

SUMMARY
Myc is a global transcriptional regulator and one of the most frequently overexpressed oncoproteins in human tumors. It is well established that activation of Myc leads to enhanced cell proliferation but can also lead to increased apoptosis. The use of animal models expressing deregulated levels of Myc has helped to both elucidate its function in normal cells and give insight into how Myc initiates and maintains tumorigenesis. Analyses of the medaka (Oryzias latipes) genome uncovered the unexpected presence of two Myc gene copies in this teleost species. Comparison of these Myc versions to other vertebrate species revealed that one gene, myc17, differs by the loss of some conserved regulatory protein motifs present in all other known Myc genes. To investigate how such differences might affect the basic biological functions of Myc, we generated a tamoxifen-inducible in vivo model utilizing a natural, fish-specific Myc gene. Using this model we show that, when activated, Myc17 leads to increased proliferation and to apoptosis in a dose-dependent manner, similar to human Myc. We have also shown that long-term Myc17 activation triggers liver hyperplasia in adult fish, allowing this newly established transgenic medaka model to be used to study the transition from hyperplasia to liver cancer and to identify Myc-induced tumorigenesis modifiers.
INTRODUCTION
The human Myc gene family, comprising MYC, MYCN and MYCL, constitutes a class of proto-oncogenes that are causally implicated in a broad spectrum of human cancers following alterations in their structure or changes in expression. One such example is Burkitt’s lymphoma, which involves a translocation of MYC leading to juxtaposition with regulatory elements of the immunoglobulin heavy- or light-chain genes (Klapproth and Wirth, 2010). Deregulated expression of MYC has also been detected in a large variety of other human cancers, including breast and colon cancer and small-cell lung carcinoma (for a review, see Vita and Henriksson, 2006).
In non-tumorigenic cells, MYC levels are low and dependent on mitogen signaling (Grandori et al., 2000), with expression being strictly controlled at both a transcriptional and a translational level (Meyer and Penn, 2008). In most human solid tumors, MYC expression is deregulated and is thought to promote tumor progression (Yokota et al., 1986). The cause of Myc deregulation can be due to retrovirus integration, insertional mutagenesis, chromosomal translocation, gene amplification, increase in MYC mRNA stability or decrease of Myc protein stability (for a review, see Meyer and Penn, 2008). Although oncogenic activation of MYC alone can cause uncontrolled cell proliferation in vitro, cellular transformation in vivo requires additional oncogenic lesions, such as activated Ras (Land et al., 1983; Jacobs et al., 1999), interaction with the anti-apoptotic activity of Bcl2 and Bcl-x (Strasser et al., 1990) or loss of the tumor suppressor p53 (Blyth et al., 1995).
The physiological function of Myc is to act as a global transcriptional regulator controlling normal cell proliferation, growth, survival and differentiation (Meyer and Penn, 2008). The prerequisite to the activation of gene transcription is the formation of a heterodimeric complex with Max through the C-terminal region of Myc, in which a basic helix-loop-helix and a leucine-zipper (bHLHZip) are present (Lüscher and Larsson, 1999). The stability of Myc is strongly linked to its phosphorylation status, with Ras controlling Myc stability by phosphorylation of two Myc residues, Thr58 and Ser62 (Sears et al., 2000), and Rho-dependent kinase phosphorylating Myc at Ser71 (Watnik et al., 2003). Additionally, Myc function is post-translationally controlled by six lysine residues, which are direct substrates for p300-mediated acetylation (Zhang et al., 2005).
To better understand Myc function, several transgenic models have been generated. Myc-null mice fail to develop normally and die at embryonic day (E)9.5-E10.5 with abnormalities of the heart, neural tube and pericardium, thereby demonstrating the crucial requirement of Myc for embryonic organ development (Davis et al., 1993). A conditional Myc-knockout mouse, using Cre-loxP technology, provided additional information regarding the role of Myc during cell cycle control and progression, and organ and body size control (Trumpp et al., 2001). Furthermore, a mouse model that enables temporal control of Myc activation after tamoxifen treatment was developed by fusing the Myc gene to the hormone-binding domain of the estrogen receptor (ER) (Eilers et al., 1989), which was used to investigate the effect of timed Myc activation in distinct tissues. This model showed that Myc activation is sufficient to induce cell cycle entry of post-mitotic keratinocytes and to block differentiation (Pelengaris et al., 1999).
Fish models that mimic human diseases such as cancer are increasingly being used, with the advantage of being able to follow well-defined stages of the disease in vivo over time (Stoletov and Klemke, 2008). Furthermore, fish are suitable for large-scale genetic and chemical compound screens to identify modifying factors that influence cancer development and progression (Mione and Trede, 2010). With respect to Myc, a zebrafish T-cell acute lymphoblastic leukemia (T-ALL) model has been generated, which uses a mouse or human Myc version, which is commonly misexpressed in leukemia. Myc is under the control of the lymphoid-cell-specific rag2 promoter and has revealed novel molecular pathways that are deregulated in T-ALL human lymphomas (Langenau et al., 2003). This model was later further optimized by conditioning Myc expression using the Cre-loxP (Langenau et al., 2005) and heat shock promoter (Feng et al., 2007) systems, and, more recently, extended to a tamoxifen-inducible version of Myc under the control of the rag2 promoter (Gutierrez et al., 2011). One possible limitation of these published models is that they have all been generated using the mouse or human gene instead of utilizing the species-specific ortholog, therefore ignoring possible species-specific functions of Myc. In addition, all published models unilaterally focus on just one species, the zebrafish.
Here we report the generation and use of an inducible medaka myc fused to a mouse ER (myc17ER) in order to obtain further insight into Myc function. The use of another species will help elucidate how genetics and metabolic differences influence Myc-driven tumorigenesis. Interestingly, we found two Myc genes in the medaka genome; one is highly conserved among vertebrates, whereas the other gene shows unique structural alterations. This discovery provided the opportunity to gain knowledge regarding the conserved Myc structure-function relationships in vertebrates. In our study we have cloned and characterized this novel Myc gene and its dose-dependent role in apoptosis and cell proliferation in vivo. Additionally, this novel Myc gene shows a potential pro-tumorigenic function in vivo, with evidence of liver hyperplasia post long-term activation.
RESULTS
Identification and cloning of a novel Myc gene
By searching available medaka databases, we identified two Myc genes, positioned at chromosomes 17 (myc17; Ensembl database identifier: ENSORLT00000008816) and on chromosome 20 (myc20; chromosome location 20:19323350-19324300). Sequence comparison with human, mouse and fugu Myc proteins (supplementary material Fig. S1) revealed a higher similarity of Myc20, as opposed to Myc17, to the other vertebrate homologs in the basic helix-loop-helix (bHLH) and DNA-binding domains (see supplementary material Tables S1, S2). Furthermore, the alignment shows that known Myc phosphorylation sites for the human protein (Adhikary and Eilers, 2005), such as glycogen synthase GSK3-targeted Thr58 and MAP-kinase-targeted Ser62, are conserved in both of the medaka Myc proteins. However, several apparent differences between the two medaka proteins were also noted. For example, Ser71, targeted by Rho-dependent kinase and related to transcriptional repression by Myc, is only conserved in Myc17 but not in Myc20 (supplementary material Fig. S2). Additionally, there are six lysine residues of human Myc that are direct substrates for acetylation by p300 (Zhang et al., 2005) and two of these are conserved in both medaka Myc genes: Lys143, within Myc homology box 1, and Lys323, within the nuclear localization signal. Three further lysine modification sites are only present in Myc20: Lys157, located next to Myc homology box 2, Lys275, not linked to any functionally important Myc domains, and Lys371, within the bHLH domain. One site is not present in either medaka copy, namely Lys317, which is located next to the nuclear localization signal in human Myc. These results indicate that essential regulatory motifs of Myc activity have been conserved in both medaka genes, but changes in the positions of post-translational regulatory sites, especially the loss of N-6 acetyllysine modification sites in Myc17, might have resulted in a functional divergence of these orthologs.
