

Abstract
The present experiments employed in vivo microdialysis to characterize the effects of commonly used endocannabinoid clearance inhibitors on basal and depolarization-induced alterations in interstitial endocannabinoid levels in the nucleus accumbens of rat brain. Compounds targeting the putative endocannabinoid transporter and hydrolytic enzymes (FAAH and MAGL) were compared. The transporter inhibitor AM404 modestly enhanced depolarization-induced increases in 2-arachidonoyl glycerol (2-AG) levels but did not alter levels of N-arachidonoyl-ethanolamide (anandamide, AEA). The transport inhibitor UCM707 did not alter dialysate levels of either endocannabinoid. The FAAH inhibitors URB597 and PF-3845 robustly increased AEA levels during depolarization without altering 2-AG levels. The MAGL inhibitor URB602 significantly enhanced depolarization-induced increases in 2-AG, but did not alter AEA levels. In contrast, the MAGL inhibitor JZL184 did not alter 2-AG or AEA levels under any condition tested. Finally, the dual FAAH/MAGL inhibitor JZL195 significantly enhanced depolarization-induced increases in both AEA and 2-AG levels. In contrast to the present observations in rats, prior work in mice has demonstrated a robust JZL184-induced enhancement of depolarization-induced increases in dialysate 2-AG. Thus, to further investigate species differences, additional tests with JZL184, PF-3845, and JZL195 were performed in mice. Consistent with prior reports, JZL184 significantly enhanced depolarization-induced increases in 2-AG without altering AEA levels. PF-3845 and JZL195 produced profiles in mouse dialysates comparable to those observed in rats. These findings confirm that interstitial endocannabinoid levels in the brain can be selectively manipulated by endocannabinoid clearance inhibitors. While PF-3845 and JZL195 produce similar effects in both rats and mice, substantial species differences in JZL184 efficacy are evident, which is consistent with previous studies.
Keywords: Endocannabinoid transporter, FAAH, in vivo microdialysis, MAGL, nucleus accumbens, endogenous cannabinoid system
The endocannabinoid (eCB) system is a relatively novel neurotransmitter system that is important in the regulation of a range of physiological and behavioral processes such as pain sensation,1 inflammation,2 memory and learning,3 metabolism and energy homeostasis,4 executive functioning,5 stress and anxiety,6 and reward and motivational processes.7 The eCB system consists of at least two G-protein coupled receptors, denoted CB1 and CB2 receptors, and several endogenous ligands including N-arachidonoyl-ethanolamide (anandamide, AEA) and 2-arachidonoyl glycerol (2-AG).8−11
AEA and 2-AG levels in the brain are strictly regulated by synthesis and degradation. As opposed to classical neurotransmitters, eCBs are thought to be primarily synthesized “on demand” following a rise in intracellular calcium and/or activation of Gq/11-coupled receptors such as group I metabotropic glutamate receptors.12−14 Because AEA and 2-AG may each be generated via multiple synthetic pathways, the development of biosynthetic inhibitors has been challenging and at present no reasonably selective inhibitors are available. Although some compounds such as tetrahydrolipstatin (THL) can inhibit DAGLα activity and effectively reduce 2-AG synthesis in cell culture systems,15 these compounds also inhibit a number of other serine hydrolases16 and therefore do not exhibit sufficient selectivity for studies employing systemic administration. Accordingly, it is presently not viable to manipulate in vivo brain eCB levels through modulation of biosynthetic mechanisms. After synthesis and release, eCB signaling is terminated by reuptake into both neurons and glia followed by intracellular hydrolysis. The existence of an active transporter that facilitates reuptake of AEA and/or 2-AG from the synaptic cleft continues to be vigorously debated (refs (17 and 18), but see ref (19)). In contrast, the hydrolytic enzymes that provide the primary clearance routes for AEA (fatty acid amide hydrolase, FAAH) and 2-AG (monoacylglycerol lipase, MAGL) have been cloned and are well characterized.20−23 The development of pharmacological tools to manipulate eCB levels through inhibition of eCB transport or FAAH/MAGL activity has allowed initial elucidation of the distinctions and similarities in the influence of AEA and 2-AG on various behavioral and physiologicial functions.10,24−26
Characterization of the efficacy and pharmacologic profile of eCB clearance inhibitors has primarily been performed using ex vivo models, often combining data showing inhibitor-induced changes in brain tissue eCB content with behavioral observations.1,26−31 Relatively little in vivo data has been gathered characterizing the temporal profile and magnitude of eCB accumulation in the brain extracellular space following clearance inhibition. The available in vivo data are generally consistent with the pharmacologic profiles observed using ex vivo models,29,32,33 though the relative magnitude of the observed eCB changes often differs substantially between ex vivo and in vivo approaches.29,34 This may result from several factors that uniquely influence these distinct approaches for brain eCB quantification (for review, see ref (34)), and for this reason there is value in gathering comparative data in each experimental construct. In addition, the majority of studies characterizing eCB clearance inhibitor effects have been conducted in mice and it is possible that species differences in compound efficacy exist.35
Therefore, our aim in the current experiments was to characterize the effects of several commonly used eCB reuptake and hydrolysis inhibitors on interstitial eCB levels using in vivo brain microdialysis in rats. The experiments were conducted in the nucleus accumbens (NAc), a brain region where we have previously measured chemically induced changes in extracellular eCB levels.29,34,36−38 For comparative reference, additional experiments with selected FAAH, MAGL, and dual FAAH/MAGL inhibitors were performed in mice. The intent of these evaluations was to provide qualitative cross-species comparison of inhibitor efficacy rather than quantitative comparisons of dose efficacy.
Results and Discussion
A variety of compounds with varying degrees of selectivity and efficacy have been developed for the inhibition of eCB uptake and/or hydrolysis. In general, compounds were selected for the present study based on the number and variety of published reports employing each inhibitor in an effort to provide a behavioral and/or physiological context for the observed effects on interstitial eCB levels. We examined the effects of systemic administration of the putative eCB transporter inhibitors UCM707 and AM404, the FAAH inhibitors URB597 and PF-3845, the MAGL inhibitors URB602 and JZL184, and the dual FAAH/MAGL inhibitor JZL195 on interstitial eCB levels in the NAc of rats and mice using in vivo microdialysis. Because eCB synthesis and/or release are dependent on neuronal activation,39−41 we evaluated inhibitor effects on both unperturbed baseline eCB levels and stimulated levels induced by neuronal depolarization (produced by delivery of a high K+/Ca2+ perfusate). The effects of this manipulation on dialysate eCB content are blocked by coperfusion with the Na+ channel blocker tetrodotoxin,29,33,34,39 confirming that high K+/Ca2+ perfusates increase dialysate eCB levels in an impulse-dependent manner.
In total, nine groups of rats were used to test the in vivo effects of eCB clearance inhibition. For each drug treatment group, mean dialysate concentrations for AEA and 2-AG prior to drug administration are depicted in Table 1. Statistical analyses showed no significant differences between the different treatment groups for either eCB (AEA: F8,47 = 1.95, NS; 2-AG: F8,47 = 1.45, NS). Subsequently, all data were expressed as a percentage of the average baseline concentration to facilitate evaluation of drug treatment effects and depolarization.
