

Abstract
There have been numerous efforts to incorporate dioxygen into chemical processes because of its economic and environmental benefits. The conversion of dioxygen to water is one such example, having importance in both biology and fuel cell technology. Metals or metal complexes are usually necessary to promote this type of reaction and several systems have been reported. However, mechanistic insights into this conversion are still lacking, especially the detection of intermediates. Reported herein is the first example of a monomeric manganese(II) complex that can catalytically convert dioxygen to water. The complex contains a tripodal ligand with two urea groups and one carboxyamidopyridyl unit—this ligand creates an intramolecular hydrogen-bonding network within the secondary coordination sphere that aids in the observed chemistry. The manganese(II) complex is five-coordinate with an N4O primary coordination sphere; the oxygen donor comes from the deprotonated carboxyamido moiety. Two key intermediates were detected and characterized: a peroxo-manganese(III) species (1). The formulation of 1 was based on spectroscopic and analytical data, including an X-ray diffraction analysis. Reactivity studies showed dioxygen was catalytically converted to water in the presence of reductants, such as diphenylhydrazine and hydrazine. Water was confirmed as a product in greater than 90% yield. A mechanism was proposed that is consistent with the spectroscopy and product distribution, in which the carboxyamido group switches between a coordinated ligand and a basic site to scavenge protons produced during the catalytic cycle. These results highlight the importance of incorporating intramolecular functional groups within the secondary coordination sphere of metal-containing catalysts.
Introduction
The ability to control the reactivity of molecular oxygen has important consequences in a variety of chemical processes.1 There are many practical benefits to using O2: it is inexpensive, plentiful, and environmentally benign, making it an attractive terminal oxidant in chemical syntheses.2 Dioxygen has also been linked to improvements in energy production, specifically in its use in H2/O2 fuel cells. With an overall reduction potential of 1.23 V vs. NHE, the oxidation of H2 by dioxygen produces a relatively large amount of energy.3 However, a limiting factor governing power efficiencies in fuel cells is the O2 reduction process.4 One of the challenges associated with improving this process is replacing the expensive platinum-containing catalysts that are currently being used to reduce dioxygen with cheaper and more abundant metals, such as mid-to-late first-row transition metals.5 Aerobic organisms use this approach by employing heme/copper oxidases, such as cytochrome c oxidase (CcO): these enzymes catalyze the highly selective reduction of O2 to H2O in the final stages of respiration.6 This highly exothermic process is coupled with the production of ATP, and thus is involved in regulating energy transfer in aerobic organisms.
There are several examples of synthetic systems that catalytically reduce O2 to H2O;4,5 these include heme/copper oxidase structural models,7 cofacial diporphyrins, bimetallic porphyrin-corrole dyads,8 and various mono- and dimeric complexes.9,10 However, there are few studies in which intermediates formed during the reductive process have been characterized. The identity of reaction intermediates is often sought because they are helpful in elucidating the mechanisms and understanding the factors required for efficient and complete reduction of O2 to H2O.11 Notable examples include a synthetic CcO model that traps an iron-superoxo intermediate,12 an O2-derived peroxo species derived from a cofacial dicobaltporphyrin,8 and a vanadium(IV)-salen complex bearing a terminal oxo ligand that is formed during the conversion of dioxygen to water.13 In all cases, only a single intermediate was observed along the reaction pathway.
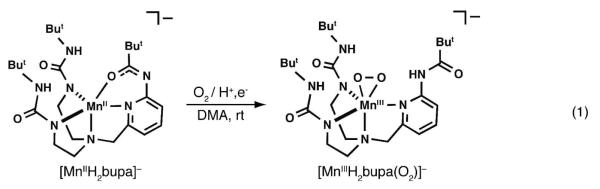
We recently showed that treating the MnII complex, [MnIIH2bupa]−, with dioxygen produced a new complex that was formulated as [MnIIIH3bupa(O2)]−,14,15 establishing that a monomeric Mn-peroxo complex can be prepared directly from dioxygen (eq 1). The peroxo ligand is contained within a hydrogen bonding (H-bonding) cavity that we suggested aided in the formation of this complex. In this report we further describe the chemistry of [MnIIIH3bupa(O2)]− and show that it converts to another manganese(III) species (1) with a terminal oxygen-containing ligand within the cavity. These two complexes served as key intermediates in the reduction of dioxygen, leading to the development of a catalytic cycle for the conversion of dioxygen to water at room temperature using 1,2-diphenylhydrazine (DPH) or hydrazine as the reductant.
 |
Experimental Section
General Methods
All reagents were purchased from commercial sources and used as received, unless otherwise noted. Solvents were purged with argon and dried over columns containing Q-5 and molecular sieves. 1,2-Diphenylhydrazine was recrystallized from petroleum ether, dried under vacuum, and stored under an inert atmosphere. 18O2 (99 atom % 18O) was purchased from ICON Isotopes (Summit, NJ). Elemental analysis was accomplished at Robertson Microlit Laboratories (Madison, NJ). The preparations of H5bupa and K[MnIIH2bupa] followed literature methods.14
Preparative Methods
Preparation of [MnIIIH3bupa(O2)]−
The original preparation for this complex has been reported14 and is repeated here for completeness. Method A: A solution of K[MnIIH2bupa] (78 mg, 130 μmol) in 3 mL anhydrous DMA was treated with dry O2 (3.2 mL, 130 μmol) and stirred for 30 min. Method B: A solution of K[MnIIH2bupa] (78 mg, 130 μmol) and diphenylhydrazine (12 mg, 65 μmol) in 3 mL anhydrous DMA was treated with dry O2 (3.2 mL, 130 μmol) and stirred for 10 min. λmax/nm (DMA, ε, M−1cm−1): 670 (160), 490 nm (sh); EPR (DMF, 10 mM, 11 K): g = 8.2, a = 57 G, D = −2.0(5) cm−1 and E/D = 0.13(3); ν(MnIIIH3bupa(16O–16O)): 885 cm−1, ν(MnIIIH3bupa(18O–18O)): 837 cm−1; MS (ESI-): Exact mass calcd for C25H43N7O3Mn16O2, 576.2706; Found, 576.2703; Exact mass calcd for C25H43N7O3Mn18O2, 580.2792; Found, 580.2794; FTIR (Nujol, ν, cm−1): 3345 (NH), 3172 (NH), 1599 (C=O), 1544 (C=O), 979, 904, 847, 803, 770, 722, 635.