To further assess the evolutionary relationship of the Myc genes of medaka to other vertebrates, phylogenetic comparison of the two medaka sequences to human (Homo sapiens), mouse (Mus musculus), Xenopus (Xenopus tropicalis), Tetraodon (Tetraodon nigroviridis), fugu (Takifugu rubripes), stickleback (Gasterosteus aculeatus) and zebrafish (Danio rerio) Myc genes was performed (Fig. 1). These analyses revealed that both of the medaka Myc genes nested perfectly within the vertebrate Myc family and not within either the MycN or MycL gene families. Moreover, the phylogenetic tree revealed that the medaka copy present on chromosome 20 clusters closely with the sequences from other vertebrates at the expected position, in line with other fish Myc genes. By contrast, the medaka copy on chromosome 17 seems to be unique and it branches off relative early (box in Fig. 1), indicating that myc17 of medaka is a highly divergent or even aberrant gene version. Conserved synteny analyses of the surrounding regions in the genome of both medaka Myc genes to the human MYC region on chromosome Hsa8 and to other fish species, showed that myc17 and myc20 lie in regions that are highly evolutionarily conserved (supplementary material Fig. S2, and Tables S2, S3). This can be interpreted that there is a common origin of the two Myc genes by fish-specific genome duplication. Calculation of Ka/Ks values in relation to the human sequence indicated that myc17 is under neutral selection (branching value 0.95), whereas myc20 is under positive selection (branching value 1.27).
Fig. 1.

Unrooted phylogenetic tree of Myc family genes. Amino acid sequences were aligned by the maximum likelihood method. Numbers on nodes are bootstrap values out of 100 iterations.
In summary, these results strongly suggest that medaka myc17 is an evolutionarily old, divergent Myc gene version, harboring possible divergent functions from other vertebrate Myc genes.
Comparative analyses of myc17 and myc20
To exclude that myc17 is a non-expressed non-functional pseudogene, expression of both medaka Myc genes was analyzed by in situ hybridization and qPCR (supplementary material Fig. S3). Expression of both genes was detected in all developmental stages analyzed (stage 10, 18, 21, 24, 30, 34 and 38). Despite a more or less ubiquitous expression pattern of both genes, some differences of spatial expression could be observed between the two paralogs. For example, a prominent expression domain of myc20 at stages 20 and 28 in midbrain structures (arrows in supplementary material Fig. S3G,H) was detected. Additional qPCR experiments showed myc17 and myc20 expression during all stages of development, with the highest levels at stages 4 and 10 (supplementary material Fig. S3I). Expression in different adult organs was observed, with high gene expression detected in the eyes, liver and testes (supplementary material Fig. S3J). This indicates that both Myc copies, present on medaka chromosomes 17 and 20, are expressed during development in a comparable pattern, but differ in subtle expression domains.
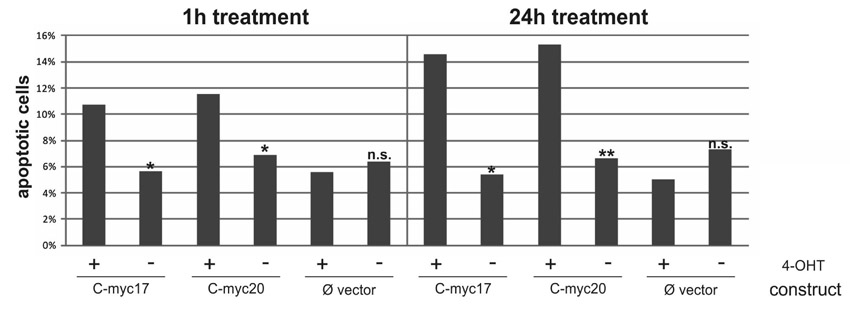
To initially look into possible functional differences between myc17 and myc20, we tested their effects on apoptosis induction in fish cells. The coding region of myc17 and myc20 lacking the stop codons were cloned and fused to the hormone-binding domain of the mouse ER, which enables induction of the transcription factor at a chosen time point by addition of tamoxifen. We transiently transfected constructs containing ER-coupled versions of both genes into A2 cells and investigated apoptotic rates (supplementary material Fig. S4). After tamoxifen treatment, the percentage of apoptotic cells was comparable between myc17ER- and myc20ER-transfected cells, indicating similar functional properties.
Generation of a tamoxifen-inducible myc17 transgenic medaka line
To analyze the cellular functions of the novel myc17 gene and to compare these with human MYC, we established an inducible in vivo model in medaka. For ubiquitous expression of the construct, the cytoskeletal actin promoter of Xenopus borealis was utilized (Lakin et al., 1993; Thermes et al., 2002). Stable integration of the construct into the medaka genome resulted in two independent transgenic lines.
Transgene expression in both lines was determined by qPCR. Only in a few tissues, such as gills and muscle, did line 1 show raised expression of the transgene, e.g. a sevenfold higher transgene expression in the gills when compared with the other organs examined (Fig. 2A). In line 2, brain, eyes, gills and muscle had at least sixfold increased expression when normalized to the liver. Direct comparison of both lines indicated that line 2 has an overall higher myc17ER RNA expression than line 1 (normalized Ct value differences of tissue samples from line 1 compared with line 2 are given in supplementary material Fig. S5). To test whether the protein is produced in both lines, western blot analyses were conducted. In both lines the fusion protein could be detected by using either an antibody directed against the mouse ER (Fig. 2B) or against human Myc (data not shown). Further studies confirmed that, in both lines, the transgene is highly expressed in nearly all organs compared with the endogenous gene (supplementary material Fig. S6A) and that tamoxifen treatment does not result in raised gene expression rates of either endogenous myc17 or the myc17ER transgene (supplementary material Fig. S6B).
Fig. 2.
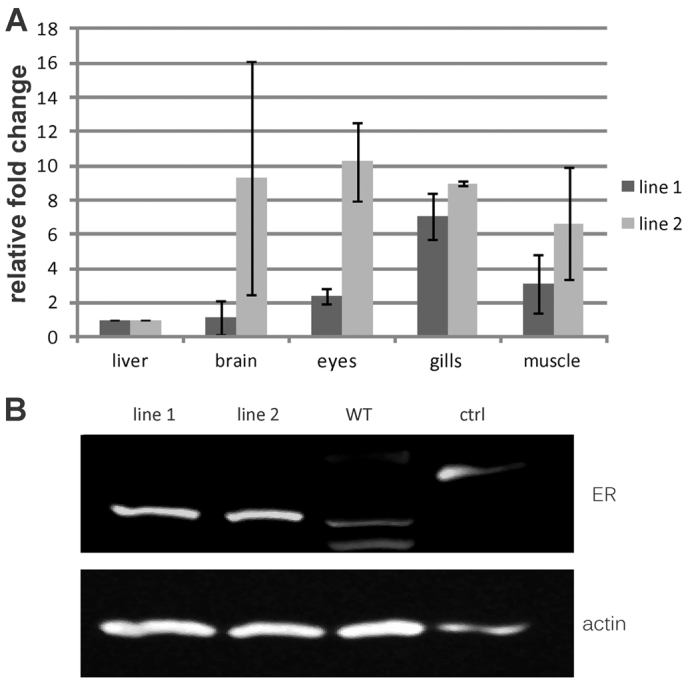
Expression of the myc17ER transgene in two medaka transgenic lines. (A) Real-time PCR analysis of the myc17ER transgene from liver, brain, eyes, gills and muscle from adult fish. Levels from the transgene were normalized against ef1a1 in each tissue. Histograms with T-bars indicate the mean standard deviation based on triplicate assays. (B) Western blot analysis of the Myc17ER fusion proteins. Protein samples extracted from pooled wild type (WT) and two transgenic fish lines were reacted with anti-ER antibody (30 hatched embryos per line). Protein extract containing human MycER (95 kDa), which has a higher molecular weight than medaka Myc17 (85 kDa), was used as control (crtl). Smaller and weaker bands in wild type control are due to unspecific antibody binding.
