

Abstract
The behavior of the nitroxide spin labels 1-oxyl-4-bromo-2,2,5,5-tetramethylpyrroline (R5a) and 1-oxyl-2,2,5,5-tetramethylpyrroline (R5) attached at a phosphorothioate-substituted site in a DNA duplex is modulated by the DNA in a site- and stereospecific manner. A better understanding of the mechanisms of R5a/R5 sensing of the DNA microenvironment will enhance our capability to relate information from nitroxide spectra to sequence-dependent properties of DNA. Towards this goal, electron paramagnetic resonance (EPR) spectroscopy and molecular dynamics (MD) simulations were used to investigate R5 and R5a attached as Rp and Sp diastereomers at phosphorothioate pSC7 of d(CTACTGpSC7Y8TTAG). d(CTAAAGCAGTAG) (Y = T or U). X-band continuous-wave EPR spectra revealed that the dT8 to dU8 change alters nanosecond rotational motions of Rp-R5a, but produces no detectable differences for Sp-R5a, Rp-R5, and Sp-R5. MD simulations were able to qualitatively account for these spectral variations and provide a plausible physical basis for the R5/R5a behavior. The simulations also revealed a correlation between DNA backbone BI/BII conformations and R5/R5a rotational diffusion, thus suggesting a direct connection between DNA local backbone dynamics and EPR-detectable R5/R5a motion. These results advance our understanding of how a DNA microenvironment influences nitroxide motion and the observed EPR spectra. This may enable use of R5/R5a for a quantitative description of the sequence-dependent properties of large biologically relevant DNA molecules.
Keywords: DNA, nitroxide, EPR, molecular dynamics, stereospecificity
Introduction
Physical properties of DNA, such as helix geometry, flexibility, water and ion binding, and electrostatics have a critical impact on biological functions of gene expression and regulation. Sequence-dependent deviations from the ideal B-form in DNA duplexes play essential roles in DNA recognition by proteins and small molecules.1-4 For example, DNA sequence-dependent mechanics (i.e., deformability, bending) and local shape variations provide a code to guide nucleosome binding in eukaryotic genomes,5-7 which may modulate the activity of transcription factors and other DNA-binding proteins and profoundly affect gene expression. However, knowledge of the sequence dependence of the structure and flexibility of DNA has yet to reach a level that allows rational design and manipulation of functions.2,3 Thus, experimental and computational characterizations of the conformational and dynamic heterogeneity of free DNA duplexes are of significance.2,4
Site-directed spin labeling (SDSL) is a biophysical tool for investigating local environments in bio-macromolecules.8-11 In SDSL, a chemically stable nitroxide radical is covalently attached at specific sites of a protein, DNA or RNA. The nitroxide behavior, which is revealed by electron paramagnetic resonance (EPR) spectroscopy, provides structural and dynamic information on the parent molecule. SDSL requires a small amount of sample (~50 μM in 5 μl) and can be applied to characterize high molecular weight systems under physiological conditions. In nucleic acid studies, SDSL has been used to deduce the structural state of individual nucleotides, to reveal local motions, and to monitor conformational changes in nucleic acids and protein/nucleic acid complexes.11-17
We have developed a phosphorothioate labeling scheme18,19 in which a nitroxide probe (R5 or R5a, Figure 1A) is attached via a flexible R-C-S-P linkage at a Rp or Sp phosphorothioate (Figure 1B) that is chemically substituted at a specific nucleotide in a DNA or RNA strand. R5 and R5a minimally perturb the native duplex conformation of DNA20-22 and RNA,23,24 and have been used to monitor RNA/RNA interactions,18 to study motions of an RNA element within a large folded ribozyme,24 and to measure nanometer distances and conformation in nucleic acids.20,23,25 We have also shown that X-band continuous-wave (cw) EPR spectra of R5a and R5, which reports on nanosecond rotational motions of the nitroxide pyrroline ring, vary within a DNA duplex in a sequence-dependent and stereospecific fashion.21,22 This indicates that R5a and R5 can act as sensors to reveal sequence-dependent properties of DNA duplexes.
Figure 1.
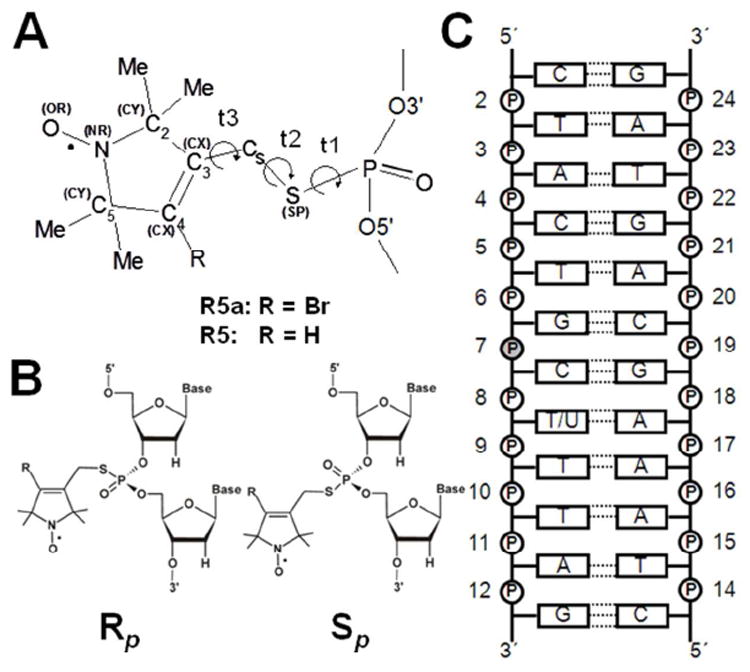
(A) Structure of nitroxide labels (R5: R=H; R5a: R=Br). The torsional angles of the DNA-label linker are defined as t1 (O5′-P-S-Cs), t2 (P-S-Cs-C3) and t3 (S-Cs-C3-C2). Symbols in parentheses indicate non-standard atom types30 used in the AMBER force field. (B) Absolute Rp and Sp configurations of the labeled phosphorothioate. (C) DNA duplex used in the study. The phosphorothioate-substituted and labeled nucleotide (pSC7) is shaded. Nucleotide 8 of the DNA was dT or dU. pSC7 was labeled with R5a or R5 as the Rp or Sp diastereoisomer.
To understand how the local DNA environment governs the rotational behavior of R5a/R5 labels, in this work we used a combination of EPR spectroscopy and molecular dynamics (MD) simulations to examine how the labels respond to a DNA sequence change: namely the removal of a methyl group in the major groove of a DNA duplex. The results suggest that specific interactions between the label and DNA bases, which depend on the conformational preferences of the label and DNA backbone, can modulate rotational motions of the label to an extent detectable by cw-EPR spectroscopy. These results are a key step forward in developing R5 and R5a as spectroscopic probes for examining intrinsic sequence-dependent features in biologically relevant DNA molecules.
