

Abstract
Summary: CalMaTe calibrates preprocessed allele-specific copy number estimates (ASCNs) from DNA microarrays by controlling for single-nucleotide polymorphism-specific allelic crosstalk. The resulting ASCNs are on average more accurate, which increases the power of segmentation methods for detecting changes between copy number states in tumor studies including copy neutral loss of heterozygosity. CalMaTe applies to any ASCNs regardless of preprocessing method and microarray technology, e.g. Affymetrix and Illumina.
Availability: The method is available on CRAN (http://cran.r-project.org/) in the open-source R package calmate, which also includes an add-on to the Aroma Project framework (http://www.aroma-project.org/).
Contact: arubio@ceit.es
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Several analytical pipelines for identifying total copy number (TCN) events from DNA microarrays are available. However, certain types of genomic alterations cannot be detected from TCNs, e.g. copy neutral LOH events. The identification of such events can be key to our biological understanding of cancer development and our ability to set up a personalized treatment plan (Albertson et al., 2003).
Genotyping microarrays (Affymetrix Inc., 2007; Peiffer et al., 2006) quantify not only TCNs but also allele-specific copy numbers (ASCNs), which are necessary to identify CN states such as copy neutral loss of heterozygosity LOH. ASCNs are the CN estimates of each allele variant (here A and B) at a particular (bi-allelic) single-nucleotide polymorphism (SNP). For the purpose of displaying ASCNs along the genome and also for detecting CN changes, ASCNs are often represented by their TCNs and B-allele fractions (BAFs) (Bengtsson et al., 2010). For instance, for diploid SNPs in a normal region, the expected TCN is 2 and expected BAFs are 0 (AA), 1/2 (AB) or 1 (BB). In a region of copy neutral LOH, the expected TCN is 2 and the expected BAFs are 0 or 1. For a single-copy gain, expected TCN is 3 and expected BAFs are 0, 1/3, 2/3 and 1. In Figure 1, observed TCNs and BAFs are displayed along the genome for a normal region, a gain and a region of copy neutral LOH.
Fig. 1.

TCNs (panels a and b) and BAFs (panels c and d) in Chr. 2 from ovarian tumor TCGA-23-1027 before CalMaTe (panels a and c) and after CalMaTe (panels b and d) based on Affymetrix GenomeWideSNP_6 data. Ditto for Illumina Human1M-Duo data is in Figure S4
Based on these type of data, segmentation methods (Chen et al., 2011; Olshen et al., 2011; Staaf et al., 2008; Van Loo et al., 2010) identify regions of constant CN state. Their performances depend greatly on the signal-to-noise ratios (SNRs) of TCN and BAF (Bengtsson et al., 2010), which in turn depend on the preprocessing method used, e.g. dChip (Lin et al., 2004), CN5 (Affymetrix Inc., 2007), CRMA v2 (Bengtsson et al., 2009), ACNE (Ortiz-Estevez et al., 2010) and ‘Illumina’ (Peiffer et al., 2006). Some of these methods perform better than others. It would be favorable to borrow strength between them, but in practice it is impossible because the preprocessing methods are developed specifically for a given SNP platform or chip generation. In addition, methods may also be proprietary, making it infeasible for researchers to improve upon them. A more sustainable solution for improving SNRs is to instead develop normalization methods that operate on the ASCN output of the aforementioned methods. For matched tumor-normal SNP microarray experiments, Bengtsson et al. (2010) provide the platform-independent TumorBoost method, which significantly improves the BAFs of the tumor. Here, we propose CalMaTe (for Calibration Matrix T), which to our knowledge is the only ASCN processing pipeline that is open source, cross-platform and that does not require matched normals.
2 METHOD AND RESULTS
CalMaTe is a platform-independent multi-array method that controls for SNP-specific systematic variation by modeling the crosstalk between alleles in bi-allelic SNPs as explained next. Non-polymorphic loci (on recent SNP array platforms) are normalized by a robust average across samples.
CalMaTe SNP model. The main assumption of CalMaTe is that cross-hybridization between alleles is linear and possibly different between SNPs but preserved across samples. Consider a SNP j=1,…,J, and let Hcj be the 2×I matrix with column vectors (CAij, CBij)T of the unobserved true ASCNs across all samples i=1,…,I. The corresponding observed ASCNs Hj can then be modeled as
| (1) |
where Wj is an unknown 2×2 crosstalk matrix shared by all samples, and εj is a 2×I error matrix. This model and its estimation are outlined below and explained in more detail in the Supplementary Materials.
Estimating the crosstalk. Wj can be estimated from a set of normal samples (‘R’) for which the ASCNs (genotypes) are known, e.g.
where (2, 0)T, (1, 1)T and (0, 2)T correspond to genotypes AA, AB and BB. The set of possible states in Hcj,R is discrete and small, which is why it is possible to estimate ŵj. There is no such constraint on Hcj, which is key when analyzing non-homogeneous samples such as tumors. Given genotypes Hcj,R and observed Hj,R, an estimate ŵj is obtained by robustly solving Equation (1) for Wj. CalMaTe calls the genotypes from Hj,R using a naive genotyping algorithm. To minimize the impact of batch effects (Scharpf et al., 2011), the reference samples are ideally in the same batch as the other samples. If normal samples are unavailable, all samples can be used as a reference. As long as the majority of the reference samples are normal at any given SNP (not necessarily the same samples for all SNPs), the robustness of the estimator warrants a good ŵj estimate. We recommend to use 6 or more reference samples (see also Supplementary Materials).
Calibration of ASCNs. Given an estimate ŵj, the calibrated ASCNs, 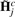 , are obtained from Equation (1) as the back-transformation
, are obtained from Equation (1) as the back-transformation 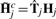 , where
, where 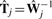 is the 2×2 calibration matrix.
is the 2×2 calibration matrix.
Results. CalMaTe was applied to the TCGA-ovarian cancer dataset ((alias?)). The DNA was hybridized to Affymetrix GenomeWideSNP_6 arrays and ASCNs were estimated using CRMA v2. In Figure 1, such ASCNs are shown as TCNs and BAFs before and after CalMaTe. CalMaTe improves the SNRs, as confirmed by extensive ROC analysis (Fig. 2 and Supplementary Materials), which makes it easier for segmentation methods to distinguish between different CN states. Similar improvements are achieved for ASCNs from dChip as well as ASCNs originating from Illumina, as shown in the Supplementary Materials.
Fig. 2.