Functionality of the Myc17ER fusion protein
To test whether the inducible myc17ER system works in the transgenic medaka lines, dissociated fibroblast cells from tail fins were cultured for 5 days followed by a 24-hour treatment with 4-hydroxytamoxifen (4-OHT). Immunofluorescence with an anti-ER antibody of these cells showed nuclear translocation of the fusion protein only in the presence of 4-OHT, confirming the functionality of the inducible myc17ER system in medaka (Fig. 3A). Differences in nuclear:cytoplasm fluorescence intensity enabled us to quantify nuclear translocation (Fig. 3B) and revealed for both lines a similarly high rate of nuclear translocation of the Myc17ER protein. In comparison to the wild-type control, line 1 showed a tenfold difference between nuclear:cytoplasmic signal after 4-OHT treatment, whereas, in line 2, the difference was only 2.7-fold.
Fig. 3.

Functionality activity of the transgene product of myc17ER transgenic lines. (A) Immunofluorescence of myc17ER line 2 in medaka fin primary cell culture in the presence or absence of 4-OHT using anti-ER antibody and nuclear counterstaining with Hoechst. Scale bar: 10 μm. (B) Quantification of mean intensity differences (n=7 cells per treatment). Each cell was measured at two independent regions in the nucleus and the cytoplasm. Mean values were subtracted to calculate cytoplasmic to nuclear difference. Student’s t-tests show no significant differences between different −4-OHT controls (line 1 compared with WT: P=0.10; line 2 compared with WT: P=0.36). (C) Direct myc17 target gene expression (ODC1, CBX3, CCT5, EIF3S8 and MTLL1) in different organs via qPCR analysis after 4-OHT treatment. Relative fold change levels for each gene were normalized against ef1a1 in each tissue separately. Histograms with T-bars indicate the mean standard deviation based on duplicate assays.
Investigation of transcriptional functionality in the transgenic lines was done by looking for expression of five target genes that are upregulated by human Myc (Wagner et al., 1993; Dang et al., 2006; Mao et al., 2003; Zeller et al., 2003): ornithine decarboxylase 1 (odc1), chromobox homolog 3 (cbx3), chaperonin containing TCP1, subunit 5 (cct5), translation initiation factor 3, subunit 8 (eif3s8) and methyltransferase-like 1 (metll1). Expression of the medaka orthologs was determined in different adult tissues from myc17ER lines after 4-OHT treatment. This revealed induced expression in almost all investigated tissues compared with control samples (Fig. 3C). Differences between the two established lines became obvious, with tissue samples of line 2 always showing a stronger transcriptional induction of the five target genes, compared with line 1.
In summary, the nuclear translocation of Myc17ER and induction of target gene expression after 4-OHT treatment showed the suitability of the transgenic lines for further functional experiments.
In vivo function of novel Myc17
We then investigated whether ectopic expression of myc17ER leads to increased cell proliferation in vivo, one hallmark function of human Myc. Distinctly higher BrdU incorporation was detected in 4-OHT-treated medaka of both transgenic lines in comparison with EtOH-treated control fish (Fig. 4A). Quantification of BrdU incorporation after Myc induction (Fig. 4B) revealed that line 1, which has a lower transgene expression level, had a higher rate of proliferation compared with line 2. Approximately 3% of liver cells in line 1 were proliferating after Myc activation, whereas, in line 2, only 2% of all cells were BrdU positive. An even higher difference between the proliferation potential of both lines was detected in gills.
Fig. 4.

Cell proliferation after myc17 activation in vivo. (A) BrdU staining on sections of liver (upper panels) and gills (lower panels) in the presence or absence of 4-OHT. Scale bar: 10 μm. (B) Quantification of BrdU-positive cells in liver (left graphic) and gills (right graphic) in both myc17ER lines. Histograms with T-bars indicate the mean percentage of BrdU-positive cells of two independent experiments with their corresponding standard deviation. C N indicates average cell number counted from five different sections.
In contrast to the proliferative potential, primary cells from line 2 had a higher rate of apoptosis after 4-OHT treatment in vitro, compared with line 1 (Fig. 5A,B) and this was also evident in the liver and gills in vivo (Fig. 6A–C). In primary fibroblasts derived from medaka fins, 4-OHT-treated cells from line 1 showed an almost three times higher apoptotic rate when compared with EtOH-treated cells. For line 2, the apoptotic rate was more than five times higher. A similar observation was made when looking at adult tissues from transgenic fish. For example, livers from line 2 fish had around four times more TUNEL-positive cells after treatment with tamoxifen, whereas the amount of apoptotic cells in the gills was around 14 times higher in tamoxifen-treated fish when compared with non-treated gills. In summary, these experiments show that, in both transgenic lines, Myc17ER can induce proliferation and apoptosis, with the differences in the potency of both lines affecting the induction effects. Line 1 shows a higher rate of proliferative cells, whereas, in line 2, the apoptotic phenotype is more prevalent.
Fig. 5.
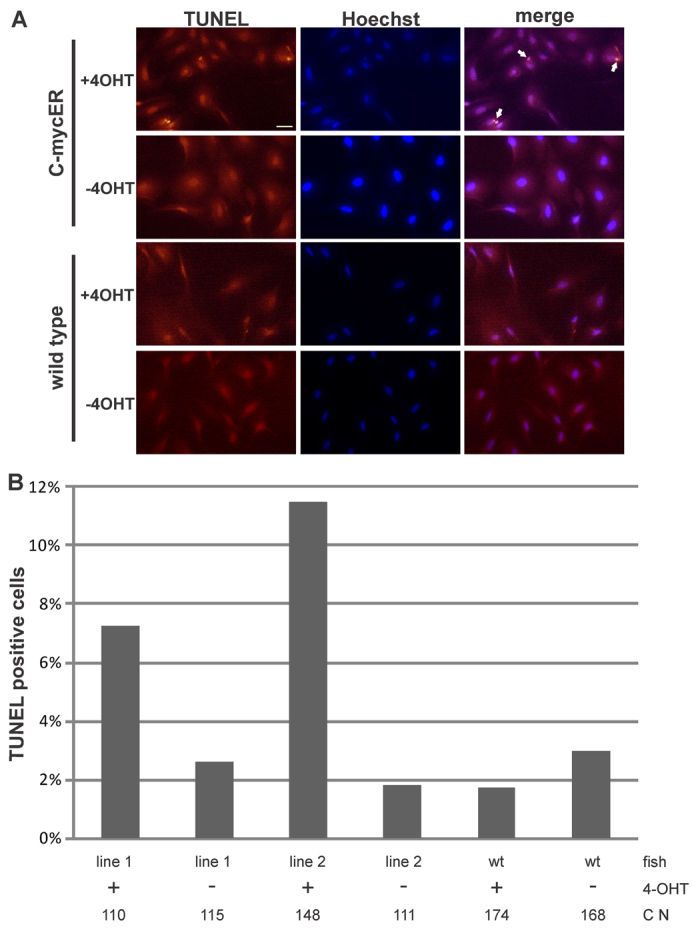
Effects of myc17 activation on cell death in vitro. (A) Detection of apoptosis in primary cell culture (arrows indicate TUNEL-positive cells). Cells were treated for 5 hours with 4-OHT before fixation. Nuclei were counterstained with Hoechst. Scale bar: 100 μm. (B) Quantification of the percentage of apoptotic cells in primary cultures from both myc17ER lines and wild-type fish in the presence or absence of 4-OHT in the media. C N indicates total cell number for each assay.
Fig. 6.

Induction of apoptosis after myc17 activation in vivo. (A) Detection of apoptosis by TUNEL assays in liver and gill sections from adult fish. Fish were treated for 24 hours with 4-OHT. DNA fragmentation is visible as red spots colocalizing with nuclei, which are stained with Hoechst. Scale bar: 10 μm. (B) Quantification of the percentage of apoptotic cells in liver (left panel) and gills (right panel) of both myc17ER lines in the presence or absence of 4-OHT. C N indicates total cell number for each assay.
We conducted long-term Myc activation experiments of adults of both myc17ER lines to investigate potential pathological changes. Histological investigations showed no prominent differences in organs of treated fish in comparison to untreated fish, except for liver. Hyperplasia of liver cells was very prominent in histological sections from both myc17ER line 1 and line 2 when compared with either wild-type fish treated or not with 4-OHT (Fig. 7). We counted approximately a twofold increase of cells per section in MycER fish after 4-OHT treatment (Fig. 7E). This result indicates that long-term activation of Myc17ER can lead to tissue hyperplasia in vivo.
