

Background: Hyperelastosis cutis in horses is caused by a homozygous mutation in cyclophilin B (PPIB).
Results: This mutation changes protein-protein interactions of CypB and delays folding of collagen.
Conclusion: Mutant CypB has PPIase activity, but the synthesized collagen contains less post-translational modifications of lysine residues.
Significance: CypB has other important functions besides its PPIase activity. The formation of these interactions is crucial for a correct biosynthesis of collagen.
Keywords: Biosynthesis, Collagen, Molecular Chaperone, Post-translational Modification, Prolyl Isomerase, Protein Complexes, Protein Folding, Protein-Protein Interactions, Osteogenesis Imperfecta
Abstract
The rate-limiting step of folding of the collagen triple helix is catalyzed by cyclophilin B (CypB). The G6R mutation in cyclophilin B found in the American Quarter Horse leads to autosomal recessive hyperelastosis cutis, also known as hereditary equine regional dermal asthenia. The mutant protein shows small structural changes in the region of the mutation at the side opposite the catalytic domain of CypB. The peptidylprolyl cis-trans isomerase activity of the mutant CypB is normal when analyzed in vitro. However, the biosynthesis of type I collagen in affected horse fibroblasts shows a delay in folding and secretion and a decrease in hydroxylysine and glucosyl-galactosyl hydroxylysine. This leads to changes in the structure of collagen fibrils in tendon, similar to those observed in P3H1 null mice. In contrast to cyclophilin B null mice, where little 3-hydroxylation was found in type I collagen, 3-hydroxylation of type I collagen in affected horses is normal. The mutation disrupts the interaction of cyclophilin B with the P-domain of calreticulin, with lysyl hydroxylase 1, and probably other proteins, such as the formation of the P3H1·CypB·cartilage-associated protein complex, resulting in less effective catalysis of the rate-limiting step in collagen folding in the rough endoplasmic reticulum.
Introduction
Hyperelastosis cutis (HC),3 also known as hereditary equine regional dermal asthenia, is an autosomal recessive skin fragility syndrome observed in Quarter Horses and related breeds (1, 2). Affected horses appear normal at birth, but by 2 years of age, most present a progressive picture of dermal seroma/hematoma formation, ulceration, and poorly resolving wounds with skin sloughing. Development of wounds on the dorsal thorax is frequently associated with initial training under saddle and renders most affected horses unusable for riding (1, 3). Because the condition is heritable, progressive, and essentially untreatable, most affected horses are electively euthanized (1). Clinical similarity between HC and the Ehlers-Danlos syndrome has been recognized (1, 4–6). The Ehlers-Danlos syndromes are a group of genetically heterogeneous connective tissue fragility syndromes that result from mutations in genes encoding various collagen types, enzymes that modify collagens, and other critical components of the dermal extracellular matrix (7–10). Mutations affecting enzymes involved in post-translational modification of collagens have been associated with autosomal recessive forms of Ehlers-Danlos syndrome and with similar recessive syndromes in animals (7, 9, 11). Recently, a homozygosity mapping approach identified a strong association between homozygosity for a nonsynonymous single nucleotide polymorphism (SNP) in the cyclophilin B (PPIB) gene and the HC phenotype, suggesting that this SNP might be causal (12). However, functional studies supporting this observation and describing a disease mechanism have not been published. This report confirms the mutation in CypB in a larger study and provides possible disease mechanisms.
The biosynthesis of collagens involves a large number of post-translational modifications in which a multitude of proteins in the rough endoplasmic reticulum (rER) are involved (7). After the translocation of the nascent procollagen chains into the rER and cleavage of the signal peptide, proline residues become 4-hydroxylated by prolyl 4-hydroxylase. 4-Hydroxylation of the Yaa position Pro residues in the repetitive Gly-Xaa-Yaa sequence of collagens increases the stability of the triple helix. Prolyl 4-hydroxylase requires unfolded procollagen chains as a substrate. Premature association of chains is prevented by chaperones such as protein-disulfide isomerase, BiP/GRP78, GRP94, and HSP47 and collagen-modifying enzymes until the biosynthesis of the individual chains is completed. Further post-translational modifications are the 3-hydroxylation of a single Pro residue in the Xaa position (Pro-986 in the α1-chain of type I collagen, designated the A1 site in (14)) by the protein complex P3H1·CRTAP·CypB (13, 14), the hydroxylation of lysine residues by lysyl hydroxylases and glycosylation. The A3 site (Pro-707) in the α1-chain of horse tendon type I collagen is partially 3-hydroxylated, a modification not found in the α1-chain of human bone type I collagen (14). Chain selection and trimer formation is determined by the association of carboxyl-terminal propeptides in fibrillar collagens. Triple helix formation proceeds from this nucleus toward the amino-terminal end in a zipper-like fashion (15). The rate-limiting step is the cis-trans isomerization of peptide bonds. This step is catalyzed by peptidylprolyl cis-trans isomerases (PPIase, cyclophilins and FK506-binding proteins) (16–18). Studies with the PPIase inhibitors cyclosporin A and FK506 indicate that cyclophilin B is the prime candidate for catalysis of collagen triple helix formation in the rER (17, 18). Because the triple helix of procollagens is only marginally stable, it was proposed that the folding of procollagens inside cells requires special chaperones (19), with HSP47 and FKBP65 (20) as potential candidates.
The functional importance of collagen chaperones and modifying enzymes is shown by the consequences of mutations in these proteins (7). The P3H1·CRTAP·CypB complex has recently been shown to play an important part in procollagen biosynthesis (21). Mutations in P3H1 in humans lead to a severe osteogenesis imperfecta (OI) phenotype (22). The CRTAP, P3H1, and CypB knock-out mice (23–25) and human mutations in CRTAP (26, 27) and CypB (28) also show severe OI phenotypes. CypB also interacts with the P-domain of calnexin, calreticulin, and calmegin (29, 30), with HSP47 (31) and with protein-disulfide isomerase (32). These proteins are also part of the machinery that is required for procollagen biosynthesis. In this report, we show that a mutation in cyclophilin B is involved in disturbances of the biosynthesis of procollagens.
EXPERIMENTAL PROCEDURES
Candidate Gene Approach
A candidate gene approach was used to identify the causative mutation in HC-affected horses. Candidate genes were selected based on previous association with Ehlers-Danlos phenotypes in man and mouse models or an involvement in collagen biosynthesis or post-translational modification (7–9, 33–39). Mammalian mRNA sequences of candidate genes or relevant partial sequences from whole genome studies/genome trace archives were obtained from the GenBankTM data base. Available sequences were aligned, and regions of conservation across species were identified using the software program Megalign (DNASTAR, Madison, WI). Conserved sequences were used to design primers for amplification of homologous sequences from equine cDNA. Candidates examined included the following: COL5A1; COL5A2; COL1A2; decorin; lumican; mimecan (osteoglycin); fibromodulin; dermatopontin; SPARC (osteonectin); thrombospondin 2; tenascin Xb; lysyl hydroxylase I, II, and III; prolyl 4-hydroxylase α-subunit; lysyl oxidase; lysyl oxidase-like I, II, III, and IV; D4 sulfotransferase; P4HB (protein-disulfide isomerase); PPIB; and SERPINH I (HSP47). ADAMTS2, encoding procollagen I N-proteinase was not studied, as retention of amino-terminal propeptides of type I procollagen was not observed in previous biochemical studies of HC-affected skin (5). Compelling candidate mutations were not identified in any gene other than PPIB; therefore, the details of these experiments are not shown.
Genomic DNA and Tissue Samples
HC cases were diagnosed based on established phenotypic criteria (1, 4, 40) at the Cornell University Equine Hospital, the Mississippi State University Equine Hospital, or by referring veterinarians familiar with the disorder. Full-thickness dermal punch biopsies were collected from HC-affected and normal control horses for establishment of skin fibroblast cultures (Animal Care and Use Protocol 1987-0194, Cornell University). Blood and hair samples from referral HC cases, from related horses, and from unrelated clinically normal control horses were provided by owners, breeders, and referring veterinarians. Genomic DNA was isolated from blood or serum using the QIAamp blood mini kit (Qiagen, Valencia, CA) or from hair roots using the DNeasy blood and tissue kit (Qiagen) with modifications described in the user-provided protocol available from the manufacturer. The initial sample set consisted of DNA samples from 70 HC-affected horses, 60 unaffected relatives, 500 clinically normal adult Quarter Horses from the general population, and 50 horses of diverse breeds (Arabians, Thoroughbreds, Standardbreds, Morgans, Tennessee Walking Horses, Rocky Mountain Gaited Horses, Hanoverians, various European Warmblood Horses, Mustangs, Belgians, Percherons, and ponies). Once a functional effect of the candidate mutation on collagen biosynthesis was documented, generalized DNA testing of registered Quarter Horses and horses with shared Quarter Horse lineage was undertaken, resulting in submission of 5322 samples, 85 of which were from symptomatic HC-affected horses and 5237 of which were from clinically normal horses. Tissue samples for biochemical analyses were collected from HC-affected and normal control horses immediately following euthanasia, snap-frozen in liquid nitrogen, and stored at −70 °C.
Preparation of Equine RNA and cDNA
Skin fibroblast cultures were prepared from dermal punch biopsies collected and minced in Dulbecco's phosphate-buffered saline containing 100 μg/ml penicillin/streptomycin, 100 μg/ml kanamycin, and 750 ng/ml amphotericin B (Invitrogen). Tissue fragments were transferred to DMEM containing 10% fetal bovine serum and antibiotics/antimycotics as described, monitored for outgrowth, and passaged at ∼90% confluency. Skin fibroblasts from mild, intermediate, and severely HC-affected horses and from a clinically normal Thoroughbred horse were used to generate cDNA libraries for initial screening. Total RNA was isolated using the TRIzol Reagent (Invitrogen) according to the manufacturer's recommendations. Reverse transcriptions were performed using the 3′-RACE kit (Invitrogen) according to the protocol provided by the manufacturer.