Preparation of 1
A solution of K[MnIIH2bupa] (147 mg, 252 μmol) in 7 mL anhydrous DMA was treated with excess dry O2 and stirred for 30 min. The solution was concentrated under reduced pressure to a solid (4.5 h), washed with Et2O, then washed with THF until the filtrate became colorless.16 The resulting green solid was redissolved in ~3 mL DMA and recrystallized by vapor diffusion with Et2O. The green crystals were filtered, washed with Et2O, and dried under vacuum to 49 mg (32%). Anal. Calcd (found) for K[MnIIIH3bupa(O)].2DMA, C33H61N9O4KMn; C, 51.20 (51.46); H, 7.96 (8.07); N, 16.27 (16.06); MS (ES-): Exact mass calcd for C25H43N7O3Mn16O, 560.2757; Found, 560.2762. Exact mass calcd for C25H43N7O3Mn18O, 562.2785; Found, 562.2800. λmax/nm (DMA, ε, M−1cm−1): 675 (360); μeff = 4.85 μBM.
Preparation of [MnIIH2bupa]− from 1
Complex 1 (28 mg, 47 μmol) was treated with 2.4 mL of a DPH solution in DMA (9.8 × 10−3 M, 24 μmol) and stirred for 5 h. The reaction was analyzed to confirm the formation of [MnIIH2bupa]−: MS (ES-), mass calcd for C25H42N7O3Mn, 543.0. Found 543.0. X-band EPR (DMA, perpendicular-mode, 4 K) g = 5.7, 1.9, 1.3 (Figure S1). These values agree with those of authentic [MnIIH2bupa]− made by an independent route.14 The reaction was then quenched by the addition of ~3 mL H2O. The organic compounds were extracted with Et2O (5 × 4 mL) and concentrated to a solid (4.5 mg). 1H NMR spectroscopy determined the solid was composed of 90% azobenzene and 10% DPH.17
Reactions of [MnIIH2bupa]− with 10 equiv of DPH and O2
Various equivalents of O2 were added to the reaction of [MnIIH2bupa]− and 10 equiv of DPH. The experiments for each amount of O2 were done at least three times (Table S1). In a typical experiment, a solution of K[MnIIH2bupa] (12 mg, 21 μmol) and DPH (38 mg, 210 μmol) in 3 mL anhydrous DMA was treated with a known amount of dry O2 (e.g., 1.5-5.0 mL, T = 297 K, P = 0.99 atm) via a gas tight syringe and stirred for 2 h. The reaction was quenched by the addition of ~5 mL H2O. The organic compounds were extracted with Et2O (5 × 5 mL). The Et2O washings were concentrated to a solid and relative amounts of DPH and azobenzene were determined using 1H NMR spectroscopy.
Quantification of Water Production
For reactions with DPH, the amount of water produced was determined by two methods: 1H NMR spectroscopy and Karl Fischer titration. For the NMR experiments, the relative amounts of products (water and azobenzene) and unreacted DPH were determined from the integration of each species and reported as percentages based on the relative mole ratios and referenced to the residual DMSO found in each sample. In all NMR experiments the yield of azobenzene was at least 95%. Background 1H NMR spectra of the DMSO-d6 used in each experiment were taken just prior to the measurements to determine the residual water present in the solvent—this amount of water was subtracted from the reported values. Karl Fischer titration results are reported as experimentally determined values corrected for background water. Each method was done in triplicate and values in the text are reported as the average values (Tables S2 & S3). In a typical experiment: a solution of K[MnIIH2bupa] (11 mg, 19 μmol) and DPH (69 mg, 0.38 mmol) in 2.00 mL anhydrous DMSO-d6 prepared under argon was treated with an excess of dry O2 and stirred for 90 min. The reaction mixture was allowed to stand without stirring for at least 15 min and the liquid was decanted (to separate it from a brown solid formed during the reaction).18 The sample was divided into equal parts and one sample was analyzed by 1H NMR spectroscopy and the other by Karl Fischer titration. All transfers were done anaerobically. 1H NMR analysis: Found 93% H2O, 97% azobenzene, and 3% DPH. Karl Fischer titration: theoretical yield of water based on yield of azobenzene was 2800 ppm H2O. Found: 3000 ppm (background corrected value: 2600 ppm H2O (93%)). Control Experiments. Karl Fischer titration experiments: a 0.2295 g sample containing O2 in DMSO was analyzed to give 430 ppm of H2O.19 This amount was considered background levels of water and subtracted from the experimental value determined above. Reactions using hydrazine were done in a similar manner with the amount of water determined by 1H NMR spectroscopy, using toluene as the internal standard (Table S4).
Physical Methods
Electronic absorbance spectra were recorded with a Cary 50 spectrophotometer using a 1.00 mm quartz cuvette or an 8453 Agilent UV-Vis spectrometer equipped with an Unisoku Unispeks cryostat. 1H NMR spectra were recorded on a Bruker DRX500 spectrometer. Electron paramagnetic resonance (EPR) spectra were collected using a Bruker EMX spectrometer equipped with an ER041XG microwave bridge, an Oxford Instrument liquid He quartz cryostat, and a dual-mode cavity (ER4116DM) or a Bruker ESP300 spectrometer equipped with an Oxford ESR910 cryostat and a bimodal cavity (Bruker ER4116DM). Mass spectrometry was done on a Waters LCT Premier mass spectrometer operated in negative ion electrospray mode. Karl Fischer titrations were performed on a Mettler Toledo Karl Fischer coulometer model DL39.
Crystallography
The single crystal X-ray diffraction study of 1 was carried out on a Bruker Platform D8 CCD diffractometer equipped with Mo Kα radiation (λ = 0.71073). A 0.45 × 0.25 × 0.10 mm dark green plate was affixed to a nylon cryoloop using oil (Paratone-n, Exxon) and mounted in the cold stream of the diffractometer (Kryo-Flex, Bruker-AXS). Data were collected in a nitrogen gas stream at 150(2) K using φ and ω scans. Crystal-to-detector distance was 60 mm and exposure time was 80 s/frame using a scan width of 0.5°. Data collection, reduction, structure solution, and refinement were performed using the Bruker APEX2 program package.20 All available reflections to 2ϴmax = 50° were harvested and corrected for Lorentz and polarization factors with Bruker SAINT.21 Reflections were then corrected for absorption, interframe scaling, and other systematic errors with SADABS 2008/1.22 The structure was solved by direct methods and refined on F2 by full-matrix least-squares techniques with the Bruker SHELXTL package.23 All non-hydrogen atoms were refined using anisotropic thermal parameters. All hydrogen atoms were included at idealized positions. Crystal, data collection, and refinement parameters for 1 are shown in Table S5.