Table 1. Mean Baseline Nucleus Accumbens Dialysate Concentrations (nM ± SEM) of AEA and 2-AG in Rats Per Drug Treatment Group.
| drug treatment | n | AEA (nM) | 2-AG (nM) |
|---|---|---|---|
| vehicle | 6 | 3.34 ± 1.32 | 8.14 ± 1.55 |
| UCM707 10 mg/kg | 6 | 1.94 ± 0.35 | 7.80 ± 1.29 |
| AM404 1 mg/kg | 7 | 2.10 ± 0.14 | 9.44 ± 1.08 |
| URB597 0.3 mg/kg | 6 | 2.16 ± 0.37 | 11.02 ± 1.36 |
| PF-3845 0.3 mg/kg | 6 | 1.63 ± 0.25 | 7.78 ± 1.71 |
| PF-3845 10 mg/kg | 6 | 1.25 ± 0.19 | 10.51 ± 2.49 |
| URB602 10 mg/kg | 7 | 3.31 ± 1.38 | 7.05 ± 1.24 |
| JZL184 10 mg/kg | 6 | 0.73 ± 0.13 | 6.10 ± 0.67 |
| JZL195 10 mg/kg | 6 | 1.18 ± 0.29 | 8.21 ± 0.95 |
Delivery of a High K+/Ca2+ Perfusate Does Not Induce Substantial Changes in NAc Dialysate eCB Levels in Vehicle-Treated Rats
Initial studies characterized the effects of neuronal depolarization (probe perfusion with aCSF containing a high concentration of KCl (90 mM) and CaCl2 (10 mM) for 45 min) on eCB release in the NAc of vehicle-treated rats. As shown in Figure 1a, applying an ionic pulse did not significantly affect dialysate eCB levels in vehicle-treated rats (AEA: F14,70 = 0.92, NS; 2-AG: F14,70 = 1.04, NS). These findings stand in contrast with previous rat microdialysis studies that observed significant increases in dialysate eCB levels after perfusion with similar ionic solutions.33,39 This distinction may reflect regional differences in depolarization-induced stimulation of eCB production as each of these experiments was performed in different brain regions [NAc (present study), dorsal striatum,39 and hypothalamus33]. However, a more likely cause of these differential observations relates to procedural issues. The prior studies employed microdialysis probes with larger areas of active membrane along with much higher perfusate flow rates, likely resulting in delivery of greater amounts of K+ and Ca2+ than achieved in the present experiment. In addition, factors related to the lipid nature of eCBs may affect their sampling by microdialysis in a manner that counters the efficacy of this method for indexing rapid and/or small changes in eCB formation. For example, several “chaperone” mechanisms have been theorized for eCB transport through the aqueous synaptic space including associations with exosomes and fatty acid binding proteins. These putative eCB–chaperone complexes may be sufficiently large so as to impede their diffusion across the dialysis membrane, thereby diminishing eCB sampling efficiency and blunting the perceived magnitude of depolarization-induced increases in interstitial eCB levels. The differential magnitude of depolarization-induced increases dialysate eCB versus monoamine or acetylcholine levels (up to 160% of baseline versus 300–1000% of baseline, respectively; see ref (34) for discussion) is consistent with this possibility. However, it is also possible that dialysis measures of depolarization-induced increases in eCB formation are countered by avid eCB clearance mechanisms that effectively mask stimulation-induced increases in eCB formation.
Figure 1.

Effects of eCB transporter inhibition on depolarization-induced increases in rat dialysate eCB levels. Effects of systemic administration of vehicle (1 mL/kg; a) or the eCB transport inhibitors UCM707 (10 mg/kg; b) and AM404 (1 mg/kg; c) on extracellular AEA and 2-AG levels prior to, during, and following application of an ionic pulse. Arrows indicate time of drug administration, while the shaded area from t = 0–45 min indicates that time that the ionic pulse was locally applied to the tissue. Data depict mean ± SEM per time point. Per treatment group n = 6–7 rats were included in the analyses. #p < 0.05 versus mean baseline 2-AG value.
eCB Transporter Inhibitors Modestly Enhance Depolarization-Induced Increases in 2-AG without Affecting AEA Levels in Rat NAc Dialysates
Cellular eCB uptake may occur through various mechanisms, including simple rate-limited diffusion across membranes, lipid raft-mediated endocytosis and through a putative membrane transporter.18,42,43 Although the existence of an eCB transporter is still vigorously debated,17,18 systemic administration of putative eCB transporter-inhibitors such as AM404 and UCM707 increases AEA and to a lesser degree 2-AG levels in bulk brain tissue40,44 and produces a variety of behavioral effects.30,31,45,46
The present data demonstrate that AM404 (1 mg/kg; Figure 1b) produces more robust effects on depolarization-induced increases in dialysate eCB levels than does UCM707 (10 mg/kg; Figure 1c). Specifically, in AM404-pretreated rats, depolarization significantly increased dialysate levels of both AEA and 2-AG (AEA: F18,108 = 2.16, p < 0.01; 2-AG: F18,108 = 4.14, p < 0.001), though post hoc analyses confirmed significant enhancement of only 2-AG levels from between t = 45 and 150 min. Importantly, we have observed similarly selective AM404 effects on 2-AG versus AEA with higher inhibitor doses (3 mg/kg; data not shown). In contrast, no significant effect of depolarization was evident on AEA or 2-AG levels in UCM707-pretreated rats (AEA: F14,70 = 1.67, NS; 2-AG: F14,70 = 1.49, NS), and neither AM404 nor UCM707 significantly altered dialysate eCB levels prior to depolarization. Thus, AM404 and UCM707 doses that significantly enhance brain tissue eCB content in post-mortem analyses40,44 produce nonsignificant effects on baseline interstitial eCB levels, and AM404 (but not UCM707) only modestly enhances depolarization-induced increases in interstitial 2-AG content as indexed by in vivo microdialysis. Similar conclusions are drawn from comparisons of area under the curve (AUC) data for the UCM707 and AM404 groups versus vehicle controls (Figure 6), as no significant differences were found for either compound, despite a trend toward AM404-related enhancement of depolarization-induced increases in 2-AG (UCM707: AEA: t10 = −0.83, NS; 2-AG: t10 = −0.90, NS; AM404: AEA: t11 = −0.04, NS; 2-AG: t11 = −1.50, p = 0.08).
Figure 6.

Comparison of the effects of eCB clearance inhibition on depolarization-induced alterations in rat and mouse dialysate eCB levels. Shown are the AUC data summarizing the effects of systemic administration of various eCB clearance inhibitors on extracellular AEA and 2-AG levels in rats (a) and mice (b) during and following application of an ionic pulse (t = 0–150 min). Compounds evaluated in both species included vehicle, PF-3845 (FAAH inhibitor; 0.3 mg/kg), JZL184 (MAGL inhibitor; 10 mg/kg), and JZL195 (dual FAAH/MAGL inhibitor; 10 and 20 mg/kg in rats and mice, respectively). Additional compounds evaluated in rats only include URB597 (FAAH inhibitor; 0.3 mg/kg), URB602 (MAGL inhibitor; 10 mg/kg), and the eCB transporter inhibitors UCM707 (10 mg/kg) and AM404 (1 mg/kg). Data depict mean ± SEM for each treatment (n = 6–10 each). *p < 0.05 and **p < 0.005 versus data from vehicle-treated controls.
The modest effects of these uptake inhibitors on dialysate eCB levels are somewhat surprising in light of the robust effects of these compounds on post-mortem brain tissue eCB content. Although the modest changes in dialysate eCB content may result in part from inefficient lipid recovery by microdialysis (see discussion in prior section), far more pronounced changes in in vivo dialysate eCB levels have been observed following other manipulations including FAAH/MAGL inhibition (see below and refs (29, 33, and 39)) and other pharmacological challenges,32,36 indicating that the subtle effects of AM404 and UCM707 in the present studies do not result from a limited “dynamic range” of microdialysis eCB sampling. It is possible that more robust effects would be evident with higher doses of each transporter inhibitor, though administration of a 3-fold higher dose of AM404 has been found to produce comparable effects as reported here (ref (32) and unpublished observations). In addition to inhibiting the putative eCB transporter, AM404 and UCM707 also interact with various other targets including TRPV1 and CB2 (for discussion, see ref (47)). The influence of these off-target actions on eCB biosynthesis and/or clearance is not well characterized, though it is possible these interactions influence the overall effects of these compounds on interstitial eCB levels. Prior studies suggest that, by occluding the putative eCB transporter AM404, UCM707 and related compounds attenuate eCB release.48,49 While this action would be expected to diminish interstitial eCB levels, no evidence of this effect was found in the present or prior microdialysis experiments evaluating the effects of eCB transport inhibitors.32 The relatively greater effects of AM404 on 2-AG versus AEA is surprising in light of evidence that this compound inhibits FAAH activity (see ref (43) for discussion) and the function of a recently described catalytically silent FAAH-1 variant that facilitates AEA (but not 2-AG) translocation into cells in in vitro assays.50 Because selective FAAH inhibition by URB597 and PF-3845 potently enhances dialysate AEA levels (see below), the present observations suggest that at the tested dose AM404 does not induce sufficient FAAH inhibition to influence interstitial AEA levels. These findings are consistent with prior microdialysis studies demonstrating that AM404 selectively potentiates alcohol-induced increases in NAc 2-AG without affecting alcohol-induced alterations in NAc AEA.32
FAAH Inhibition Selectively Potentiates Depolarization-Induced Increases in Dialysate AEA levels from Rat NAc
FAAH has been identified as the enzyme primarily responsible for the hydrolytic clearance of AEA with minimal effects on 2-AG clearance.10,20,21 Consistent with this, the present data demonstrate that the selective FAAH inhibitors URB597 (0.3 mg/kg; Figure 2a) and PF-3845 (0.3 mg/kg; Figure 2b) specifically enhance depolarization-induced increases in dialysate AEA but not 2-AG levels (URB597: AEA: F14,70 = 7.17, p < 0.001; 2-AG: F14,70 = 0.61, NS; PF-3845: AEA: F18,90 = 6.31, p < 0.001; 2-AG: F18,90 = 1.41, NS). Post hoc analyses showed significant elevations in AEA levels relative to baseline evident from t = 15 to 90 min and t = 15 to 150 min for URB597 and PF-3845, respectively (preliminary findings with URB597 are also found in ref (34)). It is noteworthy that while depolarization-induced increases in dialysate AEA were sustained beyond the ionic pulse delivery in PF-3845 treated rats, AEA levels declined back toward baseline following cessation of the pulse in URB597 treated rats. It is possible this reflects the shorter duration of FAAH inhibition exhibited by URB597 versus PF-3845.28 AUC analyses (Figure 6) further illustrate the selective effects of these compounds on AEA versus 2-AG as both compounds significantly increased dialysate AEA but not 2-AG levels relative to those observed in vehicle-treated controls (URB597: AEA: t10 = −2.80, p < 0.01; 2-AG: t10 = 0.41, NS; PF-3845: AEA: t10 = −2.32, p < 0.05; 2-AG: t10 = −0.13, NS).