Sequencing of Horse CypB
A partial coding sequence for CypB was amplified using the consensus primers 1120 and 1121 (supplemental Table 1). The 302-bp product was purified with the QIAquick PCR purification kit (Qiagen) and sequenced directly. 3′-RACE was carried out using the gene-specific primer 1123 (supplemental Table 1) and Abridged Universal Amplification Primer (Invitrogen). 5′-RACE was carried out using the 5′-RACE kit (Invitrogen) and gene-specific primers 1121, 1127, and 1129 (supplemental Table 1), according to the manufacturer's protocol. RACE products were cloned with the Zero Blunt TOPO cloning kit (Invitrogen) for sequencing. A, additional 3′ sequence of the PPIB 5′-untranslated region was predicted from equine whole genome sequence trace archives G836P6336FK4.T0 and G836P62568RE15.T0 when they became available. Introns were amplified using primer pairs in flanking exons of equine PPIB (supplemental Table 1), and the resulting products were sequenced directly or cloned with the Zero Blunt TOPO cloning kit for sequencing.
Genotyping
A 254-bp product representing the relevant region of the first exon and flanking intron of PPIB was amplified from hair root genomic DNA using primers 1130 and 1186 (supplemental Table 1). Alternatively, a 987-bp product (exon 1 to proximal exon 2) was amplified from genomic DNA prepared from blood or serum using primers 1130 and 1155 (supplemental Table 1). Following amplification, 10 μl of each reaction was treated with 2 units of shrimp alkaline phosphatase (Promega, Madison, WI) and 20 units of exonuclease I (United States Biochemical Corp., Cleveland, OH) for 45 min at 37 °C, followed by inactivation of the enzymes at 80 °C for 20 min. Automated sequencing with primer 1178 (supplemental Table 1) was used to determine genotypes.
Automated Sequencing
Automated sequencing was performed by the Cornell Bioresource Center Sequencing Facility using BigDye version 3.1 cycle sequencing chemistry (Applied Biosystems, Foster City, CA) and analyzed on 3730 × 1 DNA analyzers with 50-cm capillary arrays (Applied Biosystems). Sequence traces were analyzed using the Sequencher 4.6 program (Gene Codes Corp., Ann Arbor, MI).
Expression Plasmids
To facilitate construction of plasmids expressing wild-type and mutant equine CypB, each open reading frame (ORF) was amplified from cDNA using primers 1130 and 1124 and cloned using the Zero Blunt TOPO cloning kit. Sequences of resulting plasmids, pNJW2462 (wild type) and pNJW2460 (mutant), were verified. PPIB ORFs were amplified from pNJW2462 and pNJW2460 using, respectively, primer pairs 1136/1137 and 1138/1137 containing EcoRI sites at the 5′ end and BamHI sites after the stop codon at the 3′ end, and cloned into EcoRI/BamHI-digested pAS2-1 to yield pNJW2467 (wild type) and pNJW2479 (mutant) CypB expressors. The sequence of each construct was verified.
Expression and Purification of Wild-type and Mutant Cyclophilin B
DNA encoding wild-type and mutant CypB, without the signal peptide sequence, was isolated from pAS2-1 by PCR using primers containing an NcoI site at the 5′ end and a SalI site after the stop codon at the 3′ end. That DNA was inserted between the NcoI and SalI restriction sites of a pET30b(+) expression vector (Invitrogen). The expression vectors were transformed into Escherichia coli BL21(DE3), grown at 37 °C to an A of 0.6 at 600 nm, and expression was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside. After incubation at 30 °C overnight, the cells were harvested by centrifugation and resuspended in B-PER (Thermo Scientific). Insoluble material was removed by centrifugation, and proteins in the soluble fraction were precipitated with ammonium sulfate at a final concentration of 30% (w/v). After an hour, the sample was centrifuged to remove the precipitated materials. The supernatant was passed through a 0.22-μm filter and applied onto a Co2+-chelating column. After washing with 20 mm sodium phosphate buffer, pH 7.5, containing 0.5 m NaCl and 0.05 m imidazole (10 column volumes), CypB was eluted with 20 mm sodium phosphate buffer, pH 7.5, containing 0.5 m NaCl and 0.5 m imidazole. The fractions containing CypB were dialyzed into enterokinase cleavage buffer (50 mm Tris/HCl buffer, pH 8.0, containing 1 mm CaCl2 and 0.1% Tween 20). Enterokinase (0.1 unit/200-μl reaction volume) (Invitrogen) was used to cleave the His tag at 4 °C overnight, and the sample was incubated with DEAE-Sepharose (100 μl/1.5-ml reaction volume) for 1 h at room temperature to remove enterokinase. The reaction mixture was spun down, and the supernatant was applied onto a Co2+-chelating column to remove the His tag. CypB was eluted with 20 mm sodium phosphate buffer, pH 7.5, containing 0.5 m NaCl. The purified CypB was then dialyzed against different reaction buffers.
Circular Dichroism Measurements
Circular dichroism spectra were recorded on an Aviv 202 spectropolarimeter (Aviv, Lakewood, NJ) using a Peltier thermostated cell holder and a 1-mm path length cell (Starna Cells, Atascadero, CA). Protein concentrations were determined by amino acid analysis. The spectra represent the average of at least 10 scans recorded at a wavelength resolution of 0.1 nm. The proteins were measured in 10 mm sodium phosphate buffer, pH 7.5, at 4 °C. The spectra were analyzed using the CD Spectrum Deconvolution software CDnn (41).
Kinetic Measurements of PPIase Activity
Measurements of the catalytic efficiency (kcat/Km) for the cis-trans isomerization reaction were performed as described (42), based on the coupled α-chymotrypsin assay (43) with the following modifications: stock solutions of substrates were prepared in DMSO at a concentration of 2.5 mm for succinyl-Ala-Ala-Pro-Phe-methyl coumarylamide. The final DMSO concentration in the assay was 0.352% for kcat/Km measurements. Kinetic measurements were made at 5 °C to minimize the nonenzymatic isomerization reaction in 35 mm HEPES/NaOH buffer, pH 7.8. Final substrate and chymotrypsin concentrations were 8.8 and 12.8 μm, respectively. Fluorescence changes were monitored at 380 nm with a stopped-flow spectrophotometer (HiTech). The assay started with mixing chymotrypsin and substrate and progression curves was analyzed by fitting to a second-order exponential-decay function with Origin (OriginLab Corp., Northampton, MA). Values for kcat/Km were calculated according to kcat/Km = (kobs − ku)/[E], where ku is the rate constant for the uncatalyzed isomerization reaction, and kobs is the rate constant for the catalyzed reaction in the presence of an enzyme concentration [E].
Amino Acid Analysis
Acid- and base-hydrolyzed collagen was analyzed as reported previously (25).
Refolding of Collagens Monitored by Optical Rotatory Dispersion
pN type III collagen (type III collagen containing the amino-terminal propeptide) was extracted from fetal bovine skin as described previously (15). The refolding of pN type III collagen was monitored at 365 nm using a 241MC polarimeter (PerkinElmer Life Sciences) with a 10-cm path length thermostatted cell. The temperature was controlled by two circulating water baths (RCS, Lauda Division, Brinkmann Instruments) and measured with a digital thermometer (Omega Engineering, Inc., Stamford, CT) and a thermistor inserted into the cell. Both the temperature and the ORD signals were recorded and digitized on an HP9070A measurement and plotting system (Hewlett-Packard) connected to an IBM-compatible computer. The pN type III collagen (0.125 μm) in 50 mm Tris/HCl buffer, pH 7.5, containing 0.2 m NaCl was denatured for 10 min at 45 °C after stabilizing for 5 min at 25 °C and then refolded at 25 °C.
In Cellulo Folding of Type I Collagen in Wild-type and Mutant Horse Cells
The method followed was described previously (17, 44, 45) but with the following changes. Adult horse skin fibroblasts were grown to confluence and trypsinized. Cells were counted, and numbers were equalized between affected and control prior to beginning. Approximately 5 × 107 cells/ml were resuspended and equilibrated in serum-free labeling medium (DMEM from Invitrogen) at 37 °C, and 1 ml of 1 mCi/ml [2,3,4,5-3H]proline (PerkinElmer Life Sciences) and 100 μl of 10 mCi/ml [35S]methionine (Amersham Biosciences) were added. After 4 min, the cells were centrifuged at 1000 × g, and the supernatant was discarded. The cell pellet was rapidly resuspended in 5 ml of chase medium (serum-free DMEM) and suspension was aliquoted to an appropriate number of tubes. The cells were lysed after various periods of chase time by the addition of 0.1 mg/ml trypsin, 0.25 mg/ml chymotrypsin, 0.1% Nonidet P-40, and 10 mm EDTA. As these additions were made, the samples were vortexed then incubated at 20 °C for 2 min. The digestion was stopped by addition of 1× SDS-PAGE sample buffer with 0.01 m DTT and heating rapidly to boiling by immersion in an oil bath at 140 °C for 2 min. All samples were subjected to SDS-PAGE on 7% acrylamide gels, dried, and exposed to film for autoradiography.