Results and Discussion
Design Criteria and Dioxygen Binding.14
The ligand [H2bupa]3− provides an N4O donor set when coordinated to a metal ion. The nitrogen donors are from the monodeprotonated urea groups, the pyridine ring, and the apical nitrogen atom. The appended carboxyamido group supplies the oxygen donor that completes the primary coordination sphere. In our synthetic preparations, one urea NH group on each arm has to be deprotonated in order for the compound to correctly bind metal ions. The carboxyamido group was also deprotonated because it is more acidic than the urea moieties.24 Structural and analytical studies on [MnH2bupa]− showed that the carboxyamido group coordinates through the oxygen atom, a binding mode that has been observed in other systems (eq 1).25,26 The resulting Mn(II) complex, [MnIIH2bupa]− has a distorted trigonal bipyramidal coordination geometry, with the urea groups forming the scaffolding of a cavity surrounding the bound oxygen atom.
The addition of O2 to a DMA solution of [MnIIH2bupa]− at room temperature resulted in the formation of the peroxomanganese(III) complex, [MnIIIH3bupa(O2)]− in 1 h (eq 1, Figure 1A).14 The time required to form [MnIIIH3bupa(O2)]− was decreased to 10 min in the presence of 0.5 equiv of DPH, which was converted to azobenzene in 95% yield.27 Optical, vibrational, and EPR spectral measurements support the assignment of a peroxomanganese(III) species.14,28,29 The binding mode of dioxygen in [MnIIIH3bupa(O2)]− is still uncertain, yet mass spectral data showed a large ion peak at a mass-to-charge ratio (m/z) of 576.2703, a shift of 33 mass-units for the peak associated with [MnIIH2bupa]−.30 The mass and calculated isotopic distribution pattern corresponds to the addition of [HO2]− to the manganese(II) starting complex (calc’d, 576.2706). Moreover, [MnIIIH3bupa(O2)]− oxidatively deformylated cyclohexanecarboxaldehyde and cyclopentanecarboxaldehyde to cyclohexanone or cyclopentanone in yields of 94% and 88%, respectively. This type of reactivity is also found for other η2-peroxometal complexes, including peroxomaganese(III) species.31 Take together, these results suggest that the green species observed after O2 addition is a side-on bound peroxo-manganese( III) complex.32 Our studies also showed that production of [MnIIIH3bupa(O2)]− was enhanced by the addition of DPH, which provided the necessary additional electrons and protons through the homolytic cleavage of its N—H bonds (BDENH = 78 kcal/mol).33 We propose that the electron assisted in the reduction of the dioxygen to the peroxo ligand and the proton was used in protonation of carboxamido group of the [H3bupa]2− ligand.
Figure 1.

Electronic absorbance spectra illustrating the conversion of [MnIIH2bupa]− (⋯) to [MnIIIH3bupa(O2)]− (- - -) (A) and [MnIIIH3bupa(O2)]− to 1 (—) (B) measured in DMSO at room temperature. Inset is the absorbance spectrum of purified 1 measured in DMA at room temperature. Spectra in A were recorded every 10 min and those in B were collected every 20 min. The concentration of [MnIIH2bupa]− was 2.1 mM.
Further Reactivity of [MnIIIH3bupa(O2)]−
Monitoring the absorbance of [MnIIIH3bupa(O2)]− at room temperature indicated that the complex converts to another green species within 5 h (Figures 1B and S2).34 This new complex was prepared in bulk quantities by treating [MnIIH2bupa]− with excess dioxygen and isolated after washing with THF (total reaction time was approximately 5 h). The potassium salt of the complex was isolated in pure form by recrystallization from DMA/ether to afford green crystals in 32% yield.16 Complex 1 has a visible absorbance band at λmax = 677 nm (ε = 360 M−1cm−1, inset in Figure 1B) and its X-band EPR spectra are silent in both parallel and perpendicular modes. Although EPR inactive, 1 does exhibit a room temperature effective magnetic moment of 4.85 μBM, determined by Evan’s method,35 indicative of a Mn(III) complex with an S = 2 ground state.36 The negative-mode ESI-MS of 1 prepared from 16O2 exhibited a prominent ion peak at a mass-to-charge ratio (m/z) of 560.2757 (Figure 2A), a decrease of 16 mass-units from the peak associated with [MnIIIH3bupa(O2)]−.37 The mass and calculated isotopic distribution pattern correspond to the loss of an oxygen atom from [MnIIIH3bupa(O2)]− (calc’d, 560.2762; Figure 2B). Furthermore, when 1 was prepared from 18O2, the molecular ion peak shifts by 2 mass-units (Figure 2A) to a m/z of 562.2785 (calc’d, 562.2800; Figure 2B).
Figure 2.

Negative-mode ESI mass spectra of isolated 1 (A) using 16O2 (black) and 18O2 (light blue) and their calculated values (B).
Structural Analysis of 1
Single crystals suitable for X-ray diffraction were obtained by diffusing Et2O into a DMA solution of 1. The complex crystallized with four independent, but nearly identical, anions in the asymmetric unit. Average values for selected bond lengths and angles are found in Table 1 and the molecular structure for one of the anions is shown in Figure 3. In agreement with our other measurements, the molecular structure of 1 revealed that the peroxo ligand had been cleaved to produce a new ligand containing a single oxygen-atom (O1). The complex is five-coordinate with a distorted trigonal bipyramidal molecular structure, in which the urea and pyridyl nitrogen atoms form the trigonal plane. The position of O1 is trans to the apical nitrogen atom (N1) with a Mn1—O1 bond distance of 1.822(4) Å and a N1—Mn1—O1 bond angle of 175.34(17)° (Table 1).
Table 1.
Selected bond distances and angles for 1.a
| Distance (Å) | |
|---|---|
| Mn1—O1 | 1.822(4) |
| Mn1—N1 | 2.043(5) |
| Mn1—N2 | 2.169(4) |
| Mn1—N3 | 2.022(4) |
| Mn1—N4 | 2.010(5) |
| N5—C7 | 1.338(7) |
| C7—O2 | 1.248(7) |
| O1⋯N5 | 2.654(5) |
| O1⋯N6 | 2.775(5) |
| O1⋯N7 | 2.773(5) |
| Angle (deg) | |
| N1—Mn1—O1 | 175.34(17) |
| N1—Mn1—N2 | 79.63(18) |
| N1—Mn1—N3 | 82.13(19) |
| N1—Mn1—N4 | 82.94(18) |
| N2—Mn1—N3 | 117.36(17) |
| N2—Mn1—N4 | 114.09(18) |
| N3—Mn1—N4 | 122.23(19) |
| O1—Mn1—N2 | 95.80(17) |
| O1—Mn1—N3 | 99.56(18) |
| O1—Mn1—N4 | 99.66(18) |
Average values are reported for the four anions in the asymmetric unit cell.
Figure 3.

Thermal ellipsoid diagram of 1. Only one of the four anions found in the asymmetric unit is shown. The ellipsoids are drawn at the 50% probability level and hydrogen atoms are omitted for clarity.