Figure 2.

Effects of systemic administration of the FAAH inhibitors URB597 (0.3 mg/kg; a) and PF-3845 (0.3 and 10 mg/kg depicted in b and c, respectively) on extracellular AEA and 2-AG levels prior to, during, and following application of an ionic pulse. Arrows indicate time of drug administration, while the shaded area from t = 0 to 45 min indicates that time that the ionic pulse was locally applied to the tissue. Data depict mean ± SEM per time point. Per treatment group n = 6–7 rats were included in the analyses. *p < 0.05 versus mean baseline AEA values.
A 0.3 mg/kg dose was selected for initial tests, as this is the most commonly employed URB597 dose for inducing significant behavioral effects27,46,51−55 and robust enhancement of brain tissue AEA content in rats.55 However, most in vivo PF-3845 studies have been conducted in mice and have typically employed a 10 mg/kg dose. Thus, for comparison with this literature and subsequent microdialysis tests in mice (see below), we also characterized the effects of a higher PF-3845 dose. As shown in Figure 2c, 10 mg/kg PF-3845 significantly potentiated dialysate AEA levels without altering levels of 2-AG in these same samples (PF-3845: AEA: F18,90 = 6.05, p < 0.001; 2-AG: F18,90 = 1.47, NS). Interestingly, this higher PF-3845 dose increased AEA levels even prior to the administration of the ionic pulse (F8,40 = 8.22, p < 0.001, significant at t = −60 to 0 min) with significant increases observed from t= −60 to 0 min. Similar increases in nonstimulated AEA levels have been observed in rat hypothalamic microdialysates following administration of higher URB597 doses than employed in the present study.33 While depolarization-induced increases in dialysate AEA were somewhat greater in animals given 10 mg/kg versus 0.3 mg/kg PF-3845, the subtlety of the dose difference is in general agreement with the similar elevations in bulk tissue AEA content produced by 1 versus 10 mg/kg PF-3845.28 Collectively, these findings suggest that while alterations in nonstimulated interstitial AEA levels require relatively high FAAH inhibitor doses, the impact of FAAH inhibition on accumulation of stimulated AEA production is achieved at relatively low doses.
MAGL Inhibition Selectively Augments Depolarization-Induced Increases in Dialysate 2-AG Levels from Rat NAc
Whereas FAAH is primarily responsible for the in vivo hydrolysis of AEA, MAGL is the main enzyme hydrolyzing 2-AG in the brain.22,23 Here, we tested the effects of two selective MAGL inhibitors, URB602 (10 mg/kg; Figure 3a) and JZL184 (10 mg/kg; Figure 3b). Results demonstrate that only the former compound affects eCB levels following application of the ionic pulse. Overall analyses indicated that URB602 affected extracellular 2-AG levels (F14,84 = 5.23, p < 0.001), with post hoc analyses showing significant enhancement (∼2-fold) of 2-AG levels from baseline between t = 90 min and t = 150 min. These results are somewhat surprising given that URB602 is generally thought to be unsuited for systemic injection due to low potency.56,57 Further, despite in vitro evidence that URB602 is not selective for 2-AG versus AEA hydrolysis,58 no significant change in dialysate AEA levels from predrug baseline were evident despite a significant overall effect of time (F14,84 = 1.99, p < 0.05). Accordingly, our results are in line with an initial report demonstrating URB602 selectivity for MAGL versus FAAH.1 Interestingly, the observed magnitude of URB602-induced enhancement of stimulated 2-AG levels in rats is similar to that reported for the more potent MAGL inhibitor JZL184 in mice.29 However, in contrast to these prior observations in mice, JZL184 failed to alter dialysate 2-AG levels in rats. While there was a significant overall effect of time on dialysate 2-AG levels in JZ184-treated rats (AEA: F18,90 = 1.36, NS; 2-AG: F18,90 = 2.46, p < 0.01), post hoc analyses failed to reveal any significant deviation from predrug baseline even under depolarization conditions. The differential efficacies of URB602 and JZL184 in rats are further underscored by evaluation of AUC measures (Figure 6; URB602: AEA: t11 = 1.50, NS; 2-AG: t11 = −2.59, p < 0.05; JZL184: AEA: t10 = 1.04, NS; 2-AG: t10 = 1.51, NS). The distinct effects of JZL184 in rats (present study) and mice (ref (29) and text below) likely result from the ∼10-fold lower activity of JZL184 against rat MAGL as compared with mouse and human MAGL.35 Nonetheless, recent studies have reported significant behavioral effects of moderate JZL184 doses in rats (8–10 mg/kg59,60), though these effects were observed in a different rat strain (Sprague–Dawley) and with a different drug vehicle59 or administration route60 than used here. Despite the diminished activity of JZL184 on rat MAGL, in vitro estimates suggest this compound should be a substantially more potent MAGL inhibitor than URB602.35,58 Accordingly, it is possible that URB602-induced potentiation of depolarization-induced increases in dialysate 2-AG result from MAGL-independent effects of this compound.
Figure 3.

Effects of systemic administration of the MAGL inhibitors URB602 (10 mg/kg; a) and JZL184 (10 mg/kg; b) on extracellular AEA and 2-AG levels prior to, during, and following application of an ionic pulse. Arrows indicate time of drug administration, while the shaded area from t = 0–45 min indicates that time that the ionic pulse was locally applied to the tissue. Data depict mean ± SEM per time point. Per treatment group n = 6–7 rats were included in the analyses. #p < 0.05 versus mean baseline 2-AG values.
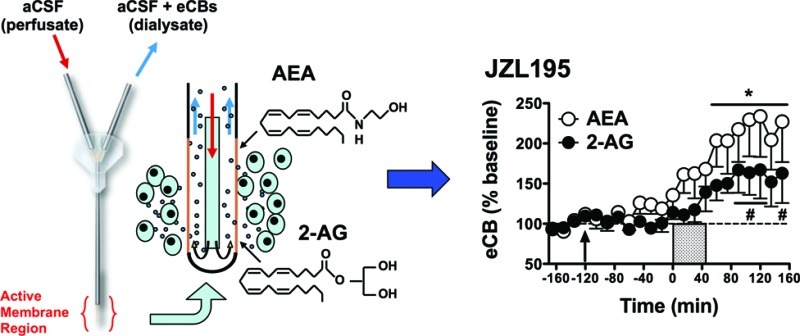
Dual FAAH-/MAGL Inhibition Enhances Depolarization-Induced Increases in Levels of Both AEA and 2-AG in Rat NAc Dialysates
Distinct behavioral and physiological effects have been ascribed to AEA and 2-AG signaling, and interactive effects of these eCBs on some aspects of behavior have been reported.26 Recently, JZL195 has been characterized as a dual inhibitor of FAAH and MAGL activity resulting in robust elevations in brain tissue levels of both AEA and 2-AG.26 Consistently, JZL195 (10 mg/kg) induced similar effects on interstitial AEA and 2-AG levels in the present study (Figure 4). JZL195 significantly enhanced depolarization-induced increases in dialysate levels of both AEA and 2-AG (AEA: F18,90 = 5.21, p < 0.001; 2-AG: F18,90 = 3.82, p < 0.001) and post hoc analyses indicated significant enhancement of AEA levels over baseline from t = 60–150 min. The effects of JZL195 on dialysate 2-AG were less pronounced, with significant elevations observed only at t = 90–120 min and t = 150 min. Differential JZL195 efficacy on AEA versus 2-AG was also reflected in the AUC data (Figure 6) with significant inhibitor effects evident on AEA levels but only a trend toward increased 2-AG levels relative to vehicle-treated controls (AEA: t10 = −3.31, p < 0.01; 2-AG: t10 = −1.73, p = 0.06). Interestingly, JZL195 significantly increased AEA (F8,40 = 2.55, p < 0.05, significant at t = 0 min) but not 2-AG (F8,40 = 1.84, p = 0.10) levels prior to depolarization. This differential efficacy of JZL195 to increase AEA versus 2-AG levels in rats is not surprising considering that the chemical structure of this dual-inhibitor is based on the active modalities of PF-3845 and JZL184,26 with the latter compound being ineffective in rats (Figure 3). For this reason, it is notable that JZL195 produced more robust elevations in rat dialysate 2-AG levels than JZL184.
