2 ± 3.7 pg/min/mg), PTGS-1−/− (20.9 ± 4.2 pg/min/mg), and PTGS-2−/− mice (18.3 ± 4.0 pg/min/mg). No significant differences were observed between the groups.
FIG. 4.

Toxicity of STZ in PTGS-deficient mice. A: Kaplan-Meier survival analysis of STZ-treated PTGS-deficient mice. Wild-type mice (CTRL) were treated with SC-236 or vehicle 1 day before single application of STZ (200 mg/kg BW). PTGS-1−/− and PTGS-2−/− were only treated with STZ (200 mg/kg BW). Mice were kept under standard conditions for 10 days. Data are shown as mean ± SEM; n = 10. B: Plasma elimination of STZ. Wild-type mice were treated with vehicle or SC-236 24 and 4 h before application of STZ. PTGS-2−/− were only treated with STZ (200 mg/kg BW). Plasma samples were collected 30 min, 60 min, and 12 h later. Amount of STZ was determined by LC-ESI-MS/MS. Data are shown as mean ± SEM; n = 5. No significant differences between the groups were observed at the indicated time points.
Metabolism of STZ.
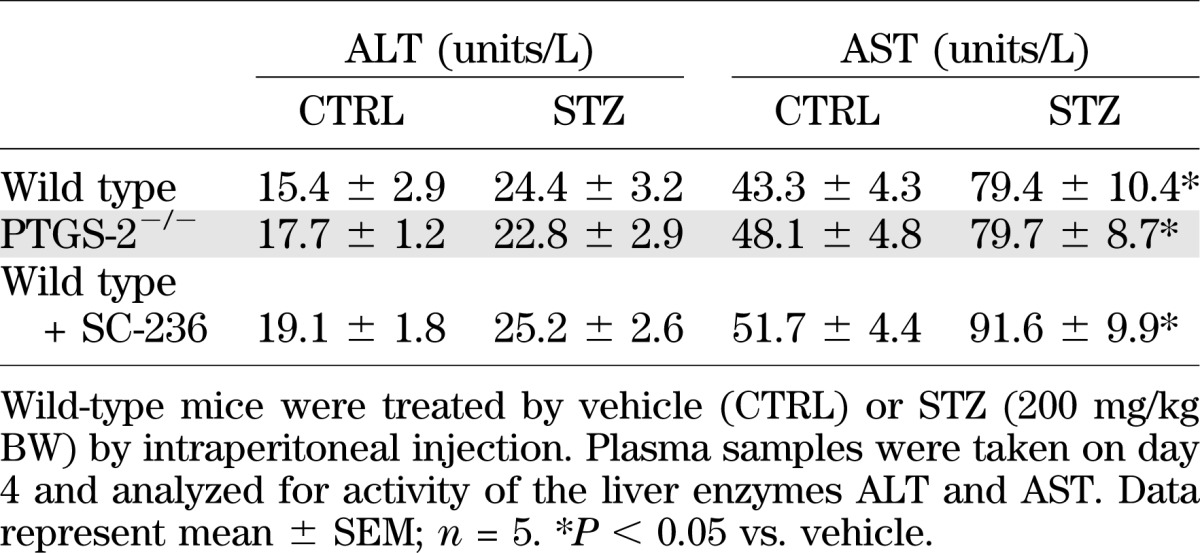
In order to clarify if differences in STZ metabolism may be responsible for enhanced toxicity, we studied STZ kinetics. STZ (200 mg/kg BW) was injected at time point 0, and plasma levels of STZ were determined at time points 30 min, 1 h, and 12 h postapplication. In all three study groups, control, PTGS-2−/−, and mice treated with SC-236, we observed comparable declines in plasma STZ levels (Fig. 4B), indicating that prolonged half-life of STZ does not contribute to enhanced effects of STZ in PTGS-2–deficient mice. Moreover, we analyzed plasma activity of the liver enzymes ALT and AST to evaluate whether differences in liver toxicity of STZ may account for the strong response of PTGS-2−/− mice toward STZ. Although activity of ALT was similar in treated and untreated groups, higher AST levels were observed after STZ application (Table 3). However, there were no significant differences between wild-type, PTGS-2−/−, and SC-236–treated wild-type mice. The increase in AST activity was in good accordance with a previous work on liver toxicity of STZ (25).
