

Abstract
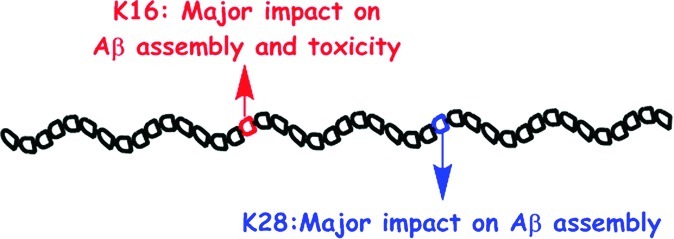
A combination of hydrophobic and electrostatic interactions is important in initiating the aberrant self-assembly process that leads to formation of toxic oligomers and aggregates by multiple disease-related proteins, including amyloid β-protein (Aβ), whose self-assembly is believed to initiate brain pathogenesis in Alzheimer’s disease. Lys residues play key roles in this process and participate in both types of interaction. They also are the target of our recently reported molecular tweezer inhibitors. To obtain further insight into the role of the two Lys residues in Aβ assembly and toxicity, here we substituted each by Ala in both Aβ40 and Aβ42 and studied the impact of the substitution on Aβ oligomerization, aggregation, and toxicity. Our data show that each substitution has a major impact on Aβ assembly and toxicity, with significant differences depending on peptide length (40 versus 42 amino acids) and the position of the substitution. In particular, Lys16→Ala substitution dramatically reduces Aβ toxicity. The data support the use of compounds targeting Lys residues specifically as inhibitors of Aβ toxicity and suggest that exploring the role of Lys residues in other disease-related amyloidogenic proteins may help understanding the mechanisms of aggregation and toxicity of these proteins.
Keywords: Alzheimer’s disease, amyloid, oligomers, aggregation, lysine, neurotoxicity
Introduction
Alzheimer’s disease (AD) is a progressive, age-related, neurodegenerative disorder, which gradually impairs cognitive abilities and leads to dementia and death. Amyloid plaques, neurofibrillary tangles, neurite dystrophy, synapse loss, and neurodegeneration in the cerebral cortex and hippocampus are pathologic hallmarks of AD.1 This pathological process is believed to occur as a result of self-assembly of amyloid β-protein (Aβ) into neurotoxic oligomers.2
Several lines of evidence suggest that a combination of hydrophobic and electrostatic interactions plays a key role in both Aβ oligomerization and its further aggregation into amyloid fibrils. Solid-state NMR studies have shown that in fibrils Aβ molecules are organized in parallel β-sheets, in which each monomer is folded into two β-strands connected by a turn. Hydrophobic interactions contribute to the stability of the β-sheets, whereas the turn is stabilized by a salt-bridge between the side chains of Asp23 and Lys28.3−5 A similar turn in monomeric Aβ has been reported to be the first structural element observed in Aβ folding and was proposed to nucleate Aβ folding and assembly,6 a suggestion that was supported by multiple experimental and computational studies.7−16 These data suggest that Lys28 may be a target for therapy. In line with this idea, recently Lys28 and residues in its vicinity have been reported to be a common binding site for inhibitors of Aβ40 toxicity.17
The other Lys residue in Aβ, Lys16, is adjacent to the central hydrophobic cluster (CHC, residues 17–21), a key region in Aβ fibrillogenesis,18,19 yet evidence for its involvement in Aβ assembly or toxicity is scarce. Lys16 has been reported to be involved in salt-bridges in certain fibrillar structures of Aβ,4,20,21 whereas other studies suggested that it was predominantly exposed to the solvent22,23 and thus available for interaction with other monomers, cell membranes, or potential inhibitors.24 Membrane permeabilization and perturbation has been suggested as a common toxic mechanism for amyloidogenic protein aggregates,25 including Aβ.26−28 Lys residues are likely mediators of membrane perturbation because they can participate in both electrostatic interactions with negatively charged phospholipid headgroups and hydrophobic interactions between the lipid hydrocarbon and the n-butylene moiety in the Lys side chain.29−33 Thus, removal of positively charged side chains from Aβ through triple substitution of Arg5, Lys16, and Lys28 for Ala resulted in significant loss of Aβ40 fibril toxicity in human embryonic kidney (HEK293) cells.34
Our recent investigation of the molecular tweezer (MT) CLR01 showed that the compound effectively inhibited the aggregation of nine amyloidogenic proteins, including Aβ40 and Aβ42, and rescued cultured cells from the toxic effect of oligomers formed by these proteins.35 CLR01 was found to prevent formation of oligomers recognized by the oligomer-specific antibody A1136 and to stabilize nontoxic Aβ oligomers by binding predominantly to the two Lys residues already in monomeric Aβ.35,37 CLR01 binds specifically to Lys residues by utilizing a combination of hydrophobic interactions between the Lys butylene and the side arms of the MT, together with Coulombic attraction between the ε-NH3+ group of Lys and the phosphates of CLR01, suggesting that both types of interactions the Lys residues normally would participate in are effectively blocked. To gain further insight into the consequences of blocking the interactions involving the Lys residues in Aβ, here we substituted each one separately and studied the effect of the substitution on the assembly and toxicity of Aβ40 and Aβ42.