Figure 4.

Effects of systemic administration of the dual FAAH-/MAGL inhibitor JZL195 (10 mg/kg) on extracellular AEA and 2-AG levels prior to, during, and following application of an ionic pulse. Arrows indicate time of drug administration, while the shaded area from t = 0–45 min indicates that time that the ionic pulse was locally applied to the tissue. Data depict mean ± SEM per time point. Per treatment group n = 6–7 rats were included in the analyses. * and # p < 0.05 versus mean baseline AEA and 2-AG values, respectively.
Comparative Experiments in Mice Confirm Species Differences in the Efficacy of JZL184
In light of the negative effect of JZL184 on dialysate 2-AG in rats and lesser effects of JZL195 on dialysate 2-AG versus AEA in rats, we performed a series of comparative studies evaluating the effects of JZL184 and JZL195 on AEA and 2-AG levels in microdialysates collected from the NAc of mice. For a full comparison, the effects of PF-3845 were also evaluated in mice. The average baseline dialysate AEA and 2-AG concentrations for each treatment group are shown in Table 2, and there were no significant between group differences (AEA: F3,26 = 2.72, NS; 2-AG: F3,26 = 0.72, NS). These experiments in mice employed a slightly modified ionic pulse protocol (probe perfusion with aCSF containing a high concentration of KCl (150 mM) and CaCl2 (10 mM) for 90 min as performed previously29,34). In contrast to our observations in rats, but consistent with our prior observations in mice,29 we observed a significant depolarization-induced increase in dialysate 2-AG levels in vehicle-treated mice (Figure 5a; AEA: F18,90 = 1.82, p < 0.05; 2-AG: F18,90 = 2.84, p < 0.001). Post hoc analyses revealed that dialysate 2-AG significantly differed from baseline at t = 60 min, while AEA levels did not differ from baseline at any point despite indication of significant changes in repeated measures over time. It is likely that the significant effects of depolarization in mice, but not rats, result from the more aggressive depolarization conditions in mice (e.g., more prolonged delivery of a perfusate containing somewhat higher K+ levels than employed in rats) that were employed to compensate for the lesser area of active dialysis membrane in these animals.
Table 2. Mean Baseline Nucleus Accumbens Dialysate Concentrations (nM ± SEM) of AEA and 2-AG in Mice Per Drug Treatment Group.
| drug treatment | n | AEA (nM) | 2-AG (nM) |
|---|---|---|---|
| vehicle | 6 | 0.62 ± 0.10 | 6.34 ± 1.52 |
| PF-3845 10 mg/kg | 8 | 0.55 ± 0.12 | 7.15 ± 1.37 |
| JZL184 10 mg/kg | 6 | 0.53 ± 0.07 | 4.86 ± 0.59 |
| JZL195 20 mg/kg | 10 | 0.36 ± 0.04 | 6.00 ± 0.91 |
Figure 5.

Effects of systemic administration of vehicle (0.1 mL/kg; a), the FAAH inhibitor PF-3845 (10 mg/kg; b), the MAGL inhibitor JZL184 (10 mg/kg; c), and the dual FAAH/MAGL inhibitor JZL195 (20 mg/kg; d) on extracellular AEA and 2-AG levels prior, during, and following application of an ionic pulse. Arrows indicate time of drug administration, while the shaded area from t = 0 to 90 min indicates that time that the ionic pulse was locally applied to the tissue. Data depict mean ± SEM per time point. Per treatment group n = 6–10 mice were included in the analyses. * and # p < 0.05 versus mean baseline AEA and 2-AG values, respectively.
Similar to our observations in rats, PF-3845 (10 mg/kg) induced robust effects on AEA release (Figure 5b; F18,126 = 4.60, p < 0.001), which increased significantly following depolarization at t = 15–105 min as well as t = 135 and 150 min. Depolarization also enhanced dialysate 2-AG levels in PF-3845-treated mice (F18,126 = 2.57, p < 0.005), though the magnitude of this effect was not greater than that observed in vehicle-treated controls. A selective PF-3845-induced enhancement of dialysate AEA levels was confirmed in AUC analyses that demonstrate significant enhancement of dialysate AEA but not 2-AG levels as compared with vehicle-treated controls (Figure 6; AEA: t12 = −2.81, p < 0.01; 2-AG: t12 = −0.39, NS). Similar to the observations in rats, PF-3845 also significantly increased AEA levels prior to the delivery of the ionic pulse (F8,56 = 5.97, p < 0.001, significant at t = −75–0 min) without altering dialysate 2-AG.
In contrast to our observations in rats, but consistent with our prior observations in mice,29,34 pretreatment with JZL184 (10 mg/kg) induced a significant enhancement of depolarization-induced increases in dialysate 2-AG levels (Figure 5c; F18,90 = 6.89, p < 0.001) with dialysate 2-AG levels returning toward baseline following the ionic pulse. This postdepolarization reduction in dialysate 2-AG is surprising given the long half-life of JZL184-induced MAGL inhibition (>12 h; ref (29)) and the fact that similar postdepolarization reductions in dialysate 2-AG were not evident in rats following URB602 pretreatment. It is possible that the postdepolarization reduction in interstitial 2-AG levels in JZL184-treated mice is mediated by 2-AG clearance through ABHD6 and/or ABHD12,23 which hydrolyze 2-AG but are not robustly inhibited by JZL184 at the presently employed dose.29 Regardless of mechanism, the present and prior data29,34 suggest that while the effects of stimulated 2-AG production on interstitial 2-AG levels are enhanced in JZL184-treated mice, the persistence of increased interstitial 2-AG may be more short-lived than predicted by measures of 2-AG content in bulk tissue extracts.29,34 Although significant fluctuations in AEA levels were also observed over time in JZL184-treated mice (F18,90 = 2.27, p < 0.01), post hoc tests failed to identify any significant changes in AEA levels from baseline. Selective JZL184-induced effects on dialysate 2-AG levels were confirmed by AUC analyses that demonstrated significant enhancement of dialysate 2-AG but not AEA levels as compared with those in vehicle-treated controls (Figure 6; AEA: t10 = −1.14, NS; 2-AG: t10 = −3.84, p < 0.005). In contrast to the effects of PF-3845 on nonstimulated levels of AEA, there was no significant effect of JZL184 on baseline dialysate 2-AG levels prior to depolarization either in the present or prior studies.29
Finally, as expected based on the similar IC50 values of JZL195 for inhibiting mouse FAAH and MAGL,26 administration of this dual FAAH/MAGL inhibitor (20 mg/kg) significantly enhanced depolarization-induced increases in dialysate levels of both AEA and 2-AG in mice (Figure 5d; AEA: F18,162 = 4.72, p < 0.001; 2-AG: F18,162 = 3.10, p < 0.001). AUC measures confirmed a significant enhancement of dialysate AEA levels relative to vehicle-treated controls (Figure 6; t14 = −4.56, p < 0.001), and while augmentation of dialysate 2-AG was also evident this effect did not quite reach significance (t14 = −1.42, p = 0.09). Interestingly, JZL195 significantly increased dialysate levels of both AEA and 2-AG prior to neuronal depolarization (AEA: F8,72 = 3.26, p < 0.005, significant at t = −45 min and t = 0 min; 2-AG: F8,72 = 4.02, p < 0.001, significant at t = −30–0 min). Because the JZL195 dose employed was somewhat higher than that for JZL184, this observation suggests that, as with AEA, nonstimulated 2-AG levels may be enhanced by sufficiently high MAGL inhibitor doses. However, while statistically significant, this predepolarization effect of JZL195 was modest in comparison to PF-3845-induced increases in baseline AEA. More complete dose evaluations will be required in future studies to elucidate possible differences in the effect of hydrolytic clearance inhibition on nonstimulated AEA and 2-AG in the brain interstitial space.