Ligand O1 resides within the H-bonding cavity and appears to form H-bonds with the urea NH groups of the tripodal ligand based on O1⋯N distances of less than 2.8 Å. Note that the carbonyl oxygen of the carboxyamido arm is no longer bound to the manganese center as was found in [MnIIH2bupa]−. Instead, the carboxyamido group is positioned such that the carbonyl group is located outside the cavity; this configuration places the amido group within the interior of the cavity. The O1⋯N5 distance of 2.654(5) Å is indicative of a strong hydrogen-bonding interaction and is significantly shorter than the O1⋯N distances involving the urea groups. In fact, this is the shortest O⋯N we have observed in any of our Mn—O(H) complexes containing intramolecular H-bonding networks.38 Unfortunately, the quality of the structure prevented us from locating the positions of the hydrogen atoms during refinement.
Two limiting tautomers are often used to describe the H-bonding around a Mn—O unit such as that in 1: 1) the carboxyamido nitrogen is protonated and forms a hydrogen bond to an oxo ligand (Figure 4A), and 2) the carboxyamido arm remains deprotonated and forms a hydrogen bond to a hydroxo ligand (Figure 4B). However, comparison of 1 with the related MnIII—oxo and MnIII—OH complexes, [MnIIIH3buea(O)]2− and [MnIIIH3buea(OH)]−,15 suggested that the best description of 1 is a hybrid species between an oxo and a hydroxo complex (Figure 4C). The structure of 1 is similar to these two complexes except that [H3buea]3− is a symmetrical ligand containing three anionic urea arms. The Mn1—O1 bond distance of 1.822(4) Å found in 1 is nearly at the midpoint between the Mn—O bond lengths found for the [MnIIIH3buea(O)]2− (1.780(5) Å) and [MnIIIH3buea(OH)]− (1.872(2) Å).38d Moreover, the N—C(O) bond distances of the carboxyamido units can be used to indicate the protonation state of the ligand.39 When protonated the average N—C bond distance is 1.367(5) Å, and when deprotonated, the bond distance decreases to 1.307(2) Å because of increased double bond character. For 1, this bond length is 1.338(7) Å, again suggesting that 1 is a hybrid between being a MnIII—oxo and a MnIII—hydroxo complex.
Figure 4.

Possible structures to describe the H-bond between the carboxyamido group and the oxomanganese unit in 1.
The molecular structure of 1 illustrates the advantages of having different types of H-bond donors within the secondary coordination sphere of metal complexes. The urea groups in 1 offer permanent H-bond donors that can interact with oxygen-containing ligands within the cavity. This finding is based, in part, on the relatively high pKa values of ureas (> 27 in DMSO), making them difficult to deprotonate.40 Furthermore, the structure of 1 agrees with our previous work that showed urea groups form intramolecular H-bonding networks around terminal oxo ligands, producing a series of MIII/IV—oxo complexes (MIII/IV = Fe, Mn).38 In contrast, carboxyamido groups are significantly more acidic (pKa ~ 22), leading to relatively facile protonation/deprotonation at N5A and a stronger H-bond to O1. The short O1⋯N5A distance suggests a more symmetric H-bond is formed than with the urea groups, which we propose has an effect on the reactivity of 1.
Further Reactivity of 1
Treating 1 with 0.5 equiv of DPH regenerated [MnIIH2bupa]− with concomitant production of H2O (eq 2). The X-band EPR and ESI mass spectra of the resulting solution showed clean conversion to [MnIIH2bupa]− (Figures S1A,B) with no other manganese products detected. The only other product found was azobenzene,
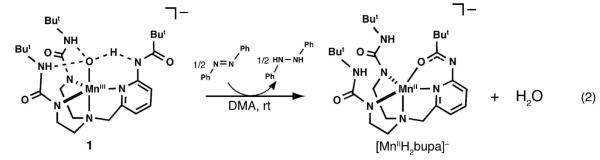 |
which was generated in 90% yield as determined by optical (Figure S1E) and 1H NMR spectroscopies. The reactivity of 1 is different than that observed for MnIII—O(H) complexes prepared with similar H-bonding tripodal ligands. The oxomanganese(III) complex, [MnIIIH3buea(O)]2− reacts with DPH to form a MnII—OH complex ([MnIIH3buea(OH)]2−) and azobenzene,38c,41,42 yet no other products were found even in the presence of excess DPH. Attempts to produce water via protonation of the hydroxo ligand in [MnIIH3buea(OH)]2− led to complex decomposition.43 The hydroxomanganese(III) complex [MnIIIH3buea(OH)]− does not react with DPH at room temperature, again suggesting that water cannot be produced in these types of complexes. The difference in reactivity between these two types of systems is attributed to the carboxyamido group in the ligand [H2bupa]3−, which we propose has a key functional role in water production and the conversion of 1 to [MnIIH2bupa]− (see below).
Mechanistic Implications and Catalysis
The regeneration of [MnIIH2bupa]− from 1 suggested that [MnIIH2bupa]− could be used to catalytically reduce O2 to H2O. A catalytic cycle consistent with our observations is shown in Figure 5 and highlights the role of the carboxyamido group. Treating K[MnIIH2bupa] with O2 presumably generates a superoxo adduct, which we have been unable to detect, even at lower temperatures (less than - 50°C). We propose that this transient species converts to the observable MnIII–peroxo complex via homolytic X—H bond cleavage (X = C or N). Although formally a hydroperoxo species, our data indicate that a peroxo species is formed (vide supra). We suggest that the proton resides on the carboxyamido group, causing its carbonyl to no longer bind to the metal center and instead, rotate in such a way as to form an intramolecular H-bond. This structural change is supported by the molecular structure of 1. Proton-coupled electron transfer (PCET) leads to homolytic cleavage of the O—O bond in [MnIIIH3bupa(O2)]−, producing the first equivalent of H2O. The identity of the transient manganese species that leads to the initial equivalent of water is still unknown. We favor the formation of a MnIII(OH)2 complex that can deprotonate the carboxyamido group to afford an equivalent of H2O and 1, a hybrid MnIII—O(H) complex that has strong intramolecular H-bonds.44 Homolytic cleavage of a N—H bond in DPH by 1 produced the second equivalent of H2O, whose release from the complex is aided by the rebinding of the carboxyamido group to afford the starting MnII complex.
Figure 5.

Proposed catalytic cycle for the four-electron reduction of O2 to H2O, with [MnH2bupa]− as the catalyst. The amount of DPH required for each step is shown within the boxes.