Results
Secondary Structure Kinetics of WT and Lys→Ala Aβ Alloforms
Circular dichroism (CD) spectroscopy was used to determine the initial secondary structure and its temporal change in Aβ40, Aβ42 and their Ala-substituted alloforms during assembly. The three Aβ40 analogues and Aβ42 initially produced spectra characterized by a minimum at 196–198 nm, consistent with primarily a statistical coil conformation, and each gradually transformed into to a spectrum characterized by a minimum centered at 215–218 nm and a maximum at 196–197 nm indicating formation of β-sheet (Figure 1). [Ala16]Aβ42 also produced an initial spectrum characteristic of primarily a statistical coil but upon incubation, two inflections/minima developed at 208 and 216 nm, suggesting formation of a mixture of α-helical and β-turn/β-sheet conformations (hereafter referred to as β-sheet). The initial spectrum of [Ala28]Aβ42 did not show a minimum at 198 nm and rather was characterized by three inflections, at 198, 204, and 226 nm. With incubation, the secondary structure of [Ala28]Aβ42 changed gradually until the final spectrum showed a minimum at 212 nm and an inflection at 226 nm, suggesting that in addition to development of β-sheet, irregular conformations were present to a substantial extent at all stages of the aggregation reaction (Figure 1).
Figure 1.

Time-dependent changes in CD spectra of Aβ40, Aβ42 and their Lys→Ala analogues. 60 μM of each Aβ analogue was incubated in 10 mM phosphate buffer, pH 7.4, at 25 °C with constant agitation. CD spectra were acquired for each peptide at the indicated time points.
To allow quantitative comparison of the contribution of α-helical, β-sheet, and disordered elements during the conformational transition of the six Aβ analogues, the spectra were deconvoluted. The temporal change in each structural element is shown in Figure 2 and the initial and final secondary structure content calculated for each peptide are summarized in Table 1.
Figure 2.

Deconvolution of CD spectra. Each of the spectra was deconvoluted using the program CONTIN/LL with IBASIS = 1 to yield the relative contribution of statistical coil, α-helix, and β-sheet/β-turn conformation.58 The data show mean ± SD of three independent experiments.
Table 1. Initial and Final Secondary Structure Content in Aβ Analogues Calculated by Deconvolution of CD Spectra Using CONTIN/LL.
| α-helix (%) |
β-sheet/β-turn (%) |
statistical coil (%) |
||||
|---|---|---|---|---|---|---|
| peptide | initial | final | initial | final | initial | final |
| Aβ40 | 3 ± 2 | 0 | 23 ± 3 | 62 ± 6 | 74 ± 1 | 37 ± 5 |
| [Ala16]Aβ40 | 5 ± 1 | 0 | 20 ± 8 | 72 ± 6 | 75 ± 10 | 27 ± 6 |
| [Ala28]Aβ40 | 7 ± 4 | 0 | 21 ± 3 | 61 ± 3 | 72 ± 5 | 38 ± 3 |
| Aβ42 | 10 ± 6 | 0 | 37 ± 5 | 70 ± 2 | 52 ± 1 | 29 ± 2 |
| [Ala16]Aβ42 | 11 ± 5 | 60 ± 12 | 22 ± 8 | 2 ± 2 | 66 ± 4 | 38 ± 11 |
| [Ala28]Aβ42 | 14 ± 5 | 8 ± 8 | 15 ± 5 | 49 ± 19 | 72 ± 10 | 43 ± 11 |
Aβ40 analogues showed similar initial secondary structure content comprising 3–7% α-helix, 20–23% β-sheet, and 72–75% statistical coil. The contribution of α-helix decreased to zero within the first 24 h in all cases. In WT Aβ40, this was accompanied by an increase in the β-sheet content and a decrease in statistical coil, which were complete by 24 h. The conformational transition of [Ala16]Aβ40 followed a similar trend but was substantially slower than that of WT Aβ40 and appeared to be near completion only by 72 h. The final β-sheet content in [Ala16]Aβ40, 72 ± 6%, was notably higher than in WT Aβ40, 62 ± 6%, or [Ala28]Aβ40, 61 ± 3%. The conformational transition of [Ala28]Aβ40 was substantially slower than those of WT Aβ40 or [Ala16]Aβ40 and appeared incomplete at 72 h.
Aβ42 showed substantially higher initial β-sheet content (37 ± 5%) than all other peptides. Within 24 h, the initial 10 ± 6 α-helix content decreased to 0 whereas the β-sheet content increased to 70 ± 2% with concomitant decrease in statistical coil from 52 ± 1% to 29 ± 2%. These values were comparable to those of [Ala16]Aβ40, whereas all other Aβ analogues had lower final β-sheet content and higher statistical coil content. The initial conformation of [Ala16]Aβ42 comprised 10 ± 5% α-helix, 22 ± 8% β-sheet, and 66 ± 4% statistical coil. In contrast to all other peptides, the conformational transition of this variant involved a decrease in both statistical coil and β-sheet (to 38 ± 11% and 2 ± 2%, respectively) while the α-helix content increased to 60 ± 12%. This transition was substantially slower than any of the other analogues and appeared incomplete by 216 h. In [Ala28]Aβ42, a 10–15% α-helix content appeared unchanged for 240 h, whereas the β-sheet content increased from 15 ± 5% to 49 ± 19 and the statistical coil content decreased from 72 ± 10% to 43 ± 11% during this period. This was the highest final statistical coil content observed for any of the analogues. The conformational transition of [Ala28]Aβ42 was slow and reached a plateau only after 168 h.
Morphological Analysis
To examine the effect of the Lys→Ala substitutions on Aβ fibril structure, we examined the morphology of the native and substituted peptides by electron microscopy (EM) (Figure 3). Immediately after dissolution, all the peptides showed small quasi-globular or amorphous morphology as reported previously38 (data not shown), whereas at the end of the aggregation reaction, all the analogues showed fibrillar morphology (Figure 3). To complement the qualitative morphological characterization, we measured the fibril diameter and length using ImageJ.39 The average diameter distribution is shown under each micrograph and the quantitative analysis is summarized in Table 2.
Figure 3.