While quantitative comparison of pharmacological efficacy across species is not possible with the present data, our findings are indicative of species-specific efficacy of JZL184 but not PF-3845 or JZL195 for augmenting interstitial eCB levels in brain. Given the frequent use of rats as an experimental model, particularly for studying higher-order behaviors, the current findings strongly advocate for the development of selective MAGL inhibitors with similar efficacy across species.61,62
Concluding Remarks
The present results demonstrate that extracellular levels of AEA and 2-AG can be independently manipulated using selective FAAH and MAGL inhibitors, or in concert using a dual FAAH and MAGL inhibitor. By comparison, inhibitors of the putative eCB transporter produce only modest enhancement of interstitial eCB levels in the rat NAc. In addition to uptake through a membrane-associated transporter, eCB uptake may occur via lipid raft-mediated endocytosis and simple diffusion through membranes.18,42,43 Accordingly, the subtle effects produced by the transporter inhibitors AM404 and UCM707 suggest that this route of cellular uptake does not exert dominant control in the maintenance of interstitial eCB levels under basal conditions or the presently employed conditions for neuronal depolarization. In contrast, the relatively greater effects produced by FAAH and MAGL inhibition underscore the primary influence of these hydrolytic clearance routes for AEA and 2-AG in the control of interstitial eCB levels in vivo. It should be noted, however, that tests with more selective uptake transporter inhibitors and thorough evaluations of dose-dependency are necessary to confirm the relative influence of these various clearance mechanisms in the in vivo regulation of brain eCB levels.
The present results add to previous observations demonstrating more robust effects of eCB clearance inhibition on brain eCB levels indexed by post-mortem measures of brain tissue lipid content versus in vivo microdialysis measures of interstitial lipid levels.1,26,29,32−34,40,44,55 Several distinctions between these eCB sampling techniques may contribute to these dissimilar profiles, including but not limited to regional sampling specificity, increased eCB production associated with animal sacrifice and post-mortem tissue handling, and the potential for multiple eCB pools in tissue.34 With particular regard to regional specificity, it is worth noting that most evaluations of eCB clearance inhibition on post-mortem brain tissue eCB content have analyzed whole-brain extracts, while the present data reflect specific effects in the nucleus accumbens. While somewhat distinct effects of FAAH inhibition on interstitial eCB levels are evident in samples collected from the nucleus accumbens (present study), dorsal striatum,39 and hypothalamus,33 parametric studies on regional differences in the effects of eCB clearance inhibitors have not been performed. Although caveats are associated with lipid collection by brain microdialysis (for discussion, see ref (34)) it is likely this approach indexes signaling-relevant changes in eCB levels in a manner that is temporally aligned with behavioral and/or physiological challenges. Although differences in effect magnitude exist between brain tissue extracts and microdialysis eCB measures, both methods have demonstrated higher levels of 2-AG versus AEA in various brain regions and both approaches have been used to demonstrate the selective influence of MAGL and FAAH activity on brain 2-AG and AEA content, respectively. However, because of the large number of factors influencing eCB measures made by both approaches, conservatism is warranted when interpreting quantitative eCB measures by either approach.
The limited cross-species comparison performed here highlights possible species differences in the effects produced by pharmacological agents targeting eCB clearance mechanisms (e.g., JZL184). However, in view of reported significant behavioral effects produced by JZL184 and eCB transport inhibitors in rats,30,31,45,46,59,60 it may be argued that even increases in brain 2-AG signaling that are not indexed by microdialysis sampling are sufficient for inducing meaningful alterations in behavior. Finally, the present findings suggest that while eCB clearance inhibition potently augments the effects of stimulated eCB production (e.g., neuronal depolarization or other stimulus), unperturbed interstitial eCB levels are not robustly altered by modest attenuation of clearance mechanisms. Accordingly, the behavioral effects of moderate eCB clearance inhibition may be preferentially evident in limited circumstances in which eCB production is stimulated. This may allow eCB clearance inhibitors to facilitate region-specific increases in brain eCB signaling as a result of distinct stimulus-induced eCB production. Accordingly, as therapeutic agents eCB clearance inhibitors may induce fewer unwanted behavioral effects than exogenous CB1 receptor agonists that induce widespread CB1 receptor activation.
Methods
Subjects
Male Wistar rats (Charles River, Wilmington, MA) weighing 250–300 g at the beginning of the experiment were housed three/cage in the temperature (22 ± 2 °C) and humidity (60 ± 15%) controlled vivarium on a 12 h light/dark cycle (lights off at 10am). Male C57Bl/6J mice at 6–10 weeks old (TSRI colony, San Diego, CA), weighing 20–30 g at the beginning of the experiment were housed five per cage in the temperature (22 ± 2 °C) and humidity (60 ± 15%) controlled vivarium on a 12 h light/dark cycle (lights off at 9am). All animals were given 1 week to acclimatize to the conditions of the vivarium. Throughout the experiments, animals had ad libitum access to food and water. All procedures were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee of The Scripps Research Institute.
Drugs
4-Nitrophenyl-4-(dibenzo[d][1,3]dioxol-5-yl(hydroxy)methyl)piperidine-1-carboxylate (JZL184), 4-nitrophenyl-4-(3-phenoxybenzyl)piperazine-1-carboxylate (JZL195), and N-(pyridin-3-yl)-4(3-(5-(trifluoromethyl)pyridin-2-yloxy)benzyl)piperdine-1-carboxamide (PF-3845) were synthesized as previously described.26,28,29N-(3-Furanylmethyl)-5Z,8Z,11Z,14Z-eicosatetraenamide (UCM707), N-(4-hydroxyphenyl)-arachidonoyl amide 4-HPA (AM404), (3′-(aminocarbonyl)[1,1′-biphenyl]-3-yl)-cyclohexylcarbamate (URB597), [1,1′-biphenyl]-3-yl-carbamic acid, cyclohexyl ester (URB602), as well as the chromatographic standards anandamide (AEA), 1(3)-arachidonoylglycerol (1-AG), 2-arachidonoyl glycerol (2-AG), and (S)-(+)-arachidonyl-2′-hydroxy-1′-propylamide (S-2 methanandamide) were purchased from Cayman Chemical (Ann Arbor, MI). All drugs were dissolved in vehicle (ethanol/emulphor/aline, 1:1:18). Drugs were freshly prepared on the test day and administered intraperitoneally (i.p.) in a volume of 1 mL/kg for rats and 0.1 mL/kg for mice. Drug doses were based on literature.26,28,31,46,55,57,59
Surgery
For rat studies, animals were anesthetized with 1.5–2.0% isoflurane vapors and mounted on a stereotaxic apparatus for implantation of a unilateral microdialysis guide cannula (14 mm, MAB 6.14.IC, SciPro, Sanborn, NY) aimed at the nucleus accumbens shell (NAc) using the following coordinates from bregma:63 anteroposterior (AP) + 1.60; mediolateral (ML) ± 0.8 mm; and dorsoventral (DV) −5.70 mm. The guide cannula was secured using skull screws and cranioplastic cement. Animals had at least 7 days to recover after the surgery. At 12–15 h prior to the initiation of sample collection (described below), animals were briefly anesthetized with isoflurane. A microdialysis probe with 2 mm active length polyethyl sulfone dialysis membrane and 15 kDa MW cutoff (MAB 6.14.2) was inserted and secured to the previously implanted guide cannula.
For mice studies, as previously published,29 the animals were anesthetized with 1.5–2.0% isoflurane vapors and mounted on a stereotaxic apparatus for implantation of a unilateral microdialysis probe (1 mm active membrane, 15 kDa MW cutoff MAB 6.14.1, also see below). The probe was implanted 12–15 h before sample collection and did not require the use of a guide cannula. The probe was aimed at the NAc using the following coordinates from mouse bregma:64 AP + 1.50; ML ± 0.8 mm; and DV −5.00 mm. The probe was secured using skull screws and Den-Mat cement (Den-Mat, Santa Maria, CA).
