

Abstract
A two-stage synthesis of lactams in flow is described. Thus, a keto alkyl halide is displaced in a microwave-assisted, continuous flow organic synthesis format (MACOS) to generate a reactive alkyl azide. Without isolation, a flowed solution containing this azide is then combined with TFA to afford a lactam.
Keywords: Microwave, Continuous Flow, Schmidt Reaction, keto-azide, lactam
It is now well appreciated that flow techniques offer much in the area of fine organic synthesis.[1] In particular, they permit the utilization of even highly reactive intermediates in a relatively safe setting and offer a convenient means of preparing large quantities of compounds by extending the time of the flow reaction (“scaling out”) and thus avoiding issues with “scaling up” of batch reactions. Since some alkyl azides are known to pose potential explosion hazards, the adoption of flow techniques for alkyl azide reactions is attractive. Previously, the in situ generation of azides and their utilization in Curtius rearrangement,[2] Staudinger aza-Wittig,[3] and 1,3-dipolar cycloaddition[4] chemistries have been reported.[5] In this communication, we report the use of microwave-assisted flow reaction conditions in the context of the intramolecular Schmidt reaction of alkyl azides, a useful method for the generation of lactams.[6]
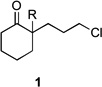
We used keto-chloride 1 as a model substrate for developing the two-step MACOS sequence depicted in Scheme 1.[7] Using a syringe pump an equimolar DMF solution of N,N,N,N-tetrabutylammonium azide (TBAA) and the halide was flowed (10 µL/min) through a glass capillary positioned inside the reaction chamber of a microwave reactor set to operate at 300W. The solution exited the chamber and reached a connection point joined to a second glass capillary that was also positioned inside the microwave chamber. Trifluoroacetic acid (20 µL/min) was introduced to the flowing stream at the connection point using a second syringe pump. The combined reaction mixture reentered the microwave chamber through the second capillary and was collected upon exiting. To prevent possible in-line cavitation the experiment was performed under 70 PSI argon pressure, which was applied through a needle inlet into the receiving vial.
Scheme 1.

General Reaction Sequence
Conversion to the lactam was determined by extractive workup and examination of the derived products by 1H NMR. While reasonable conversion (~70%) was attained, improvement was clearly required. In addition, the elimination of dead volume from the system was desired to faciliate yield determination.
Several modifications were introduced that improved the outcome. The replacement of DMF by CH3CN (AN) afforded complete conversion of the intermediate azide to the lactam, even at reduced power (125W), presumably because AN does not attenuate the acidity of TFA.[8] The reaction mixture was loaded into a sample loop made of PEEK tubing that was introduced into the beginning of the flow path. The first syringe pump delivered only solvent, removing any possibility that reactants would remain in the system uncollected. Instead of using fragile glass capillaries within the microwave chamber, mechanically-robust, flexible fused silica tubing (700 µm dia) was used (see supporting information for a schematic diagram). A reduced pressure of 40 PSI was both effective in preventing cavitation and resulted in less laborous flow. To improve the throughput, the flow rate was increased to 50 µL/min from each syringe pump. The residence time was calculated to be about 60 sec for the azide displacement and 30 sec for conversion to the lactam, assuming that the length of the irradiated flow path was 2 – 3 cm (ca. 45 – 50 µL irradiated volume). Given the absence of direct instrumental measurement, the reaction temperature was estimated to be no greater than 105 – 110 °C based upon the calculated boiling point of the solvent at 40 PSI.
Two other modifications were introduced to assist in reaction development. One was the insertion of a valve into the flow path that enabled sampling of the effluent from the azide forming step before it was combined with TFA. The other was the provision of a room temperature path outside the microwave for the effluent after combination with TFA (Figure 1). Using the modifed MACOS system, the reaction of keto chloride 1 provided lactam 3 in 69% yield (Table 1, entry 1). A slightly improved 76% yield of lactam 3 was obtained when the combined flow stream was redirected outside of the microwave chamber (Table 1, entry 2). This was the only substrate that behaved this way. In both cases conversion was 100%. These became the standard conditions of first resort.
Figure 1.

Top view of the MACOS apparatus valves. A is the three-way valve that allows in-line determination of azide formation and B is the four-way valve through which the TFA enters the reaction stream and also allows for diversion of the reaction stream away from the microwave chamber. See Supporting Information for a full experimental schematic.
Table 1.