Morphology of Aβ40, Aβ42, and their Lys→Ala analogues. Electron micrographs were obtained for each analogue at the end of the aggregation reaction. Fibril diameter was measured at 50 randomly selected locations. The respective diameter histograms are shown below each micrograph. The histograms are fitted to a single Gaussian distribution. Scale bar: 100 nm.
Table 2. Quantitative Analysis of Morphological Features of the Aβ Analogues.
| peptide | fibril length (nm) | average diameter (nm) | nonfibrillar structures |
|---|---|---|---|
| Aβ40 | >2000 | 11 ± 3 | |
| [Ala16]Aβ40 | >2000 | 9 ± 2 | few, d = 15–20 nm |
| [Ala28]Aβ40 | 325 ± 170 | 16 ± 6 | few, d = 20–60 nm |
| Aβ42 | >2000 | 8 ± 3 | |
| [Ala16]Aβ42 | >2000 | 11 ± 3 | abundant, d = 45 ± 15 nm |
| [Ala28]Aβ42 | 213 ± 100 | 16 ± 6 |
Aβ40 and [Ala16]Aβ40 showed long unbranched fibrils. A few globular structures were observed for [Ala16]Aβ40 but not for WT Aβ40, in agreement with the slower conformational transition kinetics observed for the Ala16 variant. The fibrils of [Ala28]Aβ40 were thicker, shorter, branched, and ragged compared to WT Aβ40 or [Ala16]Aβ40, and were observed together with nonfibrillar structures, consistent with the CD data showing that the conformational transition of this variant was incomplete (Figure 3). In contrast to the narrow diameter distribution of WT Aβ40 and [Ala16]Aβ40, wide diameter distributions were observed for [Ala28]Aβ40 fibrils (Figure 3) and for the nonfibrillar [Ala28]Aβ40 aggregates (Table 2).
Similar to the Aβ40 analogues, Aβ42 and [Ala16]Aβ42 formed long unbranched fibrils. A unique feature of [Ala16]Aβ42 was abundant “decoration” of the fibrils by quasi-globular structures, 45 ± 15 nm in diameter. These structures might contain predominantly α-helical [Ala16]Aβ42, suggesting that the spectra shown in Figure 1 represent a mixture of two distinct populations, one predominantly α-helical and the other comprising typical β-sheet-rich fibrils, which is underrepresented in the deconvolution analysis in Figure 2. The fibrils of [Ala28]Aβ42 had a wide diameter distribution and were relatively short compared to those of WT Aβ42 or [Ala16]Aβ42, similar to the effect of the Lys28→Ala substitution on Aβ40.
Oligomer Size Distribution
Next, we asked whether the Lys→Ala substitutions affected the initial Aβ oligomer size distribution. To answer this question, we photochemically cross-linked freshly prepared solutions of each peptide using photochemical cross-linking of unmodified proteins (PICUP)40 and fractionated the resulting oligomers by SDS-PAGE (Figure 4). Uncross-linked control samples were run side-by-side with the cross-linked ones. Uncross-linked Aβ40 analogues migrated with an apparent mobility consistent with monomer. Low abundance dimer bands also were observed in all cases. Cross-linked Aβ40 had an oligomer size distribution comprising monomer through hexamer (Figure 4A), in which the dimer and trimer bands had the highest intensities, as described previously.41 The oligomer size distributions of [Ala16]Aβ40 and [Ala28]Aβ40 were similar to that of WT Aβ40 with slightly lower dimer and trimer abundance. Peculiarly, the Lys→Ala substitutions, particularly Lys28→Ala, appeared to decrease the silver-staining of all the bands relative to WT Aβ40, even though the total amount of peptide loaded in each lane was carefully controlled for and was the same for all peptides (Figure 4A). Densitometric analysis, in which each band was normalized to the total intensity of all the bands in its own lane, showed that the differences in relative abundance of each oligomer among the three Aβ40 analogues were insignificant (Figure 4B).
Figure 4.

Oligomer size distribution of Aβ analogues. (A) 60 μM of each peptide was cross-linked using PICUP, fractionated by SDS-PAGE and silver-stained. Positions of molecular weight standards are shown on the left. The gels are representative of three independent experiments. (B) Densitometric analysis of the oligomer size distributions. The data show mean ± SEM of three independent experiments.
Uncross-linked Aβ42 produced predominantly a monomer band and a broad trimer/tetramer band (Figure 4A), which previously has been reported to be an SDS-induced artifact.42,43 The oligomer size distribution of cross-linked Aβ42 oligomers was characterized by Gaussian-like oligomer abundance between tetramer and heptamer, with a maximum at pentamer and hexamer (paranuclei), consistent with previous reports.38,44 In the oligomer size distribution of [Ala16]Aβ42, the dimer and trimer abundance was significantly lower than the corresponding WT Aβ42 bands and the characteristic abundance maxima of paranuclei were shifted to tetramer and pentamer (Figure 4B). The highest oligomer observed for [Ala16]Aβ42 was hexamer, as opposed to WT Aβ42 for which low abundance heptamer and octamer also were observed. The oligomer size distribution of [Ala28]Aβ42 was similar to that of WT Aβ42, except that the tetramer band was not observed, and similarly to [Ala16]Aβ42, hexamer was the highest oligomer observed (Figure 4). Thus, the Lys→Ala substitutions had little impact on the initial oligomer size distributions of Aβ40 but each substitution changed the oligomer size distribution of Aβ42 significantly.
Cell Viability
To assess the impact of the Lys→Ala substitutions on the neurotoxic activity of Aβ, we added each peptide at concentrations ranging from 1–100 μM to differentiated PC-12 cells and measured LDH release following 48 h of incubation (Figure 5). Aβ40 showed negligible cell death up to 10 μM, 16 ± 2% cell death at 30 μM, and 53 ± 3% cell death at 100 μM. In contrast, [Ala16]Aβ40 was not toxic at all the concentrations studied. Similar to WT Aβ40, [Ala28]Aβ40 did not show toxicity up to 10 μM, whereas at 30 μM it caused 32 ± 3% cell death. The toxicity seemed to plateau at this level and did not increase at 100 μM, possibly due to limited solubility, though we did not observe precipitation or particulate material at this concentration.