Materials and Methods
DNA labeling, purification, and diastereomer separation
The DNA duplexes d(CTACTGpSC7T8TTAG).d(CTAAAGCAGTAG) and d(CTACTGpSC7U8TTAG).d(CTAAAGCAGTAG) (pS = phosphorothioate) were used in the study (Figure 1C). These duplexes are referred to as dT8 and dU8, respectively. All DNA oligonucleotides, including those with a phosphorothioate modification, were synthesized by solid-phase chemical synthesis and obtained from Integrated DNA Technologies (Coralville, IA). R5a or R5 were attached to the phosphorothioate-modified DNA strands following reported protocols.21 The labeled DNA oligonucleotides were purified using anion-exchange HPLC, 21,22,26 which eliminates failed oligonucleotide fragments from DNA synthesis, removes excess spin labels, and separates Rp and Sp phosphorothioate diastereomers. The purities of the labeled Rp and Sp oligonucleotides were determined to be >98% by running anion-exchange HPLC on purified samples.22 Diastereomer configurations were assigned as reported.26 The HPLC fractions containing purified labeled DNA were desalted using a G-25 Sephadex column. Desalted oligonucleotides were lyophilized, suspended in 10-15 μL of water and stored at −20°C. The final concentration of labeled deoxyoligonucleotides was determined by UV absorption at 260 nm. 21,22
EPR spectroscopy
Spin-labeled DNA oligonucleotides were assembled with the complementary strand following a reported protocol.21,22 The final EPR sample (~10-15 μL) contained 100 mM NaCl, 50 mM Tris-HCl (pH=7.5), 34% (w/w) sucrose, and 20-40 μM labeled DNA duplex. EPR samples were placed in glass capillaries (1.0 mm I.D. × 1.2 mm O.D., Vitrocom, Inc. Mountain Lakes, NJ) that were sealed on one end. cw-EPR spectra were acquired on a X-band Bruker EMX spectrometer equipped with a high-sensitivity cavity (ER 4119HS, Bruker Biospin, Inc., Billerica, MA) using a 2 mW incident microwave power and field modulation of 1-2 G at 100 kHz. Sample temperature was maintained at 25°C using a liquid nitrogen variable temperature control setup. All EPR spectra were baseline corrected and normalized to the same number of spins using software kindly provided by the Hubbell group at UCLA. In some cases EPR spectra revealed features from a residual amount of a detached spin probe (< 3%). These features were manually subtracted from the reported spectra.21,22
EPR spectra fitting
Spectral fitting was carried out as previously described22 using a MATLAB-based EPRLL program suite27 that implements the Microscopic Order Macroscopic Disorder (MOMD) model.28 The g and A tensor, the diffusion tilt angle, the number of director orientations, and dimensions of the basis set were fixed at previously reported values.22 Variable fitting parameters (Supplemental Table S1) included: R̄(i) and N, spherical components of the rotational diffusion rate tensor (and N=R∥/R⊥, where R∥ and R⊥ are the respective rate constants for rotations parallel and perpendicular to the nitroxide principal diffusion axis); (ii) c20, the coefficient of an ordering potential ( , where θ is an instantaneous angle relating the director to the nitroxide diffusion frame); and (iii) Δ(0), the Gaussian inhomogeneous broadening factor. From the ordering potential, an order parameter, S20, was computed as . Errors for the fitting parameters were obtained as previously described.22
Structure building for molecular dynamics simulations
Input structures for the molecular dynamics simulations were built from the NMR structure of the dT8 duplex (model 1 of 1CS2.PDB),29 with either R5a or R5 added using the NASNOX algorithm.19,30 In this procedure, a non-bridging oxygen atom of the phosphodiester of nucleotide 7 (OA and OB for the Sp and Rp diastereoisomers, respectively) was substituted by a sulfur atom with a P-S bond of 1.99 Å, but otherwise the experimental geometry of the duplex was retained. Eight structures were built for each combination of label (R5 or R5a), chirality (Rp or Sp) and DNA sequence (dT8 or dU8) with the label conformation set at t1=t2=t3=180° (see Figure 1A for the definition of torsional angles t1, t2 and t3). Two more sets of eight structures each were built with starting conformations of t1=60° (Sp) or -60° (Rp), t2=180°, t3=0°; and t1=60° (Sp) or -60° (Rp), t2=180°, t3=120°. Overall, 24 structures were used as starting points for molecular dynamics simulations of 10 ns each. Based on the results of these simulations, a fourth set with t1=180°, t2=120°, t3=180° was used for additional simulation of selected combinations of the chirality, label and DNA sequence.
Parameters for the 1-oxyl-4-bromo-2,2,5,5-tetramethylpyrroline (R5a) label
A previously defined geometry for the R5 label30 was maintained for R5a, and the Br atom was added to position 4 of the pyrroline ring with a bond length of 1.94 Å (see Figure 1A for atom nomenclature). Further parameterizations were based on those for the “br” atom type in the General AMBER ForceField (GAFF):31 CX-br bond kstr = 269.6 kcal/(mol·Å2), req = 1.897 Å (based on GAFF CA-br); CX-CX-br and CY-CX-br angles kθ = 63.0 kcal/(mol·rad2), θ0 = 123.55° (force constant from GAFF, geometry from Price et al.30). Atomic point charges (mE) for the R5a label were P +601; O5′ -327; O3′ -327; O -344; S -129; CS (HS) +33 (+57); C2 +58; C3 -54; N1 +84; C4 +34; Br -87; C5 +74; O1 -345; C2-Me (H-Me) -88 (+46); C5-Me (H-Me) -83 (+46).
Molecular dynamics simulations
Each labeled duplex was solvated in a periodic box of TIP3P water molecules with the box extending 12 Å from the extremes of the solute in the ±x, ±y and ±z directions. This gave a box of approximate dimensions 60 × 60 × 75 Å that contained 6000 to 6500 water molecules (the exact number depended on the starting structure for the simulation). The labeled duplexes were neutralized by addition of 21 sodium ions and the system was minimized for 1000 steps (500 steps of steepest decent, followed by 500 steps of conjugate gradient minimization). Simulations were run using the AMBER parm99 force field with a time step of 0.002 ps, a residue-based cut-off of 8 Å, and the particle-mesh Ewald method for electrostatic interactions.32 SHAKE was applied to all hydrogen atoms and the target pressure was 1 atm. Equilibration of the solvent molecules was achieved in an initial simulation of 5,000 steps (100 ps) with position restraint of all solute atoms (except for sodium ions) with a force constant of 20 kcal/(molÅ2), during which the temperature of the system was raised linearly from 0 to 298 K over the first 2,500 steps (50 ps). Following the 100-ps solute-restrained period, the restraint on the solute atoms was removed and a further 100 ps of equilibration was performed at 298 K. This was followed by a 10 ns simulation for data collection, during which structures were collected every 0.1 ps.
Three simulations of 10 ns each were also performed for the unlabeled DNA duplex, starting from PDB ID 1CS2 (model 1) and using an identical solvation and equilibration procedure to that for the labeled duplexes. For the unlabeled helix, the structure after 10 ns was resolvated and subjected to re-equilibration to generate a second 10 ns trajectory. The structure after 20 ns was used similarly as a starting point for the third simulation. Results from all simulations were visualized using WebLab Viewer (Molecular Simulations Inc., San Diego, CA) and geometrical data were obtained using AMBER (PTRAJ) and CURVES 5.2.33
Results
EPR spectra reveal that the dT8 to dU8 change affects rotational motion of Rp-R5a only
In this work, R5a and R5 were attached to nucleotide 7 located at the center of the dT8 or dU8 duplex (Figure 1C), and Rp and Sp diastereomers were separated to >98% pure (see Methods). R5a or R5 labeling has been previously shown to minimally perturb the native B-DNA conformation of the dT8 duplex.20-22 The dU8 duplex, which differs from dT8 only by a thymine C5-methyl group residing in the DNA major groove, is expected to maintain a B-form conformation with or without R5/R5a attachment. This is indeed the case observed in MD simulations.
Figure 2 shows EPR spectra of R5a and R5 obtained at 25°C. The dT8 to dU8 change results in a small but reproducible change in Rp-R5a spectra. The Rp-R5a-dU8 spectrum has narrower lines compared to that of Rp-R5a-dT8, indicating an increase in R5a mobility upon removal of the dT8 C5-methyl group. No detectable spectral differences were observed for Sp-R5a, Rp-R5, and Sp-R5 (Figure 2), which suggests that changing dT8 to dU8 has no measurable effect on nanosecond rotations of these probes. We also note that the Rp-R5 spectra are broader than those of Sp-R5 (Figure 2). This is consistent with previously reported Rp-R5a and Sp-R5a behavior,22 which has been attributed to the Rp-diastereomer positioning the nitroxide towards the major groove of the DNA duplex, while the Sp-diastereomer positions the nitroxide towards the solvent.
Figure 2.
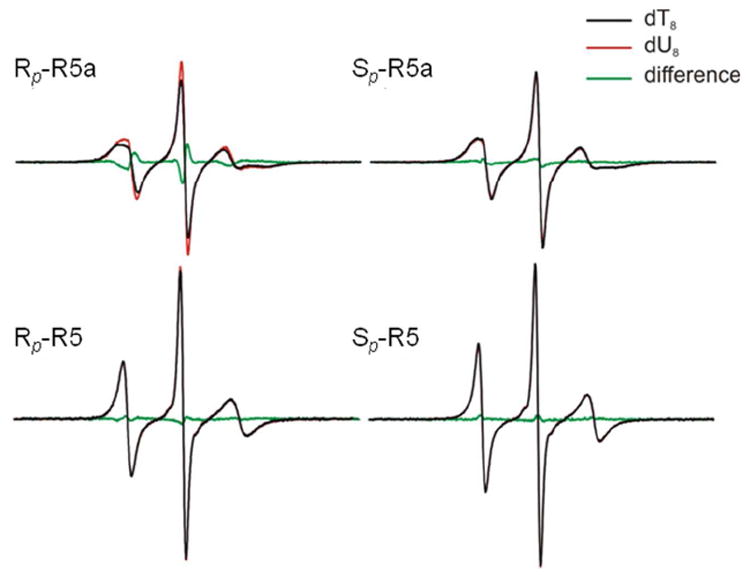
EPR spectra obtained at 25°C. Black lines represent spectra obtained in the d(CTACTGpSC7T8TTAG).d(CTAAAGCAGTAG) duplexes (dT8), and red lines represent those in the d(CTACTGpSC7U8TTAG).d(CTAAAGCAGTAG) duplexes (dU8). Green lines represent the corresponding difference spectra.