Fig. 7.
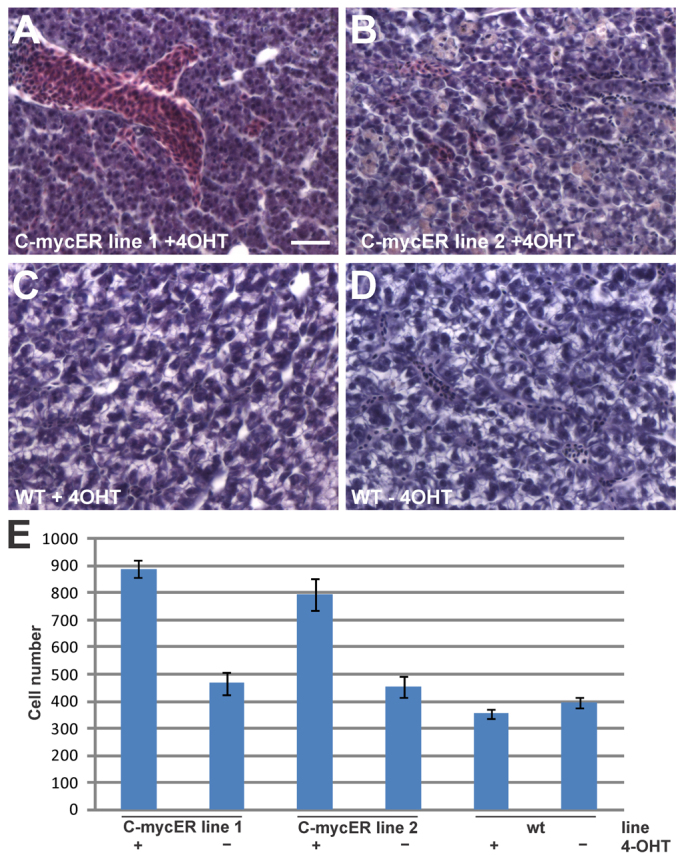
Constant myc17 activation triggers liver cell hyperplasia in vivo. (A-D) Images show hematoxylin- and eosin-stained sections of liver from adult transgenic fish from line 1 (A), line 2 (B) and wild type (C) treated for 4 weeks with 4-OHT or non-treated wild type (D). Scale bar: 20 μm. (E) Cell number per section (87,410 μm2) in both myc17ER lines and in wild-type fish treated or not treated with 4-OHT.
DISCUSSION
Evolution of myc genes in medaka
In the present study, we identified and characterized a previously unknown Myc gene in vertebrates. The amino acid sequence alignments indicated that the two medaka Myc genes are orthologs of human MYC (Fig. 1). In the ancient lineage leading to the teleosts fish of today, the whole genome had duplicated. Searches in the available sequenced teleost genomes revealed that only the zebrafish genome has kept two Myc copies along with medaka (Schreiber-Agus et al., 1993). Their evolutionary origin from the fish-specific genome duplication (Meyer and Schartl, 1999) was clearly established from synteny analysis comparing myc17 and myc20 with the human MYC locus on chromosome 8q24 and the corresponding fugu locus, which revealed a high degree of conservation between these species (supplementary material Fig. S2, and Tables S2, S3). The phylogenetic analysis also indicated that both medaka Myc genes are bona fide members of the Myc gene family. However, myc17 differs significantly from other vertebrate Myc genes because it branches off very basal in the phylogenetic tree (box in Fig. 1), whereas both zebrafish genes come out closely within the teleost branch. This indicated a high degree of diversification of this gene copy. Unless the presence of an extra gene product is advantageous, two genes with identical functions are unlikely to be stably maintained in the genome (Nowak et al., 1997). Duplicated genes can be maintained when they differ in some aspects of their functions, which can for example occur by sub-functionalization. During this process each gene copy adopts parts of the functions of their parental genes (Zhang, 2003). Another (non-exclusive) possibility is that one of the two versions adopts a novel function. Our studies of Myc17 indicate that, after the duplication process of the locus, the structure, and to a minor extent the expression, of the medaka Myc genes changed but the essential Myc functions are maintained, even after significant structural and amino acid changes. Further experiments will reveal whether the two medaka Myc genes have compensating or overlapping functions or whether they differ in more subtle aspects of their overall function. Observations of additionally duplicated Myc gene copies in carp and goldfish have been recently reported and confirm our phylogenetic conclusion (Marandel et al., 2012).
Conservation of functional motifs of medaka Myc genes
By comparing amino acid sequences of the human DNA-binding domain of Myc with Myc17 and Myc20 we observed 92% and 100% similarities, suggesting that both copies retained the ability to bind to DNA. Changes in eight positions of the bHLH domain of Myc17 are exclusively detected in this gene version and open up the possibility for changed DNA-motif binding specificity (De Masi et al., 2011). However, the leucine-zipper, which is responsible for the interaction with Max, corresponds only 50% and 68% in Myc20 and Myc17, respectively, to human Myc (supplementary material Table S1). This suggests that, although Myc20 shares a higher degree of conservation with other vertebrate Myc proteins, it is likely that the unique Myc17 is also able, or perhaps more likely, to interact with Max for activation of gene transcription. This is supported by induced expression of known Myc target genes after Myc17ER activation (Fig. 3C). As in humans, medaka homologs of odc1, cbx3, cct5, eif3s8 and metll1 were upregulated after 4-OHT treatment in the Myc17ER fish.
Additionally, the protein alignment indicated the loss or change of essential residues in Myc17 that are responsible for post-translational regulation of Myc function and stability (indicated as yellow arrows in supplementary material Fig. S1). These changes did not ablate the transcriptional function of Myc17 in vivo, but might result in altered potential of Myc17 function during cell transformation by influencing the half-life of Myc17 (Spencer and Groudine, 1991). In human cancers, besides the deregulated expression of MYC, stabilized versions of Myc act as main reasons for malignant transformation and cancer progression (Junttila and Westermarck, 2008). Phosphorylation (Westermarck, 2010) and ubiquitylation (Gregory and Hann, 2000) are key regulators of Myc turnover. In addition to these modifications, acetylation of Myc by histone acyltransferases, such as p300/CBP, Tip60 and GN5 (Vervoorts et al., 2003; Patel et al., 2004) is crucial for protein stabilization. Our analysis indicates that Myc17 has lost a number of these conserved modification sites, hinting to reduced stability by reducing acetylation and increasing the turnover rate. Interestingly, Myc20 displays a higher rate of residue conservation compared with Myc17, pointing to differences in post-transcriptional regulation and, subsequently, different protein stability rates between the two medaka orthologs. To initially study the functional differences of both genes, we performed transient transfections of myc17 and myc20 into A2 cells. These experiments show no differences in the ability to drive cells into apoptosis (supplementary material Fig. S4), indicating functional conservation between the two medaka gene versions in this context.
A correlation with known tumor-related human single nucleotide polymorphism (SNP) changes in MYC, for example mutations in the Pro57 and Pro59 residues found in Burkitt’s lymphoma (Bhatia, 1993), indicated that there are a number of conserved residues that are unchanged in the medaka proteins. Other residues that are not conserved, such as Glu39 and Asn86, are found in both versions of Myc in medaka. Functional consequences of these changed residues in fish have not been investigated so far.