Secretion Rate Assay
Primary adult horse skin fibroblasts at an early passage number (P1) were grown to confluence, trypsinized, and counted. Cells were adjusted to equalize cell number across different cell types and then resuspended in serum-free medium and recounted. Cells were preincubated in labeling medium (methionine-free DMEM without FBS) but with ascorbate at a final concentration of 50 μg/ml at 37 °C with gentle shaking for at least 20 min. Cells were briefly centrifuged; the medium was removed, replaced with 10 ml of labeling medium containing 1 mCi of [2,3,4,5-3H]proline (90 Ci/mmol) and 1 mCi of [35S]methionine (1175 Ci/mmol), and incubated with shaking at 37 °C for 30 min. Cells were then spun out briefly, resuspended in chase medium (standard DMEM with cold methionine and ascorbate), and aliquoted for separate time points (i.e. 0, 15, 30, and 60 min and 2 h) Cells were incubated at 37 °C with shaking, removed at each time point, and spun out at high speed. The medium was moved to a labeled fresh tube, and the cell pellet and medium were frozen for each time point. After thawing, 1 ml of lysis buffer (1% Nonidet P-40 and 10 mm EDTA in PBS) was added to each cell pellet. Cell lysates and medium were precipitated with ammonium sulfate (200 mg per 1 ml of volume added to each tube and incubated 2–3 h with shaking at 4 °C). Precipitates were centrifuged at high speed for 20–30 min at 4 °C, and the pellet was resuspended in 0.5 m acetic acid with 0.4 mg/ml pepsin. Samples were digested overnight at 4 °C with shaking. Pepsinized collagen extracts were precipitated with 0.7 m NaCl (final concentration) at 4 °C and centrifuged at high speed for 30 min. Pellets were resuspended in 0.5 n acetic acid and SDS-PAGE sample buffer, run on 6–8% acrylamide gels, dried, and exposed to film for autoradiography.
Immunofluorescence Localization of CypB in Horse Fibroblasts
Light microscopic immunohistochemical procedures were performed as described previously (13). Briefly, horse dermal fibroblasts were grown on chamber slides in DMEM containing 10% fetal bovine serum and 1% penicillin/streptomycin/fungizone. Cells were used between passages 8 and 10. Fibroblasts were washed with ice-cold phosphate-buffered saline (PBS) and then fixed in 4% paraformaldehyde for 1 h. Cells were washed again in PBS and then permeabilized with PBS and 0.1% Triton X-100 for 30 min at room temperature. Blocking was done for 1 h at room temperature with 2% goat serum. Rabbit anti-CypB polyclonal antibody (Affinity Bioreagents, Thermo Scientific) was diluted 1:100, added to cells, and incubated overnight at 4 °C. Cells were washed in PBS and incubated in goat anti-rabbit antibody labeled with Alexa 488 (Invitrogen) for 30 min. After washing in PBS and covering in Prolong Gold antifade reagent (Invitrogen), the cells were imaged on a Leica confocal microscope.
Electron Microscopy
Freshly obtained tissues from a 20-month-old HC horse and a 24-month-old control horse were fixed in cacodylate-buffered 1.5% glutaraldehyde, 1.5% paraformaldehyde containing 0.05% tannic acid (w/v), then rinsed, exposed to 1% osmium tetroxide, and then dehydrated in a grade series of ethanol to 100%. Fixed tissues were rinsed in propylene oxide and infiltrated and embedded in Spurr's epoxy. 80-nm ultrathin sections were mounted on Formvar-coated single hole slot grids and stained in ethanolic uranyl acetate followed by Reynold's lead citrate. Stained sections were examined using a FEI Tecnai G2 electron microscope operated at 120 kV and photographed using either a FEI Eagle 2K camera or an AMT 2K camera. Magnifications were calibrated using a grating replica (Ted Pella catalogue no. 603).
Collagen Digestion and MS Analysis
Pepsinized collagen was resuspended in 70% formic acid and digested overnight at room temperature with cyanogen bromide (Sigma) to generate fragments for further analysis. The digested solution was lyophilized and then subjected to sieve chromatography on tandem Superose 12 columns using the ÄKTA FPLC system (GE Healthcare) to separate the fragments in 0.1 m sodium acetate buffer, pH 4.5. The appropriate cyanogen bromide peptides containing the putative 3-hydroxyproline sites were selected based on fragment size and time of elution and were subjected to further digestion with sequencing grade trypsin (Promega) in 100 mm ammonium bicarbonate at 37 °C overnight. Additionally, pepsinized type I collagen extracted from tissues was run on one-dimensional SDS-PAGE, and the α1 and α2 gel bands were subjected to in-gel digestion with trypsin using a protocol similar to that described by Shevchenko et al. (46). Digestion conditions were 13 ng/μl Promega trypsin in 10 mm ammonium bicarbonate at 37 °C for 18 h. Prior to analysis, digested proteins were desalted and purified using Oasis C18 SPE columns (Waters, Billerica, MA). LC-MS/MS was performed on a Q-TOF micro mass spectrometer (Waters, Billerica, MA) equipped with an electrospray ionization source coupled to a Waters CapLC HPLC system. Chromatographic separation took place by gradient elution using a 75 μm × 100 mm × 3 μm Atlantis dC18 analytical column (Waters, Billerica, MA). Data were collected with the MassLynx (version 4.1) data acquisition software (Waters, Billerica, MA) and processed using Mascot Distiller (Matrix Software, London, UK). Analysis was performed in survey scan mode. Tryptic peptides were identified from MS/MS spectra by a Mascot data base search against the NCBI nonredundant data base (peptide tolerance 0.5 Da, MS/MS tolerance 0.5 Da).
Pulldown Assay Using Chicken rER Fraction
Purified recombinant WT and mutant horse CypB (0.8 mg) were immobilized on CNBr-activated Sepharose (GE Healthcare). Chicken rER fraction was prepared following a previously published protocol (13, 47) with slight modifications. The pellet of enriched rER vesicles (3 g) was resuspended by 10 ml of TBS buffer containing 1 mm CaCl2, 0.1% (v/v) Tween 20, and EDTA-free protease inhibitor mixture (Roche Applied Science). The extract was centrifuged for 1 h at 40,000 rpm, and 1.5 ml of supernatant was incubated with 200 μl of gelatin Sepharose, non-protein-coupled, horse WT-coupled, and horse mutant-coupled CNBr-activated Sepharose for 2 h at 4 °C. After washing with the same TBS buffer, Sepharose beads were mixed with 100 μl of SDS sample buffer and boiled at 100 °C for 5 min to elute binding proteins. Eluted fractions were run on a NuPAGE BisTris 4–12% gradient gel (Invitrogen) under reducing conditions, and the gel was stained with GelCode Blue Stain Reagent (Thermo Scientific). The intensities of stained protein bands were calculated by ImageJ, and the ratios of unique interacted protein between horse WT and mutant CypB Sepharose were represented using this intensity following the equation: ratio = ((WTI) − (blankI))/((MutI)− (blankI)), where I is intensity of protein band. Proteins were identified by mass spectrometry as described above.
Expression and Purification of Recombinant P-domain of Human Calreticulin
DNA encoding the P-domain of human calreticulin (residues 189–288 of mature protein) was isolated using the “Mammalian Gene Collection” (MGC) full-length, Clone ID 3898550 (Invitrogen) as the template. The PCR product contained a thrombin cleavage site after an EcoRI site at the 5′ end and an XhoI site after the stop codon at the 3′ end. This DNA was inserted between the EcoRI and XhoI restriction sites of the pET30a(+) expression vector (Invitrogen). The expression vectors were transformed into E. coli BL21(DE3) and grown at 37 °C to an absorbance of 0.6 at 600 nm, and expression was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside. After incubation at 20 °C overnight, the cells were harvested by centrifugation and resuspended in Tris base B-PER (Thermo Scientific) containing 1 mm CaCl2. Insoluble material was removed by centrifugation, and proteins in the soluble fraction were precipitated with ammonium sulfate at a final concentration of 30% (w/v). After overnight incubation at 4 °C, the sample was centrifuged, and the supernatant was passed through a 0.22-μm filter and applied onto a Co2+-chelating column. After washing with 5 column volumes of equilibrium buffer (20 mm HEPES buffer, pH 7.5, containing 1.0 m NaCl, 20 mm imidazole, and 1 mm CaCl2), the P-domain was eluted with elution buffer (20 mm HEPES buffer, pH 7.5, containing 1.0 m NaCl, 500 mm imidazole, and 1 mm CaCl2). The fractions containing the P-domain were dialyzed into thrombin cleavage buffer (50 mm Tris/HCl buffer, pH 8.0, containing 1 mm CaCl2 and 0.1% Tween 20). Thrombin (1 unit/ml reaction volume) (BD Biosciences) was used to cleave the His tag at 4 °C overnight, and the sample was dialyzed into equilibrium buffer. This solution was applied onto a Co2+-chelating column to remove the His tag. The P-domain passed through the Co2+-chelating column, and the flow-through fraction was dialyzed against 20 mm triethanolamine/HCl buffer, pH 7.5, containing 1 mm CaCl2 and 20 mm NaCl. The solution was loaded onto a HiTrapQ XL column (GE Healthcare) and washed with 20 ml of the same buffer. The elution was done with a 60-ml gradient of 20 mm triethanolamine/HCl buffer, pH 7.5, containing 1 mm CaCl2 from 20 mm NaCl to 500 mm NaCl. The peak containing pure P-domain was pooled and dialyzed against different reaction buffers for further experiments.
Surface Plasmon Resonance Analysis
Surface plasmon resonance experiments were carried out using a BIAcore X instrument (GE Healthcare). Purified P-domain of human calreticulin was immobilized at a concentration of about 0.3 ng/mm2 (300 resonance units) on a CM5 sensor chip by amide coupling. The experiments were carried out at a flow rate of 10 μl/min and 20 °C in HBS-P buffer (10 mm HEPES buffer, pH 7.4, containing 150 mm NaCl and 0.005% Surfactant P20) containing 1 mm CaCl2 and 0.15% DMSO. A stock solution of cyclosporin A (40 mm) was prepared in DMSO, and the preincubation of cyclosporin A with recombinant WT and HC horse CypB was at 4 °C for 1 h. The final DMSO and cyclosporine concentration in the assay were 0.15% and 30 times higher concentration than that of CypB, respectively.