In Situ Intramolecular Schmidt Reaction of Known Substrates[a]
| Entry | Halide | Product | Yield (%) |
Recovery (%) (Azide/Halide) |
||
|---|---|---|---|---|---|---|
 |
 |
|||||
| 1 | 1a | R = H | 3a | R = H | 69 | – |
| 2 | 1a | R = H | 3a | R = H | 76[b] | – |
| 3 | 1b | R = CO2Et | 3b | R = CO2Et | 68 | 5/6 |
| 4 | 1c | R = Ph | 3c | R = Ph | 5 | 60/15 |
 |
 |
|||||
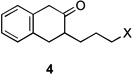
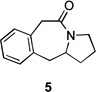
| 5 | 4a | X = Cl | 5 | 60 | –/23 | |
| 6 | 4b | X = Br | 5 | 61 | – | |
 |
 |
|||||
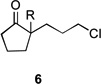
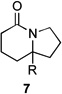
| 7 | 6a | R = H | 7a | R = H | 8 | 37/19 |
| 8 | 6b | R = CO2Et | 7b | R = CO2Et | – | 66/16 |
 |
 |
|||||
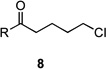
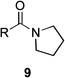
| 9 | 8a | R = Me | 9a | R = Me | 25 | 36/14 |
| 10 | 8b | R = CH2CO2Et | 9b | R = CH2CO2Et | – | 44/7 |
Conditions: 1. Halide 2M in CH3CN, NBu4N3 (1.1 equiv.), 125 W MWI, 50 µL/min. 2. TFA (3 mL, excess) 50 µL/min, 40 PSI back pressure.
Second step of the sequence performed outside of the microwave cavity.
Application of these conditions to other substrates afforded the variable results collected in Table 1. In some cases, the identities of the recovered byproducts suggested possible improvements. For example, reaction of β-tetralone derived chloride 4a afforded lactam 5 in 60% yield with 23% of the recovered chloride but no azide (Table 1, entry 5). This result suggested that the yield-limiting step was the halide displacement, so we tried the corresponding bromide 4b. An increase in the yield of 5 was obtained and no bromide was recovered, although some decomposed material was isolated (Table 1, cf. entries 5 and 6). Marginal increases in Schmidt reaction conversion were observed starting with the respective azides of 6b and 8a when the flow rate was slowed to 10 µL/min and the power increased from 125 W to 300 W (results not shown). Overall, these results were in line with previously observed reactivity trends of these compounds.[6]
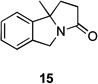
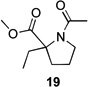
Several previously unexamined Schmidt reaction sequences were carried out to further define the scope of the MACOS method (Table 2). As before, both chloride- and bromide-containing substrates gave useful results. Cyclobutanones, which have only occasionally been examined in the context of this reaction,[6a,c] gave mixed results. Thus, while tricyclic lactam 15 was obtained from the Schmidt reaction of cyclobutanone 14, cyclobutanone 16 cleanly afforded azide 17 in high yield (Table 2, entry 6). This conversion, previously unobserved in our hands, could occur via initial azide displacement followed by rapid acid-promoted isomerization, affording the α,β-unsaturated ketone. Unsaturated ketones do not undergo the Schmidt reaction under these conditions.[9] α-Carboxyketone 18 was found to give <5% of lactam 19, which is consistent with the known lower reactivities associated with esters (Table 2, entry 7).[6c] One substrate produced a mixture of lactams. Thus, the reaction of 22 afforded a 1:3 mixture of lactams 23 and 24 (Table 2, entry 9), in contrast to the result shown for a corresponding spirocyclic halide substrate (Table 2, entry 8).[10]
Table 2.
In Situ Intramolecular Schmidt Reaction of New Substrates[a]
| Entry | Halide | Product | Yield (%) |
Recovery (%) (Azide/Halide) |
|
|---|---|---|---|---|---|
 |
 |
||||
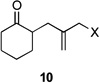
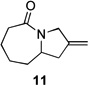
| 1 | 10a | X = Cl | 11 | 51 | –/14 |
| 2 | 10b | X = Br | 11 | 53 | – |
 |
 |
||||
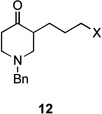
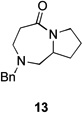
| 3 | 12a | X = Cl | 13 | 77[b] | –/11 |
| 4 | 12b | X = Br | 13 | 61[b] | – |
| 5 | 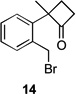 |
 |
54 | – | |
| 6 | 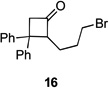 |
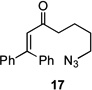 |
83 | – | |
| 7 | 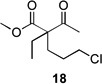 |
 |
trace | 66/– | |
| 8 |  |
 |
67 | – | |
| 9 | 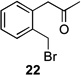 |
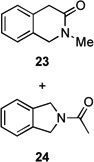 |
40 (23:24 = 3:1) |
33/– | |
Conditions: 1. Substrate 2M in CH3CN, 1.1 equiv. NBu4N3, 300 W MWI, 50 µL/min. 2. TFA (3 mL, excess) 50 µL/min, 40 PSI back pressure.
Reaction was performed at 125 W.