To further characterize rotational motions of R5a, spectral simulations were carried out using the Microscopic Order Macroscopic Disorder (MOMD) model, which describes the nitroxide motion within the macromolecular environment as an anisotropic rotational diffusion constricted by an ordering potential.27 Simulations yield two key parameters: (i) R̄, which describes the rate of nitroxide rotational motions; and (ii) S20, which provides a measure of local ordering. The simulations gave best-fit spectra that match the corresponding measured spectra very well (Figure 3A). The dT8 to dU8 change was found to increase Rp-R5a mobility, with a faster rate (larger R̄) and lower ordering (smaller S20) obtained for Rp-R5a-dT8 (Figure 3B). For Sp-R5a, neither rate nor order parameters are affected by the change from dT8 to dU8 (Figure 3B). This is in complete agreement with conclusions drawn from direct lineshape comparisons described above.
Figure 3.
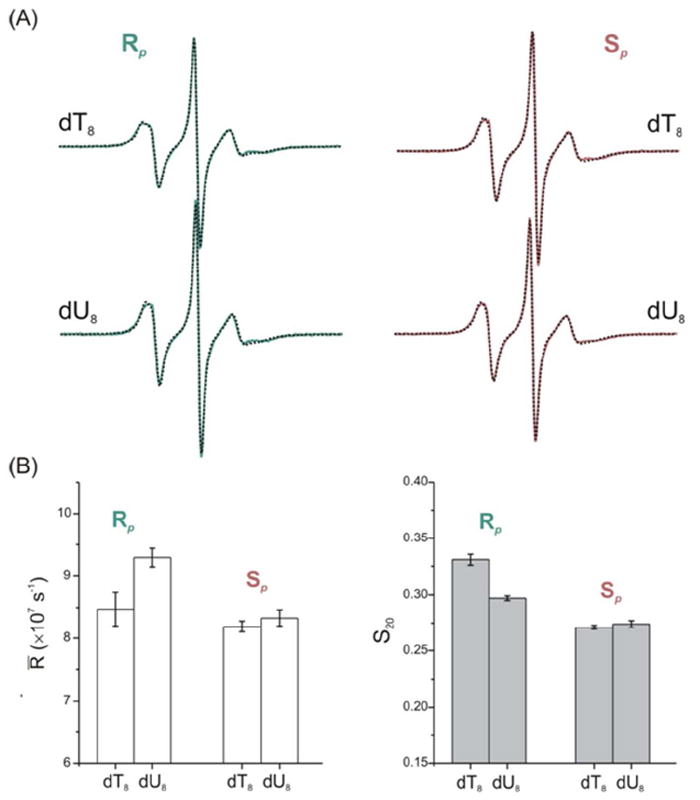
R5a spectral simulations. (A) Overlays of measured (solid lines) and simulated (dotted lines) spectra. (B) Comparisons of nitroxide motional parameters obtained from simulations. Additional details on simulation parameters are given in Table S1.
In summary, EPR spectra reveal that the dT8 to dU8 change gives rise to increased mobility for Rp-R5a, but has no detectable effects on Sp-R5a, Rp-R5, and Sp-R5. These observations are accounted for by MD simulations described below.
MD simulations qualitatively capture observed features of nitroxide rotational behavior in response to the dT8 to dU8 change
A total of 40 simulations of 10 ns each were performed in explicit solvent using AMBER on the dT8 and dU8 duplexes carrying R5a or R5 as the Rp or Sp phosphorothioate diastereomer of nucleotide 7 (Figure 1). For each sample set, defined by diastereomer configuration (Rp or Sp), label identity (R5a or R5), and DNA sequence (dT8 or dU8), the results were obtained by analyzing multiple simulations that differed only in the starting conformation of the label, defined by torsion angles t1 (O5′-P-S-Cs), t2 (P-S-Cs-C3), and t3 (S-Cs-C3-C2) around bonds connecting the nitroxide pyrroline ring to the DNA (Figure 1A). Details of the simulations are reported in Tables S2 and S3 for Rp and Sp labels, respectively. In each simulation, the energy of the system was stable, and the DNA duplex structure was retained, as confirmed by analysis of helical parameters and base pair hydrogen bonds (data not shown).
Variations of the t1, t2 and t3 torsion angles reflect rotational motions of the nitroxide with respect to the DNA, which is measured by the cw-EPR spectra described above. Torsion angle distributions from all simulations are summarized in Table 1 (see also Tables S2 and S3). For the Rp labels, the MD data show that the dT8 to dU8 change yields significant changes only for Rp-R5a, with occupancy of the t1 gauche+ (g+) conformer being 46.6 ± 24.3% for Rp-R5a-dT8 vs. 17.7 ± 9.7% for Rp-R5a-dU8 (Tables 1 and S2). This suggests that the dT8 to dU8 change alters the rotational behavior of Rp-R5a, but has no apparent effect on Rp-R5. This is in agreement with the EPR data (Figures 2 and 3).
Table 1.
Label conformer distributions in molecular dynamics simulations of DNA duplexes of sequence d(CTACTGpSC7Y8TTAG).d(CTAAAGCAGTAG) (Y = T or U) labeled with a 1-oxyl-4-bromo-2,2,5,5-tetramethylpyrroline (R5a) or a 1-oxyl-2,2,5,5-tetramethylpyrroline (R5) radical at pSdC7.
| Label | Chirality | dY8 | n a | t1 (%) b,c | t2 (%) b,c | t3 (%) b,d | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| g+ | t | g- | g+ | t | g- | +100° | -100° | ||||
| R5a | Rp | dT8 | 8 | 46.6±24.3 | 11.6±13.5 | 41.8±27.7 | 0.5±0.7 | 98.9±2.3 | 0.6±1.8 | 64.4±39.6 | 35.6±39.6 |
| R5a | Rp | dU8 | 6 | 17.7±9.7 | 23.9±16.3 | 58.4±25.4 | 2.9±4.4 | 93.6±6.0 | 3.5±4.1 | 57.5±33.7 | 42.5±33.7 |
| R5 | Rp | dT8 | 3 | 18.5±4.9 | 23.8±9.5 | 57.6±13.0 | 8.8±6.5 | 72.7±5.7 | 18.5±1.0 | 52.8±7.9 | 47.2±7.9 |
| R5 | Rp | dU8 | 3 | 14.2±8.8 | 16.8±9.7 | 68.9±4.8 | 13.3±6.9 | 67.0±17.1 | 19.7±21.6 | 51.3±10.1 | 48.7±10.1 |
| R5a | Sp | dT8 | 8 | 48.6±9.7 | 48.7±9.3 | 2.6±1.4 | 5.8±4.7 | 88.7±8.2 | 5.5±7.0 | 69.1±35.8 | 31.1±35.9 |
| R5a | Sp | dU8 | 6 | 37.0±14.2 | 59.0±14.9 | 3.9±2.1 | 4.9±7.9 | 91.0±7.2 | 4.2±4.6 | 60.1±35.5 | 39.9±35.5 |
| R5 | Sp | dT8 | 3 | 52.7±19.5 | 45.3±18.2 | 2.0±1.7 | 21.2±9.3 | 59.7±3.7 | 19.1±12.6 | 46.3±20.6 | 53.7±20.6 |
| R5 | Sp | dU8 | 3 | 38.8±18.2 | 58.8±17.3 | 2.4±1.3 | 8.9±4.7 | 58.0±5.6 | 33.1±8.9 | 54.0±6.9 | 46.0±6.9 |
Number of simulations of 10 ns each.
Torsion angles are defined in Figure 1A. Average and standard deviations are calculated from the corresponding individual 10-ns simulations.
g+: 0-120°; t: 120-240°; g-: 240-360°
Torsion angle t3 adopts values centered on +100° and -100°. Each distribution was determined based on conformations with t3 > 0° and t3 < 0°, respectively, for a torsional angle range from +180° to -180°.
For the Sp labels, the sequence change from dT8 to dU8 results in no significant changes of all torsion angle distributions except for t2 in Sp-R5, in which the average occupancy of g+ and gauche- (g-) conformers differ beyond one standard deviation (Tables 1 and S3). Examination of MD data for Sp labels revealed no discernable nitroxide/DNA interaction that is dependent on the dT8 to dU8 change, since the Sp labels are directed away from the DNA helix and into the solvent.22,30 This is consistent with the absence of a difference between the spectra of the Sp labels with the dT8 and dU8 duplexes (Figure 2).
In the following text, only the MD data for the Rp labels are further analyzed to explore the physical basis for the two key features observed in the EPR spectra: (i) difference between Rp-R5a-dT8 and Rp-R5a-dU8; and (ii) the lack of a detectable difference between Rp-R5-dT8 and Rp-R5-dU8.