TABLE 3.
Liver enzyme activity
Combined inhibition of PTGER2 and PTGER4 increases diabetogenic effect of STZ.
Based on our observation that PGE2 represents the major prostanoid synthesized by pancreatic tissue and that PGE2 is induced several-fold after STZ application, we questioned whether distinct PTGERs may be involved in the modulation of STZ action. First, using PCR methodology, we studied PTGER expression. Figure 5A demonstrates that all four types of PGE2 receptors, PTGER1, PTGER2, PTGER3, and PTGER4, are expressed in pancreatic tissue and islets of mice, albeit the signal for PTGER2 mRNA appears to be weaker.
FIG. 5.

Effects of STZ in PTGER–deficient mice. A: Expression of PTGER mRNA in mouse pancreas and islets. RNA was prepared from the pancreas and islets of wild-type mice, and mRNA expression for PGE2 receptor types PTGER1, PTGER2, PTGER3, and PTGER4 was determined by PCR. As a control, β-actin was included. A representative analysis from three experiments is shown. B: Hyperglycemic effect of STZ in PTGER-deficient mice. Data are shown as mean ± SEM; n = 6. *P < 0.05 vs. vehicle; #P < 0.05 vs. STZ-treated PTGER2−/−. C: Effect of STZ on plasma insulin concentration in PTGER2- and PTGER4-deficient mice. Data are shown as mean ± SEM; n = 6. *P < 0.05 vs. vehicle; #P < 0.05 vs. STZ-treated PTGER2−/−. D: Kaplan-Meier survival analysis in PTGER2−/− and PTGER4−/− mice after application of STZ. In comparison, data obtained for PTGS-2−/− mice are included. Mice were kept under standard conditions for 10 days. Data are shown as mean ± SEM; n = 10. Wild-type (CTRL), PTGER1−/−, PTGER2−/−, PTGER3−/−, and PTGER4−/− mice were treated by a single injection of STZ (200 mg/kg BW), and on day 4, blood glucose or insulin was measured. To block PTGER2 and PTGER4 signaling pathways, we treated PTGER2−/− mice with PTGER4-selective antagonist ONO-AE3-208 (25 mg/kg) 1 day before STZ application and then daily.
In the next approach, we investigated diabetogenic effects of STZ in PTGER-deficient mice. Basal blood glucose concentrations were similar in control and PTGER-deficient mice (Fig. 5B), and STZ application caused hyperglycemia in all genetic mouse lines, not different from STZ-treated control mice (Fig. 5B). Due to the fact that both PTGER2 and PTGER4 mediate the activation of adenylate cyclase, we intended to block the PTGER2/PTGER4 signaling pathway by a combination of genetic and pharmacological interventions. Therefore, PTGER2−/− mice were treated with selective PTGER4 antagonist ONO-AE3-208 (25 mg/day/kg BW). Under this experimental setting, STZ application caused a significantly higher rise in blood glucose concentration compared with STZ-treated PTGER2−/− mice alone (Fig. 5B), which was quite similar to PTGS-2−/− mice (Fig. 2).
These observations were reflected by differences in plasma insulin concentrations. Whereas insulin levels in STZ-treated PTGER2−/− and PTGER4−/− mice were not different from STZ-treated control mice, pretreatment of PTGER2−/− mice with PTGER4 antagonist ONO-AE3-208 resulted in a significantly stronger decline in circulating insulin concentration, indicating stronger toxicity of STZ on β-cells (Fig. 5C). After long-term observation, Kaplan-Meier analysis revealed that STZ-treated PTGER2−/− and PTGER4−/− mice displayed a similar survival rate as control mice. However, when PTGER2−/− mice were treated with PTGER4 antagonist ONO-AE3-208, STZ significantly reduced survival (P < 0.0001) (Fig. 5D). PTGER4 antagonist alone has had no influence on mouse survival (data not shown).
PTGER2/PTGER4 agonists protect against STZ toxicity in PTGS-2−/− and wild-type mice.