In Vivo Microdialysis
Experimental in vivo microdialysis was performed as previously described.32,36 Briefly, following implantation, the microdialysis probe was perfused overnight with freshly prepared artificial CSF (aCSF) composed of 145 mM NaCl, 2.8 mM KCl, 1.2 mM MgCl2, 1.2 mM CaCl2, 5.4 mM d-glucose, 0.25 mM ascorbic acid (pH 7.2 – 7.4) at a low flow rate (0.1 μL/min) to allow for the re-equilibration of neurotransmitter levels. Animals were single housed following implantation of the microdialysis probe for the remainder of the experiment.
Two hours prior to baseline sampling, the aCSF solution was replaced with an aCSF solution containing 30% (w/v) hydroxypropyl-β-cyclodextrin, and the flow rate was increased to 0.6 μL/min. As previously described,36 inclusion of hydroxypropyl-β-cyclodextrin substantially increases the recovery of endocannabinoids (eCBs) by microdialysis. Dialysis samples were collected every 15 min and stored on dry ice during the experiment, and then at −70 °C until analysis for eCB content using liquid chromatography coupled with mass spectrometry. For all experiments, four samples were used as a preinjection baseline. The rats and mice were then injected with a specific drug or vehicle. For rats injected with vehicle, URB597, URB602, and UCM707, postinjection baseline sample collection continued for 1 h, whereas for vehicle-injected mice and rats as well as mice injected with PF-3845, JZL184, JZL195, and AM404 sample collection continued for 2 h. Following the postinjection baseline sample collection period, the aCSF solution was switched to an ionic pulse aCSF solution with a high concentration of KCl (90 mM) and CaCl2 (10 mM). The ionic pulse aCSF solution was designed to mimic depolarization conditions that are thought to stimulate endocannabinoid synthesis and/or release.33,39 For rats, after 45 min, the perfusate was switched back to regular aCSF, and sample collection continued for another 105 min (postpulse baseline). In mice, as previously described,29,34 probes were instead perfused with an ionic pulse solution (150 mM KCl and CaCl2 10 mM) for 90 min to compensate for the smaller probe size. Consequently, in mice, after switching back the perfusate to regular ACSF postpulse baseline sampling continued only for 1 h.
Liquid Chromatography/Mass Spectrometry Analysis of Dialysate Endocannabinoid Content
Sample analysis of AEA, 2-AG, and 1-AG was performed as previously described32,36 using liquid chromatography coupled with mass spectrometry. Briefly, 6 μL of microdialysate was mixed with 6 μL of 50 mM S-2 methanandamide internal standard and subsequently loaded onto a precolumn (1 × 10 mm, Haisil HL C18 5 μm, Higgins Analytical Inc., Mountain View, CA). The loaded precolumn was washed for 4 min using 10% MeOH (v/v) mobile phase delivered at 47 μL/min to remove hydrophilic species and especially the hydroxypropyl-β-cyclodextrin from the sample. Next, mobile phase flow through the precolumn was reversed using a switch valve, and analytes were washed off the precolumn and delivered to the analytical column (0.5 × 150 mm, Haisil HL C18 3 μm; Higgins Analytical Inc.) using an 80% MeOH (v/v) isocratic mobile phase delivered at 9 μL/min. The analytical column was connected to an 1100MSD mass spectrometer (Agilent Technologies, Santa Clara, CA) that was run in positive selected ion monitoring mode to enhance detection of low-abundance eCB, and hence, mass/charge ratios used were as follows: AEA (370.3 (M + 1Na), 2-AG and 1-AG (401.3 (M + 1Na), and S-2 methanandamide (384.3 (M + 1Na). Quantification was achieved using daily generated external calibration curves constructed from three standard concentrations (each in duplicate). Under these conditions, the limit of detection was ∼0.1 nM for AEA and 2-AG.
Histology
After all the microdialysis samples were collected, the animals were disconnected from the microdialysis perfusion system, deeply anesthetized with CO2, and subsequently sacrificed. The brains were then collected, frozen, and cut in 50 μm coronal sections to verify probe placement. Only animals with correct cannula placement were included in the analyses.
Statistics
All data were analyzed using NCSS2007 version 07.1.18 (NCSS, LLC., Kaysville, UT). Group differences in baseline dialysate AEA and 2-AG concentrations were evaluated using one-way ANOVAs with drug treatment as between-subjects factor. Subsequently, per treatment dialysate AEA and 2-AG levels were transformed to percentages of mean baseline dialysate concentration (set at 100%) for evaluation of changes in dialysate eCB content following drug injection as performed by ANOVA with repeated measures over time. In case of significant overall effects, a Bonferroni multiple comparison test was used for post hoc comparisons. In addition, AUC measures for AEA and 2-AG were calculated for each animal by subtracting 100 from each percentage of baseline data point and summing all data points collected from the onset of the ionic pulse on (t = 0–150 min). AUC data was analyzed using Student’s t tests. The level of probability for statistically significant effects was set at 0.05. All graphs were produced using GraphPad Prism version 5.02 for Windows (GraphPad Software, San Diego).
Acknowledgments
This is manuscript number 21707 from The Scripps Research Institute. The authors wish to thank David Stouffer for technical assistance with the microdialysate measures.
Glossary
Abbreviations
- 1-AG
1(3)-arachidonoyl glycerol
- 2-AG
2-arachidonyl glycerol
- AEA
N-arachidonoyl-ethanolamide (anandamide)
- AUC
area under the curve
- eCB
endocannabinoid
- FAAH
fatty acid amide hydrolase
- i.p.
intraperitoneally
- MAGL
monoacylglycerol lipase
- NAc
nucleus accumbens
Author Contributions
J.W. and C.I. contributed equally to the gathering of primary data and manuscript preparation. B.C. provided several of the FAAH and MAGL inhibitors (PF-3845, JZL184, JZL195) and provided editorial comment. T.J.D., A.N.M.S., and T.P. also provided editorial comment and experimental guidance. L.H.P. oversaw all aspects of experimental conduct, data evaluation, and manuscript preparation.
This work was supported by NIDA grant PO1 DA017259 (Project P.I., L.H.P.)
The authors declare no competing financial interest.
Author Contributions
∥ Authors contributed equally to this manuscript.