Myc17 plays a role in apoptosis and proliferation
It is known from higher vertebrates that activation of Myc leads to cell proliferation through its ability to activate expression of genes involved in cell cycle progression (Steiner et al., 1995), its repression of CDK inhibitors (Staller et al., 2001) and also its involvement in chromatin remodeling (Amati et al., 2001). Deregulated Myc activation might also lead to cell growth arrest and subsequent apoptosis under certain conditions (Evan et al., 1992). To test the ability of Myc17 to perform established Myc functions, we generated two transgenic medaka lines expressing an ER-coupled version of the gene under the ubiquitously expressed β-actin promoter. Interestingly, the two independently generated medaka lines displayed differences in the amount of transgene expression (Fig. 2A). With respect to Myc17ER protein, we could detect protein production in both lines during early larval development, but future experiments are necessary to monitor how the observed differences in mRNA expression quantifiably translate into protein levels. Interestingly both transgenic lines showed clearly distinguishable functional differences after activation (Figs 4–6). Line 1, which had a lower myc17ER transgene expression, showed a raised proliferative rate when compared with line 2. Conversely, line 2 had a higher transgene expression level and seemed to induce a higher apoptotic rate than line 1 (Figs 5, 6). A comparable observation was obtained by Murphy et al. in mice, where the activation of Myc under the weak Rosa26 promoter triggered proliferation, whereas the more powerful rat insulin promoter, when driving Myc expression, led to apoptosis (Murphy et al., 2008). This indicated that cellular fate decisions are obviously taken depending on different threshold levels of Myc. Our work supports this view because the ability to induce higher proliferation or higher apoptosis rates was related to the differences in the level of transgene expression in the two medaka lines, which were otherwise genetically identical.
A pro-tumorigenic role for Myc17?
Although a link between Myc and cancer is well established, the molecular and cellular mechanisms of Myc-mediated transformation are not yet fully understood (Wang et al., 2011). Myc is thought to contribute to tumorigenesis by deregulating gene expression and abrogating cell cycle checkpoints, thereby driving uncontrolled cellular growth and promoting cell proliferation. Additional effects on cellular adhesion, angiogenesis, metabolism and genomic instability have been described and are key elements of tumor progression (Lutz et al., 2002). Nevertheless, Myc-induced cellular alterations can lead to apoptosis and senescence as a mechanism to protect the organism from proliferation of damaged or abnormal cells, which could be progenitors of cancer cells (Evan et al., 1992). In an oncogenic background, with additional mutations that activate anti-apoptotic signals, Myc can lead to neoplasms and subsequently to malignant transformation (Nilsson and Cleveland, 2003). For example, Myc was shown to collaborate with loss of p53 (Blyth et al., 2000) or Arf (Jacobs et al., 1999) in mouse lymphomagenesis.
To further decipher Myc functions and cell fate decisions, genetic models utilizing inducible protein versions have been established in higher vertebrates and recently in zebrafish. Two examples from transgenic mice are an inducible activated Myc version in mouse epidermis (Pelengaris et al., 1999) and in adult mouse pancreatic β-cells (Pelengaris et al., 2002). Both mouse models greatly differ in the potential of inducing apoptosis and proliferation, and in their dependence on additional mutations. Although laboratory fish systems offer several experimental advantages in comparison to mice, e.g. the possibility to perform large scale screens and in vivo visualization of tumor progression, a similar inducible mycER system, like the one presented in this work, has been established only recently for lymphomas (Gutierrez et al., 2011). Prior to this study, mouse or human Myc was used in the zebrafish to induce T-ALL (Langenau et al., 2003). The examination of molecular pathways activated in response to Myc overexpression closely resembled the most common subclass of human leukemia (Langenau et al., 2004). Lymphoid malignancy in zebrafish also requires additional events of transformation such as tal1 and lmo2 deregulation. Gutierrez et al. showed that ablation of Myc activation results in regressions of T-ALL and tumor regression depending on Akt-Pten singling (Gutierrez et al., 2011). In contrast to these published zebrafish models, our work presents a more general tumorigenesis model to induce Myc function in organs other than blood, in an evolutionarily divergent laboratory fish species by using a species-specific MYC ortholog.
As a first indication of cellular transformation caused by Myc activation, hyperplasia, but not tumor formation, was observed in livers of Myc17ER transgenic medaka individuals after 4-OHT treatment. Several toxicological studies implicate the effects of tamoxifen application alone on impaired organ development in fish. Sun et al. showed that embryonic treatment for 14 days with more than 125 μg/l tamoxifen results in hatching problems in medaka and in liver toxicity at high doses of 625 μg/l tamoxifen in a 21-day treatment period (Sun et al., 2007). In contrast to these experiments, our incubations were conducted using 4-OHT, the active, less-toxic metabolite of tamoxifen. Additionally, the final concentrations in our experiments were doses of 304 μg/l 4-OHT, and were therefore rather low. Besides changed toxicology properties, 4-OHT differs to tamoxifen by several biochemical properties. For example, 4-OHT shows a raised affinity to the ERs and a greater potency to suppress breast cancer cell growth (Wakeling and Slater, 1980; Coezy et al., 1982). We could not detect 4-OHT toxicological side effects after long-term application. Additionally, only treatment of transgenic Myc17 lines with 4-OHT resulted in hyperplasia, whereas wild-type control fish treated with 4-OHT for the same time did not show any changes in cell number (Fig. 7).
On the basis of the zebrafish and mouse studies mentioned above, we can hypothesize that myc17 medaka would need an additional anti-apoptotic event to function as a viable tumor model. In accordance with our findings in medaka, sustained overexpression of Myc in the liver of transgenic mice led to cancer only after 12 months (Santoni-Rugiu et al., 1996). In zebrafish, constantly expressed oncogenic Kras, an upstream factor of Myc in the MAP-kinase signaling pathway (Gupta and Davis, 1994; Jin et al., 2004), was sufficient to drive liver tumorigenesis (Nguyen et al., 2011). These fish exhibit a similar histological phenotype as observed in our myc17 medaka.
In summary, the established medaka model we have developed utilizing a teleost Myc gene opens up the possibility to decipher the transition from hyperplasia to liver cancer. Medaka can be easily manipulated to produce double transgenic individuals lacking anti-apoptotic factors – for example, p53 mutants (Taniguchi et al., 2006) – and is also suitable for large-scale genetic and chemical compound screens to identify anticancer agents. Additionally, it facilitates the investigation of fate decision and their consequences during early steps of tumorigenesis and development in closer detail.
METHODS
Animal maintenance
Wild-type medaka embryos (Oryzias latipes) of the carbio strain (WLC# 2674) were raised as described by Kirchen and West (Kirchen and West, 1976) under standard laboratory conditions at 26°C. Embryos were staged by morphological characteristics according to Iwamatsu et al. (Iwamatsu et al., 2004). All procedures involving experimental animals were performed in compliance with local animal welfare laws, guidelines and policies.
Cloning of myc17 constructs and qPCR
Newly identified medaka myc17 and myc20 coding sequences have been submitted to GenBank (myc17: GenBank-nr JN542547; myc20: GenBank-nr JN634762). Medaka myc17 (ENSEMBL database identifier: ENSORLG00000007021) and myc20 sequences were PCR amplified from pooled medaka organ cDNA using ReproFast polymerase (Genaxxon) with primers inserting a HindIII restriction site downstream and a BamHI site upstream of the gene (for primer sequences, see supplementary material Table S3). Using those restriction sites, myc17 was further sub-cloned into the vector pDNA3. The mouse ER was subsequently cloned in frame to Myc17 from pWZL-neo-ERG525R (Schulte et al., 2008) using BamHI and EcoRI restriction sites. Additionally, the myc17 sequence was subsequently replaced by the full-length cloned myc20 sequence. The myc17ER construct was then subcloned into pI-SceI containing the cytoskeletal-actin promoter of Xenopus borealis for ubiquitous expression in the entire embryo (Lakin et al., 1993) and an SV40-poly A and named myc17ER I-SceI plasmid.
Generation of transgenic lines
Medaka transgenics were generated using Meganuclease (I-SceI) technology (Thermes et al., 2002). One-cell-stage embryos were injected with the following solution: 10 ng/μl of myc17ER I-SceI plasmid, 1× Tango buffer (Fermentas), 0.35 U/μl of I-SceI (Fermentas). Injection solution was incubated for 30 minutes at 37°C. Before injection, 0.1% phenol red and 0.2% Fitc-dextran (53471; Sigma-Aldrich) were added to the solution. DNA solution was directly injected through the chorion into the cytoplasm of the first cell. Injected embryos were raised to sexual maturity followed by sibling screening (for primers, see supplementary material Table S4) for transgene integration.