The detrimental impact of PTGER2/PTGER4 blockade led us to the question of whether PTGER2 and PTGER4 agonists may exert a protective role in our model. Therefore, we treated PTGS-2−/− mice with a combination of selective PTGER2 agonist ONO-AE1-259-01 and selective PTGER4 agonist ONO-AE1-329 (each 40 μg/day/kg BW). Application of PTGER2/PTGER4 agonists significantly suppressed an STZ-induced rise in blood glucose concentration in PTGS-2−/− mice (Fig. 6A). To answer the question of whether enhanced PGE2 levels would protect from STZ toxicity also in wild-type mice, we treated mice with PTGER2/PTGER4 agonists (each 40 μg/day/kg BW). Although these animals exhibited elevated blood glucose concentrations, STZ caused a significantly lower increase compared with vehicle-treated mice (Fig. 6A). Moreover, treatment with PTGER2/PTGER4 agonists significantly reduced mortality of STZ-treated PTGS-2−/− mice (P < 0.0001) (Fig. 6B). By day 6, all PTGS-2−/− mice had died, whereas 75% of PTGS-2−/− mice treated with PTGER agonists survived until day 10.
FIG. 6.

Protective effects of PTGER2/PTGER4 agonists. A: Effect of PTGER2 and PTGER4 agonists on STZ-induced hyperglycemia in PTGS-2−/− and wild-type mice (CTRL). Mice were treated for 3 days, starting 24 h before STZ application (200 mg/kg BW), with a combination of PTGER2-selective agonist ONO-AE1-259-01 (40 ng/kg) and PTGER4-selective agonist ONO-AE1-329 (40 ng/kg) (PTGER-Ag). On day 4, blood glucose concentrations were measured by glucose meter. Data are shown as mean ± SEM; n = 6. *P < 0.05 vs. untreated controls; #P < 0.05 vs. STZ-treated PTGS-2−/−; $P < 0.05 vs. STZ-treated CTRL. B: Effect of PTGER2 and PTGER4 agonists on survival of STZ-treated PTGS-2−/− mice. PTGS-2−/− mice were treated 1 day before STZ application (200 mg/kg BW) and then daily with a combination of PTGER2-selective agonist ONO-AE1-259-01 (40 ng/kg) and PTGER4-selective agonist ONO-AE1-329 (40 ng/kg) (PTGER-Ag) or vehicle. Mice were kept under standard conditions for 10 days. Data are shown as mean ± SEM; n = 10.
DISCUSSION
In the current study, we disclosed enhanced diabetogenic and toxic potential of STZ when PTGS-2 activity is missing and present first evidence for a role of the PGE2 and PTGER2/4 pathway in protection against STZ-induced diabetes in mice. This conclusion is based on four main findings. First, PGE2 is the major prostanoid formed by normal and STZ-treated pancreatic tissue. Second, extinction of PTGS-2 activity either by gene deletion or by selective enzyme inhibition potentiates diabetogenic effect of STZ. Third, in mice with a blocked PTGER2/PTGER4 signaling pathway, STZ exerts similar toxicity as in PTGS-2−/− mice. Fourth, in mice missing PTGS-2 activity and in wild-type mice, effects of STZ can be reduced by PTGER2 and PTGER4 agonists.
Regarding the ability of pancreatic tissue to synthesize prostanoids, only limited information is available. Using 14C-arachidonic acid, TXB2 and 6-keto-PGF1α were observed as main conversion products in rat pancreatic tissue, whereas TXB2 increased and 6-keto-PGF1α decreased after STZ application (26). We accurately determined prostanoid formation from arachidonic acid by mouse pancreatic tissue using LC-MS/MS methodology and identified PGE2 as the main product formed, followed by 6-keto-PGF1α. STZ administration raised PGE2 synthesis threefold. In human islets, mRNA for PTGS-1 and PTGS-2 was detected (12,27), but in Syrian hamster islets, only PTGS-2 mRNA has been detected (28). Our experiments demonstrate that both PTGS-1 and PTGS-2 are present in mouse pancreas and in islets. However, only PTGS-2 mRNA increased due to STZ, and the PTGS-2–selective inhibitor SC-236, but not the PTGS-1–selective inhibitor SC-560, reduced STZ-enhanced PGE2 synthesis significantly. These findings allocate PTGS-2 a preferred role in prostanoid synthesis in the STZ diabetes model.