RESULTS
Genetic Analysis
An anchored PCR approach was used to clone the CypB cDNA from normal and HC-affected horses. Comparison of the resulting 1032-bp assembled sequence with mammalian sequences available in GenBankTM suggested that the 5′-RACE product may have been artifactually truncated due to local high GC content. Two SNPs resulting in amino acid substitutions were identified in cDNAs derived from horses exhibiting mild, moderate, and severe HC phenotypes (cds 17A→G), specifying substitution of glycine for glutamic acid 6 in the endoplasmic reticulum targeting sequence, and (cds 115G→A), specifying substitution of arginine for glycine 39 (glycine 6 in the mature peptide), were homozygous in affected horses (supplemental Fig. 1). Sequencing of PPIB exon 1 in additional normal Quarter Horses showed that (cds 17A→G) SNP occurred in a subset of normal Quarter Horses, and this, coupled with the observation that CypB expression (Western blot, supplemental Fig. 2) and localization in HC fibroblasts were indistinguishable from wild type, suggested that it was unrelated to the development of HC. Amplification and sequencing of the four introns of PPIB failed to reveal additional SNPs. A genotyping assay for the (cds 115G→A) SNP (supplemental Table 1) was implemented for testing larger numbers of horses. This SNP was not identified in horses of unrelated breeds, all of which were homozygous wild type. Samples from 500 clinically normal control Quarter Horses identified 46 horses heterozygous for this SNP and 454 horses homozygous for the wild-type allele, suggesting a carrier frequency of 9.2% in this population.
In the initial sample set, 15 parent/HC-affected offspring trios were available for genotyping. All 30 clinically normal parents of 15 affected offspring were heterozygous for the (cds 115G→A) SNP, consistent with the previously established autosomal recessive pattern of inheritance of the disorder (1, 2). χ2 analysis demonstrated that the observed number of affected offspring homozygous for the (cds 115G→A) SNP (15) differed significantly (p < 0.001) from that expected (3.75) for matings of heterozygous parents. The likelihood of all 15 affected horses being homozygous for this allele based on binomial probability was p = 9.3 × 10−10. The 55 additional affected horses available for study were also shown to be homozygous for the (cds 115G→A) SNP, whereas 19 unaffected relatives were heterozygous for this SNP and 11 were homozygous wild type.
Generalized genotyping of registered American Quarter Horses and horses of shared Quarter Horse lineage was undertaken, resulting in submission of 5322 samples from 85 symptomatic HC-affected horses and 5237 clinically normal horses. All 85 symptomatic horses were homozygous for the (cds 115G→A) SNP. Among clinically normal horses, 906 were heterozygous for this SNP, whereas 4331 were homozygous for the wild-type allele, suggesting a 17.3% carrier frequency in the population tested. This frequency is higher than previous estimates of 1.8–6.5% for the general Quarter Horse population (2, 12, 48), as well as normal control data from this study. This difference in carrier frequency between the population tested and the general population is a consequence of horses from bloodlines in which HC was known to segregate representing the majority of the testing caseload. Horses exhibiting consanguinity to a specific influential stallion (or one of his full siblings) in the American Quarter Horse Registry were previously shown to be at risk for development of HC (1). Genotyping data are summarized in supplemental Table 2.
Biochemical Analysis
The causative mutation in cyclophilin B is located in the sixth residue of the mature protein. The mutation of the glycine to an arginine residue was suggested to act as a nuclear translocation signal (49). In this study it was suggested that CypB acts as a chaperone for the nuclear retranslocation of proteins and that the amino-terminal end is functionally important. However, immunofluorescent localization of CypB in normal and HC horse fibroblasts shows little difference in the distribution. CypB is mainly localized in the rER of these cells (supplemental Fig. 3).
To determine the effects of the Gly-6 to Arg-6 mutation, we expressed normal and HC horse CypB in E. coli with a removable His tag, because the mutation is near the amino-terminal end of CypB, and the amino- and carboxyl-terminal ends are in close proximity as determined by x-ray crystallography (50–52). Fig. 1 shows an SDS-polyacrylamide gel of purified normal and HC CypB before (Fig. 1A) and after the removal of the His tag (Fig. 1B).
FIGURE 1.

SDS-PAGE of normal and HC horse CypB. Normal and HC horse CypB expressed in E. coli were eluted from a chelating column by 0.5 m imidazole. Lanes 1 and 3 in A are normal and HC, respectively. The His tags of purified CypBs were cleaved off by enterokinase. Lanes 2 and 4 in A were normal and HC CypB after removal of the His tag by enterokinase, respectively. B shows the purified CypB used in these studies. Lanes 1 and 2 were normal and HC CypB, respectively. These samples were run on SDS-polyacrylamide gel under reducing conditions and stained with GelCode Blue Stain Reagent. * and ** in A indicate dimers of CypB before and after His tag cleavage, respectively.
The PPIase activity of normal and HC CypB was compared with two assays, and only minor differences were found. The catalytic efficiency (kcat/Km) values for the peptide succinyl-Ala-Ala-Pro-Phe-methyl coumarylamide are 13,900 ± 1350 for normal and 11,200 ± 1800 mm−1 s−1 for HC CypB, respectively (Fig. 2). The PPIase activity was also tested on the refolding of pN-type III collagen. Very little difference was found between the normal and HC CypB, indicating that the mutation does not cause a loss of PPIase activity with collagen as a substrate (Fig. 3).
FIGURE 2.

PPIase activity of normal and HC horse CypB. The PPIase activity of normal and HC horse CypB was measured in 35 mm HEPES/NaOH buffer, pH 7.8, at 3 °C using succinyl-Ala-Ala-Pro-Phe-methyl coumarylamide as substrate. The activity was monitored by fluorescence at 380 nm generated by methyl coumarylamide after cleavage with chymotrypsin. A, in the absence (blue) and presence of 0.0018 μm (green), 0.007 μm (red) and 0.018 μm (black) normal horse CypB. B, in the absence (blue) and presence of 0.008 μm (green), 0.014 μm (red) and 0.019 μm (black) HC horse CypB.
FIGURE 3.

Refolding of pN-type III collagen in the presence of normal and HC horse CypB. pN-type III collagen was denatured for 10 min at 45 °C and then refolded at 25 °C for 75 min in the absence (black) and presence of 0.125 μm normal (red) or 0.125 μm HC horse CypB (blue). Refolding was monitored by optical rotatory dispersion at 365 nm. The increased slope of the linear refolding phase indicates catalysis of cis-trans isomerization. The final concentration of pN-type III collagen was 0.125 μm.
Fig. 4 shows a comparison of the wild-type and mutant horse CypB CD spectra, indicating that the secondary structure is unchanged.4 We have determined the crystal structures of wild-type and HC horse CypB. The wild-type structure is very similar to the structure of human CypB (51). The main difference is in the first β-sheet that is shorter and the loss of flexibility of the first few residues in the mutated structure. These changes are on the opposite side of the catalytic site of CypB and therefore consistent with the retention of PPIase activity as determined.
FIGURE 4.

Circular dichroism spectra of normal and HC horse CypB. Normal and HC horse CypBs were measured at 4 °C in 10 mm phosphate buffer, pH 7.4. The concentration of normal and HC horse CypB was 0.15 mg/ml. The filled and open circles are normal and HC horse CypB, respectively.
Analysis of Collagens
Besides the skin phenotype described in the Introduction, we also observed differences in the collagen fibrils of tendon in the HC horse. The tendon fibrils have varying diameters, with a higher proportion of very small fibrils compared with the wild-type horse tendon (Fig. 5). These small diameter fibrils are seen in longitudinal sections to arise via a general disorganization in the tendon fibrils whereby many large diameter fibrils split into a group of smaller diameter fibrils. A general disorganization in the tendon fibrils is seen in longitudinal sections.
FIGURE 5.

Electron micrographs of wild-type and HC horse tendon. Tendons are shown in cross-section and longitudinal section. A, cross-section of wild-type horse tendon, and B, HC horse tendon. C, longitudinal section of wild-type horse tendon, and D, HC horse tendon. A graph of the fibril number as a function of the fibril diameter is shown in E.
The folding of type I procollagen in the rER of HC horse cells is significantly delayed. The appearance of chymotrypsin/trypsin-resistant α-chains, a measure of fully triple helical type I procollagen, does not occur until 12.5–15 min of the pulse-chase experiment in HC horse cells. In normal horse cells, α-chains can already be detected at 7.5–10 min. As both experiments were done with an identical number of cells and the same amount of radioisotope, it is evident that less procollagen was synthesized in HC fibroblasts (Fig. 6). The migration of the α-chains from HC fibroblasts seems a little faster than that of normal fibroblasts, possibly indicating a lesser degree of post-translational modification in HC fibroblasts. In agreement with the slower folding, a delay in secretion is observed in tendon fibroblasts from HC horses (Fig. 7). The faster migration of the α-chains of type I collagen is also observed in collagen extracted from skin (supplemental Fig. 4).
FIGURE 6.

Rate of folding of type I procollagen in normal and HC horse dermal fibroblasts. Rate of folding of type I procollagen in normal and HC cultured dermal fibroblasts is shown as a function of time in minutes. Type I procollagen is fully folded when it is resistant to trypsin/chymotrypsin digestion and with the appearance of the appropriate size α1 and α2 bands as shown on the autoradiograph.