Western blot
Total protein was prepared from hatched medaka embryos. Embryos were lysed in lysis buffer [20 mM HEPES (pH 7.8), 500 mM NaCl, 5 mM MgCl2, 5 mM KCl, 0.1% deoxycholate, 0.5% Nonidet-P40, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 200 μM Na3VO4, 1 mM PMSF and 100 mM NaF]. A total of 50 μg of protein lysate was separated by SDS/PAGE and transferred to nitrocellulose according to standard western blotting protocols. Primary antibodies used were anti-ER (1:500; Santa Cruz) and anti-β-actin (1:10,000; Santa Cruz). Peroxidase-coupled anti-mouse (1:3000; Thermo Scientific) and anti-rabbit (1:10,000; Bio-Rad) were used as secondary antibodies.
qPCR
Total RNA was isolated from adult medaka fish using peqGOLD TriFast (peqlab). First-strand cDNA was synthesized using RevertAid First Strand cDNA Synthesis Kit (Fermentas). For qPCR detection, a 241 bp region of the myc17ER fusion was amplified using primers flanking the fusion point (supplementary material Table S4). Ct values were relatively calculated to the housekeeping gene ef1a1. All qPCR experiments were performed in a MyiQ single-color real-time PCR detection system (Bio-Rad).
4-OHT treatment for myc17 induction
4-OHT (H7904; Sigma) was dissolved in ethanol at a final stock concentration of 10 mM and kept in the dark at 4°C. To induce myc17 activity for short-term experiments, transgenic adult fish were kept once in fish water containing 1 μM of 4-OHT in the dark for 48 hours continuously. Long-term tamoxifen treatments were conducted as constant chemical treatments of fish over 4 weeks with changes of fish water three times a week. Each long-term experiment included four individuals from each transgenic line, two males and two females, at an age between 10 and 12 months. To induce myc17 activity in primary cell culture, L-15 medium was mixed with 1 μM of 4-OHT for 24 hours.
Whole mount in-situ hybridization
RNA in situ hybridization was performed according to standard protocols described previously (Hauptmann and Gerster, 1994). For signal improvement, embryos were incubated after coloration in 100% methanol overnight at −20°C prior to rehydration in PBST, mounting and imaging. Sense controls were performed as a negative control for each antisense probe separately.
Primary cell culture
Pectoral medaka fins were cut from narcotized fish from both myc17 lines and wild type and immersed in PBS containing 0.4% sodium hypochlorite for 30 seconds. Tissues were rinsed three times in PBS and minced into small fragments. PBS was replaced by Leibovitz L-15 medium (Invitrogen) supplemented with 10% fetal bovine serum (PAA), 2 mM glutamine (PAA), 100 U/ml penicillin (Invitrogen) and 100 μg/ml streptomycin (Invitrogen). Tissue fragments were cultivated in a gelatin-coated μ-slide (eight-well; IBIDI) containing 300 μl of the culture medium. The culture was incubated at 28°C for 5 days and fixed with 2% formaldehyde.
Proliferation, apoptosis and nuclear translocation assays
For the in vivo analysis of BrdU incorporation and apoptosis detection, liver and gills from adult fish were fixed for 2 hours in 10% neutral buffered formalin. For in vivo and primary cell apoptosis detection, TUNEL assay was performed using ApopTag Red In Situ Apoptosis Detection Kit (Millipore) according to the manufacturer’s protocol. For detection of proliferating cells, adult fish were bathed in a solution of 1 g/l BrdU (Sigma) in aquarium water for 5 hours before dissection of organs. BrdU assay was performed using anti-BrdU antibody (1:250; Serotec) and Alexa-Fluor-568-conjugated goat anti-rat IgG secondary antibody (1:250; Invitrogen). Nuclear translocation assays were performed using anti-ER antibody (Santa Cruz) and Alexa-Fluor-594-conjugated goat anti-rabbit IgG (Invitrogen). Nuclei were counterstained with Hoechst 33342 fluorescent stain (Invitrogen). Imaging was done with a Nikon Eclipse Ti confocal microscope and NIS-Elements imaging software.
Transfection experiments for myc17 and myc20 comparison were performed in A2 cells (Kuhn et al., 1979). Cell number counting was performed in a Neubauer chamber, whereas apoptosis staining in these experiments was performed using an anti-cleaved-caspase-3 antibody (1:400; Cell Signaling) for immunfluorescence.
Phylogenetic methods and alignments
Alignments were generated applying BioEdit and ClustalW (Thompson et al., 1994). Maximum likelihood analysis was performed using Mega 5.0 software according to a JTT model of amino acid substitutions. Confidence of each node was confirmed by 1000 bootstrap replicates. Analysis of Myc gene synteny was performed utilizing the Ensembl database and the Synteny database (Catchen et al., 2009).
Histological methods
For histology, adult medaka fish of approximately 10 months of age were overanesthetized with ethyl 3-aminobenzoate methanesulfonate (MS-222, tricane; Sigma-Aldrich). Specimens were fixed in 4% paraformaldehyde prior to decalcification and embedding. Paraffin-embedded fish were sectioned into 5 μm thick sections, fixed on polylysine-coated slides (Mentzel) and stained with Mayer’s hematoxylin and eosin. Images were taken using a Zeiss Axiophot microscope with an attached AxioCam MRc camera and the Zeiss AxioVision software.
TRANSLATIONAL IMPACT.
Clinical issue
The transcription factor Myc is a central regulator of many cellular processes, including growth, proliferation, apoptosis, metabolism and differentiation, and its expression is deregulated in a wide variety of human cancers. Although the role of Myc in cancer has still not been fully elucidated, it is known that the enhanced cell proliferation and apoptosis triggered by Myc activation is related to its function in tumor progression. Studying the function of this highly conserved transcription factor in animal models has contributed to understanding its molecular function.
Results
In this paper, the authors identify and functionally characterize a novel, unusual vertebrate Myc gene. Analyses of the medaka genome reveal two Myc genes, one of which lacks protein-regulatory regions that are conserved in all other known versions of the gene. The authors establish a tamoxifen-inducible version of this unusual myc17 gene and produce two independent myc17 transgenic medaka lines with different levels of transgene expression. Studies of these lines reveal that Myc17 is transcriptionally functional and can drive apoptosis and proliferation in vivo in a manner similar to the conventional, more highly conserved version of Myc. Additionally, they report that long-term activation of myc17 results in liver hyperplasia, but no pathology in other tissues.
Implications and future directions
Given that the two myc17 transgenic medaka lines established here express different levels of the transgene, they can be further used to examine cell fate decisions between apoptosis and proliferation following Myc activation. In addition, future work will address whether loss of strict cell cycle control can lead to tumor induction in the liver, and whether changes in Myc expression levels influence tumor progression. Owing to the small size of medaka, these models (and their future modifications) will be advantageous for high-throughput drug screening and toxicology studies; furthermore, this small fish is ideal for imaging tumorigenic processes in real time. Finally, comparing myc17 with the conventional, more highly conserved version of the gene might reveal subtle differences that provide new insight into the evolution and function of vertebrate Myc genes.
Supplementary Material
Acknowledgments
The authors thank Toni Wagner for cloning help and in situ probes and Daniel Murphy for his critical comments to the manuscript. The pWZL-neo-ERG525R vector was a kind gift from Martin Eilers. We thank Anita Hufnagel for her help with cell culture experiments. We thank Shannon Graver for English writing editing.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
L.A.M. performed the experiments and prepared the manuscript; C.S. performed the cloning; and D.L. and M.S. developed the concept and edited the manuscript.