Compared with control mice, in PTGS-2−/− mice, STZ caused a much stronger destruction of pancreatic β-cells associated with a dramatic fall in circulating insulin and rise in blood glucose. A similar effect of STZ was observed in mice treated with the selective PTGS-2 inhibitor SC-236, which indicates that PTGS-2 activity (and products thereof) suppresses diabetogenic potential of STZ. Of note, the effect of STZ in PTGS-1−/− mice is indistinguishable from control mice, pointing toward a specific involvement of the PTGS-2/prostanoid pathway. Even though our data indicate a fall in PGE2 as an amplifying mechanism for STZ action, inhibition of the PTGS pathway may cause a shift of released arachidonic acid to the epoxygenase and/or lipoxygenase pathways, thereby rising their metabolites, which may modulate STZ effects. Still it is contrarily discussed whether these enzymes are able to use the available arachidonic acid pool (29,30), however, such a shift would matter. For example, 12-HETE, the product of 12-lipoxygenase, has been shown to reduce the viability of cultured human islets (31), and mice deficient in 12-lipoxygenase exhibited increased resistance to low-dose STZ–inflicted damage. Yet no data are available regarding the role of 12-HETE after high-dose STZ application as used in the current study. Further, 5-lipoxygenase has also been shown to modulate pancreatic function (32,33). Due to the complexity of arachidonic acid metabolism, complete detection of all metabolites appears to be a difficult challenge. At least we exclude a prominent role for the 5-lipoxygenase pathway in our model because we observed no difference in the pancreatic formation of its product 5-HETE by wild-type or PTGS-deficient mice. Our data also do not point toward altered STZ metabolism as a reason for enhanced STZ toxicity. Degradation of STZ was similar in wild-type and PTGS knockout mice, and liver toxicity of STZ was not dependent on PTGS expression. Therefore, regarding STZ action on the pancreas, our data present clear evidence for a protective role of PTGS-2. Interestingly a recent work on STZ-induced neurodegeneration in rats suggested a deleterious role of PTGS-2 (34). An explanation for this difference could be that the PTGS-2/prostanoid pathway is acting differentially and organ dependent.
Application of a high dose of STZ causes rapid death of pancreatic β-cells and resembles cytotoxic-based pathogenesis of type 1 diabetes, whereas application of multiple low doses of STZ is regarded to reflect less the cytotoxic but more the autoimmune-based pathogenesis (24). In addition to the high-dose model, we also observed in the low-dose model an exaggerated increase in the hyperglycemic effect of STZ, but only in the early phase of pathogenesis. Most likely in the later phase, STZ-induced diabetes pathology is ruled by immune processes independent of, or at least less dependent on, PTGS-2.
Under conditions resembling type 1 diabetes, the role of PGE2 is undefined, although there is some evidence to suggest that PGE2 may play a protective role. Recently it has been shown in NOD mice, a model for autoimmune diabetes including insulitis, that decreased PGE2 levels caused higher incidence of diabetes (35). In this work, a suppressive function on the generation of cytokines such as TNFα was attributed to PGE2 (35). The role of PGE2 in chemically induced noninsulitis models of type 1 diabetes is unclear, but some evidence is given that PGE2 also acts protective. PGE2 suppressed alloxan-induced cytotoxicity in rat insulinoma cells (36), and recently it has been shown that intraperitoneal application of PGE2 abrogated alloxan-induced diabetes in rats (37). Albeit alloxan and STZ express different modes of toxicity, alloxan via generation of reactive oxygen species (ROS) and STZ via DNA alkylation (38), we suggest that in both models, PGE2 acts protective on β-cells in a similar yet unidentified way.