Rp-R5a and Rp-R5 show different t2 and t3 rotational behavior
Representative traces for torsion angles t1, t2 and t3 for Rp-R5a-dT8 and Rp-R5-dT8 are shown in Figure 4. A clear feature of Rp-R5a-dT8 is that motions at t2 and t3 are limited on the nanosecond timescale. Throughout all simulations, including the one with the nitroxide starting conformation deliberately set at t2 = 120°, t2 was fixed in the trans (t) conformation (Figure 4B, Tables 1 and S2). In comparison, t2 for Rp-R5-dT8 also favors a t conformation (Figure 4E, Tables 1 and S2), but deviations from t occur on the nanosecond timescale. Clearly the Br atom in R5a is restricting this motion.
Figure 4.
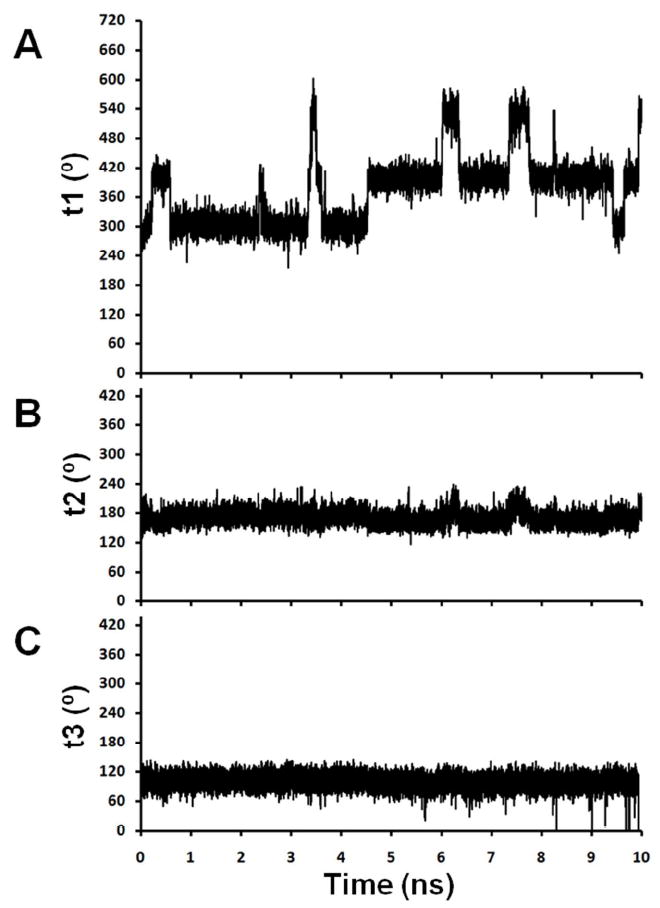
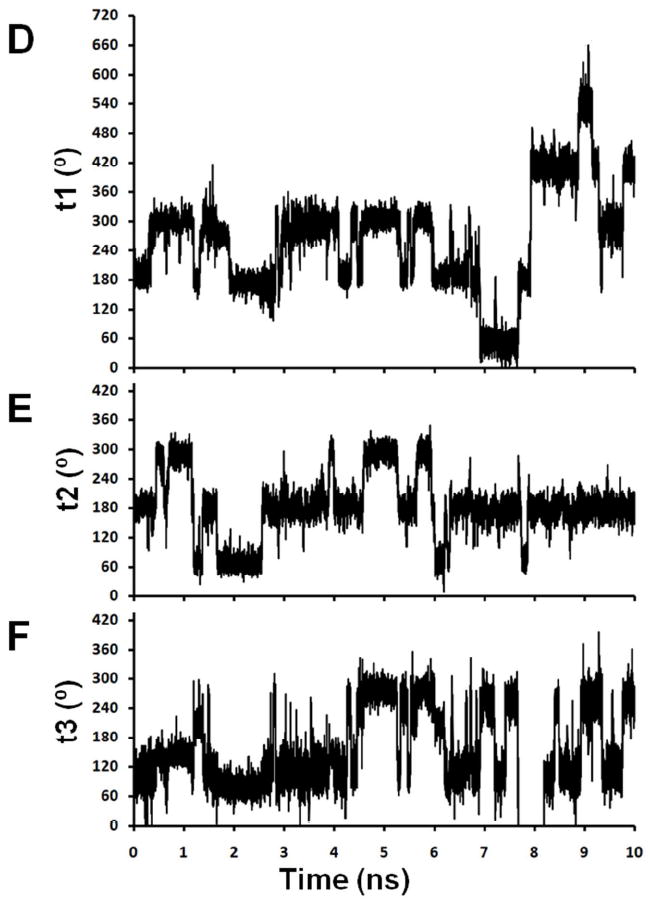
Sample traces of torsional motions for Rp-R5a-dT8 (A, t1; B, t2; C, t3; simulation #1) and Rp-R5-dT8 (D, t1; E, t2; F, t3; simulation #13). The t1 traces (A, D) are shown over a range of 0-720° with occupancy of g+ (60° or 420°), t (180° or 540°), and g- (300° or 660°). In F, the trace between 7.5 and 8.5 ns reflects t3 motion from +260° through a 360° turn to -100° (an equivalent conformation with a numerical value off the scale of the y-axis) and then a reverse 360° turn back to +260°, with only brief occupancy of the +100° conformer during each full turn.
For t3, the trace of Rp-R5a-dT8 in Figure 4C shows persistence of a conformation around +100°. However, analysis of all data for Rp-R5a-dT8 (Tables 1 and S2) shows that t3 adopts two conformations centered around +100° and -100° (+260°), respectively, with infrequent transitions between these two positions. Among the eight 10-ns Rp-R5a-dT8 simulations, 4 had no formal t3 transition, and 4 gave only one transition (data not shown). In these two conformations, the pyrroline ring of R5a is positioned at the same location, but the peripheral functional groups (i.e., 4-Br, C2 and C5 methyls) are oriented differently with respect to the DNA. Over all Rp-R5a-dT8 simulations, occupancy of the two t3 conformations is approximately equal (Table 1). Similar +100° and -100° conformations are seen for Rp-R5-dT8, but with more frequent transitions between them (Figure 4F). The additional motion may reflect the greater mobility of t2, although there is no clear correlation between the t2 and t3 torsional angles for Rp-R5-dT8 (Figure 4E, F).
The characteristics of t2 and t3 described for R5 and R5a occurred in all simulations regardless of diastereomeric configuration (Rp and Sp) and DNA sequence (dT8 and dU8) (Table 1). Therefore, they appear to be inherent properties of the respective labels. The MD results show that the presence of the 4-Br group limits rotations about bonds connecting the pyrroline ring to the DNA. The t2 and t3 motions in Rp-labels are similar for the dT8 and dU8 duplexes (Table 1), but limitations on t2 and t3 reduce the available conformations of Rp-R5a. This is an important factor in accounting for the sequence-dependent EPR spectral changes observed for Rp-R5a, but not for Rp-R5 (see below).
Interactions between Rp-R5a and DNA account for sequence-dependent changes in EPR spectra
For Rp-R5a, the t1 torsion samples the canonical g+, t, and g- conformations (Figure 4A), with the dT8 to dU8 change significantly decreasing the t1 g+ occupancy (Table 1). Examples of Rp-R5a-dT8 t1 g+ conformations with t3 = +100° or -100° are shown in Figures 5A and 5B, respectively. In both conformations, the nitroxide pyrroline ring is proximal to the dT8 C5-methyl group in a face-on position, suggesting a direct interaction between these two entities. We also note that in Figures 5A and 5B the dG6-pSdC7 dinucleotide linkage adopts a BII conformation (defined as ε/ζ: g-/t; ε-ζ ~ +90°),34 whereas all simulations started with dG6-pSdC7 in a BI conformation (ε /ζ: t/g-; ε-ζ ~ -90°).34 The relationship between the nitroxide t1 torsion and dG6-pSdC7 dinucleotide conformation will be further examined later.
Figure 5.