FUNDING
This work was supported by a grant of the German Excellence Initiative to the Graduate School of Life Sciences (GSLS), University of Würzburg (to L.A.M.); by the University of Würzburg funding program for Open Access Publishing; and by the DFG Transregio 17-Ras-dependent pathways in human cancer.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.008730/-/DC1
REFERENCES
- Adhikary S., Eilers M. (2005). Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 6, 635–645 [DOI] [PubMed] [Google Scholar]
- Amati B., Frank S. R., Donjerkovic D., Taubert S. (2001). Function of the c-Myc oncoprotein in chromatin remodeling and transcription. Biochim. Biophys. Acta 1471, 135–145 [DOI] [PubMed] [Google Scholar]
- Bhatia K., Huppi K., Spangler G., Siwarski D., Iyer R., Magrath I. (1993). Point mutations in the c-Myc transactivation domain are common in Burkitt’s lymphoma and mouse plasmacytomas. Nat. Genet. 5, 56–61 [DOI] [PubMed] [Google Scholar]
- Blyth K., Terry A., O’Hara M., Baxter E. W., Campbell M., Stewart M., Donehower L. A., Onions D. E., Neil J. C., Cameron E. R. (1995). Synergy between a human c-myc transgene and p53 null genotype in murine thymic lymphomas: contrasting effects of homozygous and heterozygous p53 loss. Oncogene 10, 1717–1723 [PubMed] [Google Scholar]
- Blyth K., Stewart M., Bell M., James C., Evan G., Neil J. C., Cameron E. R. (2000). Sensitivity to myc-induced apoptosis is retained in spontaneous and transplanted lymphomas of CD2-mycER mice. Oncogene 19, 773–782 [DOI] [PubMed] [Google Scholar]
- Catchen J. M., Conery J. S., Postlethwait J. H. (2009). Automated identification of conserved synteny after whole-genome duplication. Genome Res. 19, 1497–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coezy E., Borgna J. L., Rochefort H. (1982). Tamoxifen and metabolites in MCF7 cells: correlation between binding to estrogen receptor and inhibition of cell growth. Cancer Res. 42, 317–323 [PubMed] [Google Scholar]
- Dang C. V., O’Donnell K. A., Zeller K. I., Nguyen T., Osthus R. C., Li F. (2006). The c-Myc target gene network. Semin. Cancer. Biol. 16, 253–264 [DOI] [PubMed] [Google Scholar]
- Davis A. C., Wims M., Spotts G. D., Hann S. R., Bradley A. (1993). A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 7, 671–682 [DOI] [PubMed] [Google Scholar]
- De Masi F., Grove C. A., Vedenko A., Alibés A., Gisselbrecht S. S., Serrano L., Bulyk M. L., Walhout A. J. (2011). Using a structural and logics systems approach to infer bHLH-DNA binding specificity determinants. Nucleic Acids Res. 39, 4553–4563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers M., Picard D., Yamamoto K. R., Bishop J. M. (1989). Chimaeras of Myc oncoprotein and steroid receptors cause hormone-dependent transformation of cells. Nature 340, 66–68 [DOI] [PubMed] [Google Scholar]
- Evan G. I., Wyllie A. H., Gilbert C. S., Littlewood T. D., Land H., Brooks M., Waters C. M., Penn L. Z., Hancock D. C. (1992). Induction of apoptosis by c-myc protein in fibroblasts. Cell 69, 119–128 [DOI] [PubMed] [Google Scholar]
- Feng H., Langenau D. M., Madge J. A., Quinkertz A., Gutierrez A., Neuberg D. S., Kanki J. P., Look A. T. (2007). Heat-shock induction of T-cell lymphoma/leukaemia in conditional Cre/lox-regulated transgenic zebrafish. Br. J. Haematol. 138, 169–175 [DOI] [PubMed] [Google Scholar]
- Grandori C., Cowley S. M., James L. P., Eisenman R. N. (2000). The Myc/Max/Mad network and the transcriptional control of the cell behavior. Annu. Rev. Cell Dev. Biol. 16, 653–699 [DOI] [PubMed] [Google Scholar]
- Gregory M. A., Hann S. R. (2000). c-Myc proteolysis by the ubiquitin-proteasome pathway: stabilization of c-Myc in Burkitt’s lymphoma cells. Mol. Cell. Biol. 20, 2423–2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S., Davis R. J. (1994). MAP kinase binds to the NH2-terminal activation domain of c-Myc. FEBS Lett. 353, 281–285 [DOI] [PubMed] [Google Scholar]
- Gutierrez A., Grebliunaite R., Feng H., Kozakewich E., Zhu S., Guo F., Payne E., Mansour M., Dahlberg S. E., Neuberg D. S., et al. (2011). Pten mediates Myc oncogene dependence in a conditional zebrafish model of T cell acute lymphoblastic leukemia. J. Exp. Med. 208, 1595–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauptmann G., Gerster T. (1994). Two-color whole-mount in situ hybridization to vertebrate and Drosophila embryos. Trends Genet. 10, 266. [DOI] [PubMed] [Google Scholar]
- Iwamatsu T. (2004). Stages of normal development in medaka Orysias latipes. Mech. Dev. 121, 605–618 [DOI] [PubMed] [Google Scholar]
- Jacobs J. J. L., Scheijen B., Voncken J.-W., Kieboom K., Berns A., van Lohuizen M. (1999). Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes Dev. 13, 2678–2690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z., Gao F., Flagg T., Deng X. (2004). Tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone promotes functional cooperation of Bcl2 and c-Myc through phosphorylation in regulating cell survival and proliferation. J. Biol. Chem. 279, 40209–40219 [DOI] [PubMed] [Google Scholar]
- Junttila M. R., Westermarck J. (2008). Mechanisms of MYC stabilization in human malignancies. Cell Cycle 7, 592–596 [DOI] [PubMed] [Google Scholar]
- Kirchen R. V., West W. R. (1976). The Japanese Medaka: Its Care and Development. North Carolina: Carolina Biological Supply Company [Google Scholar]
- Klapproth K., Wirth T. (2010). Advances in the understanding of MYC-induced lymphomagenesis. Br. J. Haematol. 149, 484–497 [DOI] [PubMed] [Google Scholar]
- Kuhn C., Vielkind U., Anders F. (1979). Cell cultures derived from embryos and melanoma of poeciliid fish. In Vitro 15, 537–544 [DOI] [PubMed] [Google Scholar]
- Lakin N. D., Boardman M., Woodland H. R. (1993). Determination of the sequence requirements for the expression of a Xenopus borealis embryonic/larval skeletal actin gene. Eur. J. Biochem. 214, 425–435 [DOI] [PubMed] [Google Scholar]
- Land H., Parada L. F., Weinberg L. A. (1983). Tumorigenic conversion of primary embryo fibloblasts require at least two cooperating oncogenes. Nature 304, 596–602 [DOI] [PubMed] [Google Scholar]
- Langenau D. M., Traver D., Ferrando A. A., Kutok J. L., Aster J. C., Kanki J. P., Lin S., Prochownik E., Trede N. S., Zon L. I., et al. (2003). Myc-induced T cell leukemia in transgenic zebrafish. Science 299, 887–890 [DOI] [PubMed] [Google Scholar]
- Langenau D. M., Ferrando A. A., Traver D., Kutok J. L., Hezel J.-P. D., Kanki J. P., Zon L. I., Look A. T., Trede N. S. (2004). In vivo tracking of T cell development, ablation, and engraftment in transgenic zebrafish. Proc. Natl. Acad. Sci. USA 101, 7369–7374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenau D. M., Feng H., Berghmans S., Kanki J. P., Kutok J. L., Look A. T. (2005). Cre/lox-regulated transgenic zebrafish model with conditional myc-induced T cell acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 102, 6068–6073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher B., Larsson L.-G. (1999). The basic region/helix-loop-helix/leucine zipper domain of Myc proto-oncoproteins: Function and regulation. Oncogene 18, 2955–2966 [DOI] [PubMed] [Google Scholar]
- Lutz W., Leon J., Eilers M. (2002). Contributions of Myc to tumorigenesis. Biochim. Biophys. Acta 1602, 61–71 [DOI] [PubMed] [Google Scholar]
- Mao D. Y. L., Watson J. D., Yan P. S., Barsyte-lovejoy D., Khosravi F., Wong W. W., Farnham P. L., Huang T. H., Penn L. Z. (2003). Analysis of Myc bound loci identified by CpG island arrays shows that Max is essential for Myc-dependent repression. Curr. Biol. 13, 882–886 [DOI] [PubMed] [Google Scholar]