FIGURE 7.
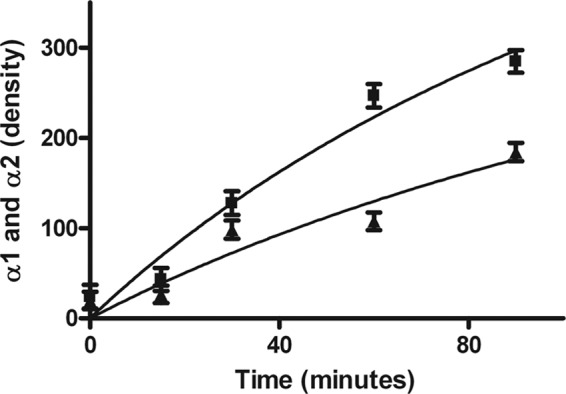
Rate of secretion of type I collagen in normal (square) and HC horse dermal fibroblasts (triangle). Graphs represent the combined density of the α1 and α2 collagen bands using ImageJ software on autoradiographs as a function of time.
These results indicate that the HC CypB is not as efficient in catalyzing the folding of procollagens in the rER of fibroblasts. A delay in folding can lead to overmodification of the collagen α-chains. However, the amino acid composition of type I collagen from skin and tendon of HC horses shows a significant decrease in hydroxylysine and glucosyl-galactosyl-hydroxylysine (Table 1). This is unusual as in other cases of a delay in folding, such as the P3H1 null mouse type I collagen, an increase in hydroxylysine was observed (25). The 3-hydroxylation of type I collagen was analyzed in normal and HC tendon. Fig. 8 shows that the mutation in CypB leads to only minor changes in the 3-hydroxylation of type I collagen. The A1 and A3 sites of type I collagen (14) were analyzed from the tendon and skin of normal and HC horses. This is in contrast to the results observed in fibroblasts of humans that lack CypB (28) and in the CypB null mice (24), where a significant reduction in 3-hydroxylation occurred. These results also show a tissue-specific difference of the extent of 3-hydroxylation of the A3 site, which is present in tendon but almost completely absent in skin.
TABLE 1.
Amino acid analysis with acid or base hydrolysis of type I collagen extracted from wild type and mutant horse tendon and skin
GGH means glucosyl-galactosylhydroxylysine.
| Skin |
Tendon |
|||||
|---|---|---|---|---|---|---|
| % Prolyl 4-hydroxylationa | % Lysyl hydroxylationa | Relative GGH contentb | % Prolyl 4-hydroxylationa | % Lysyl hydroxylationa | Relative GGH contentb | |
| Wild-type | 43.3 ± 1.1 | 12.3 ± 1.1 | 1 | 47.1 ± 0.3 | 31.1 ± 3.2 | 1 |
| HC | 45.6 ± 1.9 | 4.7 ± 0.3 | 0.48 ± 0.10 | 45.5 ± 1.0 | 25.8 ± 4.6 | 0.72 ± 0.20 |
a Data were determined by acid hydrolysis.
b Data were determined by base hydrolysis.
FIGURE 8.
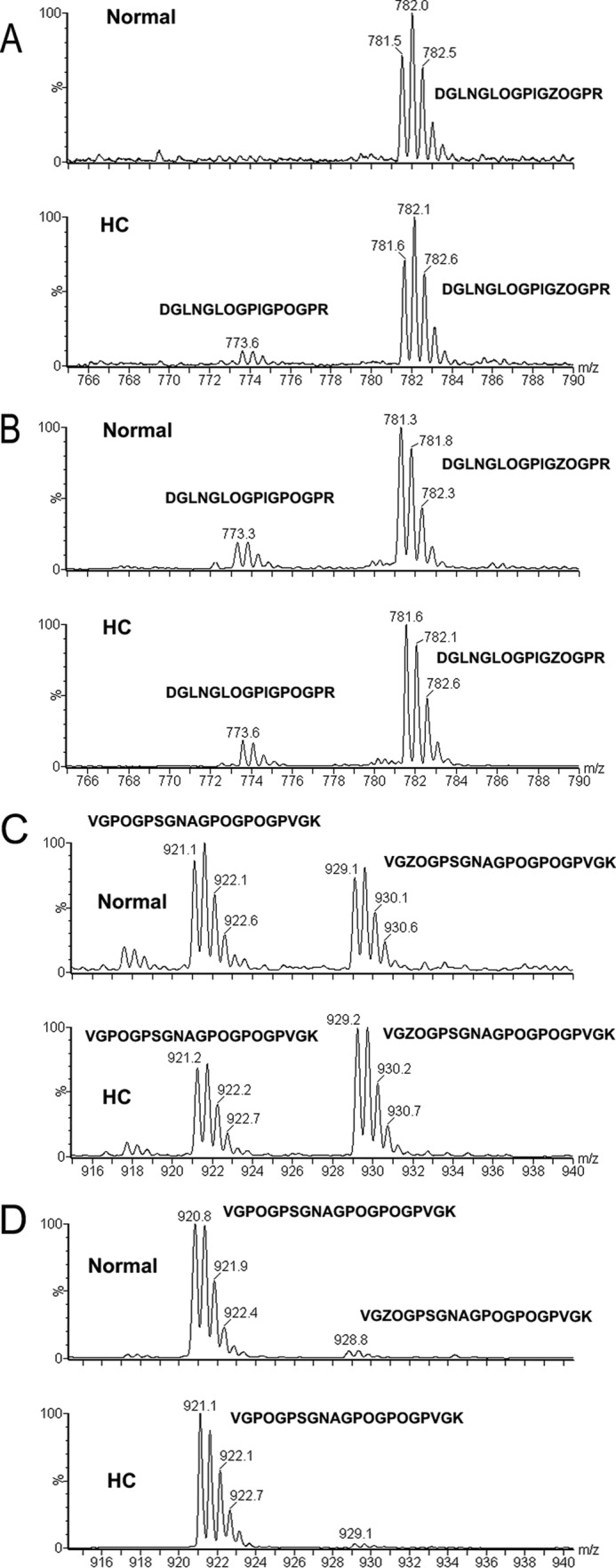
3(S)-hydroxyproline analysis in normal and HC horse tissues by mass spectroscopy. Type I collagen extracted from tendon and skin was analyzed for the 3-hydroxylation of the A1 (Pro-986) and A3 (Pro-707) sites in the α1-chain. A and C are from tendon, and B and D are from skin. A and B represent the A1 site, and C and D represent the A3 site. The corresponding peptide sequences are indicated (Z is 3(S)-hydroxyproline and O is 4(R)-hydroxyproline).
Interaction of CypB with the P-domain
The recently published structure of the interaction of the P-domain of calnexin/calreticulin with CypB (29) involves the region of CypB in which structural changes were observed. Fig. 9 shows that the structure of HC CypB interferes with the binding to the P-domain. The side chain of Lys-5 in HC CypB is mainly disordered. The side chain of the mutated Arg-6 in HC CypB predominantly occupies the position of normal Lys-5 and leads to a significant displacement of the main chain thus preventing the normal interaction with the P-domain. Binding studies were done using surface plasmon resonance detection of the P-domain of human calreticulin bound to the surface of a CM5 sensor chip. Wild-type and HC CypB were used as analytes. Fig. 10A shows that in initial experiments both proteins bind to the P-domain with biphasic kinetics. As the P-domain contains many proline residues, we suspected that binding of the active site of CypB was also observed in these measurements. The wild-type CypB shows a fast association phase that represents the interaction with the active site and a slow phase representative of the P-domain binding with about equal amplitudes. The HC CypB shows a similar rate of association to the active site and a slower rate for a second interaction. The binding isotherms for HC CypB are more complex, and no plateau was reached during the measurement. The amplitude of the fast phase is three times larger than that of the slow phase.
FIGURE 9.

Crystal structure of the region of the mutation in CypB. Stereo schematic representation of three superimposed crystal structures of horse WT CypB (green), horse HC CypB (red), and previously reported human WT CypB (white) in a complex with the proline-rich P-domain of calmegin (yellow) (Protein Data Bank 3IHC) (29). Key lysine residues that contribute to P-domain binding are shown as sticks. The side chain of Lys-5 in HC CypB is mainly disordered. The side chain of the mutated Arg-6 (shown as sticks) in HC CypB predominantly occupies the position of normal Lys-5 and leads to a significant displacement of the main chain thus preventing the normal interaction with the P-domain. The figure was prepared by using the PyMol visualization program (The PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC).
FIGURE 10.

Interactions of P-domain of human calreticulin with recombinant WT and HC horse CypB. A, P-domain of human calreticulin was immobilized on a CM5 chip and recombinant WT (red), and HC (black) horse CypB (2 μm) was injected as an analyte. B, recombinant WT and mutant horse CypB were preincubated with 30 times higher concentrations of cyclosporin A at 4 °C for 1 h and then injected onto the same CM5 chip. The following binding curves are shown: recombinant 2 μm (red), 1.5 μm (blue), 1.0 μm (green), and 0.5 μm (magenta) WT and 2 μm HC (black) horse CypB.
To eliminate the active site interaction, we added an excess of cyclosporin A to our sample to block this binding site. Fig. 10B shows that the binding of wild-type CypB was now dominated by the slow phase, and the ratio of the slower phase to the fast phase was 7 (Table 2). Under these conditions, the amplitude of the fast phase observed for HC CypB was decreased, and a new even slower phase appeared, which probably indicates an aggregation with a strange dissociation behavior. The slow phase of the wild-type CypB interaction with the P-domain of calreticulin results in a Kd value of 20 μm (Table 2), similar to the published value of 10 μm (29). HC CypB clearly shows an altered interaction with the P-domain of calnexin/calreticulin.