Figure 5.

Toxicity of Aβ40, Aβ42, and their Ala-substituted analogues. Aβ40, Aβ42, and their respective Lys→Ala analogues were incubated with differentiated PC-12 cells for 48 h, and cell death was measured using the LDH release assay. The results are an average of four to six experiments and are presented as mean ± SEM.
Aβ42 caused 18 ± 3%, 42 ± 4%, and 56 ± 2% cell death at 10, 30, and 100 μM, respectively. The Lys16→Ala substitution again caused a dramatic decrease in toxicity. [Ala16]Aβ42 showed 13 ± 2% cell death at 100 μM and no toxicity at lower concentrations. [Ala28]Aβ42 at 30 and 100 μM caused 14 ± 3% and 37 ± 3% cell death, respectively. Thus, the Lys16→Ala substitution abolished the toxicity of Aβ40 completely and suppressed the toxicity of Aβ42 by >75%. In comparison, the effect of the Lys28→Ala substitution on Aβ toxicity was modest.
Discussion
Molecular-level understanding of the mechanism of Aβ assembly and toxicity is important for developing reagents that inhibit Aβ toxicity and may lead to treatment for AD. Our study shows that removal of the combination of hydrophobic and electrostatic interactions mediated by the Lys residues in Aβ has profound effects on the assembly and the toxicity of Aβ40 and Aβ42 and sheds light on the important roles of each Lys residue.
Substitution of Lys16 by Ala in Aβ40 had a relatively moderate effect on Aβ40 assembly. The conformational transition of [Lys16]Aβ40 was slower than that of WT Aβ40 (Figure 1), yet both peptides formed similar degree of β-sheet upon incubation (Figure 2), and both gave rise to typical long and unbranched amyloid fibrils (Figure 3). The two analogues also had similar initial distributions of small oligomers (Figure 4). However, this substitution practically abolished the toxicity of Aβ40, even at a high concentration such as 100 μM, suggesting that Lys16 is directly involved in the cellular interaction of Aβ40 oligomers that mediate toxicity. Supporting this view, most FAD-linked mutations within the Aβ-encoding sequence of the app gene lead to amino acid substitutions that increase the positive charge of Aβ.45 In addition, in a series of C-terminal fragments of Aβ42 with the general formula Aβ(x–42) (x = 28–39), the only peptide showing high toxicity was Aβ(28–42), in which the N-terminal residue was Lys, whereas all the other peptides had little or no toxicity.46 In comparison to the Lys16→Ala variants, substitution of Lys28 by Ala caused substantial perturbation of the assembly process in both Aβ40 and Aβ42, and in both Aβ alloforms, toxicity was affected to a lesser extent by the Lys28→Ala than by the Lys16→Ala substitution.
Though all three Aβ40 analogues formed similar initial oligomer size distributions (Figure 4) and all three converted over time from a largely unstructured conformation to β-sheet-rich fibrils, the rate of conformational transition varied substantially in the order Aβ40 > [Lys16]Aβ40 > [Lys28]Aβ40 (Figures 1 and 2). Interestingly, the final β-sheet content in [Ala16]Aβ40 was higher than in WT Aβ40 or [Ala28]Aβ40 (Figure 2), suggesting that the substitution of Lys16 by Ala may allow extension of the β-strand encompassing the CHC region47−49 to include residues N-terminal to the CHC.
In contrast to the Aβ40 analogues, the initial oligomer size distributions of the Aβ42 analogues were distinct (Figure 4) and each analogue underwent a unique structural transition. It is difficult to directly correlate the differences among the oligomer size distribution of each analogue with conformational transition and/or aggregation kinetics,44,50−52 as these processes occur on substantially different time scales and may be governed by distinct sets of interactions. Indeed, the initial Aβ oligomer populations detected using PICUP are characterized by a primarily statistical-coil conformation. Deconvolution of the CD spectra of [Ala16]Aβ42 showed development of high α-helix content (Figures 1 and 2), as opposed to all other Aβ analogues. EM examination of this peptide at the end of the aggregation reaction showed two distinct morphologies: typical amyloid fibrils together with abundant 45 ± 15 nm diameter quasi-spherical structures (Figure 3). Presumably, the high α-helix content observed for [Ala16]Aβ42 represents predominantly these quasi-spherical structures, whereas the fibrils observed are typical, β-sheet-rich amyloid fibrils that may be underrepresented in the CD spectra because α-helical structures in proteins generally show smaller geometric distortions compared to those observed in β-sheets and turns.54,55 [Ala28]Aβ42 showed substantial contribution of irregular structural elements throughout its conformational transition and, unlike all other analogues, had an α-helical element contributing ∼10% of the CD spectrum that appeared to remain stable for 10 days.
The stark difference between the conformational transition of [Ala16]Aβ40 and [Ala16]Aβ42 suggests that direct interaction between the C-terminus and CHC regions plays an important role in early Aβ folding and assembly, particularly in Aβ42, in agreement with modeling studies.47−49 The modeling studies suggested that, in Aβ40, because the C-terminus is shorter and less hydrophobic than in Aβ42, the N-terminus competes with the C-terminus for interaction with the CHC. Thus, disruption of C-terminus–CHC contacts would be predicted to affect Aβ42 more than Aβ40, consistent with our observations.