- Marandel L., Labbe C., Bobe J., Le Bail P. Y. (2012). Evolutionary history of c-myc in teleosts and characterization of the duplicated c-myca genes in goldfish embryos. Mol. Reprod. Dev. 79, 85–96 [DOI] [PubMed] [Google Scholar]
- Meyer A., Schartl M. (1999). Gene and genome duplications in vertebrates: the one-to-four (-to-eight in fish) rule and the evolution of novel gene functions. Curr. Opin. Cell Biol. 11, 699–704 [DOI] [PubMed] [Google Scholar]
- Meyer N., Penn L. Z. (2008). Reflecting on 25 years with MYC. Nat. Rev. Cancer 8, 976–990 [DOI] [PubMed] [Google Scholar]
- Mione M. C., Trede N. S. (2010). The zebrafish as a model for cancer. Dis. Model. Mech. 3, 517–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy D. J., Junttila M. R., Pouyet L., Karnezis A., Shchors K., Bui D. A., Brown-Swigart L., Johnson L., Evan G. I. (2008). Distinct thresholds govern Myc’s biological output in vivo. Cancer Cell 14, 447–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen A. T., Emelyanov A., Koh C. H. V., Spitsbergen J. M., Lam S. H., Mathavan S., Parinov S., Gong Z. (2011). A high level of liver-specific expression of oncogenic KrasV12 drives robust liver tumorigenesis in transgenic zebrafish. Dis. Model. Mech. 4, 801–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson J. A., Cleveland J. L. (2003). Myc pathways provoking cell suicide and cancer. Oncogene 22, 9007–9021 [DOI] [PubMed] [Google Scholar]
- Nowak M. A., Boerlijst M. C., Cooke J., Smith J. M. (1997). Evolution of genetic redundancy. Nature 388, 167–171 [DOI] [PubMed] [Google Scholar]
- Patel J. H., Du Y., Ard P. G., Phillips C., Carella B., Chen C. J., Rakowski C., Chatterjee C., Lieberman P. M., Lane, et al. (2004). The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol. Cell. Biol. 24, 10826–10834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelengaris S., Littlewood T., Khan M., Elia G., Evan G. (1999). Reversible activation of c-Myc in skin: induction of a complex neoplastic phenotype by a single oncogenic lesion. Mol. Cell 3, 565–577 [DOI] [PubMed] [Google Scholar]
- Pelengaris S., Khan M., Evan G. I. (2002). Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell 109, 321–334 [DOI] [PubMed] [Google Scholar]
- Santoni-Rugiu E., Preisegger K. H., Kiss A., Audolfsson T., Shiota G., Schmidt E. V., Thorgeirsson S. S. (1996). Inhibition of neoplastic development in the liver by hepatocyte growth factor in a transgenic mouse model. Proc. Natl. Acad. Sci. USA 93, 9577–9582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber-Agus N., Horner J., Torres R., Chiu F.-C., DePinho R. A. (1993). Zebra fish myc family and max genes: differential expression and oncogenic activity throughout vertebrate evolution. Mol. Cell. Biol. 13, 2765–2775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte J. H., Horn S., Otto T., Samans B., Heukamp L. C., Eilers U. C., Krause M., Astrahantseff K., Klein-Hitpass L., Buettner R., et al. (2008). MYCN regulates oncogenic MicroRNAs in neuroblastoma. Int. J. Cancer 122, 699–704 [DOI] [PubMed] [Google Scholar]
- Sears R., Nuckolls F., Haura E., Taya Y., Tamai K., Nevins J. R. (2000). Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 14, 2501–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer C. A., Groudine M. (1991). Control of c-myc regulation in normal and neoplastic cells. Adv. Cancer Res. 56, 1–48 [DOI] [PubMed] [Google Scholar]
- Staller P., Peukert K., Kiermaier A., Seoane J., Lukas J., Karsunky H., Möröy T., Bartek J., Massagué J., Hänel F., et al. (2001). Repression of p15 INK4b expression by Myc through association with Miz-1. Cell Biol. 3, 392–399 [DOI] [PubMed] [Google Scholar]
- Steiner P., Philipp A., Lukas J., Godden-Kent D., Pagano M., Mittnacht S., Bartek J., Eilers M. (1995). Identification of a Myc-dependent step during the formation of active G1 cyclin-cdk complexes. EMBO J. 14, 4814–4826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoletov K., Klemke R. (2008). Catch of the day: zebrafish as a human cancer model. Oncogene 27, 4509–4520 [DOI] [PubMed] [Google Scholar]
- Strasser A., Harris A. W., Bath M. L., Cory S. (1990). Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature 348, 331–333 [DOI] [PubMed] [Google Scholar]
- Sun L., Zha J., Spear P. A., Wang Z. (2007). Tamoxifen effects on the early life stages and reproduction of Japanese medaka (Oryzias latipes). Environ. Toxicol. Pharmacol. 24, 23–29 [DOI] [PubMed] [Google Scholar]
- Taniguchi Y., Takeda S., Furutani-Seiki M., Kamei Y., Todo T., Sasado T., Deguchi T., Kondoh H., Mudde J., Yamazoe, et al. (2006). Generation of medaka gene knockout models by target-selected mutagenesis. Genome Biol. 7, R116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thermes V., Grabher C., Ristoratore F., Bourrat F., Choulika A., Wittbrodt J., Joly J.-S. (2002). I-SceI meganuclease mediates highly efficient transgenesis in fish. Mech. Dev. 118, 91–98 [DOI] [PubMed] [Google Scholar]
- Thompson J. D., Higgins D. G., Gibson T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trumpp A., Refaeli Y., Oskarsson T., Gasser S., Murphy M., Martin G. R., Bishop M. (2001). c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 414, 768–773 [DOI] [PubMed] [Google Scholar]
- Vervoorts J., Lüscher-Firzlaff J. M., Rottmann S., Lilischkis R., Walsemann G., Dohmann K., Austen M., Lüscher B. (2003). Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep. 4, 484–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vita M., Henriksson M. (2006). The Myc oncoprotein as a therapeutic target for human cancer. Semin. Cancer Biol. 16, 318–330 [DOI] [PubMed] [Google Scholar]
- Wagner A. J., Meyers C., Laimins L. A., Hay N. (1993). c-Myc induces the expression and activity of ornithine decarboxylase. Cell Growth Differ. 4, 879–883 [PubMed] [Google Scholar]
- Wakeling A. E., Slater S. R. (1980). Estrogen-receptor binding and biologic activity of tamoxifen and its metabolites. Cancer Treat. Rep. 64, 741–744 [PubMed] [Google Scholar]
- Wang C., Tai Y., Lisanti M. P., Liao D. J. (2011). c-Myc induction of programmed cell death may contribute to carcinogenesis: A perspective inspired by several concepts of chemical carcinogenesis. Cancer Biol. Ther. 11, 615–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watnick R. S., Cheng Y. N., Rangarajan A., Ince T. A., Weinberg R. A. (2003). Ras modulates Myc activity to repress thrombospondin-1 expression and increase tumor angiogenesis. Cancer Cell 3, 219–231 [DOI] [PubMed] [Google Scholar]
- Westermarck J. (2010). Regulation of transcription factor function by targeted protein degradation: an overview focusing on p53, c-Myc, and c-Jun. Methods Mol. Biol. 647, 31–36 [DOI] [PubMed] [Google Scholar]
- Yokota J., Tsunetsugu-Yokota Y., Battifora H., Le Fevre C., Cline M. J. (1986). Alterations of myc, myb, and rasHa proto-oncogenes in cancers are frequent and show clinical correlation. Science 231, 261–265 [DOI] [PubMed] [Google Scholar]
- Zeller K. I., Jegga A. G., Aronow B. J., O’Donnell K. A., Dang C. V. (2003). An integrated database of genes responsive to the Myc oncogenic transcription factor: identification of direct genomic targets. Genome Biol. 4, R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. (2003). Evolution by gene duplication: an update. Trends Ecol. Evol. 18, 292–298 [Google Scholar]
- Zhang K., Faiola F., Martinez E. (2005). Six lysine residues on c-Myc are direct substrates for acetylation by p300. Biochim. Biophys. Res. Commun. 336, 274–280 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}