TABLE 2.
Binding of wild type and HC CypB to the P-domain of human calreticulin
| Binding to active site (fast) |
Binding to P-domain (slow) |
Ratio of amplitudesa fast: slow | |||||
|---|---|---|---|---|---|---|---|
| ka1 | kd1 | Kd1 | ka2 | kd2 | Kd2 | ||
| (1/ms) × 10−3 | (1/s) × 102 | μm | (1/ms) × 10−3 | (1/s) × 102 | μm | ||
| WT (2 μm) | 3.56 ± 0.7 | 12.3 ± 5.4 | 33 ± 11 | 0.81 ± 0.1 | 1.9 ± 0.4 | 23.9 ± 4.7 | 1:1 |
| WT (2 μm) + CsAb | 0.02 ± 0.001 | 0.0004 ± 0.0001 | 0.17 ± 0.04 | 0.5 ± 0.02 | 3.6 ± 0.04 | 70 ± 3 | 1:7 |
| HC (2 μm) | 23.8 ± 5.5 | 24.7 ± 3.9 | 10 ± 2 | 0.84 ± 0.4 | 0.7 ± 0.07 | 9 ± 3.4 | 3:1 |
| WT + CsAc | 1.3 ± 0.6 | 2.4 ± 0.6 | 20.1 ± 7.4 | ||||
a Amplitudes were determined by a fit of the association kinetics.
b Data were calculated from a single concentration.
c Data were calculated by global fit from the concentration-dependent measurement in Fig. 10B in the presence of cyclosporin A (CsA).
To identify further interaction partners of CypB, both wild-type and HC CypB were bound to Sepharose beads. The beads were incubated with rER extract from chick embryos. Fig. 11 shows an SDS gel of Coomassie-stained proteins that attached to the beads. There is one band visible that only appears with wild-type CypB beads but not with HC CypB or control beads. This band was identified to contain lysyl hydroxylase 1 (LH1) by mass spectroscopy (supplemental Table 3). Although there has been no report of an interaction between CypB and LH1, this finding is interesting because of the lower levels of hydroxylysine found in type I collagen of HC horse tendon (Table 1). In contrast to these findings in HC fibroblasts and tissues, previous studies in other models concluded that slower folding of type I collagen results in an increase in hydroxylysine content (25).
FIGURE 11.
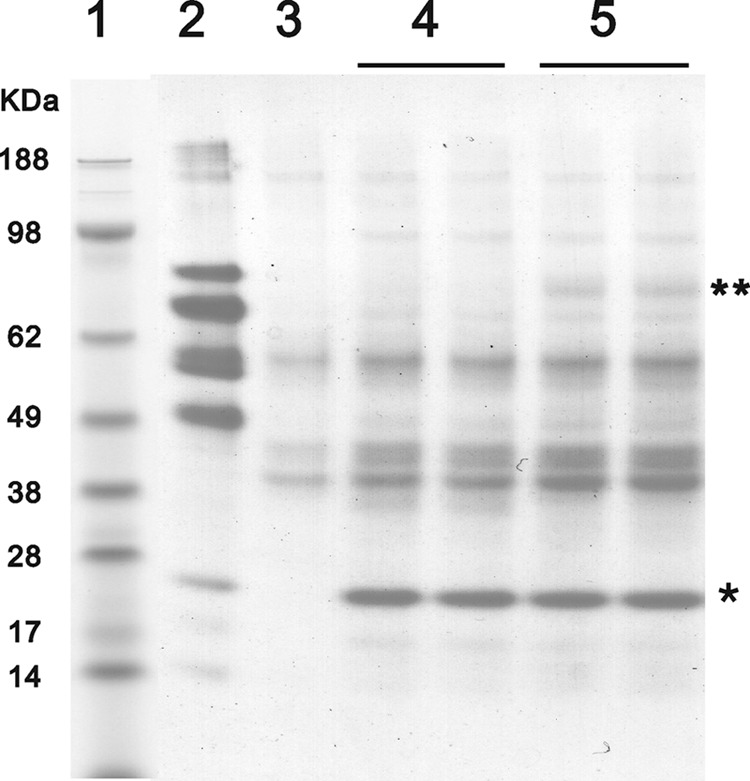
Identification of unique interacting proteins with either WT or HC CypB. Chicken rER proteins were eluted from an equal amount of horse WT and mutant CypB-coupled CNBr-activated Sepharose, nonprotein-coupled CNBr-activated Sepharose, and gelatin Sepharose by SDS sample buffer. Samples were applied to NuPAGE Novex BisTris 4–12% gradient gel (Invitrogen) under reducing conditions, and the gel was stained with GelCode Blue Stain Reagent. Lane 1 contains molecular weight markers, and lane 2–5 are elution samples from gelatin-Sepharose, nonprotein-coupled CNBr-activated Sepharose, horse HC CypB-coupled CNBr-activated Sepharose, and horse WT CypB-coupled CNBr-activated Sepharose, respectively. * and ** indicate the coupled horse CypB and uniquely interacting proteins with WT CypB, respectively.
DISCUSSION
We have confirmed a missense mutation in exon one of the equine PPIB gene and demonstrated its association with the development of hyperelastosis cutis in the Quarter Horse. These results are consistent with a previous report in which homozygosity mapping was used to reach similar conclusions (12). The (cds115G→A) mutation occurred only in the at-risk population of Quarter Horses (defined by pedigree analysis) and was not identified in horses of diverse unrelated breeds. Homozygosity for the mutation was observed in all HC-affected individuals studied (n = 155) and was not observed in clinically normal Quarter Horses (n = 5797). Transmission of the mutation was consistent with the previously established autosomal recessive pattern of inheritance and predictable pattern of inbreeding (1, 12). The mutation specifies nonconservative replacement of glycine in the sixth position of the mature CypB protein with arginine. This glycine residue shows absolute evolutionary conservation across vertebrate species and occurs in a highly conserved sequence context, making the mutation a compelling candidate as the cause of HC. However, Western blot analysis of CypB in skin fibroblasts of HC-affected horses showed expression levels indistinguishable from those of wild-type fibroblasts (supplemental Fig. 2), emphasizing the need for functional studies to confirm a causal relationship and delineate potential pathogenetic mechanisms.
The structural changes and functional consequences of this mutation in CypB are difficult to detect. From the x-ray structure of cyclophilin, it seems that the amino-terminal region is rather flexible, as it is not visible in the crystal structure (50–52). This is confirmed by the known NMR structures of cyclophilin (54, 55). The CD spectra of normal and HC horse CypB are identical, suggesting that the mutation does not affect the overall secondary structure of CypB, rather it is localized to the region of the mutation4 and that the site of the mutation in CypB blocks the interaction with the proline rich P-domain of calnexin/calreticulin and probably other protein-protein interactions. The PPIase activity against a tetra-peptide substrate of the HC CypB is slightly less than that of normal CypB. However, when the PPIase activity was tested with pN-type III collagen as a substrate, no differences in activity were detected.
Given that horses, which are homozygous in this mutation, do have a phenotype, we studied cells from normal and HC horses. The rate of folding of type I procollagen is most likely to be affected by mutations in CypB. Indeed, there is a significantly lower rate of folding in the HC cells. Although previous studies in different model systems showed that a slower rate of folding may lead to overmodifications of the procollagen chains and to a delay in secretion, surprisingly, the hydroxylysine content in HC tendon and skin was lower than in wild-type horses. A decrease in glycosylation was also observed. This can be explained by the finding that wild-type CypB interacts with LH1, whereas HC CypB does not. The mutation in HC horses, which seems to prevent this interaction, negatively impacts LH1 activity.
CypB was shown to have several other functions in cells (56–58). It also forms multiple complexes with other rER proteins, such as protein-disulfide isomerase, the P-domain of calnexin/calreticulin, and HSP47 (29–32, 59). CypB is also part of an enzyme complex that 3-hydroxylates at least one proline residue in the α-chains of type I procollagen. The P3H1·CRTAP·CypB complex does not bind all CypB in chick embryo rER extracts. There is additional CypB, which sediments as free CypB, when analyzed in sucrose gradients (23). The finding that LH1 interacts with CypB is consistent with the observed reduction of hydroxylysine in type I collagen despite the slower folding, as well as the altered ratio of urine deoxypyridinoline to pyridinoline reported in HC horses (60). Full LH1 activity seems to require the interaction with CypB. The conclusion is that CypB seems to play an important part in the collagen folding machinery, with a multitude of interactions of other proteins involved in collagen biosynthesis.
In cells from patients with mutations in P3H1 (22) and in P3H1 null mice (25), a delay in secretion and overmodifications of the α-chains were reported. Similar findings were reported for the CRTAP knock-out mouse (23). However, the slower rate of folding exposes the unfolded collagen chains for a longer period of time to the modifying enzymes. Homozygous mutations in P3H1 and CRTAP lead to a bone phenotype (OI). The loss of CypB in humans also leads to a severe form of OI (28). At present, it is not clear why these disturbances in the rate of folding primarily seem to affect the skin of the horses. Alterations in the cornea of HC horses were recently reported (53).
It is not clear at present if this is caused by CypB not being targeted to the P3H1·CRTAP·CypB complex and therefore affecting its chaperone activity or the failure of CypB to form other complexes. Further studies are required to answer that question.
Acknowledgments
We thank Kerry Maddox for amino acid analysis and the Analytical Core of Shriners Hospital for Children, Portland, OR, for the sequencing of the expression constructs and Sara Tufa for electron microscopy. We also thank Ruggero Tenni (University of Pavia) for a sample of glucosylgalactosyl hydroxylysine.