The substantially slower conformational transition kinetics of both [Ala28]Aβ40 and [Ala28]Aβ42, and the high content of irregular structures in [Ala28]Aβ42 support an important role of Lys28 in stabilizing the folding nucleus of Aβ, as suggested by Lazo et al.6 In agreement with the modeling data emphasizing an interaction between the C-terminal and CHC regions in Aβ42,47−49 our data suggest that destabilization of the turn within Aβ(21–30), which allows this interaction, by the Lys28→Ala substitution affects Aβ42 folding and assembly to a substantially larger extent than Aβ40.
Interestingly, comparison of the fibril morphology of the two Lys28→Ala variants to those of the WT or Lys16→Ala variants suggests that the rate of nucleation (kN) of the Lys28-substituted analogues is high relative to the rate of elongation (kE). Typically, a situation in which kE ≫ kN leads to formation of relatively few long fibrils, whereas abundant short fibrils are observed when kE ≪ kN, as is the case for [Lys28]Aβ40 and [Lys28]Aβ42 (Figure 3). Thus, the data suggest that the Lys28→Ala substitution facilitates nucleation, yet decreases elongation rate.
Our data suggest that disruption of the interactions mediated by the Lys residues in Aβ would suppress Aβ toxicity in two complementary ways. First, preventing such interactions perturbs the assembly process and the resulting oligomers/aggregates were found to be less toxic than those formed by WT Aβ40 or Aβ42. Second, interfering with the interaction of Lys16 with its cellular targets, presumably the cell membrane, might in itself inhibit Aβ toxicity substantially. Our CD experiments show that in Aβ42, the Lys16→Ala substitution induces formation of substantial α-helical content, which was not observed in the Aβ40 analogue. Nonetheless, the toxicity of both [Ala16]Aβ40 and [Ala16]Aβ42 was dramatically lower than the toxicity of their WT counterparts. This suggests that, in our experimental system, the secondary structure contributes little to the observed toxicity of these Aβ analogues and Lys16 by itself is a crucial player in mediating the toxicity. These results support the use of Lys-specific inhibitors, such as CLR01, as potential drugs for treatment and prevention of AD. The data also highlight the importance of the amphipathic Lys side chain in Aβ assembly and toxicity and suggest that Lys residues may have similar roles in other amyloidogenic proteins.
Methods
Peptides
Aβ40, Aβ42, [Ala16]Aβ40, and [Ala28]Aβ40 were purchased from the UCLA Biopolymers Laboratory. [Ala16]Aβ42 and [Ala28]Aβ42 were obtained from AnaSpec (Fremont, CA). Unless otherwise stated, samples of Aβ40, Aβ42, and their corresponding Lys→Ala variants were disaggregated by dissolution in 1,1,1,3,3,3,-hexafluoro-2-propanol (HFIP, Sigma, St. Louis, MO) followed by evaporation of the solvent to dryness as described previously.56 Dry peptide films were stored at −20 °C until use.
CD Spectroscopy
Dry, HFIP-treated peptide films were dissolved in a minimal volume of 60 mM NaOH followed by dilution with deionized water (18.2 MΩ produced using a Milli-Q system, Millipore, Bellerica, MA) to half the final volume and then sonicated for 1 min using a model 1510 bath sonicator (Branson, Danbury, CT). The peptides were diluted to 60 μM by adding an equal volume of 20 mM sodium phosphate buffer (PB, Sigma), pH 7.4, containing 0.02% (w/v) sodium azide (Sigma) to prevent bacterial growth. Samples were centrifuged at 16 000g using an Eppendorf model 5415C benchtop microcentrifuge (Brinkmann Instruments, Westbury, NY) for 3 min. The supernate, containing monomers and low molecular weight oligomers, was incubated at room temperature with agitation to allow peptide assembly in a 1 mm path length quartz cuvette (Hellma, Forest Hills, NY). At different time points, CD spectra were obtained using a J-810 spectropolarimeter (JASCO, Tokyo, Japan). Secondary structure content initially was calculated following buffer subtraction using the deconvolution programs Selcon3,57 CONTIN/LL,58 and CDSstr59 within the CDpro software package. CONTIN/LL consistently generated fits with the lowest root-mean-square deviations among these three programs and therefore was chosen for deconvolution of all CD spectra.
Electron Microscopy (EM)
Aliquots (10 μL) of the peptide solutions monitored by CD were spotted on glow-discharged, carbon-coated Formvar grids (Electron Microscopy Science, Hatfield, PA) at the end of the aggregation reaction, fixed with 5 μL of 2.5% (v/v) glutaraldehyde, and stained with uranyl acetate. The samples were analyzed using a CX 100 transmission electron microscope (JEOL, Peabody, MA). To evaluate the size and homogeneity of the aggregates, we measured the fibril length and fibril thickness at randomly selected points (N = 50) using ImageJ. The thickness frequency distribution was fitted to a single Gaussian distribution using Origin 6.1 (OriginLab, Northampton, MA).
Photoinduced Cross-linking of Unmodified Proteins (PICUP)
Initially, experiments were performed using samples prepared as described above in azide-free buffer with slight modifications of protocols described previously.40 Briefly, 1 μL of 6 mM tris(2,2′-bipyridyl)dichlororuthenium(II) (Sigma) and 1 μL of 120 mM ammonium persulfate (Sigma) in 10 mM PB were added to 18 μL of 60 μM peptide. The mixture was cross-linked by irradiation with visible light for 1 s, and the reaction was quenched immediately with 10 μL of Tricine sample buffer (Invitrogen, Carlsbad, CA) containing 5% β-mercaptoethanol (Sigma). Samples were analyzed using 10–20% Tris-Tricine gradient gels (Invitrogen). Peptide bands were visualized by silver staining (SilverXpress kit, Invitrogen) and analyzed by densitometry as described previously.41 The initial experiments showed that HFIP-treated Aβ40 yielded oligomer size distributions similar to those observed previously, in which aggregate-free-Aβ40 was prepared using mechanical methods,38,41 whereas the oligomer size distribution of Aβ42 was altered substantially by the HFIP treatment (Supporting Information Figure S1). To facilitate comparison with previous data, in subsequent experiments, the peptides were prepared as described above for the CD experiments but without HFIP treatment. To remove large aggregates that may be present in the preparation without HFIP treatment, the solutions were centrifuged for 10 min at 16 000g.