The proposed catalytic cycle predicted that for every equivalent of O2 reduced, four equivalents of hydrogen atoms (i.e., two equiv of DPH) are consumed and two equivalents of H2O and azobenzene are produced. These stoichiometric predictions were evaluated and our results illustrated that [MnIIH2bupa]− serves as a catalyst for the reduction of O2 to H2O at room temperature. [MnIIH2bupa]− was treated with 20.0 equiv of DPH and excess O2, producing azobenzene in 97% relative yield. The relative yields of H2O were determined by two methods: average yields of 94% and 99% were measured by 1H NMR spectroscopy and Karl-Fischer titrations, respectively (Figure S4, Tables S2 and S3). To determine the relative stoichiometry of O2 to azobenzene, 10 equiv of DPH was added to [MnIIH2bupa]− and treated with various amounts of O2 (eq. 3). The results of this study showed that two equivalents of azobenzene were produced for each equivalent of O2 consumed (Table 2). Under a large excess of DPH, we observed a turnover number of 200—it appears that the stability of the catalyst is affected by the presence of increasing amounts of water. We also found that similar results were obtained using hydrazine as the reductant (Table S4). In a typical experiment using excess dioxygen and 20 equiv of hydrazine at room temperature, water was produced in 93% percent yield. Under the same experimental conditions however, no catalysis was observed when H2 was used as the source of reducing equivalents.
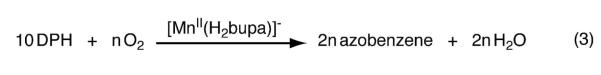 |
Table 2.
Dioxygen-dependence of azobenzene production as depicted by eq 3.
| Equivalents of O2 Consumed |
Equivalents of Azobenzene Produced |
|---|---|
| 1.0 | 2.0 |
| 2.0 | 4.0 |
| 3.0 | 5.9 |
| 4.0 | 8.1 |
| 5.0 | 9.8 |
Summary and Conclusions
This work demonstrates that a monomeric MnII complex catalyzed the reduction of dioxygen to water using DPH or hydrazine as the sacrificial reductant. Two key species produced during reduction were identified: a monomeric peroxomanganese(III) complex and a hybrid MnIII—O/OH species that is strongly H-bonded to the supporting tripodal ligand. These observations led to a proposed catalytic cycle that is supported by results from product distribution studies.
The ability of [MnIIH2bupa]− to reduce dioxygen to water is attributed, in part, to the control of the secondary coordination sphere around the catalytic metal center. It is now accepted that the secondary coordination sphere regulates aspects of function in metalloproteins and some synthetic systems.45 For instance, the selective reduction of O2 to H2O in CcO is achieved through the precise coupling of electron and proton transfers that is aided by acidic/basic amino acids and H-bonds within the secondary coordination sphere.46 Certain types of secondary-sphere interactions, such as intramolecular H-bonding networks, are also known to affect the redox potentials of metal complexes.38b-d,47 In addition, H-bonds can participate in proton relays to efficiently transfer protons to/from metal centers.48
The tripodal ligand [H2bupa]3− created a cavity around the manganese center composed of two urea groups and a carboxyamido unit. Because of their differing acidities, each functional group had a different role in catalysis. The less acidic urea groups offered permanent H-bond donors that can interact with oxygen-containing ligands within the cavity and aid in the overall stability of the complexes. The carboxyamido group served various functions: it is deprotonated in [MnIIH2bupa]− and binds to the manganese(II) ion through its oxygen atom. The carboxyamido unit is proposed to be protonated during the initial reduction steps to produce the peroxo ligand, preventing the accumulation of the more reactive hydroperoxo species. Once protonated, the carboxyamido group adopted a different conformation—one that places its NH group within the interior of the cavity—and thus served as an additional H-bond donor to the peroxo ligand. Conversion to 1 liberated the first equivalent of water; a process proposed to be facilitated by deprotonation of the carboxyamido group. The resultant complex 1 contained a single oxygen-containing ligand bonded to the Mn(III) center, which was assigned as a hybrid MnIII—O(H) unit strongly H-bonded to the carboxyamido group. We were unable to determine the exact position of the proton, a result that most probably was caused by the similarities in the pKa values of the carboxyamido group and the hydroxo/oxo ligand. Finally, additional reduction produced the second equivalent of water, causing the carboxyamido to rebind to the manganese(II) ion.
The catalytic conversion of O2 to H2O by [MnIIH2bupa]− is unusual for our complexes that bind and activate dioxygen.38 These systems reductively cleave dioxygen, producing stable MIII—O(H) complexes that cannot produce water. Many of these complexes contain intramolecular H-bond donors but lack the carboxyamido group that we suggest is essential in controlling proton transfer within a complex and water removal. Three systems have recently appeared that also show similar secondary coordination sphere effects in the reduction of dioxygen to water.9a-c Moreover, work of DuBois and Rakowski DuBois have illustrated that the placement of amines within the secondary coordination sphere provides a proton relay that is essential for H2 activation.48 These findings underscore the importance of positioning basic functionalities within the secondary coordination sphere in metal complexes for the activation of small molecules.
Supplementary Material
Acknowledgement
We thank the NIH (Grant NIH50781) for financial support and Dr. Young Jun Park for helpful advice.
Footnotes
Supporting Information Available: CIF for the X-ray experiment on K[1]·1/2DMF, tables for the yields of products and additional crystallography results, and Figures S1-S3. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Selected references: Borovik AS, Zart MK, Zinn PJ. In: Activation of Small Molecules: Organometallic and Bioinorganic Perspectives. Tolman WB, editor. Wiley-VCH; Weinheim: 2006. pp. 187–234. and references therein; de Montellano P. R. Ortiz., editor. Cytochrome P450: Structure, Mechanism, and Biochemistry. 3rd ed Kluwer Academic/Plenum Publishers; New York: 2005. Que L Jr., Tolman WB, editors. Comprehensive Coordination Chemistry II. Vol. 8. Elsevier; Oxford: 2004. Decker A, Chow MS, Kemsley JN, Lehnert N, Solomon EI. J. Am. Chem. Soc. 2006;128:4719–4733. doi: 10.1021/ja057378n. Groves JT, Han Y-Z. In: Cytochrome P-450. Structure, Mechanism and Biochemistry. de Montellano R. R. Ortiz., editor. Plenum Press; New York: 1995. pp. 3–48. (a)
- 2.(a) Sheldon RA. Green Chemistry and Catalysis. Wiley-VCH; John Wiley; Weinheim: Chichester: 2007. [Google Scholar]; (b) Doble M. Green Chemistry and Processes. Elsevier; Academic Press; Amsterdam: Burlington, MA: 2007. [Google Scholar]; Neumann R, Khenkin AM. Chem. Commun. 2006:2529–2538. doi: 10.1039/b600711m. [DOI] [PubMed] [Google Scholar]
- 3.Adler SB. Chem. Rev. 2004;104:4791–4843. doi: 10.1021/cr020724o. [DOI] [PubMed] [Google Scholar]
- 4.Recent reviews: Wang BJ. Power Source. 2005;152:1–15. Gewirth AA, Thorum MS. Inorg. Chem. 2010;49:3557–3566. doi: 10.1021/ic9022486. Feng Y, Alonso-Vante N. Phys. Stat. Sol. 2008;245:1792–1806.