This work was supported by grants from Shriners Hospitals for Children (to H. P. B. and D. R. K.) and the American Quarter Horse Association (to A. R. R.).

This article contains supplemental Figs. 1–4 and Tables 1–3.
S. P. Boudko, Y. Ishikawa, J. Nix, T. F. Lerch, M. S. Chapman, and H. P. Bächinger, manuscript in preparation.
- HC
- hyperelastosis cutis
- P3H1
- prolyl 3-hydroxylase 1
- CypB
- cyclophilin B
- PPIase
- peptidylprolyl cis-trans isomerase
- rER
- rough endoplasmic reticulum
- LH1
- lysyl hydroxylase 1
- OI
- osteogenesis imperfecta
- RACE
- rapid amplification of cDNA ends
- SNP
- single nucleotide polymorphism
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- CRTAP
- cartilage-associated protein
- cds
- coding DNA sequence.
REFERENCES
- 1. Rashmir-Raven A. M., Winand N. J., Read R. W., Hopper R. M., Ryan P. L., Poole H. M., Erb H. N. (2004) in Proceedings of the 50th Annual Convention of the American Association of Equine Practitioners, Denver, CO, December 4–8, 2004, pp. 47–50, American Association of Equine Practitioners, Lexington, KY [Google Scholar]
- 2. Tryon R. C., White S. D., Famula T. R., Schultheiss P. C., Hamar D. W., Bannasch D. L. (2005) Inheritance of hereditary equine regional dermal asthenia in Quarter Horses. Am. J. Vet. Res. 66, 437–442 [DOI] [PubMed] [Google Scholar]
- 3. Borges A. S., Conceição L. G., Alves A. L., Fabris V. E., Pessoa M. A. (2005) Hereditary equine regional dermal asthenia in three related Quarter Horses in Brazil. Vet. Dermatol. 16, 125–130 [DOI] [PubMed] [Google Scholar]
- 4. Brounts S. H., Rashmir-Raven A. M., Black S. S. (2001) Zonal dermal separation. A distinctive histopathological lesion associated with hyperelastosis cutis in a Quarter Horse. Vet. Dermatol. 12, 219–224 [DOI] [PubMed] [Google Scholar]
- 5. Hardy M. H., Fisher K. R., Vrablic O. E., Yager J. A., Nimmo-Wilkie J. S., Parker W., Keeley F. W. (1988) An inherited connective tissue disease in the horse. Lab. Invest. 59, 253–262 [PubMed] [Google Scholar]
- 6. Lerner D. J., McCracken M. D. (1978) Hyperelastosis cutis information from Vetstream Equis. J. Eq. Med. Surg. 2, 350–352 [Google Scholar]
- 7. Myllyharju J., Kivirikko K. I. (2004) Collagens, modifying enzymes and their mutations in humans, flies, and worms. Trends Genet. 20, 33–43 [DOI] [PubMed] [Google Scholar]
- 8. Faiyaz-Ul-Haque M., Zaidi S. H., Al-Ali M., Al-Mureikhi M. S., Kennedy S., Al-Thani G., Tsui L. C., Teebi A. S. (2004) A novel missense mutation in the galactosyltransferase-I (B4GALT7) gene in a family exhibiting facioskeletal anomalies and Ehlers-Danos syndrome resembling the progeroid type. Am. J. Med. Genet. 128A, 39–45 [DOI] [PubMed] [Google Scholar]
- 9. Mao J. R., Bristow J. (2001) The Ehlers-Danlos syndrome. On beyond collagens. J. Clin. Invest. 107, 1063–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Seidler D. G., Faiyaz-Ul-Haque M., Hansen U., Yip G. W., Zaidi S. H., Teebi A. S., Kiesel L., Götte M. (2006) Defective glycosylation of decorin and biglycan, altered collagen structure, and abnormal phenotype of the skin fibroblasts of an Ehlers-Danlos syndrome patient carrying the novel R270C substitution in galactosyltransferase I (β4GalT-7). J. Mol. Med. 84, 583–594 [DOI] [PubMed] [Google Scholar]
- 11. Colige A., Sieron A. L., Li S. W., Schwarze U., Petty E., Wertelecki W., Wilcox W., Krakow D., Cohn D. H., Reardon W., Byers P. H., Lapière C. M., Prockop D. J., Nusgens B. V. (1999) Human Ehlers-Danlos syndrome type VII C and bovine dermatosparaxis are caused by mutations in the procollagen I N-proteinase gene. Am. J. Hum. Genet. 65, 308–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tryon R. C., White S. D., Bannasch D. L. (2007) Homozygosity mapping approach identifies a missense mutation in equine cyclophilin B (PPIB) associated with HERDA in the American Quarter Horse. Genomics 90, 93–102 [DOI] [PubMed] [Google Scholar]
- 13. Vranka J. A., Sakai L. Y., Bächinger H. P. (2004) Prolyl 3-hydroxylase 1, enzyme characterization and identification of a novel family of enzymes. J. Biol. Chem. 279, 23615–23621 [DOI] [PubMed] [Google Scholar]
- 14. Weis M. A., Hudson D. M., Kim L., Scott M., Wu J. J., Eyre D. R. (2010) Location of 3-hydroxyproline residues in collagen types I, II, III, and V/XI implies a role in fibril supramolecular assembly. J. Biol. Chem. 285, 2580–2590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bächinger H. P., Bruckner P., Timpl R., Prockop D. J., Engel J. (1980) Folding mechanism of the triple helix in type-III collagen and type-III pN-collagen. Role of disulfide bridges and peptide bond isomerization. Eur. J. Biochem. 106, 619–632 [DOI] [PubMed] [Google Scholar]
- 16. Bächinger H. P. (1987) The influence of peptidyl-prolyl cis-trans isomerase on the in vitro folding of type III collagen. J. Biol. Chem. 262, 17144–17148 [PubMed] [Google Scholar]
- 17. Bächinger H. P., Morris N. P., Davis J. M. (1993) Thermal stability and folding of the collagen triple helix and the effects of mutations in osteogenesis imperfecta on the triple helix of type I collagen. Am. J. Med. Genet. 45, 152–162 [DOI] [PubMed] [Google Scholar]
- 18. Steinmann B., Bruckner P., Superti-Furga A. (1991) Cyclosporin A slows collagen triple-helix formation in vivo. Indirect evidence for a physiologic role of peptidyl-prolyl cis-trans isomerase. J. Biol. Chem. 266, 1299–1303 [PubMed] [Google Scholar]
- 19. Makareeva E., Leikin S. (2007) Procollagen triple helix assembly. An unconventional chaperone-assisted folding paradigm. PLoS ONE 2, e1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ishikawa Y., Vranka J., Wirz J., Nagata K., Bächinger H. P. (2008) The rough endoplasmic reticulum-resident FK506-binding protein FKBP65 is a molecular chaperone that interacts with collagens. J. Biol. Chem. 283, 31584–31590 [DOI] [PubMed] [Google Scholar]
- 21. Ishikawa Y., Wirz J., Vranka J. A., Nagata K., Bächinger H. P. (2009) Biochemical characterization of the prolyl 3-hydroxylase 1. Cartilage-associated protein·cyclophilin B complex. J. Biol. Chem. 284, 17641–17647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cabral W. A., Chang W., Barnes A. M., Weis M., Scott M. A., Leikin S., Makareeva E., Kuznetsova N. V., Rosenbaum K. N., Tifft C. J., Bulas D. I., Kozma C., Smith P. A., Eyre D. R., Marini J. C. (2007) Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat. Genet. 39, 359–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Morello R., Bertin T. K., Chen Y., Hicks J., Tonachini L., Monticone M., Castagnola P., Rauch F., Glorieux F. H., Vranka J., Bächinger H. P., Pace J. M., Schwarze U., Byers P. H., Weis M., Fernandes R. J., Eyre D. R., Yao Z., Boyce B. F., Lee B. (2006) CRTAP is required for prolyl 3-hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell 127, 291–304 [DOI] [PubMed] [Google Scholar]
- 24. Choi J. W., Sutor S. L., Lindquist L., Evans G. L., Madden B. J., Bergen H. R., 3rd, Hefferan T. E., Yaszemski M. J., Bram R. J. (2009) Severe osteogenesis imperfecta in cyclophilin B-deficient mice. PLoS Genet. 5, e1000750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vranka J. A., Pokidysheva E., Hayashi L., Zientek K., Mizuno K., Ishikawa Y., Maddox K., Tufa S., Keene D. R., Klein R., Bächinger H. P. (2010) Prolyl 3-hydroxylase 1 null mice display abnormalities in fibrillar collagen-rich tissues such as tendons, skin, and bones. J. Biol. Chem. 285, 17253–17262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Barnes A. M., Chang W., Morello R., Cabral W. A., Weis M., Eyre D. R., Leikin S., Makareeva E., Kuznetsova N., Uveges T. E., Ashok A., Flor A. W., Mulvihill J. J., Wilson P. L., Sundaram U. T., Lee B., Marini J. C. (2006) Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N. Engl. J. Med. 355, 2757–2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Marini J. C., Cabral W. A., Barnes A. M., Chang W. (2007) Components of the collagen prolyl 3-hydroxylation complex are crucial for normal bone development. Cell Cycle 6, 1675–1681 [DOI] [PubMed] [Google Scholar]