Cell Viability
Cell death was measured using the lactate dehydrogenase (LDH) release assay as described previously.46 Briefly, differentiated rat pheochromocytoma (PC-12) cells were plated at a density of 20 000 cells per well using 96-well plates in 90 μL fresh medium and incubated for 24 h. Aβ40, Aβ42, or their Lys→Ala variants were solubilized in a minimal volume of 60 mM NaOH and then diluted in F12K media to the desired concentration, added to the cells, and incubated for 48 h at 37 °C. No visible aggregates were observed in the peptide solution when added to the cells or at the end of the assay. The final concentration of NaOH in the media was <0.6 mM. Media with the same concentration of NaOH but no Aβ was used as a negative control. LDH release was measured using CytoTox ONE kits (Promega, Madison, WI) after 48 h of incubation with the respective peptides. The data are an average of three independent experiments with five wells per condition (n = 15).
Acknowledgments
We thank Dr. David Teplow for the use of his CD spectrometer and microplate reader and Dr. Huiyuan Li, Aida Attar, and Tanya Liu for useful discussions and critical reading of the manuscript.
Supporting Information Available
Cross-linking of HFIP-treated and untreated Aβ42. This material is available free of charge via the Internet at http://pubs.acs.org.
The work was supported by Grants A2008-350 from American Health Assistance Foundation and IIRG-07-58334 from Alzheimer’s Association, and by the UCLA Jim Easton Consortium for Alzheimer’s Drug Discovery and Biomarker Development.
The authors declare the following competing financial interest(s): Gal Bitan is a founder and a shareholder of Clear Therapeutics, Inc.
Supplementary Material
References
- Selkoe D. J. (2001) Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 81, 741–766. [DOI] [PubMed] [Google Scholar]
- Roychaudhuri R.; Yang M.; Hoshi M. M.; Teplow D. B. (2009) Amyloid β-protein assembly and Alzheimer disease. J. Biol. Chem. 284, 4749–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova A. T.; Ishii Y.; Balbach J. J.; Antzutkin O. N.; Leapman R. D.; Delaglio F.; Tycko R. (2002) A structural model for Alzheimer’s β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. U.S.A. 99, 16742–16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova A. T.; Leapman R. D.; Guo Z.; Yau W. M.; Mattson M. P.; Tycko R. (2005) Self-propagating, molecular-level polymorphism in Alzheimer’s β-amyloid fibrils. Science 307, 262–265. [DOI] [PubMed] [Google Scholar]
- Petkova A. T.; Yau W. M.; Tycko R. (2006) Experimental constraints on quaternary structure in Alzheimer’s β-amyloid fibrils. Biochemistry 45, 498–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazo N. D.; Grant M. A.; Condron M. C.; Rigby A. C.; Teplow D. B. (2005) On the nucleation of amyloid β-protein monomer folding. Protein Sci. 14, 1581–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borreguero J. M.; Urbanc B.; Lazo N. D.; Buldyrev S. V.; Teplow D. B.; Stanley H. E. (2005) Folding events in the 21–30 region of amyloid β-protein (Aβ) studied in silico. Proc. Natl. Acad. Sci. U.S.A. 102, 6015–6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz L.; Urbanc B.; Borreguero J. M.; Lazo N. D.; Teplow D. B.; Stanley H. E. (2005) Solvent and mutation effects on the nucleation of amyloid β-protein folding. Proc. Natl. Acad. Sci. U.S.A. 102, 18258–18263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumketner A.; Bernstein S. L.; Wyttenbach T.; Lazo N. D.; Teplow D. B.; Bowers M. T.; Shea J. E. (2006) Structure of the 21–30 fragment of amyloid β-protein. Protein Sci. 15, 1239–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.; Mousseau N.; Derreumaux P. (2006) The conformations of the amyloid-β (21–30) fragment can be described by three families in solution. J. Chem. Phys. 125, 084911. [DOI] [PubMed] [Google Scholar]
- Tarus B.; Thirumalai D.; Straub J. E. (2008) Structures and Free-Energy Landscapes of the Wild Type and Mutants of the Aβ21–30 Peptide Are Determined by an Interplay between Intrapeptide Electrostatic and Hydrophobic Interactions. J. Mol. Biol. 379, 815–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawzi N. L.; Phillips A. H.; Ruscio J. Z.; Doucleff M.; Wemmer D. E.; Head-Gordon T. (2008) Structure and dynamics of the Aβ(21–30) peptide from the interplay of NMR experiments and molecular simulations. J. Am. Chem. Soc. 130, 6145–6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant M. A.; Lazo N. D.; Lomakin A.; Condron M. M.; Arai H.; Yamin G.; Rigby A. C.; Teplow D. B. (2007) Familial Alzheimer’s disease mutations alter the stability of the amyloid β-protein monomer folding nucleus. Proc. Natl. Acad. Sci. U.S.A. 104, 16522–16527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krone M. G.; Baumketner A.; Bernstein S. L.; Wyttenbach T.; Lazo N. D.; Teplow D. B.; Bowers M. T.; Shea J. E. (2008) Effects of familial Alzheimer’s disease mutations on the folding nucleation of the amyloid β-protein. J. Mol. Biol. 381, 221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciarretta K. L.; Gordon D. J.; Petkova A. T.; Tycko R.; Meredith S. C. (2005) Aβ40-Lactam(D23/K28) models a conformation highly favorable for nucleation of amyloid. Biochemistry 44, 6003–6014. [DOI] [PubMed] [Google Scholar]