- 5.(a) Steele BCH, Heinzel A. Nature. 2001;414:345–352. doi: 10.1038/35104620. [DOI] [PubMed] [Google Scholar]; (b) Zhang J, Sasaki K, Sutter E, Adzic RR. Science. 2007;315:220–222. doi: 10.1126/science.1134569. [DOI] [PubMed] [Google Scholar]; (c) Kongkanand A, Kuwabata S, Girishkumar G, Kamat P. Langmuir. 2006;22:2392–2396. doi: 10.1021/la052753a. [DOI] [PubMed] [Google Scholar]; (d) Che G, Lakshmi BB, Fisher ER, Martin CR. Nature. 1998;393:346–349. [Google Scholar]; (e) Anson FC, Shi C, Steiger B. Acc. Chem. Res. 1997;30:579–590. [Google Scholar]
- 6.(a) Ferguson-Miller S, Babcock GT. Heme/Copper Terminal Oxidases. Chem. Rev. 1996;96:2889–2908. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]; (b) Wilkström M. Biochimica et Biophysica Acta. 2004;1655:241–247. doi: 10.1016/j.bbabio.2003.07.013. [DOI] [PubMed] [Google Scholar]; (c) Brudvig GW, Wikström M. In: Photosystem II: The Light-Driven Water:Plastoquinone Oxidoreductase. Wrdrzynski T, Satoh K, editors. Springer; The Netherlands: 2005. pp. 697–713. [Google Scholar]
- 7.(a) Collman JP, Devaraj NK, Decreau RA, Yang Y, Yan Y-L, Ebina W, Eberspacher TA, Chidsey CED. Science. 2007;315:1565–1568. doi: 10.1126/science.1135844. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Collman JP, Decreau RA. Chem. Commun. 2008:5065–5076. doi: 10.1039/b808070b. [DOI] [PubMed] [Google Scholar]; (c) Collman JP, Decreau RA, Dey A, Yang Y. Proc. Natl. Acad. Sci. USA. 2009;106:4101–4105. doi: 10.1073/pnas.0900893106. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shin H, Lee D-H, Kang C, Karlin KD. 2003;48:4077–4082. [Google Scholar]; (e) Ricard D, Didier A, L’Her M, Boitrel B. ChemBioChem. 2001;2:144–148. doi: 10.1002/1439-7633(20010202)2:2<144::AID-CBIC144>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 8.Kadish KM, Fremond L, Shen J, Chen P, Ohkubo K, Fukuzumi S, Ojaimi ME, Gros CP, Barbe J-M, Guilard R. Inorg. Chem. 2009;48:2571–2582. doi: 10.1021/ic802092n. and references therein. Kadish KM, Fremond L, Zhongping O, Shao J, Shi C, Anson FC, Burdet F, Gros CP, Barbe J-M, Guilard R. J. Am. Chem. Soc. 2005;127:5625–5631. doi: 10.1021/ja0501060. Chang CK, Liu HY, Abdalmuhdi I. J. Am. Chem. Soc. 1984;106:2725–2726.
- 9.(a) Chang CJ, Loh Z-H, Shi C, Anson FC, Nocera DG. J. Am. Chem. Soc. 2004;126:10013–10020. doi: 10.1021/ja049115j. [DOI] [PubMed] [Google Scholar]; (b) Kobayashi N, Nevin WA. Applied Organometallic Chem. 1996;10:579–590. [Google Scholar]; (c) Fulmer GR, Muller RP, Kemp RA, Goldberg KI. J. Am. Chem. Soc. 2009;131:1346–1347. doi: 10.1021/ja807936q. [DOI] [PubMed] [Google Scholar]; (d) Thorum MS, Yadav J, Gerwirth AA. Angew. Chem. Int. Ed. 2009;48:165–167. doi: 10.1002/anie.200803554. [DOI] [PubMed] [Google Scholar]; (e) Chang CJ, Baker EA, Pistorio BJ, Deng YQ, Loh ZH, Miller SE, Carpenter SD, Nocera DG. Inorg. Chem. 2002;41:3102–3109. doi: 10.1021/ic0111029. [DOI] [PubMed] [Google Scholar]; (f) Chang CJ, Deng YQ, Shi CN, Chang CK, Anson FC, Nocera DG. Chem. Commun. 2000:1355–1356. [Google Scholar]; (g) Kuwana T, Fujihira M, Sunakawa K, Osa T. J. Electroanal. Chem. Interfacial Electrochem. 1978;88:299–303. [Google Scholar]; (h) Oyaizu K, Haryono A, Yonemaru H, Tsuchida E. J. Chem. Soc. Faraday Trans. 1998;94:3393–3399. [Google Scholar]
- 10.(a) Yang JY, Bullock RM, Dougherty WG, Kassel WS, Twamley B, DuBois DL, DuBois M. Rakowski. Dalton Trans. 2010;39:3001–3010. doi: 10.1039/b921245k. [DOI] [PubMed] [Google Scholar]; (b) Ishiwata K, Kuwata S, Ikariya T. J. Am. Chem. Soc. 2009;131:5001–5009. doi: 10.1021/ja900650j. [DOI] [PubMed] [Google Scholar]; (c) Heiden ZM, Rauchfuss TB. J. Am. Chem. Soc. 2007;129:14303–14310. doi: 10.1021/ja073774p. [DOI] [PubMed] [Google Scholar]; (d) Liu Z, Anson FC. Inorg. Chem. 2001;40:1329–1333. doi: 10.1021/ic0010407. [DOI] [PubMed] [Google Scholar]; (e) Fukuzumi S, Kotani H, Lucas HR, Doi K, Suenobu T, Petersen RL, Karlin KD. J. Am. Chem. Soc. 2010;132:6874–6875. doi: 10.1021/ja100538x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosenthal J, Nocera DG. Acc. Chem. Res. 2007;40:543–553. doi: 10.1021/ar7000638. and references therein.