In summary the in situ conversion of keto-halides to azides and their conversion to lactams via the intramolecular Schmidt reaction has been demonstrated under MACOS conditions. Future work will be directed to new improvements in the flow technology and to utilizing this method for the synthesis of challenging new examples of this useful reaction.
Experimental Section
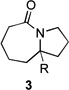
Representative Procedure for MACOS Azide displacement / Schmidt Sequence: Hexahydro-1H-pyrrolo[1,2-a]azepin-5(6H)-one 3a. The MACOS apparatus was constructed as detailed in the supporting information. A solution of chloride 1a (312 mg, 1.78 mmol, 1 equiv.) and N,N,N,N-tetrabutylammonium azide (558 mg, 1.96 mmol, 1.1 equiv.) in acetonitrile (0.89 mL, 2M with respect to the halide) was added into a 2 mL sample loop. (A 4 ± 2% material remainder due to syringe handling was typical for this procedure. Yields are uncorrected). The loop was connected to the apparatus and a luer-lock syringe containing 3 mL of acetonitrile connected to the other end and placed in the syringe pump. A second luer-lock syringe was loaded with trifluoracetic acid (3 mL, excess), connected to the second valve, and placed into the second syringe pump. Back-pressure tubes were connected to the collection vessels (crimp-sealed microwave vials) and then the microwave was turned on to 125 W (or 300 W as indicated in the text) and both pumps were set to infuse at 50 µl/min. The back-pressure was then immediately set to 40 psi and the reaction flowed over 1h. The collected material was flushed with toluene three times under vacuum and the mixture dissolved in toluene and filtered through a plug of basic alumina, further eluting with ethylacetate. The filtrate was concentrated under vacuum to afford a crude mixture of product and tetrabutylammonium salts. The crude mixture was purified via silica gel column chromatography (0.5% aqueous ammonium hydroxide, 2.5% methanol in dichloromethane) to afford 3a (189 mg, 69%) as a pale-yellow oil. Data for the product is consistent with literature values.[6b] Please see the supporting information for further experimental details.
Supplementary Material
Acknowledgements
The authors thank Erin Hirt for some initial substrate synthesis, Erin Hirt and Erik Fenster for useful discussions, David Hill for equipment support, Patrick Porubsky for mass spectroscopy, and Justin Douglas for assistance with NMR spectroscopy. We also thank the National Institute of General Medical Sciences (P5069663 and GM-49093) and the Kansas Technology Enterprise Corporation for financial support.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
References
- 1.a) Devine WG, Leadbeater NE. Arkivoc. 2011:127–143. [Google Scholar]; b) Razzaq T, Kappe CO. Chem. Asian J. 2010;5:1274–1289. doi: 10.1002/asia.201000010. [DOI] [PubMed] [Google Scholar]; c) Glasnov TN, Findenig S, Kappe CO. Chem. Eur. J. 2009;15:1001–1010. doi: 10.1002/chem.200802200. [DOI] [PubMed] [Google Scholar]; d) Benali O, Deal M, Farrant E, Tapolczay D, Wheeler R. Org. Process Res. Dev. 2008;12:1007–1011. [Google Scholar]; e) Hernando J, Leton P, Matia MP, Novella JL, Alvarez-Builla J. Fuel. 2007;86:1641–1644. [Google Scholar]; f) Baxendale IR, Hayward JJ, Ley SV. Comb. Chem. High T. Scr. 10:802–836. doi: 10.2174/138620707783220374. [DOI] [PubMed] [Google Scholar]; g) Glasnov TN, Kappe CO. Macromol. Rapid Commun. 2007;28:395–410. [Google Scholar]; h) Paulus RM, Erdmenger T, Becer CR, Hoogenboom R, Schubert US. Macromol. Rapid Commun. 2007;28:484–491. [Google Scholar]; i) Baxendale IR, Griffiths-Jones CM, Ley SV, Tranmer GK. Chem. Eur. J. 2006;12:4407–4416. doi: 10.1002/chem.200501400. [DOI] [PubMed] [Google Scholar]; j) Jachuck RJJ, Selvaraj DK, Varma RS. Green Chem. 2006;8:29–33. [Google Scholar]; k) Bagley MC, Jenkins RL, Lubinu MC, Mason C, Wood RJ. J. Org. Chem. 2005;70:7003–7006. doi: 10.1021/jo0510235. [DOI] [PubMed] [Google Scholar]; l) He P, Haswell SJ, Fletcher PDI. Appl. Catal. A. 2004;274:111–114. [Google Scholar]; m) Wilson NS, Sarko SR, Roth G. Org. Process. Res. Dev. 2004;8:535–538. [Google Scholar]; n) Savin KA, Robertson M, Gernert D, Green S, Hembre EJ, Bishop J. Mol. Diversity. 2003;7:171–174. doi: 10.1023/b:modi.0000006801.27748.3b. [DOI] [PubMed] [Google Scholar]