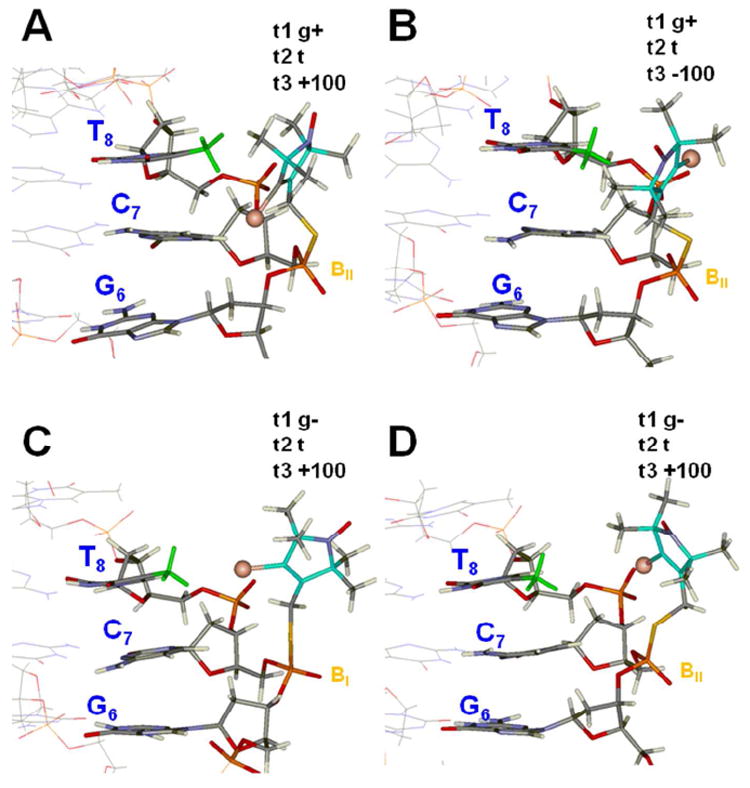
Representative structures from simulations of Rp-R5a-dT8. A. t1 g+, t2 t, t3 +100°, dG6-pSdC7 BII (simulation #1, at 5 ns, see Fig. 4A-C). B. t1 g+, t2 t, t3 -100°, dG6-pSdC7 BII (#37, at 8 ns). C. t1 g-, t2 t, t3 +100°, dG6-pSdC7 BI (#1, at 2 ns, see Fig. 4A-C). D. t1 g-, t2 t, t3 +100°, dG6-pSdC7 BII (#27, at 3.8 ns, see Fig. 6). In each structure, the dT8 methyl group is colored green, the C atoms of the pyrroline ring are colored light blue, and the bromine atom is shown as a brown sphere. The backbone conformation for the dG6-pSdC7 dinucleotide (BI or BII) is shown in orange.
The characteristics of the conformers shown in Figures 5A and 5B are maintained for the entire Rp-R5a-dT8 t1 g+ ensemble. Specifically, distributions of distances between the dT8 C5-methyl group and center of the pyrroline ring, the peripheral methyl groups of the ring, and the 4-Br atom each show predominantly one population, with an average distance of ≤ 5.1 Å in each case (Table 2). These close contacts suggest the presence of a hydrophobic interaction between the label and the dT8 C5-methyl group, which may explain the higher t1 g+ occupancy for Rp-R5a-dT8 compared to Rp-R5a-dU8.
Table 2.
Distances from the 5-methyl carbon atom of dT8 to Rp-R5a and Rp-R5 labels in key conformers found in simulations of the labeled DNA duplex d(CTACTGpSC7T8TTAG).d(CTAAAGCAGTAG).a
| Conformer b (t1,t2,t3) | DNA c | R5a | R5a d | R5a d | R5a d | R5 | R5 d | R5 d |
|---|---|---|---|---|---|---|---|---|
| Occupancy (%) | Pyrroline ring center (Å) | Closest ring methyl (Å) | Ring 4-Br (Å) | Occupancy (%) | Pyrroline ring center (Å) | Closest ring methyl (Å) | ||
| g+,t,+100 | BI | 0.0 e | --- | --- | --- | 0.0 e | --- | --- |
| g+,t,-100 | BI | 0.0 e | --- | --- | --- | 0.0 e | --- | --- |
| g+,t,+100 | BII | 32.9 | 4.9 ± 0.5 | 4.1 ± 0.5 | 4.9 ± 0.9 | 8.8 | 5.4 ± 0.7 | 4.0 ± 0.5 |
| g+,t,-100 | BII | 13.6 | 5.1 ± 0.7 | 4.3 ± 0.6 | 5.0 ± 1.2 | 9.2 | 5.3 ± 0.7 | 4.2 ± 0.6 |
| g-,t,+100 | BI | 27.9 | 6.6 ± 0.8 | 5.8 ± 0.9 | 4.2 ± 0.5 | 9.6 f | 6.9 ± 1.6 | 6.1 ± 1.9 |
| g-,t,-100 | BI | 8.3 | 6.3 ± 0.8 | 4.1 ± 0.8 | 8.9 ± 0.7 | 16.1 | 6.4 ± 0.9 | 4.2 ± 1.0 |
| g-,t,+100 | BII | 1.7 | 7.0 ± 0.8 | 6.4 ± 1.0 | 4.4 ± 0.8 | 6.6 f | 8.4 ± 1.3 | 8.1 ± 1.5 |
| g-,t,-100 | BII | 3.1 f | 8.1 ± 1.4 | 6.0 ± 1.6 | 9.6 ± 1.1 | 5.0 f | 7.7 ± 1.5 | 5.7 ± 1.7 |
| g-,g-,+100 | BI | 0.0 e | --- | --- | --- | 2.8 f | 8.9 ± 1.4 | 7.5 ± 1.6 |
| g-,g-,-100 | BI | 0.2 | 9.3 ± 1.0 | 9.4 ± 1.2 | 7.8 ± 0.9 | 2.9 f | 8.3 ± 1.3 | 8.1 ± 1.5 |
| g-,g-,+100 | BII | 0.0 e | --- | --- | --- | 10.0 | 9.7 ± 0.7 | 9.1 ± 0.8 |
| g-,g-,-100 | BII | 0.4 | 9.4 ± 0.8 | 9.0 ± 0.9 | 8.8 ± 0.8 | 2.5 | 9.4 ± 0.9 | 9.4 ± 1.0 |
Analyses carried out on 400,000 Rp-R5a-dT8 structures (eight 10-ns simulations) and 150,000 Rp-R5-dT8 structures (three 10-ns simulations). Data for all conformers are shown in Tables S4 (R5a) and S5 (R5).
g+: 0-120°; t: 120-240°; g-: 240-360°
Backbone conformation at the dG6-pSdC7 dinucleotide (see Table S6)
Average ± standard deviation of distance (Å) determined for the corresponding ensemble of conformers.
The t1 g- is another significantly occupied conformer for Rp-R5a (Table 1, Figure 5C). In this conformer, the pyrroline ring is moved away from the dT8 C5-methyl group (compare Figures 5A and 5C), with the average distance between the center of the pyrroline ring and dT8 C5-methyl becoming 6-8 Å (Table 2). At the same time, an average distance of <5.0 Å from the dT8 C5-methyl to the R5a peripheral methyl groups or 4-Br atom is observed in three of the four ensembles of t1 g- conformers (Table 2), as well as in one of the subpopulations of the fourth ensemble (g-,t,-100; BII) (Table S4). This indicates that Rp-R5a t1 g- conformers may also support interactions between the label and dT8 C5-methyl. A similar conclusion can be drawn regarding the t1 t conformers (Table S4).
Overall, upon analysis of all Rp-R5a-dT8 MD simulations, 86.8% of the conformer population was found to have one or more contacts of <5.0 Å between the dT8 C5-methyl group and the center of the Rp-R5a pyrroline, a ring methyl group, or the 4-Br atom. This suggests that a majority of the population may support direct interaction between the dT8 C5-methyl group and Rp-R5a. The dT8 to dU8 change abolishes this specific label/DNA interaction, thus resulting in the mobility changes observed in the EPR spectra of Rp-R5a-dT8 and Rp-R5a-dU8 (Figures 2 and 3).
Analysis of Rp-R5 sequence-dependent behavior
MD data reveal both similarities and differences between Rp-R5 and Rp-R5a. All major conformers of Rp-R5a are also present for Rp-R5 and maintain similar characteristics, as judged by the distances from the dT8 C5-methyl to the pyrroline ring center and methyl groups (Tables 2, S4 and S5). This suggests that interaction between the label and dT8 C5-methyl may occur for certain Rp-R5 conformers (e.g., t1 g+). However, contrary to R5a, R5 shows frequent transitions between t3 +100° and -100° (Figure 4F), as well as t2 transitions among g+, t, and g- (Figure 4E). Consequently, Rp-R5 can access many more sterically allowed conformations compared to Rp-R5a, and many of them may not have interactions between dT8 C5-methyl and the label (Tables 2 and S5). For example, in (t1 g-; t2 g-) conformers, which are largely absent in Rp-R5a but account for ~ 18% of the total Rp-R5 population (Table 2), the pyrroline ring center and methyl groups are on average >7.5 Å away from dT8 C5-methyl (Table 2). These conformers are unlikely to be involved in direct interactions between the label and the DNA. Furthermore, instead of a 4-Br atom, R5 has a 4-H (Figure 1A), which is unable to interact with dT8 C5-methyl.