- Sandberg A.; Luheshi L. M.; Sollvander S.; Pereira de Barros T.; Macao B.; Knowles T. P.; Biverstal H.; Lendel C.; Ekholm-Petterson F.; Dubnovitsky A.; Lannfelt L.; Dobson C. M.; Hard T. (2010) Stabilization of neurotoxic Alzheimer amyloid-β oligomers by protein engineering. Proc. Natl. Acad. Sci. U.S.A. 107, 15595–15600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshet B.; Gray J. J.; Good T. A. (2010) Structurally distinct toxicity inhibitors bind at common loci on β-amyloid fibril. Protein Sci. 19, 2291–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esler W. P.; Stimson E. R.; Ghilardi J. R.; Lu Y. A.; Felix A. M.; Vinters H. V.; Mantyh P. W.; Lee J. P.; Maggio J. E. (1996) Point substitution in the central hydrophobic cluster of a human β-amyloid congener disrupts peptide folding and abolishes plaque competence. Biochemistry 35, 13914–13921. [DOI] [PubMed] [Google Scholar]
- de Groot N. S.; Aviles F. X.; Vendrell J.; Ventura S. (2006) Mutagenesis of the central hydrophobic cluster in Aβ42 Alzheimer’s peptide. Side-chain properties correlate with aggregation propensities. FEBS J. 273, 658–668. [DOI] [PubMed] [Google Scholar]
- Tycko R.; Sciarretta K. L.; Orgel J. P.; Meredith S. C. (2009) Evidence for novel β-sheet structures in Iowa mutant β-amyloid fibrils. Biochemistry 48, 6072–6084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy E.; Carman M. D.; Fernandez-Madrid I. J.; Power M. D.; Lieberburg I.; van Duinen S. G.; Bots G. T. A. M.; Luyendijk W.; Frangione B. (1990) Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch-type. Science 248, 1124–1126. [DOI] [PubMed] [Google Scholar]
- Zhang S. S.; Casey N.; Lee J. P. (1998) Residual structure in the Alzheimer‘s disease peptide - Probing the origin of a central hydrophobic cluster. Folding Des. 3, 413–422. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Iwata K.; Lachenmann M. J.; Peng J. W.; Li S.; Stimson E. R.; Lu Y.; Felix A. M.; Maggio J. E.; Lee J. P. (2000) The Alzheimer’s peptide Aβ adopts a collapsed coil structure in water. J. Struct. Biol. 130, 130–141. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Krause G.; Reif B. (2005) Structure and orientation of peptide inhibitors bound to β-amyloid fibrils. J. Mol. Biol. 354, 760–776. [DOI] [PubMed] [Google Scholar]
- Dobson C. M. (2006) Protein aggregation and its consequences for human disease. Protein Pept. Lett. 13, 219–227. [DOI] [PubMed] [Google Scholar]
- Kagan B. L.; Azimov R.; Azimova R. (2004) Amyloid peptide channels. J. Membr. Biol. 202, 1–10. [DOI] [PubMed] [Google Scholar]
- Kayed R.; Sokolov Y.; Edmonds B.; McIntire T. M.; Milton S. C.; Hall J. E.; Glabe C. G. (2004) Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 279, 46363–46366. [DOI] [PubMed] [Google Scholar]
- Butterfield D. A.; Castegna A.; Lauderback C. M.; Drake J. (2002) Evidence that amyloid β-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiol. Aging 23, 655–664. [DOI] [PubMed] [Google Scholar]
- Bokvist M.; Lindstrom F.; Watts A.; Grobner G. (2004) Two types of Alzheimer’s β-amyloid (1–40) peptide membrane interactions: aggregation preventing transmembrane anchoring versus accelerated surface fibril formation. J. Mol. Biol. 335, 1039–1049. [DOI] [PubMed] [Google Scholar]
- Chauhan A.; Ray I.; Chauhan V. P. (2000) Interaction of amyloid β-protein with anionic phospholipids: possible involvement of Lys28 and C-terminus aliphatic amino acids. Neurochem. Res. 25, 423–429. [DOI] [PubMed] [Google Scholar]
- Terzi E.; Holzemann G.; Seelig J. (1994) Reversible random coil-β-sheet transition of the Alzheimer β-amyloid fragment (25–35). Biochemistry 33, 1345–1350. [DOI] [PubMed] [Google Scholar]
- Williamson M. P.; Suzuki Y.; Bourne N. T.; Asakura T. (2006) Binding of amyloid β-peptide to ganglioside micelles is dependent on histidine-13. Biochem. J. 397, 483–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertel C.; Terzi E.; Hauser N.; Jakob-Rotne R.; Seelig J.; Kemp J. A. (1997) Inhibition of the electrostatic interaction between β-amyloid peptide and membranes prevents β-amyloid-induced toxicity. Proc. Natl. Acad. Sci. U.S.A. 94, 9412–9416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiike Y.; Akagi T.; Takashima A. (2007) Surface structure of amyloid-β fibrils contributes to cytotoxicity. Biochemistry 46, 9805–9812. [DOI] [PubMed] [Google Scholar]
- Sinha S.; Lopes D. H.; Du Z.; Pang E. S.; Shanmugam A.; Lomakin A.; Talbiersky P.; Tennstaedt A.; McDaniel K.; Bakshi R.; Kuo P. Y.; Ehrmann M.; Benedek G. B.; Loo J. A.; Klärner F. G.; Schrader T.; Wang C.; Bitan G. (2011) Lysine-specific molecular tweezers are broad-spectrum inhibitors of assembly and toxicity of amyloid proteins. J. Am. Chem. Soc. 133, 16958–16969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R.; Head E.; Thompson J. L.; McIntire T. M.; Milton S. C.; Cotman C. W.; Glabe C. G. (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489. [DOI] [PubMed] [Google Scholar]