- 2.a) Baumann M, Baxendale IR, Ley SV, Nikbin N, Smith CD. Org. Biomol. Chem. 2008;6:1587–1593. doi: 10.1039/b801634h. [DOI] [PubMed] [Google Scholar]; b) Baumann M, Baxendale IR, Ley SV, Nikbin N, Smith CD, Tierney JP. Org. Biomol. Chem. 2008;6:1577–1586. doi: 10.1039/b801631n. [DOI] [PubMed] [Google Scholar]; c) Carter CF, Lange H, Ley SV, Baxendale IR, Wittkamp B, Goode JG, Gaunt NL. Org. Process Res. Dev. 2010;14:393–404. [Google Scholar]
- 3.a) Smith CJ, Smith CD, Nikbin N, Ley SV, Baxendale IR. Org. Biomol. Chem. 2011;9:1927. doi: 10.1039/c0ob00813c. [DOI] [PubMed] [Google Scholar]; b) Baxendale IR, Deeley J, Griffiths-Jones CM, Ley SV, Saaby S, Tranmer GK. Chem. Commun. 2006:2566–2568. doi: 10.1039/b600382f. [DOI] [PubMed] [Google Scholar]
- 4.a) Bogdan AR, Sach NW. Adv. Synth. Catal. 2009;351:849–854. [Google Scholar]; b) Ceylan S, Klande T, Vogt C, Friese C, Kirschning A. Synlett. 2010:2009–2013. [Google Scholar]
- 5.For recent examples of flow reactions utilizing azides directly, see: Baxendale IR, Ley SV, Mansfield AC, Smith CD. Angew. Chem., Int. Ed. 2009;48:4017–4021. doi: 10.1002/anie.200900970. Smith CD, Baxendale IR, Tranmer GK, Baumann M, Smith SC, Lewthwaite RA, Ley SV. Org. Biomol. Chem. 2007;5:1562–1568. doi: 10.1039/b703033a. Smith CD, Baxendale IR, Lanners S, Hayward JJ, Smith SC, Ley SV. Org. Biomol. Chem. 2007;5:1559–1561. doi: 10.1039/b702995k. Franckevicius V, Knudsen KR, Ladlow M, Longbottom DA, Ley SV. Synlett. 2006:889–892. Fuchs M, Goessler W, Pilger C, Kappe CO. Adv. Synth. Catal. 2010;352:323–328. Tinder R, Farr R, Heid R, Zhao R, Rarig RS, Storz T. Org. Process Res. Dev. 2009;13:1401–1406.
- 6.a) Aubé J, Milligan GL. J. Am. Chem. Soc. 1991;113:8965–8966. [Google Scholar]; b) Milligan GL, Mossman CJ, Aubé J. J. Am. Chem. Soc. 1995;117:10449–10459. [Google Scholar]; c) Mossman CJ, Aubé J. Tetrahedron. 1996;52:3403–3408. [Google Scholar]; (d) Lee H-L, Aubé J. Tetrahedron. 2007;63:9007–9015. doi: 10.1016/j.tet.2007.05.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For prior applications of MACOS technologies, see: Shore G, Yoo W-J, Li C-J, Organ MG. Chem. Eur. J. 2010;16:126–133. doi: 10.1002/chem.200902396. Ullah F, Samarakoon T, Rolfe A, Kurtz RD, Hanson PR, Organ MG. Chem. Eur. J. 2010;16:10959–10962. doi: 10.1002/chem.201001651. Shore G, Organ MG. Chem. Comm. 2008:838–840. doi: 10.1039/b715709f. Shore G, Organ MG. Chem. Eur. J. 2008;14:9641–9646. doi: 10.1002/chem.200801610. Shore G, Morin S, Mallik D, Organ MG. Chem. Eur. J. 2008;14:1351–1356. doi: 10.1002/chem.200701588. Bremner WS, Organ MG. J. Comb. Chem. 2007;9:14–16. doi: 10.1021/cc060130p. Shore G, Morin S, Organ MG. Angew. Chem. Int. Ed. 2006;45:2761–2766. doi: 10.1002/anie.200503600. Comer E, Organ MG. J. Am. Chem. Soc. 2005;127:8160–8167. doi: 10.1021/ja0512069.
- 8.Maiorov VD, Kislina IS, Voloshenko GI, Librovich NB. Russ. Chem. Bull. Int. Ed. 2000;49:1526–1530. [Google Scholar]
- 9.Reddy DS, Judd WR, Aubé J. Org. Lett. 2003;5:3899–3902. doi: 10.1021/ol0355130. [DOI] [PubMed] [Google Scholar]
- 10.Please see supporting information for synthetic routes, full experimental details, and characterization data.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.