Overall, analyses of all Rp-R5-dT8 MD simulations reveal that 40.1% of the label conformers have at least one distance <5.0 Å from the dT8 C5-methyl group to the pyrroline ring center or a ring methyl group. This is more than a two-fold reduction compared to Rp-R5a. Therefore, it is likely that Rp-R5 shows no detectable sequence-dependent spectral changes because its sensitivity to the presence of the dT8 C5-methyl is reduced by a combination of two factors: (i) fewer label functional groups making direct interactions with dT8 C5-methyl; and (ii) a smaller fraction of conformers that include these interactions. Both of these changes originate from removal of the 4-Br atom.
Correlation of t1 and DNA backbone conformations for Rp labels
An intriguing feature observed for Rp labels was the apparent dependence of the t1 value on the DNA backbone conformation at the dG6-pSdC7 dinucleotide (Figure 5 and Table 2). This behavior is further illustrated in Figure 6 using one of the Rp-R5a-dT8 simulations (simulation #27, Table S2). The t1 torsion angle shows a similar distribution to that in Figure 4A, with approximately half occupancy of g+ and g- conformations (420° and 300°, respectively, in Figure 6A). Notably, the t1 g+ can only form when dG6-pSdC7 adopts a BII conformation (Figure 6B, C). The strict association of t1 g+ with dG6-pSdC7 BII is true for both Rp-R5a and Rp-R5, since the total occupancies of t1 g+; dG6-pSdC7 BI for Rp-R5a-dT8 and Rp-R5-dT8 were only 0.02% and 0.07%, respectively, over all simulations (Tables 2, S4 and S5, Figure S1). Modeling showed that for a Rp label, the t1 g+ conformer has severe clashes of the pyrroline ring with the dG6 sugar when dG6-pSdC7 remains in the BI conformation, thus explaining the necessity of adopting the BII conformation.
Figure 6.
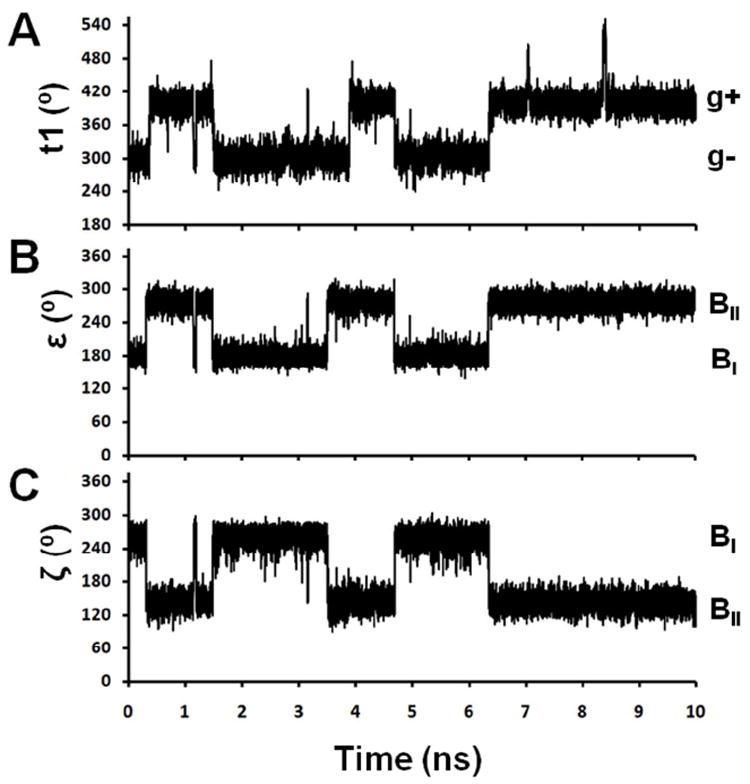
Sample traces of torsional motions for Rp-R5a-dT8 (simulation #27). A. t1. B. ε torsion angle of dG6: t conformers correspond to the BI conformation at dG6-pSdC7 and g- conformers to BII. C. ζ torsion angle of dG6: g- conformers correspond to the BI conformation and approximately t conformers to BII. See Table S6 for detailed definition of BI and BII conformations.
A control 30-ns MD simulation of the unlabeled dT8 duplex gave a 35.3%:63.7% ratio for the BI:BII equilibrium at dG6-dC7 (Table S6, Figure S2). A similar BI:BII ratio was found upon analyzing all Rp-R5a-dT8 and Rp-R5-dT8 simulations (38.0%:61.8% and 40.9%:58.3%, respectively, Table S6). This suggests that interactions between the Rp labels and DNA are not strong enough to alter the thermodynamics of the BI:BII equilibrium. In addition, analysis of Rp-R5a-dU8 traces shows approximately 52% BII occupancy, indicating the nucleotide at position 8 does not significantly influence BI/BII equilibrium at dG6-pSdC7. Close examination of the MD traces show that the t1 g+ conformation does not induce the dG6-pSdC7 BII conformation, but rather forms after a BI to BII transition at dG6-pSdC7. For example, the t1 g- to g+ transition between 3 and 4 ns in Figure 6A clearly occurs after a BI to BII transition at dG6-pSdC7 (Figure 6B,C), and the intermediate structure (i.e., t1 g-; dG6pSdC7 BII) is formed (Figure 5D).
Previously, MD simulations have observed BI/BII transitions occur in nanosecond timescale.34-37 Both MD simulation36 and NMR measurements37 have shown that a GpC dinucleotide step, at which the R5/R5a labels were attached in this study, have a significant population of the BII conformation. Overall, our results indicate that the Rp labels are exploring a naturally occurring transition of the DNA backbone to establish (additional) contact(s) between dT8 C5-methyl group and the pyrroline ring, which subsequently influences nitroxide motions and gives rise to the observed cw-EPR spectral changes. This suggests a direct connection between label motion and local DNA backbone dynamics.
Discussion
A synergistic combination of EPR and MD simulation for probing DNA local environment using spin labeling
In this work, the behavior of R5 and R5a nitroxide labels in response to a subtle DNA sequence change was examined using a combination of EPR spectroscopy and MD simulation. X-band cw-EPR spectra of labeled DNA duplexes revealed that a dT8 to dU8 change altered the nanosecond rotational motions of Rp-R5a, but produced no detectable changes for Sp-R5a, Rp-R5, and Sp-R5. MD simulations suggested that hydrophobic interactions of the label with the dT8 C5-methyl group and variations in the occupancy of label conformers that form these interactions, which account qualitatively for the observed EPR spectral changes. This demonstrates a synergistic interplay of experimental and computational approaches, with EPR measurements providing a critical validation of MD simulations, and the MD simulations revealing the molecular characteristics of the system and directing further experimental investigations (e.g., sequence changes). We note that in addition to the R5/R5a probes, a number of nitroxides have been reported to produce variable cw-EPR spectra depending on sequence and secondary structure of DNA and RNA.38-45 However, to connect the observed spectral features to molecular details at the nitroxide attachment site has been challenging. While MD simulations have been applied to investigate spin-labeled biomolecules,30,46-56 the combined EPR-mutagenesis-MD approach reported here could broadly aid further development and application of SDSL.
The results reported here also revealed issues that remain to be addressed in investigations of interactions between nitroxide labels and DNA. Specifically, force-field issues and ergodicity are always of concern in MD simulations. The force-field parameters used here have previously given results consistent with experimentally measured inter-nitroxide distances,30 and the label parameters have also been used independently by other groups.55,56 However, there were relatively large variations in torsion angle distributions between the 10-ns traces (Tables 1, S2 and S3), indicating that convergence may not have been established. This hampers in-depth analyses, such as computing transition rates between label conformers and order parameters for a given ensemble, which are required for quantitative correlations with cw-EPR spectra. We note that new parameter sets for similar spin labels57,58 and improved DNA parameterizations36,59 have been proposed, particularly for longer (microsecond) simulations, and work has been reported on generating a full EPR spectrum based on MD trajectories, particularly taking advantage of advances in simulation techniques.46-51 Thus, further improvement and extension of MD simulations may ultimately allow computation of R5/R5a spectra for direct comparison with experimental data.
Modulation of R5a/R5 motions by DNA
It has been hypothesized that site- and stereospecific behavior of R5 and R5a mainly arise from DNA sequence-dependent variations of (i) sterically allowable rotamer space; (ii) contacts/interactions between DNA and the pyrroline ring; and (iii) local DNA dynamics (flexibility).21,22 The results reported here provided details to validate this proposed framework, as well as revealing further complexity.