ROC analysis results for two change points at ∼124 Mb (left) and ∼141 Mb (right) in Figure 1. DH=|BAF−1/2| for heterozygous SNPs
3 CONCLUSIONS
CalMaTe normalizes ASCNs from any technology and preprocessing method, and without requiring matched normals. The normalized ASCNs are on average more accurate, resulting in greater SNRs along the genome This enhances the power to detect alterations such as LOH using segmentation methods. The method is readily available in an open-source open-access R package, which provides a low-level API for incorporating CalMaTe in third-party solutions, as well as a high-level API for the Aroma Project framework (http://www.aroma-project.org/) making it possible to analyze an unlimited number of arrays.
Funding: HB & PN were supported by NCI grant U24 CA126551.
Conflict of Interest: none declared.
Supplementary Material
Footnotes
† All authors contributed equally.
REFERENCES
- Affymetrix Inc. Genome-Wide Human SNP Nsp/Sty 6.0 User Guide. Santa Clara: Affymetrix Inc. Rev 1; 2007. [Google Scholar]
- Albertson D.G., et al. Chromosome aberrations in solid tumors. Nat. Genet. 2003;34:369–376. doi: 10.1038/ng1215. [DOI] [PubMed] [Google Scholar]
- Bengtsson H., et al. A single-array preprocessing method for estimating full-resolution raw copy numbers from all Affymetrix genotyping arrays including GenomeWideSNP 5 & 6. Bioinformatics. 2009;25:2149–2156. doi: 10.1093/bioinformatics/btp371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson H., et al. TumorBoost: normalization of allele-specific tumor copy numbers from a single pair of tumor-normal genotyping microarrays. BMC Bioinformatics. 2010;11:245. doi: 10.1186/1471-2105-11-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., et al. Estimation of parent specific DNA copy number in tumors using high-density genotyping arrays. PLoS Comput. Biol. 2011;7:e1001060. doi: 10.1371/journal.pcbi.1001060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M., et al. dChipSNP: significance curve and clustering of SNP-array-based loss-of-heterozygosity data. Bioinformatics. 2004;20:1233. doi: 10.1093/bioinformatics/bth069. [DOI] [PubMed] [Google Scholar]
- Olshen A.B., et al. Parent-specific copy number in paired tumor-normal studies using circular binary segmentation. Bioinformatics. 2011;27:2038–2046. doi: 10.1093/bioinformatics/btr329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Estevez M., et al. ACNE: a summarization method to estimate allele-specific copy numbers for Affymetrix SNP arrays. Bioinformatics. 2010;26:1827–1833. doi: 10.1093/bioinformatics/btq300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiffer D., et al. High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 2006;16:1136. doi: 10.1101/gr.5402306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharpf R.B., et al. A multilevel model to address batch effects in copy number estimation using SNP arrays. Biostatistics. 2011;12:33–50. doi: 10.1093/biostatistics/kxq043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staaf J., et al. Segmentation-based detection of allelic imbalance and loss-of-heterozygosity in cancer cells using whole genome SNP arrays. Genome Biol. 2008;9:R136. doi: 10.1186/gb-2008-9-9-r136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Loo P., et al. Allele-specific copy number analysis of tumors. Proc. Natl Acad. Sci. USA. 2010;107:16910. doi: 10.1073/pnas.1009843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.