- 28. van Dijk F. S., Nesbitt I. M., Zwikstra E. H., Nikkels P. G., Piersma S. R., Fratantoni S. A., Jimenez C. R., Huizer M., Morsman A. C., Cobben J. M., van Roij M. H., Elting M. W., Verbeke J. I., Wijnaendts L. C., Shaw N. J., Högler W., McKeown C., Sistermans E. A., Dalton A., Meijers-Heijboer H., Pals G. (2009) PPIB mutations cause severe osteogenesis imperfecta. Am. J. Hum. Genet. 85, 521–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kozlov G., Bastos-Aristizabal S., Määttänen P., Rosenauer A., Zheng F., Killikelly A., Trempe J. F., Thomas D. Y., Gehring K. (2010) Structural basis of cyclophilin B binding by the calnexin/calreticulin P-domain. J. Biol. Chem. 285, 35551–35557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang J., Herscovitz H. (2003) Nascent lipidated apolipoprotein B is transported to the Golgi as an incompletely folded intermediate as probed by its association with network of endoplasmic reticulum molecular chaperones, GRP94, ERp72, BiP, calreticulin, and cyclophilin B. J. Biol. Chem. 278, 7459–7468 [DOI] [PubMed] [Google Scholar]
- 31. Smith T., Ferreira L. R., Hebert C., Norris K., Sauk J. J. (1995) Hsp47 and cyclophilin B traverse the endoplasmic reticulum with procollagen into pre-Golgi intermediate vesicles. A role for Hsp47 and cyclophilin B in the export of procollagen from the endoplasmic reticulum. J. Biol. Chem. 270, 18323–18328 [DOI] [PubMed] [Google Scholar]
- 32. Horibe T., Yosho C., Okada S., Tsukamoto M., Nagai H., Hagiwara Y., Tujimoto Y., Kikuchi M. (2002) The chaperone activity of protein-disulfide isomerase is affected by cyclophilin B and cyclosporin A in vitro. J. Biochem. 132, 401–407 [DOI] [PubMed] [Google Scholar]
- 33. Ameye L., Young M. F. (2002) Mice deficient in small leucine-rich proteoglycans. Novel in vivo models for osteoporosis, osteoarthritis, Ehlers-Danlos syndrome, muscular dystrophy, and corneal diseases. Glycobiology 12, 107R–116R [DOI] [PubMed] [Google Scholar]
- 34. Bradshaw A. D., Puolakkainen P., Dasgupta J., Davidson J. M., Wight T. N., Helene Sage E. (2003) SPARC-null mice display abnormalities in the dermis characterized by decreased collagen fibril diameter and reduced tensile strength. J. Invest. Dermatol. 120, 949–955 [DOI] [PubMed] [Google Scholar]
- 35. Chakravarti S., Magnuson T., Lass J. H., Jepsen K. J., LaMantia C., Carroll H. (1998) Lumican regulates collagen fibril assembly: skin fragility and corneal opacity in the absence of lumican. J. Cell Biol. 141, 1277–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Danielson K. G., Baribault H., Holmes D. F., Graham H., Kadler K. E., Iozzo R. V. (1997) Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J. Cell Biol. 136, 729–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kyriakides T. R., Zhu Y. H., Smith L. T., Bain S. D., Yang Z., Lin M. T., Danielson K. G., Iozzo R. V., LaMarca M., McKinney C. E., Ginns E. I., Bornstein P. (1998) Mice that lack thrombospondin 2 display connective tissue abnormalities that are associated with disordered collagen fibrillogenesis, an increased vascular density, and a bleeding diathesis. J. Cell Biol. 140, 419–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mao J. R., Taylor G., Dean W. B., Wagner D. R., Afzal V., Lotz J. C., Rubin E. M., Bristow J. (2002) Tenascin-X deficiency mimics Ehlers-Danlos syndrome in mice through alteration of collagen deposition. Nat. Genet. 30, 421–425 [DOI] [PubMed] [Google Scholar]
- 39. Takeda U., Utani A., Wu J., Adachi E., Koseki H., Taniguchi M., Matsumoto T., Ohashi T., Sato M., Shinkai H. (2002) Targeted disruption of dermatopontin causes abnormal collagen fibrillogenesis. J. Invest. Dermatol. 119, 678–683 [DOI] [PubMed] [Google Scholar]
- 40. White S. D., Affolter V. K., Bannasch D. L., Schultheiss P. C., Hamar D. W., Chapman P. L., Naydan D., Spier S. J., Rosychuk R. A., Rees C., Veneklasen G. O., Martin A., Bevier D., Jackson H. A., Bettenay S., Matousek J., Campbell K. L., Ihrke P. J. (2004) Hereditary equine regional dermal asthenia (“hyperelastosis cutis”) in 50 horses. Clinical, histological, immunohistological, and ultrastructural findings. Vet. Dermatol. 15, 207–217 [DOI] [PubMed] [Google Scholar]
- 41. Böhm G., Muhr R., Jaenicke R. (1992) Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng. 5, 191–195 [DOI] [PubMed] [Google Scholar]
- 42. Harrison R. K., Stein R. L. (1990) Mechanistic studies of peptidyl prolyl cis-trans isomerase. Evidence for catalysis by distortion. Biochemistry 29, 1684–1689 [DOI] [PubMed] [Google Scholar]
- 43. Fischer G., Bang H., Mech C. (1984) Determination of enzymatic catalysis for the cis-trans-isomerization of peptide binding in proline-containing peptides. Biomed. Biochim. Acta 43, 1101–1111 [PubMed] [Google Scholar]
- 44. Bruckner P., Eikenberry E. F. (1984) Formation of the triple helix of type I procollagen in cellulo. Temperature-dependent kinetics support a model based on cis in equilibrium trans isomerization of peptide bonds. Eur. J. Biochem. 140, 391–395 [DOI] [PubMed] [Google Scholar]
- 45. Bruckner P., Eikenberry E. F., Prockop D. J. (1981) Formation of the triple helix of type I procollagen in cellulo. A kinetic model based on cis-trans isomerization of peptide bonds. Eur. J. Biochem. 118, 607–613 [DOI] [PubMed] [Google Scholar]
- 46. Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 47. Zeng B., MacDonald J. R., Bann J. G., Beck K., Gambee J. E., Boswell B. A., Bächinger H. P. (1998) Chicken FK506-binding protein, FKBP65, a member of the FKBP family of peptidylprolyl cis-trans isomerases, is only partially inhibited by FK506. Biochem. J. 330, 109–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tryon R. C., Penedo M. C., McCue M. E., Valberg S. J., Mickelson J. R., Famula T. R., Wagner M. L., Jackson M., Hamilton M. J., Nooteboom S., Bannasch D. L. (2009) Evaluation of allele frequencies of inherited disease genes in subgroups of American Quarter Horses. J. Am. Vet. Med. Assoc. 234, 120–125 [DOI] [PubMed] [Google Scholar]
- 49. Rycyzyn M. A., Reilly S. C., O'Malley K., Clevenger C. V. (2000) Role of cyclophilin B in prolactin signal transduction and nuclear retrotranslocation. Mol. Endocrinol. 14, 1175–1186 [DOI] [PubMed] [Google Scholar]
- 50. Mikol V., Kallen J., Pflügl G., Walkinshaw M. D. (1993) X-ray structure of a monomeric cyclophilin A-cyclosporin A crystal complex at 2.1 Å resolution. J. Mol. Biol. 234, 1119–1130 [DOI] [PubMed] [Google Scholar]
- 51. Mikol V., Kallen J., Walkinshaw M. D. (1994) X-ray structure of a cyclophilin B/cyclosporin complex. Comparison with cyclophilin A and delineation of its calcineurin-binding domain. Proc. Natl. Acad. Sci. U.S.A. 91, 5183–5186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhao Y., Ke H. (1996) Crystal structure implies that cyclophilin predominantly catalyzes the trans to cis isomerization. Biochemistry 35, 7356–7361 [DOI] [PubMed] [Google Scholar]
- 53. Mochal C. A., Miller W. W., Cooley A. J., Linford R. L., Ryan P. L., Rashmir-Raven A. M. (2010) Ocular findings in Quarter Horses with hereditary equine regional dermal asthenia. J. Am. Vet. Med. Assoc. 237, 304–310 [DOI] [PubMed] [Google Scholar]
- 54. Ottiger M., Zerbe O., Güntert P., Wüthrich K. (1997) The NMR solution conformation of unligated human cyclophilin A. J. Mol. Biol. 272, 64–81 [DOI] [PubMed] [Google Scholar]
- 55. Spitzfaden C., Braun W., Wider G., Widmer H., Wüthrich K. (1994) Determination of the NMR solution structure of the cyclophilin A-cyclosporin A complex. J. Biomol. NMR 4, 463–482 [DOI] [PubMed] [Google Scholar]
- 56. Bukrinsky M. I. (2002) Cyclophilins. Unexpected messengers in intercellular communications. Trends Immunol. 23, 323–325 [DOI] [PubMed] [Google Scholar]
- 57. Swanson S. K., Born T., Zydowsky L. D., Cho H., Chang H. Y., Walsh C. T., Rusnak F. (1992) Cyclosporin-mediated inhibition of bovine calcineurin by cyclophilins A and B. Proc. Natl. Acad. Sci. U.S.A. 89, 3741–3745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Watashi K., Ishii N., Hijikata M., Inoue D., Murata T., Miyanari Y., Shimotohno K. (2005) Cyclophilin B is a functional regulator of hepatitis C virus RNA polymerase. Mol. Cell 19, 111–122 [DOI] [PubMed] [Google Scholar]
- 59. Meunier L., Usherwood Y. K., Chung K. T., Hendershot L. M. (2002) A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins. Mol. Biol. Cell 13, 4456–4469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Swiderski C., Pasquali M., Schwarz L., Boyle C., Read R., Hopper R., Ryan P., Rashmir-Raven A. (2006) Proceedings of the 24th Annual American College of Veterinary Internal Medicine Forum, Louisville, KY, May 31–June 3, 2006, p. 756, American College of Veterinary Internal Medicine, Lakewood, CO [Google Scholar]