Regarding sterically allowable rotamers, MD simulations clearly demonstrated that steric exclusion from the DNA is a dominant factor that dictates the label behavior. For example, for Rp-R5, while g+, t, and g- conformations are accessible to both t1 and t2, certain t1, t2 combinations (for example, g+, g+; g+, g-; and g-, g+) have <1% occupancy (Table S5), since they position the pyrroline ring within the steric volume of the DNA. In addition, when the DNA backbone at dG6-pSdC7 adopts a BI conformation, the t1 g+ conformer is hardly occupied for Rp-R5a or Rp-R5 (Tables 2, S4, and S5, Figure S1) because of severe clashes between the pyrroline ring and the dG6 sugar.
In terms of contacts/interactions between DNA and the pyrroline ring, simulations reveal that certain conformers of Rp labels (e.g., t1 g+) position the dT8 C5-methyl group within close proximity to the pyrroline ring and ring methyl groups, indicating probable hydrophobic contacts. For Rp-R5a, the 4-Br atom may further strengthen this label/DNA interaction, while at the same time increasing the fraction of these interaction-capable conformers by reducing the total number of allowed label conformers (Tables 2 and S4). This qualitatively accounts for the spectral differences between Rp-R5a-dT8 and Rp-R5a-dU8 (Figures 2 and 3). On the other hand, the absence of 4-Br in Rp-R5 reduces the percentage of interaction-enabled conformers and eliminates interactions between the DNA and the label C4 functionality (Tables 2 and S5), resulting in a lack of detectable spectral changes between Rp-R5-dT8 and Rp-R5-dU8 (Figure 2).
Last but not least, a connection between EPR detected R5/R5a motions to local nanosecond DNA backbone dynamics is demonstrated by the covariation of the dG6-pSdC7 BII and R5/R5a t1 g+ conformations (Table 2, Figures 6 and S1). In previous studies, BI to BII transitions in DNA duplexes have been reported to occur on a nanosecond timescale in a sequence-dependent manner, with the dG-dC dinucleotide step favoring substantial occupancy of the BII conformation.34-37 The naturally-occurring nanosecond BI/BII transition at dG6-pSdC7 results in expanded sterically allowable rotamer space for the labels, allowing additional DNA-nitroxide contacts that influence nitroxide motions to an extent detectable by cw-EPR (i.e., Rp-R5a-dT8 vs. Rp-R5-dU8). In future studies, it should be very interesting to examine effects on R5/R5a motions from other modes of DNA motions (e.g., sugar puckering, base breathing).
Implications for use of R5a/R5 to probe dynamics and conformations of nucleic acids
Understanding of sequence-dependent coupling between DNA and R5/R5a will significantly improve our ability to extract information from these probes. For instance, spectral sensitivity of Rp-R5a to the dT->dU mutation may be further explored to probe sequence-dependent BI/BII transition, which may play a role in protein/DNA recognition.37,60-63 Interestingly, NMR37 and MD36 studies on BI/BII distributions for all 16 dinucleotide steps have revealed an asymmetric preference for BII within dinucleotide base-pair units (e.g., a difference between GpA and TpC37). These findings draw a parrallel with asymmetric R5a/R5 EPR lineshapes previously obtained within dinucleotide base-pair units.21,22 This provides additional support to a link between DNA local backbone motion and R5a/R5 behavior.
Information on coupling between DNA and R5/R5a will also improve modeling of allowable nitroxide conformers and enable more accurate interpretation of inter-nitroxide distances. We have developed an algorithm, referred to as NASNOX,19,30 for rapid computation of distances between labels at two nucleotides in a DNA20 or RNA structure.23 This algorithm depends on prediction of the conformational distribution of each label. From a computational perspective, MD simulations demand signficant computer resources and are difficult to apply for direct computation of multiple sets of inter-label distances.30 Therefore, we envision a strategy in which the details of the label conformational distribution obtained from MD simulations of model systems are incorporated empirically in a simple conformer search model, thus enabling efficient and accurate calculations of inter-label distances in the context of determination of nucleic acid folds.
We note that computational approaches, including MD simulations, have been an important tool in characterizing sequence-dependent conformational and dynamic heterogeneity of free DNA duplexes,36,64-67 but experimental validation of conclusions drawn from these analyses remains challenging. The combined experimental (EPR spectroscopy, DNA mutagenesis) and computational (MD simulations) approach reported in this work paves the way for an in-depth understanding of the behavior of R5a/R5 at a given DNA micro-environment. This is a key step forward towards the use of R5a/R5 to examine intrinsic sequence-dependent conformational and dynamic heterogeneity in large biologically relevant DNA molecules under physiological conditions.
Supplementary Material
Acknowledgments
We gratefully acknowledge support from NIH (GM069557, PZQ), NSF (MCB 054652, PZQ) and a fellowship from Hashemite University (Jordan) to MH. We thank Dr. Kálmán Hideg (University of Pécs, Pécs, Hungary) for providing reactive precursors of the R5 and R5a probes. Computation for the work described in this paper was supported by the University of Southern California Center for High-Performance Computing and Communications (www.usc.edu/hpcc).
Footnotes
Supporting Information Available
Tables S1 to S6 and Figures S1 and S2 are provided as supplemental information. This information is available free of charge via the Internet at http://pubs.acs.or
References
- 1.Lawson CL, Berman HM. Indirect Readout of DNA Sequence by Proteins. In: Correll CC, Rice PA, editors. Protein-Nucleic Acid Interactions. Royal Society of Chemistry; Cambridge: 2008. p. 66. [Google Scholar]
- 2.Rohs R, West SM, Liu P, Honig B. Curr Opin Struct Biol. 2009;19:171. doi: 10.1016/j.sbi.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rohs R, Jin X, West SM, Joshi R, Honig B, Mann RS. Annu Rev Biochem. 2010;79:233. doi: 10.1146/annurev-biochem-060408-091030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Egli M, Pallan PS. Curr Opin Struct Biol. 2010;20:262. doi: 10.1016/j.sbi.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Segal E, Fondufe-Mittendorf Y, Chen L, Thastrom A, Field Y, Moore IK, Wang JP, Widom J. J Nature. 2006;442:772. doi: 10.1038/nature04979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peckham HE, Thurman RE, Fu Y, Stamatoyannopoulos JA, Noble WS, Struhl K, Weng Z. Genome Res. 2007;17:1170. doi: 10.1101/gr.6101007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rohs R, West SM, Sosinsky A, Liu P, Mann RS, Honig B. Nature. 2009;461:1248. doi: 10.1038/nature08473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hubbell WL, Cafiso DS, Altenbach C. Nat Struct Biol. 2000;7:735. doi: 10.1038/78956. [DOI] [PubMed] [Google Scholar]
- 9.Fajer PG. Electron spin resonance spectroscopy labeling in proteins and peptides analysis. In: Meyers R, editor. Encyclopedia of analytical chemistry. John Wiley & Sons; Chichester: 2000. p. 5725. [Google Scholar]
- 10.Klug CS, Feix JB. Methods Cell Biol. 2008;84:617. doi: 10.1016/S0091-679X(07)84020-9. [DOI] [PubMed] [Google Scholar]
- 11.Sowa GZ, Qin PZ. Site-directed spin labeling studies on nucleic acid structure and dynamics. Prog Nucleic Acids Res Mol Biol. 2008;82:147. doi: 10.1016/S0079-6603(08)00005-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robinson BH, Mailer C, Drobny G. Annu Rev Biophys Biomol Struct. 1997;26:629. doi: 10.1146/annurev.biophys.26.1.629. [DOI] [PubMed] [Google Scholar]
- 13.Keyes RS, Bobst AM. Spin-labeled nucleic acids. In: Berliner LJ, editor. Biological Magnetic Resonance. Vol. 14. Plenum Press; New York: 1998. p. 283. [Google Scholar]