- 12.(a) Boulatov R, Collman JP, Shiryaeva IM, Sunderland CJ. J. Am. Chem. Soc. 2002;124:11923–11935. doi: 10.1021/ja026179q. [DOI] [PubMed] [Google Scholar]; (b) Collman JP, Sunderland CJ, Berg KE, Vance MA, Solomon EI. J. Am. Chem. Soc. 2003;125:6648–6649. doi: 10.1021/ja034382v. [DOI] [PubMed] [Google Scholar]; (c) Collman JP, Decrau RA, Yan Y, Yoon J, Solomon EI. J. Am. Chem. Soc. 2007;129:5794–5795. doi: 10.1021/ja0690969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Z, Anson FC. Inorg. Chem. 2001;40:1329–1333. doi: 10.1021/ic0010407. [DOI] [PubMed] [Google Scholar]
- 14.Shook RL, Gunderson WA, Greaves J, Ziller JW, Hendrich MP, Borovik AS. J. Am. Chem. Soc. 2008;130:8888–8889. doi: 10.1021/ja802775e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abbreviations: [H2bupa]3-, bis[(N’-tert-butylurealy)-N-ethyl]-(6-pivalamido-2- pyridylmethyl)-aminato; [H3buea]3-, tris(tert-butylureaylethylene)aminato.
- 16.A brown-colored manganese di-urea compound was also produced during this reaction that was soluble in THF. Its exact formulation is currently under investigation.
- 17.The amounts of the DPH and azobenzene reported are relative to each other. NMR analysis also showed that a small amount of H5bupa was present in the samples, a result of the work-up procedure–we attribute the higher than expected yield of sample to the presence of the ligand.
- 18.The brown solid is an insoluble manganese complex(es) whose identity has yet to be determined.
- 19.We found that DPH interfered with the Karl Fischer analysis and thus the controls for this method used only DMSO-d6 and O2.
- 20.APEX2. Version 2008.3-0 Bruker AXS, Inc.; Madison, WI: 2008. [Google Scholar]
- 21.SAINT Software Users Guide. Version 6.0 Bruker Analytical X-Ray Systems, Inc.; Madison, WI: 1999. [Google Scholar]
- 22.Sheldrick GM. SADABS. Version 2.10 Bruker Analytical X-Ray Systems, Inc.; Madison, WI: 2002. [Google Scholar]
- 23.Sheldrick GM. SHELXTL. Version 6.12 Bruker Analytical X-Ray Systems, Inc.; Madison, WI: 2001. [Google Scholar]
- 24.Bordwell FG. Acc. Chem. Res. 1988;21:456–463. [Google Scholar]
- 25.(a) Mareque-Rivas JC, Salvagni E, Parsons S. Dalton Trans. 2004:4185–4192. doi: 10.1039/b414223c. [DOI] [PubMed] [Google Scholar]; (b) Natale D, Mareque-Rivas JC. Chem. Commun. 2008:425–437. doi: 10.1039/b709650j. [DOI] [PubMed] [Google Scholar]; (c) Mareques-Rivas JC, de Rosales RTM, Parsons S. Dalton Trans. 2003:2156–2163. [Google Scholar]; (d) Mareques-Rivas JC, Salvagni E, de Rosales RTM, Parsons S. Dalton Trans. 2003:3339–3349. [Google Scholar]
- 26.(a) Ingle GK, Makowska-Grzyka MM, Arif AM, Berreau LM. Eur. J. Inorg. Chem. 2007:5262–5269. [Google Scholar]; (b) Rudzka K, Arif AM, Berreau LM. J. Am. Chem. Soc. 2006;128:17018–7023. doi: 10.1021/ja0601336. [DOI] [PubMed] [Google Scholar]; (c) Berreau LM, Makowska-Grzyska MM, Arif AM. Inorg. Chem. 2000;39:4390–4391. doi: 10.1021/ic001190h. [DOI] [PubMed] [Google Scholar]; (d) Szajna E, Makowska-Grzyska MM, Wasden CC, Arif AM, Berreau LM. Inorg. Chem. 2005;44:7595–7605. doi: 10.1021/ic050750f. [DOI] [PubMed] [Google Scholar]; (e) Rudzka K, Arif AM, Berreau LM. Inorg. Chem. 2005;44:7234–7242. doi: 10.1021/ic0508122. [DOI] [PubMed] [Google Scholar]; (f) Ingle GK, Makowska-Grzyska MM, Szajna-Fuller E, Sen I, Price JC, Arif AM, Berreau LM. Inorg. Chem. 2007;46:1471–1480. doi: 10.1021/ic062020t. [DOI] [PubMed] [Google Scholar]; (g) Grubel K, Fuller AL, Chambers BM, Arif AM, Berreau LM. Inorg. Chem. 2010;49:1071–1081. doi: 10.1021/ic901981y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yields for the manganese complex were determined by analysis of the EPR spectra using the program SpinCount: Golombek AP, Hendrich MP. J. Magn. Reson. 2003;165:33–48. doi: 10.1016/j.jmr.2003.07.001.
- 28.Other examples of peroxomanganese complexes: Bossek U, Weyhermüller T, Wieghardt K, Nuber B, Weiss J. J. Am. Chem. Soc. 1990;112:6387–6388. Seo MS, Kim JY, Annaraj J, Kim Y, Lee Y-M, Kim S-J, Kim J, Nam W. Angew. Chem. Int. Ed. 2007;46:377–380. doi: 10.1002/anie.200603414. Kitajima N, Komatsuzaki H, Hikichi S, Osawa M, Moro-oka Y. J. Am. Chem. Soc. 1994;116:11596–11597. Shirazi A, Goff HM. J. Am. Chem. Soc. 1982;104:6318–6322. Groves JT, Watanabe Y, McMurry TJ. J. Am. Chem. Soc. 1983;105:4489–4490. VanAtta RB, Strouse CE, Hanson LK, Valentine JS. J. Am. Chem. Soc. 1987;109:1425–1434.
- 29.(a) Groni S, Blain G, Guillot R, Policar C, Anxolabéhère -Mallart E. Inorg. Chem. 2007;46:1951–1953. doi: 10.1021/ic062063+. [DOI] [PubMed] [Google Scholar]; (b) Groni S, Dorlet P, Blain G, Bourcier RG, Anxolabéhère-Mallart E. Inorg. Chem. 2008;47:3166–3172. doi: 10.1021/ic702238z. [DOI] [PubMed] [Google Scholar]
- 30.The mass spectral data collected on samples prepared with 18O2 showed a molecular ion peak to a m/z of 580.2794 (calc’d for [MnIIIH3bupa(18O2,)]−, 580.2792).
- 31.(a) Park MJ, Lee J, Suh Y, Kim J, Nam W. J. Am. Chem. Soc. 2006;128:2630–2634. doi: 10.1021/ja055709q. [DOI] [PubMed] [Google Scholar]; (b) Seo MS, Kim JY, Annaraj J, Kim Y, Lee Y-M, Kim S-J, Kim J, Nam W. Angew. Chem. Int. Ed. 2007;46:377–380. doi: 10.1002/anie.200603414. [DOI] [PubMed] [Google Scholar]
- 32.This assignment is also consistent with other known mononuclear peroxomanganese complexes that have the peroxo ligand coordinated in an η-fashion.24b,c,f
- 32.In the absence of DPH we suggest the additional electrons and protons are provided by the homolytic C-H bond cleavage of DMA molecules. The BDECH (DMA) is 94 kcal/mol, which is substantially smaller than that estimated for BDENH of ureas (greater than 100 kcal/mol).24
- 34.This process was also followed by ESI mass spectrometry, which showed that both [MnIIIH3bupa(O2)]− and [MnIIIH3bupa(O)]− were present during the reaction (Figure S2B).