The present data give evidence that the effect of PGE2 in limiting diabetogenic potential of STZ is mediated by PTGER2 and PTGER4. Our mRNA analysis revealed that all types of PGE2 receptors, PTGER1, PTGER2, PTGER3, and PTGER4, are expressed by mouse pancreatic tissue and also by isolated islets, which is in line with a recent expression study using human islets (12). To elucidate a role of distinct PGE2 receptors for diabetogenic action of STZ, we used PTGER-deficient mice; however, enhanced STZ toxicity, as in PTGS-2–blocked mice, was not observed. Of note, PTGER2 and PTGER4 are both coupled to activation of adenylate cyclase and therefore compensation for the deletion of one receptor type by the other receptor type may be feasible. The generation of PTGER2/PTGER4 double-knockout mice is nearly impossible to achieve due to their role in fertility and fetal circulation (39). Therefore, we injected a selective PTGER4 antagonist in PTGER2−/− mice in order to disable both cAMP signaling pathways of PGE2. Under this experimental condition, STZ caused diabetic pathogenesis and toxicity quite similar to PTGS-2 deficiency. Moreover, a combination of PTGER2 and PTGER4 agonists was able to reduce diabetogenic and lethal effects of STZ in PTGS-2−/− mice and reduce hyperglycemia in wild-type mice, even though the blood glucose level was not completely normalized.
On the protective role of PGE2 via PTGER2 and PTGER4, we can only speculate. One possibility may be that PGE2 reduces the transport of STZ into β-cells, thereby minimizing the toxic potential of STZ. This mechanism would also give an explanation for the protective effect of PGE2 in alloxan-induced diabetes (37) because STZ and alloxan both gain entry into β-cells by the SLC2A2 type of glucose transporter. Modulation of SLC2A2 by PGE2 has not been studied yet. However, inhibition of PTGS-2 has been shown to augment the rate of hexose transport in myotubes by recruiting the glucose transporter SLC2A4 (40). Whether PTGS-2 inhibition may also affect SLC2A2 needs to be clarified. Another explanation may be given by an interaction of PTGER2/PTGER4 with alkylating processes by STZ (e.g., by activation of repair systems that maintain genome integrity). O6-Guanine is an important target for alkylation by STZ (41) and O6-alkylguanine-DNA alkyltransferase might therefore be an object for PGE2 action. Furthermore, STZ has been shown to generate high levels of DNA adducts, resulting in extensive poly (adenosine-5′-diphosphate-ribose) polymerase (PARP) activation and the rapid depletion of intracellular amounts of NAD+, which then causes cell death through necrosis (42). Mice deficient in PARP or treated with PARP inhibitors are protected from STZ-induced β-cell death and do not develop hyperglycemia (43,44). The interaction between PARP and PGE2 in β-cells is unknown; however, inhibition of PARP directly or indirectly by PGE2 would protect from STZ toxicity. Interestingly, PGE2 is able to cause apoptosis in fibroblasts through PTGER2 and PTGER4, a process involving cleavage of PARP (45). Very recently, PGE2 has been shown to limit chronic inflammation in autoimmune diseases by mediating the formation of lipoxin A3 (46), which facilitates the resolution of inflammation. Therefore it may be feasible that low lipoxin levels, due to suppressed PGE2 synthesis, aid STZ toxicity. Although we cannot exclude this possibility, such a mechanism appears less likely because β-cell death by high-dose STZ is related more to a cytotoxic and less to an inflammatory destructive process (47). ROS have been shown to rise after STZ application (48,49), and exaggerated ROS production under PTGS deficiency may explain enhanced STZ toxicity. However, an interaction of PTGER2/PTGER4 signaling with ROS appears unlikely for two reasons. First, in STZ-treated control mice and PTGS-2−/− mice, we did not observe any significant difference in the formation of F-isoprostanes (data not shown), known to be indicators of oxidative stress (50). Second, PTGS has been shown to cause free radical generation in the process of arachidonic acid metabolism in pancreatic tissue (49), and inhibition of PTGS-2 would therefore reduce the amount of ROS, thereby minimizing the STZ damaging effect, contrary to our observation.
In summary, our findings uncover a previously unrecognized protective role of PTGS-2–derived PGE2 in diabetogenicity by STZ. The protective function of PGE2 is mediated by the receptor types PTGER2 and PTGER4. Although the molecular mechanism of β-cell protection by PGE2 needs to be elucidated, our data offer the pharmacological option to intervene in type 1 diabetes progression by use of PTGER2 and PTGER4 agonists.
ACKNOWLEDGMENTS
T.M. is employed by the Minase Research Institute, ONO Pharmaceuticals, Osaka, Japan. No other potential conflicts of interest relevant to this article were reported.