- 14.Krstić I, Endeward B, Margraf D, Marko A, Prisner T. Structure and Dynamics of Nucleic Acids. In: Drescher M, Jeschke G, editors. EPR Spectroscopy, Applications in Chemistry and Biology. Vol. 321. Springer; Berlin / Heidelberg: 2011. p. 159. [DOI] [PubMed] [Google Scholar]
- 15.Reginsson GW, Schiemann O. Biochem Soc Trans. 2011;39:128. doi: 10.1042/BST0390128. [DOI] [PubMed] [Google Scholar]
- 16.Sigurdsson ST. Pure Appl Chem. 2011;83:677. [Google Scholar]
- 17.Nguyen P, Qin PZ. WIREs: RNA. 2012;3:62. doi: 10.1002/wrna.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qin PZ, Butcher SE, Feigon J, Hubbell WL. Biochemistry. 2001;40:6929. doi: 10.1021/bi010294g. [DOI] [PubMed] [Google Scholar]
- 19.Qin PZ, Haworth IS, Cai Q, Kusnetzow AK, Grant GPG, Price EA, Sowa GZ, Popova A, Herreros B, He H. Nat Protocols. 2007;2:2354. doi: 10.1038/nprot.2007.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cai Q, Kusnetzow AK, Hubbell WL, Haworth IS, Gacho GPC, Van Eps N, Hideg K, Chambers EJ, Qin PZ. Nucleic Acids Res. 2006;34:4722. doi: 10.1093/nar/gkl546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Popova AM, Kálai T, Hideg K, Qin PZ. Biochemistry. 2009;48:8540. doi: 10.1021/bi900860w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Popova AM, Qin PZ. Biophys J. 2010;99:2180. doi: 10.1016/j.bpj.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cai Q, Kusnetzow AK, Hideg K, Price EA, Haworth IS, Qin PZ. Biophys J. 2007;93:2110. doi: 10.1529/biophysj.107.109439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grant GPG, Boyd N, Herschlag D, Qin PZ. J Am Chem Soc. 2009;131:3136. doi: 10.1021/ja808217s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X, Tung C-S, Sowa GZ, Hatmal MM, Haworth IS, Qin PZ. J Am Chem Soc. 2012;134:2644. doi: 10.1021/ja2093647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grant GPG, Popova A, Qin PZ. Biochem Biophys Res Commun. 2008;371:451. doi: 10.1016/j.bbrc.2008.04.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Earle KA, Budil DE. Calculating Slow-motion ESR Spectra of Spin-Labeled Polymers. In: Schlick S, editor. Advanced ESR Methods in Polymer Reaserch. John Wiley and Sons; New York: 2006. p. 53. [Google Scholar]
- 28.Meirovitch E, Nayeem A, Freed JH. J Phys Chem B. 1984;88:3454. [Google Scholar]
- 29.Leporc S, Mauffret O, Tevanian G, Lescot E, Monnot M, Fermandjian S. Nucleic Acids Res. 1999;27:4759. doi: 10.1093/nar/27.24.4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Price EA, Sutch BT, Cai Q, Qin PZ, Haworth IS. Biopolymers. 2007;87:40. doi: 10.1002/bip.20769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. J Comput Chem. 2004;25:1157. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 32.Darden T, York D, Pedersen L. J Chem Phys. 1993;98:10089. [Google Scholar]
- 33.Lavery R, Sklenar H. J Biomol Struct Dyn. 1988;6:63. doi: 10.1080/07391102.1988.10506483. [DOI] [PubMed] [Google Scholar]
- 34.Hartmann B, Piazzola D, Lavery R. Nucleic Acids Res. 1993;21:561. doi: 10.1093/nar/21.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perez A, Luque FJ, Orozco M. J Am Chem Soc. 2007;129:14739. doi: 10.1021/ja0753546. [DOI] [PubMed] [Google Scholar]
- 36.Lavery R, Zakrzewska K, Beveridge D, Bishop TC, Case DA, Cheatham T, 3rd, Dixit S, Jayaram B, Lankas F, Laughton C, Maddocks JH, Michon A, Osman R, Orozco M, Perez A, Singh T, Spackova N, Sponer J. Nucleic Acids Res. 2010;38:299. doi: 10.1093/nar/gkp834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heddi B, Oguey C, Lavelle C, Foloppe N, Hartmann B. Nucleic Acids Res. 2010;38:1034. doi: 10.1093/nar/gkp962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okonogi TM, Alley SC, Reese AW, Hopkins PB, Robinson BH. Biophys J. 2000;78:2560. doi: 10.1016/S0006-3495(00)76800-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edwards TE, Okonogi TM, Robinson BH, Sigurdsson ST. J Am Chem Soc. 2001;123:1527. doi: 10.1021/ja005649i. [DOI] [PubMed] [Google Scholar]
- 40.Gannett PM, Darian E, Powell J, Johnson EM, Mundoma C, Greenbaum NL, Ramsey CM, Dalal NS, Budil DE. Nucleic Acids Res. 2002;30:5328. doi: 10.1093/nar/gkf634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cekan P, Jonsson EO, Sigurdsson ST. Nucleic Acids Res. 2009;37:3990. doi: 10.1093/nar/gkp277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cekan P, Sigurdsson ST. J Am Chem Soc. 2009;131:18054. doi: 10.1021/ja905623k. [DOI] [PubMed] [Google Scholar]
- 43.Smith AL, Cekan P, Brewood GP, Okonogi TM, Alemayehu S, Hustedt EJ, Benight AS, Sigurdsson ST, Robinson BH. J Phys Chem B. 2009;113:2664. doi: 10.1021/jp808260b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krstic I, Frolow O, Sezer D, Endeward B, Weigand JE, Suess B, Engels JW, Prisner TF. J Am Chem Soc. 2010;132:1454. doi: 10.1021/ja9077914. [DOI] [PubMed] [Google Scholar]
- 45.Shelke SA, Sigurdsson ST. Nucleic Acids Res. 2011 doi: 10.1093/nar/gkr1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robinson BH, Slusky LJ, Auteri FP. J Chem Phys. 1992;96:2609. [Google Scholar]
- 47.Stoica I. J Phys Chem B. 2003;108:1771. [Google Scholar]
- 48.Beier C, Steinhoff H-J. Biophys J. 2006;91:2647. doi: 10.1529/biophysj.105.080051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Budil DE, Sale KL, Khairy KA, Fajer PG. J Phys Chem A. 2006;110:3703. doi: 10.1021/jp054738k. [DOI] [PubMed] [Google Scholar]
- 50.DeSensi SC, Rangel DP, Beth AH, Lybrand TP, Hustedt EJ. Biophys J. 2008;94:3798. doi: 10.1529/biophysj.107.125419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sezer D, Freed JH, Roux B. J Am Chem Soc. 2009;131:2597. doi: 10.1021/ja8073819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sale K, Song L, Liu YS, Perozo E, Fajer P. J Am Chem Soc. 2005;127:9334. doi: 10.1021/ja051652w. [DOI] [PubMed] [Google Scholar]
- 53.Fajer MI, Li H, Yang W, Fajer PG. J Am Chem Soc. 2007;129:13840. doi: 10.1021/ja071404v. [DOI] [PubMed] [Google Scholar]
- 54.Piton N, Mu Y, Stock G, Prisner TF, Schiemann O, Engels JW. Nucl Acids Res. 2007;35:3128. doi: 10.1093/nar/gkm169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ding F, Layten M, Simmerling C. J Am Chem Soc. 2008;130:7184. doi: 10.1021/ja800893d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Galiano L, Ding F, Veloro AM, Blackburn ME, Simmerling C, Fanucci GE. J Am Chem Soc. 2009;131:430. doi: 10.1021/ja807531v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Houriez Cl, Ferreì N, Siri D, Masella M. J Phys Chem B. 2009;113:15047. doi: 10.1021/jp906828v. [DOI] [PubMed] [Google Scholar]
- 58.Stendardo E, Pedone A, Cimino P, Cristina Menziani M, Crescenzi O, Barone V. Phys Chem Chem Phys. 2010;12:11697. doi: 10.1039/c001481h. [DOI] [PubMed] [Google Scholar]
- 59.Perez A, Marchan I, Svozil D, Sponer J, Cheatham TE, 3rd, Laughton CA, Orozco M. Biophys J. 2007;92:3817. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wecker K, Bonnet MC, Meurs EF, Delepierre M. Nucleic Acids Res. 2002;30:4452. doi: 10.1093/nar/gkf559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heddi B, Foloppe N, Bouchemal N, Hantz E, Hartmann B. J Am Chem Soc. 2006;128:9170. doi: 10.1021/ja061686j. [DOI] [PubMed] [Google Scholar]
- 62.Rene B, Masliah G, Antri SE, Fermandjian S, Mauffret O. J Phys Chem B. 2007;111:4235. doi: 10.1021/jp0683115. [DOI] [PubMed] [Google Scholar]
- 63.Tian Y, Kayatta M, Shultis K, Gonzalez A, Mueller LJ, Hatcher ME. J Phys Chem B. 2009;113:2596. doi: 10.1021/jp711203m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Olson WK, Gorin AA, Lu XJ, Hock LM, Zhurkin VB. Proc Natl Acad Sci U S A. 1998;95:11163. doi: 10.1073/pnas.95.19.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lankas F, Sponer J, Langowski J, Cheatham TE., 3rd Biophys J. 2003;85:2872. doi: 10.1016/S0006-3495(03)74710-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fujii S, Kono H, Takenaka S, Go N, Sarai A. Nucleic Acids Res. 2007;35:6063. doi: 10.1093/nar/gkm627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goni JR, Perez A, Torrents D, Orozco M. Genome Biol. 2007;8:R263. doi: 10.1186/gb-2007-8-12-r263. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.