- 35.Evans DF. J. Chem. Soc. 1959:2003–2005. [Google Scholar]
- 36.Drago R. Physical Methods in Inorganic Chemistry. 2nd ed Surfside Scientific Publishers; Gainesville, FL: 1992. Chapter 11. [Google Scholar]
- 37.A spectrum with a larger m/z range is shown in Figure S2A.
- 38.(a) Hammes BS, Young VG, Borovik AS. Angew. Chem., Int. Ed. 1999;38:666–669. doi: 10.1002/(SICI)1521-3773(19990301)38:5<666::AID-ANIE666>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]; (b) MacBeth CE, Golombek AP, Young VG, Jr., Yang C, Kuczera K, Hendrich MP, Borovik AS. O2 Science. 2000;289:938–941. doi: 10.1126/science.289.5481.938. [DOI] [PubMed] [Google Scholar]; (c) Gupta R, Borovik AS. J. Am. Chem. Soc. 2003;125:13234–13242. doi: 10.1021/ja030149l. [DOI] [PubMed] [Google Scholar]; (d) MacBeth CE, Gupta R, Mitchell-Koch KR, Young VG, Lushington GH, Thompson WH, Hendrich MP, Borovik AS. J. Am. Chem. Soc. 2004;126:2556–2567. doi: 10.1021/ja0305151. [DOI] [PubMed] [Google Scholar]; (e) Borovik AS. Acc. Chem. Res. 2005;38:54–61. doi: 10.1021/ar030160q. [DOI] [PubMed] [Google Scholar]; (f) Shook RL, Borovik AS. Chem. Commun. 2008:6095–6107. doi: 10.1039/b810957e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.All N—C(O) carboxyamido bond distances found in H5bupa and its corresponding FeII and MnII complexes with [H2bupa]3- are given in Table S6.
- 40.This is based on the pKa of urea (see reference 24).
- 41.For a general reference on this type of reaction see, Limberg C. Angew. Chem. Int. Ed. 2003;42:5932–5954. doi: 10.1002/anie.200300578. and references therein.
- 42.(a) Parsell TH, Yang M-Y, Borovik AS. J. Am. Chem. Soc. 2009;131:2762–2763. doi: 10.1021/ja8100825. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Parsell TH, Behan RK, Hendrich MP, Green MT, Borovik AS. J. Am. Chem. Soc. 2006;128:8728–8729. doi: 10.1021/ja062332v. [DOI] [PubMed] [Google Scholar]
- 43.All attempts to prepare [MnIIH3buea(OH2)]− were unsuccessful.
- 44.Another possibility is a MnIV–oxo complex, which are known to form from dioxygen activation. In fact, we have preliminary evidence that 1 can be chemically oxidized to a high-spin MnIV complex that has spectroscopic properties similar to those of the previously reported [MnIVH3buea(O)]−.42b However, we have not detected this type of complex during the conversion of O2 to H2O.
- 45.Recent references: Shook RL, Borovik AS. Inorg. Chem. 2010;49:3646–3660. doi: 10.1021/ic901550k. Marshall NM, Garner DK, Wilson TD, Gao Y-G, Robinson H, Nilges MJ, Lu Yi. Nature. 2009;462:113–116. doi: 10.1038/nature08551. Yeung N, Lin Y-W, Gao Y-G, Russell BS, Lei L, Miner KD, Robinson H, Lu Y. Nature. 2009;462:1079–1082. doi: 10.1038/nature08620. Miller AF. Acc. Chem. Res. 2008;41:501–510. doi: 10.1021/ar700237u. Yikilmaz E, Porta J, Grove LE, Vahedi-Faridi A, Bronshteyn Y, Brunold TC, Borgstahl GEO, Miller AF. J. Am. Chem. Soc. 2007;129:9927–9940. doi: 10.1021/ja069224t. Jackson TA, Brunold TC. Acc. Chem. Res. 2004;37:461–470. doi: 10.1021/ar030272h.
- 46.(a) Yoshikawa S. In: Biological Inorganic Chemistry. Bertini I, Gray HB, Stiefel EI, Valentine JS, editors. University Science Books; Sausalito: 2007. pp. 413–426. [Google Scholar]; (b) Gennis RB. Biochim. Biophys. Acta. 1998;1365:241–248. doi: 10.1016/s0005-2728(98)00095-4. [DOI] [PubMed] [Google Scholar]; (c) Michel H. Biochemistry. 1999;38:15129–15140. doi: 10.1021/bi9910934. [DOI] [PubMed] [Google Scholar]
- 47.(a) Mareque-Rivas JC, Hinchley SL, Metteua L, Parson S. Dalton Trans. 2006:2316–2322. doi: 10.1039/b516234c. [DOI] [PubMed] [Google Scholar]; (b) Suzuki N, Higuchi T, Urano Y, Kikuchi K, Uekusa H, Ohashi Y, Uchida T, Kitagawa T, Nagano T. J. Am. Chem. Soc. 1999;121:11571–11572. [Google Scholar]; (c) Tani F, Matsu-ura M, Nakayama S, Ichimura M, Nakamura N, Naruta Y. J. Am. Chem. Soc. 2001;123:1133–1142. doi: 10.1021/ja003430y. [DOI] [PubMed] [Google Scholar]; (d) Mareque-Rivas JC, Prabaharan R, Torres R, Prabaharan R, de Rosales R. Torres Martin, Metteau L, Parsons S. Dalton Trans. 2004:2800–2807. doi: 10.1039/B407790C. [DOI] [PubMed] [Google Scholar]; (e) Mukherjee J, Lucas RL, Zart MK, Powell DR, Day VW, Borovik AS. Inorg. Chem. 2008;47:5780–5786. doi: 10.1021/ic800048e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilson AD, Newell RH, McNevin MJ, Muckerman JT, DuBois M. Rakowski, DuBois DL. J. Am. Chem. Soc. 2006;128:358–366. doi: 10.1021/ja056442y. Wilson AD, Shoemaker R, Miedaner A, Muckerman JT, DuBois DL, DuBois M. Rakowski. Proc. Natl. Acad. Sci. U.S.A. 2007;104:6951–6956. doi: 10.1073/pnas.0608928104. DuBois M Rakowski, DuBois DL. Chem. Soc. Rev. 2009;38:62–72. doi: 10.1039/b801197b. and references therein.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.