A.V. and A.G. designed and conducted experiments and performed data analyses. N.K. contributed to the animal experiments (survival rate). N.F.B. contributed to experimental work (STZ determination). S.N. and T.M. provided reagents and contributed to discussion. R.M.N. designed research, analyzed data, and wrote the manuscript. R.M.N. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
We thank the animal care staff (Goethe University Frankfurt) for excellent technical assistance.
REFERENCES
- 1.Van Dyke K, Jabbour N, Hoeldtke R, Van Dyke C, Van Dyke M. Oxidative/nitrosative stresses trigger type I diabetes: preventable in streptozotocin rats and detectable in human disease. Ann N Y Acad Sci 2010;1203:138–145 [DOI] [PubMed] [Google Scholar]
- 2.Hober D, Sauter P. Pathogenesis of type 1 diabetes mellitus: interplay between enterovirus and host. Nat Rev Endocrinol 2010;6:279–289 [DOI] [PubMed] [Google Scholar]
- 3.Mathis D, Vence L, Benoist C. Beta-cell death during progression to diabetes. Nature 2001;414:792–798 [DOI] [PubMed] [Google Scholar]
- 4.Smyth EM, Grosser T, Wang M, Yu Y, FitzGerald GA. Prostanoids in health and disease. J Lipid Res 2009;50(Suppl.):S423–S428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luo P, Wang MH. Eicosanoids, β-cell function, and diabetes. Prostaglandins Other Lipid Mediat 2011;95:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol 1998;38:97–120 [DOI] [PubMed] [Google Scholar]
- 7.Robertson RP. Dominance of cyclooxygenase-2 in the regulation of pancreatic islet prostaglandin synthesis. Diabetes 1998;47:1379–1383 [DOI] [PubMed] [Google Scholar]
- 8.Heitmeier MR, Kelly CB, Ensor NJ, et al. Role of cyclooxygenase-2 in cytokine-induced beta-cell dysfunction and damage by isolated rat and human islets. J Biol Chem 2004;279:53145–53151 [DOI] [PubMed] [Google Scholar]
- 9.Luo C, Kallajoki M, Gross R, et al. Cellular distribution and contribution of cyclooxygenase COX-2 to diabetogenesis in NOD mouse. Cell Tissue Res 2002;310:169–175 [DOI] [PubMed] [Google Scholar]
- 10.Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem 2000;69:145–182 [DOI] [PubMed] [Google Scholar]
- 11.Narumiya S. Molecular diversity of prostanoid receptors; subtypes and isoforms of prostaglandin E receptor. Adv Exp Med Biol 1997;400A:207–213 [DOI] [PubMed] [Google Scholar]
- 12.Persaud SJ, Muller D, Belin VD, et al. The role of arachidonic acid and its metabolites in insulin secretion from human islets of langerhans. Diabetes 2007;56:197–203 [DOI] [PubMed] [Google Scholar]
- 13.Han X, Chen S, Sun Y, Nadler JL, Bleich D. Induction of cyclooxygenase-2 gene in pancreatic beta-cells by 12-lipoxygenase pathway product 12-hydroxyeicosatetraenoic acid. Mol Endocrinol 2002;16:2145–2154 [DOI] [PubMed] [Google Scholar]
- 14.Metz SA, Robertson RP, Fujimoto WY. Inhibition of prostaglandin E synthesis augments glucose-induced insulin secretion is cultured pancreas. Diabetes 1981;30:551–557 [DOI] [PubMed] [Google Scholar]
- 15.Meng ZX, Sun JX, Ling JJ, et al. Prostaglandin E2 regulates Foxo activity via the Akt pathway: implications for pancreatic islet beta cell dysfunction. Diabetologia 2006;49:2959–2968 [DOI] [PubMed] [Google Scholar]
- 16.Zawalich WS, Zawalich KC, Yamazaki H. Divergent effects of epinephrine and prostaglandin E2 on glucose-induced insulin secretion from perifused rat islets. Metabolism 2007;56:12–18 [DOI] [PubMed] [Google Scholar]
- 17.Like AA, Rossini AA. Streptozotocin-induced pancreatic insulitis: new model of diabetes mellitus. Science 1976;193:415–417 [DOI] [PubMed] [Google Scholar]