Funding Statement
National Institutes of Health, United States
References
- Hohmann A. G.; Suplita R. L.; Bolton N. M.; Neely M. H.; Fegley D.; Mangieri R.; Krey J. F.; Walker J. M.; Holmes P. V.; Crystal J. D.; Duranti A.; Tontini A.; Mor M.; Tarzia G.; Piomelli D. (2005) An endocannabinoid mechanism for stress-induced analgesia. Nature 435, 1108–1112. [DOI] [PubMed] [Google Scholar]
- Holt S.; Comelli F.; Costa B.; Fowler C. J. (2005) Inhibitors of fatty acid amide hydrolase reduce carrageenan-induced hind paw inflammation in pentobarbital-treated mice: comparison with indomethacin and possible involvement of cannabinoid receptors. Br. J. Pharmacol. 146, 467–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsicano G.; Wotjak C. T.; Azad S. C.; Bisogno T.; Rammes G.; Cascio M. G.; Hermann H.; Tang J.; Hofmann C.; Zieglgansberger W.; Di Marzo V.; Lutz B. (2002) The endogenous cannabinoid system controls extinction of aversive memories. Nature 418, 530–534. [DOI] [PubMed] [Google Scholar]
- Maccarrone M.; Gasperi V.; Catani M. V.; Diep T. A.; Dainese E.; Hansen H. S.; Avigliano L. (2010) The endocannabinoid system and its relevance for nutrition. Annu. Rev. Nutr. 30, 423–440. [DOI] [PubMed] [Google Scholar]
- Pattij T.; Wiskerke J.; Schoffelmeer A. N. (2008) Cannabinoid modulation of executive functions. Eur. J. Pharmacol. 585, 458–463. [DOI] [PubMed] [Google Scholar]
- Hill M. N.; Gorzalka B. B. (2009) The endocannabinoid system and the treatment of mood and anxiety disorders. CNS Neurol. Disord.: Drug Targets 8, 451–458. [DOI] [PubMed] [Google Scholar]
- Solinas M.; Goldberg S. R.; Piomelli D. (2008) The endocannabinoid system in brain reward processes. Br. J. Pharmacol. 154, 369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M.; Ohno-Shosaku T.; Hashimotodani Y.; Uchigashima M.; Watanabe M. (2009) Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev. 89, 309–380. [DOI] [PubMed] [Google Scholar]
- Iversen L. (2003) Cannabis and the brain. Brain 126, 1252–1270. [DOI] [PubMed] [Google Scholar]
- Di Marzo V. (2009) The endocannabinoid system: its general strategy of action, tools for its pharmacological manipulation and potential therapeutic exploitation. Pharmacol. Res. 60, 77–84. [DOI] [PubMed] [Google Scholar]
- Astarita G.; Geaga J.; Ahmed F.; Piomelli D. (2009) Targeted lipidomics as a tool to investigate endocannabinoid function. Int. Rev. Neurobiol. 85, 35–55. [DOI] [PubMed] [Google Scholar]
- Di Marzo V. (2010) Anandamide serves two masters in the brain. Nat. Neurosci. 13, 1446–1448. [DOI] [PubMed] [Google Scholar]
- Natarajan V.; Reddy P. V.; Schmid P. C.; Schmid H. H. (1982) N-Acylation of ethanolamine phospholipids in canine myocardium. Biochim. Biophys. Acta 712, 342–355. [DOI] [PubMed] [Google Scholar]
- Bisogno T.; Howell F.; Williams G.; Minassi A.; Cascio M. G.; Ligresti A.; Matias I.; Schiano-Moriello A.; Paul P.; Williams E. J.; Gangadharan U.; Hobbs C.; Di Marzo V.; Doherty P. (2003) Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J. Cell Biol. 163, 463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T.; Cascio M. G.; Saha B.; Mahadevan A.; Urbani P.; Minassi A.; Appendino G.; Saturnino C.; Martin B.; Razdan R.; Di Marzo V. (2006) Development of the first potent and specific inhibitors of endocannabinoid biosynthesis. Biochim. Biophys. Acta 1761, 205–212. [DOI] [PubMed] [Google Scholar]
- Hoover H. S.; Blankman J. L.; Niessen S.; Cravatt B. F. (2008) Selectivity of inhibitors of endocannabinoid biosynthesis evaluated by activity-based protein profiling. Bioorg. Med. Chem. Lett. 18, 5838–5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland M. J.; Barker E. L. (2004) Anandamide transport. Pharmacol. Ther. 104, 117–135. [DOI] [PubMed] [Google Scholar]
- Glaser S. T.; Kaczocha M.; Deutsch D. G. (2005) Anandamide transport: a critical review. Life Sci. 77, 1584–1604. [DOI] [PubMed] [Google Scholar]
- Fu J., Bottegoni G., Sasso O., Bertorelli R., Rocchia W., Masetti M., Guijarro A., Lodola A., Armirotti A., Garau G., Bandiera T., Reggiani A., Mor M., Cavalli A., and Piomelli D. (2011) A catalytically silent FAAH-1 variant drives anandamide transport in neurons. Nat Neurosci., DOI: 10.1007/s13311-011-0100-y, PMID: 22270809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K.; McKinney M. K.; Cravatt B. F. (2008) Enzymatic Pathways That Regulate Endocannabinoid Signaling in the Nervous System. Chem. Rev. 108, 1687–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravatt B. F.; Giang D. K.; Mayfield S. P.; Boger D. L.; Lerner R. A.; Gilula N. B. (1996) Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 384, 83–87. [DOI] [PubMed] [Google Scholar]
- Dinh T. P.; Carpenter D.; Leslie F. M.; Freund T. F.; Katona I.; Sensi S. L.; Kathuria S.; Piomelli D. (2002) Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc. Natl. Acad. Sci. U.S.A. 99, 10819–10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankman J. L.; Simon G. M.; Cravatt B. F. (2007) A Comprehensive Profile of Brain Enzymes that Hydrolyze the Endocannabinoid 2-Arachidonoylglycerol. Chem. Biol. 14, 1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosino S.; Ligresti A.; Di Marzo V. (2009) Endocannabinoid chemical biology: a tool for the development of novel therapies. Curr. Opin. Chem. Biol. 13, 309–320. [DOI] [PubMed] [Google Scholar]
- Fowler C. J. (2008) ″The tools of the trade″ - An overview of the pharmacology of the endocannabinoid system. Curr. Pharm. Des. 14, 2254–2265. [DOI] [PubMed] [Google Scholar]
- Long J. Z.; Nomura D. K.; Vann R. E.; Walentiny D. M.; Booker L.; Jin X.; Burston J. J.; Sim-Selley L. J.; Lichtman A. H.; Wiley J. L.; Cravatt B. F. (2009) Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc. Natl. Acad. Sci. U.S.A. 106, 20270–20275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortolato M.; Mangieri R. A.; Fu J.; Kim J. H.; Arguello O.; Duranti A.; Tontini A.; Mor M.; Tarzia G.; Piomelli D. (2007) Antidepressant-like activity of the fatty acid amide hydrolase inhibitor URB597 in a rat model of chronic mild stress. Biol. Psychiatry 62, 1103–1110. [DOI] [PubMed] [Google Scholar]
- Ahn K.; Johnson D. S.; Mileni M.; Beidler D.; Long J. Z.; McKinney M. K.; Weerapana E.; Sadagopan N.; Liimatta M.; Smith S. E.; Lazerwith S.; Stiff C.; Kamtekar S.; Bhattacharya K.; Zhang Y.; Swaney S.; Van Becelaere K.; Stevens R. C.; Cravatt B. F. (2009) Discovery and Characterization of a Highly Selective FAAH Inhibitor that Reduces Inflammatory Pain. Chem. Biol. 16, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J. Z.; Li W.; Booker L.; Burston J. J.; Kinsey S. G.; Schlosburg J. E.; Pavon F. J.; Serrano A. M.; Selley D. E.; Parsons L. H.; Lichtman A. H.; Cravatt B. F. (2009) Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat. Chem. Biol. 5, 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortolato M.; Campolongo P.; Mangieri R. A.; Scattoni M. L.; Frau R.; Trezza V.; La Rana G.; Russo R.; Calignano A.; Gessa G. L.; Cuomo V.; Piomelli D. (2006) Anxiolytic-like properties of the anandamide transport inhibitor AM404. Neuropsychopharmacology 31, 2652–2659. [DOI] [PubMed] [Google Scholar]
- de Lago E.; Fernandez-Ruiz J.; Ortega-Gutierrez S.; Viso A.; Lopez-Rodriguez M. L.; Ramos J. A. (2002) UCM707, a potent and selective inhibitor of endocannabinoid uptake, potentiates hypokinetic and antinociceptive effects of anandamide. Eur. J. Pharmacol. 449, 99–103. [DOI] [PubMed] [Google Scholar]
- Alvarez-Jaimes L.; Stouffer D. G.; Parsons L. H. (2009) Chronic ethanol treatment potentiates ethanol-induced increases in interstitial nucleus accumbens endocannabinoid levels in rats. J. Neurochem. 111, 37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bequet F.; Uzabiaga F.; Desbazeille M.; Ludwiczak P.; Maftouh M.; Picard C.; Scatton B.; Le Fur G. (2007) CB1 receptor-mediated control of the release of endocannabinoids (as assessed by microdialysis coupled with LC/MS) in the rat hypothalamus. Eur. J. Neurosci. 26, 3458–3464. [DOI] [PubMed] [Google Scholar]