- Sinha S.; Du Z.; Maiti P.; Klärner F.-G.; Schrader T.; Wang C.; Bitan G.. Comparison of three amyloid assembly inhibitors – the sugar scyllo-inositol, the polyphenol epigallocatechin gallate, and the molecular tweezer CLR01. ACS Chem Neurosci 2012, not supplied. [DOI] [PMC free article] [PubMed]
- Bitan G.; Kirkitadze M. D.; Lomakin A.; Vollers S. S.; Benedek G. B.; Teplow D. B. (2003) Amyloid β-protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. U.S.A. 100, 330–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramoff M. D.; Magelhaes P. J.; Ram S. J. (2004) Image Processing with ImageJ. Biophotonics Int. 11, 36–42. [Google Scholar]
- Bitan G. (2006) Structural study of metastable amyloidogenic protein oligomers by photo-induced cross-linking of unmodified proteins. Methods Enzymol. 413, 217–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitan G.; Lomakin A.; Teplow D. B. (2001) Amyloid β-protein oligomerization: prenucleation interactions revealed by photo-induced cross-linking of unmodified proteins. J. Biol. Chem. 276, 35176–35184. [DOI] [PubMed] [Google Scholar]
- Bitan G.; Fradinger E. A.; Spring S. M.; Teplow D. B. (2005) Neurotoxic protein oligomers–what you see is not always what you get. Amyloid 12, 88–95. [DOI] [PubMed] [Google Scholar]
- Hepler R. W.; Grimm K. M.; Nahas D. D.; Breese R.; Dodson E. C.; Acton P.; Keller P. M.; Yeager M.; Wang H.; Shughrue P.; Kinney G.; Joyce J. G. (2006) Solution state characterization of amyloid β-derived diffusible ligands. Biochemistry 45, 15157–15167. [DOI] [PubMed] [Google Scholar]
- Ono K.; Condron M. M.; Teplow D. B. (2009) Structure-neurotoxicity relationships of amyloid β-protein oligomers. Proc. Natl. Acad. Sci. U.S.A. 106, 14745–14750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Monien B. H.; Fradinger E. A.; Urbanc B.; Bitan G. (2010) Biophysical characterization of Aβ42 C-terminal fragments: inhibitors of Aβ42 neurotoxicity. Biochemistry 49, 1259–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradinger E. A.; Monien B. H.; Urbanc B.; Lomakin A.; Tan M.; Li H.; Spring S. M.; Condron M. M.; Cruz L.; Xie C. W.; Benedek G. B.; Bitan G. (2008) C-terminal peptides coassemble into Aβ42 oligomers and protect neurons against Aβ42-induced neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 105, 14175–14180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanc B.; Cruz L.; Yun S.; Buldyrev S. V.; Bitan G.; Teplow D. B.; Stanley H. E. (2004) In silico study of amyloid β-protein folding and oligomerization. Proc. Natl. Acad. Sci. U.S.A. 101, 17345–17350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun S.; Urbanc B.; Cruz L.; Bitan G.; Teplow D. B.; Stanley H. E. (2007) Role of Electrostatic Interactions in Amyloid β-Protein (Aβ) Oligomer Formation: A Discrete Molecular Dynamics Study. Biophys. J. 92, 4064–4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M.; Teplow D. B. (2008) Amyloid β-protein monomer folding: free-energy surfaces reveal alloform-specific differences. J. Mol. Biol. 384, 450–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maji S. K.; Amsden J. J.; Rothschild K. J.; Condron M. M.; Teplow D. B. (2005) Conformational dynamics of amyloid β-protein assembly probed using intrinsic fluorescence. Biochemistry 44, 13365–13376. [DOI] [PubMed] [Google Scholar]
- Maji S. K.; Ogorzalek Loo R. R.; Inayathullah M.; Spring S. M.; Vollers S. S.; Condron M. M.; Bitan G.; Loo J. A.; Teplow D. B. (2009) Amino acid position-specific contributions to amyloid β-protein oligomerization. J. Biol. Chem. 284, 23580–23591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitan G.; Vollers S. S.; Teplow D. B. (2003) Elucidation of primary structure elements controlling early amyloid β-protein oligomerization. J. Biol. Chem. 278, 34882–34889. [DOI] [PubMed] [Google Scholar]
- Manavalan P.; Johnson W. C. (1983) Sensitivity of circular dichroism to protein tertiary structure class. Nature 305, 831–832. [Google Scholar]
- Sreerama N.; Woody R. W. (2000) Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 287, 252–260. [DOI] [PubMed] [Google Scholar]
- Rahimi F., Maiti P., and Bitan G. (2009) Photo-induced cross-linking of unmodified proteins (PICUP) applied to amyloidogenic peptides J. Visualized Exp. DOI: dx.doi.org/10.3791/1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreerama N.; Woody R. W. (1993) A self-consistent method for the analysis of protein secondary structure from circular dichroism. Anal. Biochem. 209, 32–44. [DOI] [PubMed] [Google Scholar]
- Sreerama N.; Venyaminov S. Y.; Woody R. W. (2000) Estimation of protein secondary structure from circular dichroism spectra: inclusion of denatured proteins with native proteins in the analysis. Anal. Biochem. 287, 243–251. [DOI] [PubMed] [Google Scholar]
- Johnson W. C. (1999) Analyzing protein circular dichroism spectra for accurate secondary structures. Proteins 35, 307–312. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.