- 18.Schnedl WJ, Ferber S, Johnson JH, Newgard CB. STZ transport and cytotoxicity. Specific enhancement in GLUT2-expressing cells. Diabetes 1994;43:1326–1333 [DOI] [PubMed] [Google Scholar]
- 19.Nüsing RM, Treude A, Weissenberger C, et al. Dominant role of prostaglandin E2 EP4 receptor in furosemide-induced salt-losing tubulopathy: a model for hyperprostaglandin E syndrome/antenatal Bartter syndrome. J Am Soc Nephrol 2005;16:2354–2362 [DOI] [PubMed] [Google Scholar]
- 20.Olliges A, Wimmer S, Nüsing RM. Defects in mouse nephrogenesis induced by selective and non-selective cyclooxygenase-2 inhibitors. Br J Pharmacol 2011;163:927–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seewaldt S, Thomas HE, Ejrnaes M, et al. Virus-induced autoimmune diabetes: most beta-cells die through inflammatory cytokines and not perforin from autoreactive (anti-viral) cytotoxic T-lymphocytes. Diabetes 2000;49:1801–1809 [DOI] [PubMed] [Google Scholar]
- 22.Schmidt R, Coste O, Geisslinger G. LC-MS/MS-analysis of prostaglandin E2 and D2 in microdialysis samples of rats. J Chromatogr B Analyt Technol Biomed Life Sci 2005;826:188–197 [DOI] [PubMed] [Google Scholar]
- 23.Maier TJ, Tausch L, Hoernig M, et al. Celecoxib inhibits 5-lipoxygenase. Biochem Pharmacol 2008;76:862–872 [DOI] [PubMed] [Google Scholar]
- 24.Kolb H. Mouse models of insulin dependent diabetes: low-dose streptozocin-induced diabetes and nonobese diabetic (NOD) mice. Diabetes Metab Rev 1987;3:751–778 [DOI] [PubMed] [Google Scholar]
- 25.Imaeda A, Kaneko T, Aoki T, Kondo Y, Nagase H. DNA damage and the effect of antioxidants in streptozotocin-treated mice. Food Chem Toxicol 2002;40:979–987 [DOI] [PubMed] [Google Scholar]
- 26.González E, Jawerbaum A, Sinner D, et al. Evolution of streptozotocin-pancreatic damage in the rat: modulatory effect of endothelins on the nitridergic and prostanoid pathway. Nitric Oxide 1999;3:459–466 [DOI] [PubMed] [Google Scholar]
- 27.Persaud SJ, Muller D, Belin VD, et al. Expression and function of cyclooxygenase and lipoxygenase enzymes in human islets of Langerhans. Arch Physiol Biochem 2007;113:104–109 [DOI] [PubMed] [Google Scholar]
- 28.Sorli CH, Zhang HJ, Armstrong MB, Rajotte RV, Maclouf J, Robertson RP. Basal expression of cyclooxygenase-2 and nuclear factor-interleukin 6 are dominant and coordinately regulated by interleukin 1 in the pancreatic islet. Proc Natl Acad Sci USA 1998;95:1788–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manigrasso MB, O’Connor JP. Accelerated fracture healing in mice lacking the 5-lipoxygenase gene. Acta Orthop 2010;81:748–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peters-Golden M, Brock TG. Intracellular compartmentalization of leukotriene biosynthesis. Am J Respir Crit Care Med 2000;161:S36–S40 [DOI] [PubMed] [Google Scholar]
- 31.Ma K, Nunemaker CS, Wu R, Chakrabarti SK, Taylor-Fishwick DA, Nadler JL. 12-Lipoxygenase products reduce insulin secretion and beta-cell viability in human islets. J Clin Endocrinol Metab 2010;95:887–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cuzzocrea S, Rossi A, Serraino I, et al. 5-Lipoxygenase knockout mice exhibit a resistance to acute pancreatitis induced by cerulein. Immunology 2003;110:120–130 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Mehrabian M, Schulthess FT, Nebohacova M, et al. Identification of ALOX5 as a gene regulating adiposity and pancreatic function. Diabetologia 2008;51:978–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dhull DK, Bhateja D, Dhull RK, Padi SS. Differential role of cyclooxygenase isozymes on neuronal density in hippocampus CA1 region of intracerebroventricular streptozotocin treated rat brain. J Chem Neuroanat 2012;43:48–51 [DOI] [PubMed] [Google Scholar]