- Buczynski M. W.; Parsons L. H. (2010) Quantification of brain endocannabinoid levels: methods, interpretations and pitfalls. Br. J. Pharmacol. 160, 423–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J. Z.; Nomura D. K.; Cravatt B. F. (2009) Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem. Biol. 16, 744–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caille S.; Alvarez-Jaimes L.; Polis I.; Stouffer D. G.; Parsons L. H. (2007) Specific alterations of extracellular endocannabinoid levels in the nucleus accumbens by ethanol, heroin, and cocaine self-administration. J. Neurosci. 27, 3695–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orio L.; Edwards S.; George O.; Parsons L. H.; Koob G. F. (2009) A role for the endocannabinoid system in the increased motivation for cocaine in extended-access conditions. J. Neurosci. 29, 4846–4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer B.; Bermudez-silva F. J.; Bilbao A.; Alvarez-jaimes L.; Sanchez-vera I.; Giuffrida A.; Serrano A.; Baixeras E.; Khaturia S.; Navarro M.; Parsons L. H.; Piomelli D.; Rodriguez de Fonseca F. (2007) Regulation of brain anandamide by acute administration of ethanol. Biochem. J. 404, 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffrida A.; Parsons L. H.; Kerr T. M.; Rodriguez de Fonseca F.; Navarro M.; Piomelli D. (1999) Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat. Neurosci. 2, 358–363. [DOI] [PubMed] [Google Scholar]
- Di S.; Boudaba C.; Popescu I. R.; Weng F. J.; Harris C.; Marcheselli V. L.; Bazan N. G.; Tasker J. G. (2005) Activity-dependent release and actions of endocannabinoids in the rat hypothalamic supraoptic nucleus. J. Physiol. 569, 751–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung K.-M.; Mangieri R.; Stapleton C.; Kim J.; Fegley D.; Wallace M.; Mackie K.; Piomelli D. (2005) Stimulation of Endocannabinoid Formation in Brain Slice Cultures through Activation of Group I Metabotropic Glutamate Receptors. Mol. Pharmacol. 68, 1196–1202. [DOI] [PubMed] [Google Scholar]
- Hermann A.; Kaczocha M.; Deutsch D. G. (2006) 2-Arachidonoylglycerol (2-AG) membrane transport: history and outlook. AAPS J. 8, E409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates M. L.; Barker E. L. (2009) Organized trafficking of anandamide and related lipids. Vitam. Horm. 81, 25–53. [DOI] [PubMed] [Google Scholar]
- de Lago E.; Petrosino S.; Valenti M.; Morera E.; Ortega-Gutierrez S.; Fernandez-Ruiz J.; Di Marzo V. (2005) Effect of repeated systemic administration of selective inhibitors of endocannabinoid inactivation on rat brain endocannabinoid levels. Biochem. Pharmacol. 70, 446–452. [DOI] [PubMed] [Google Scholar]
- Beltramo M.; Stella N.; Calignano A.; Lin S. Y.; Makriyannis A.; Piomelli D. (1997) Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science 277, 1094–1097. [DOI] [PubMed] [Google Scholar]
- Adamczyki P.; Golda A.; Mccreary A. C.; Filip M.; Przegalnski E. (2008) Activation of endocannabinoid transmission induces antidepressant-like effects in rats. J. Physiol. Pharmacol. 59, 217–228. [PubMed] [Google Scholar]
- De Lago E.; Gustafsson S. B.; Fernandez-Ruiz J.; Nilsson J.; Jacobsson S. O.; Fowler C. J. (2006) Acyl-based anandamide uptake inhibitors cause rapid toxicity to C6 glioma cells at pharmacologically relevant concentrations. J. Neurochem. 99, 677–688. [DOI] [PubMed] [Google Scholar]
- Ronesi J.; Gerdeman G. L.; Lovinger D. M. (2004) Disruption of endocannabinoid release and striatal long-term depression by postsynaptic blockade of endocannabinoid membrane transport. J. Neurosci. 24, 1673–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adermark L.; Lovinger D. M. (2007) Retrograde endocannabinoid signaling at striatal synapses requires a regulated postsynaptic release step. Proc. Natl. Acad. Sci. U.S.A. 104, 20564–20569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J.; Bottegoni G.; Sasso O.; Bertorelli R.; Rocchia W.; Masetti M.; Guijarro A.; Lodola A.; Armirotti A.; Garau G.; Bandiera T.; Reggiani A.; Mor M.; Cavalli A.; Piomelli D. (2012) A catalytically silent FAAH-1 variant drives anandamide transport in neurons. Nat. Neurosci. 15, 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayamanne A.; Greenwood R.; Mitchell V. A.; Aslan S.; Piomelli D.; Vaughan C. W. (2006) Actions of the FAAH inhibitor URB597 in neuropathic and inflammatory chronic pain models. Br. J. Pharmacol. 147, 281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock E. M.; Limebeer C. L.; Mechoulam R.; Piomelli D.; Parker L. A. (2008) The effect of cannabidiol and URB597 on conditioned gaping (a model of nausea) elicited by a lithium-paired context in the rat. Psychopharmacology 196, 389–395. [DOI] [PubMed] [Google Scholar]
- Hasanein P.; Shahidi S.; Komaki A.; Mirazi N. (2008) Effects of URB597 as an inhibitor of fatty acid amide hydrolase on modulation of nociception in a rat model of cholestasis. Eur. J. Pharmacol. 591, 132–135. [DOI] [PubMed] [Google Scholar]
- Cippitelli A.; Astarita G.; Duranti A.; Caprioli G.; Ubaldi M.; Stopponi S.; Kallupi M.; Sagratini G.; Rodriguez de Fonseca F.; Piomelli D.; Ciccocioppo R. (2011) Endocannabinoid regulation of acute and protracted nicotine withdrawal: effect of FAAH inhibition. PloS One 6, e28142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathuria S.; Gaetani S.; Fegley D.; Valino F.; Duranti A.; Tontini A.; Mor M.; Tarzia G.; Rana G. L.; Calignano A.; Giustino A.; Tattoli M.; Palmery M.; Cuomo V.; Piomelli D. (2003) Modulation of anxiety through blockade of anandamide hydrolysis. Nat. Med. 9, 76–81. [DOI] [PubMed] [Google Scholar]
- King A. R.; Duranti A.; Tontini A.; Rivara S.; Rosengarth A.; Clapper J. R.; Astarita G.; Geaga J. A.; Luecke H.; Mor M.; Tarzia G.; Piomelli D. (2007) URB602 inhibits monoacylglycerol lipase and selectively blocks 2-arachidonoylglycerol degradation in intact brain slices. Chem. Biol. 14, 1357–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comelli F.; Giagnoni G.; Bettoni I.; Colleoni M.; Costa B. (2007) The inhibition of monoacylglycerol lipase by URB602 showed an anti-inflammatory and anti-nociceptive effect in a murine model of acute inflammation. Br. J. Pharmacol. 152, 787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandevoorde S.; Jonsson K. O.; Labar G.; Persson E.; Lambert D. M.; Fowler C. J. (2007) Lack of selectivity of URB602 for 2-oleoylglycerol compared to anandamide hydrolysis in vitro. Br. J. Pharmacol. 150, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciolino N. R.; Zhou W. Y.; Hohmann A. G. (2011) Enhancement of endocannabinoid signaling with JZL184, an inhibitor of the 2-arachidonoylglycerol hydrolyzing enzyme monoacylglycerol lipase, produces anxiolytic effects under conditions of high environmental aversiveness in rats. Pharmacol. Res. 64, 226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleson E. B.; Beckert M. V.; Morra J. T.; Lansink C. S.; Cachope R.; Abdullah R. A.; Loriaux A. L.; Schetters D.; Pattij T.; Roitman M. F.; Lichtman A. H.; Cheer J. F. (2012) Endocannabinoids Shape Accumbal Encoding of Cue-Motivated Behavior via CB1 Receptor Activation in the Ventral Tegmentum. Neuron 73, 360–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naidoo V.; Karanian D. A.; Vadivel S. K.; Locklear J. R.; Wood J. T.; Nasr M.; Quizon P. M.; Graves E. E.; Shukla V.; Makriyannis A.; Bahr B. A.. Equipotent Inhibition of Fatty Acid Amide Hydrolase and Monoacylglycerol Lipase - Dual Targets of the Endocannabinoid System to Protect against Seizure Pathology. Neurotherapeutics 2012, not supplied. [DOI] [PMC free article] [PubMed]
- Cisneros J. A.; Bjorklund E.; Gonzalez-Gil I.; Hu Y.; Canales A.; Medrano F. J.; Romero A.; Ortega-Gutierrez S.; Fowler C. J.; Lopez-Rodriguez M. L. (2012) Structure-activity relationship of a new series of reversible dual monoacylglycerol lipase/fatty Acid amide hydrolase inhibitors. J. Med. Chem. 55, 824–836. [DOI] [PubMed] [Google Scholar]
- Paxinos G., and Watson C. (1998) The rat brain in stereotaxic coordinates, 4th ed., Academic Press, San Diego. [Google Scholar]
- Paxinos G., and Franklin K. B. J. (2001) The mouse brain in stereotaxic coordinates, 2nd ed., Academic Press, San Diego. [Google Scholar]