- 35.Oikawa Y, Yamato E, Tashiro F, et al. Protective role for cytosolic phospholipase A2alpha in autoimmune diabetes of mice. FEBS Lett 2005;579:3975–3978 [DOI] [PubMed] [Google Scholar]
- 36.Devi MM, Das UN. Effect of prostaglandins against alloxan-induced cytotoxicity to insulin secreting insulinoma RIN cells in vitro. Prostaglandins Leukot Essent Fatty Acids 2004;71:309–318 [DOI] [PubMed] [Google Scholar]
- 37.Sailaja Devi MM, Das UN. Effect of prostaglandins against alloxan-induced diabetes mellitus. Prostaglandins Leukot Essent Fatty Acids 2006;74:39–60 [DOI] [PubMed] [Google Scholar]
- 38.Lenzen S. The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia 2008;51:216–226 [DOI] [PubMed] [Google Scholar]
- 39.Narumiya S, FitzGerald GA. Genetic and pharmacological analysis of prostanoid receptor function. J Clin Invest 2001;108:25–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alpert E, Gruzman A, Lardi-Studler B, Cohen G, Reich R, Sasson S. Cyclooxygenase-2 (PTGS2) inhibitors augment the rate of hexose transport in L6 myotubes in an insulin- and AMPKalpha-independent manner. Diabetologia 2006;49:562–570 [DOI] [PubMed] [Google Scholar]
- 41.Drabløs F, Feyzi E, Aas PA, et al. Alkylation damage in DNA and RNA—repair mechanisms and medical significance. DNA Repair (Amst) 2004;3:1389–1407 [DOI] [PubMed] [Google Scholar]
- 42.Okamoto H. Molecular basis of experimental diabetes: degeneration, oncogenesis and regeneration of pancreatic B-cells of islets of Langerhans. BioEssays 1985;2:15–21 [Google Scholar]
- 43.Burkart V, Wang ZQ, Radons J, et al. Mice lacking the poly(ADP-ribose) polymerase gene are resistant to pancreatic beta-cell destruction and diabetes development induced by streptozocin. Nat Med 1999;5:314–319 [DOI] [PubMed] [Google Scholar]
- 44.Pieper AA, Brat DJ, Krug DK, et al. Poly(ADP-ribose) polymerase-deficient mice are protected from streptozotocin-induced diabetes. Proc Natl Acad Sci USA 1999;96:3059–3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang SK, White ES, Wettlaufer SH, et al. Prostaglandin E(2) induces fibroblast apoptosis by modulating multiple survival pathways. FASEB J 2009;23:4317–4326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan MM, Moore AR. Resolution of inflammation in murine autoimmune arthritis is disrupted by cyclooxygenase-2 inhibition and restored by prostaglandin E2-mediated lipoxin A4 production. J Immunol 2010;184:6418–6426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang SW, Taylor GE. Immune insulitis and antibodies to nucleic acids induced with streptozotocin in mice. Clin Exp Immunol 1981;43:425–429 [PMC free article] [PubMed] [Google Scholar]
- 48.Mendola J, Wright JR, Jr, Lacy PE. Oxygen free-radical scavengers and immune destruction of murine islets in allograft rejection and multiple low-dose streptozocin-induced insulitis. Diabetes 1989;38:379–385 [DOI] [PubMed] [Google Scholar]
- 49.Tabatabaie T, Vasquez-Weldon A, Moore DR, Kotake Y. Free radicals and the pathogenesis of type 1 diabetes: beta-cell cytokine-mediated free radical generation via cyclooxygenase-2. Diabetes 2003;52:1994–1999 [DOI] [PubMed] [Google Scholar]
- 50.Morrow JD, Roberts LJ. The isoprostanes: unique bioactive products of lipid peroxidation. Prog Lipid Res 1997;36:1–21 [DOI] [PubMed] [Google Scholar]