

Abstract
Most of the dioxygen on earth is generated by the oxidation of water by photosystem II (PS II) using light from the sun. This light-driven, four-photon reaction is catalyzed by the Mn4CaO5 cluster located at the lumenal side of PS II. Various X-ray studies have been carried out at cryogenic temperatures to understand the intermediate steps involved in the water oxidation mechanism. However, the necessity for collecting data at room temperature, especially for studying the transient steps during the O–O bond formation, requires the development of new methodologies. In this paper we report room temperature X-ray diffraction data of PS II microcrystals obtained using ultrashort (< 50 fs) 9 keV X-ray pulses from a hard X-ray free electron laser, namely the Linac Coherent Light Source. The results presented here demonstrate that the ”probe before destroy” approach using an X-ray free electron laser works even for the highly-sensitive Mn4CaO5 cluster in PS II at room temperature. We show that these data are comparable to those obtained in synchrotron radiation studies as seen by the similarities in the overall structure of the helices, the protein subunits and the location of the various cofactors. This work is, therefore, an important step toward future studies for resolving the structure of the Mn4CaO5 cluster without any damage at room temperature, and of the reaction intermediates of PS II during O–O bond formation.
Keywords: manganese, oxygen-evolving complex
Photosystem II (PS II) is the only known biological system that has the unique capability of utilizing visible light for the oxidation of water into molecular oxygen (reaction 1) (1–4).
| [1] |
PS II is a large protein complex embedded in the thylakoid membranes of cyanobacteria, algae, and plants (5). It is composed of at least 20 protein subunits and about 100 cofactor molecules, including the antenna and reaction center chlorophylls (Chl), pheophytins, cytochromes, carotenoids, quinones, lipids, and the Mn4CaO5 cluster. Light absorption and excitation of the P680 reaction center of PS II leads to charge separation across the thylakoid membrane, resulting in the formation of the positively charged Chl-based radical cation 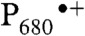 and the negatively charged quinone radical anion
and the negatively charged quinone radical anion 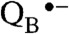 .
. 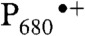 oxidizes the Mn4CaO5 cluster of the oxygen-evolving complex (OEC) via a tyrosine residue, YZ. The Mn4CaO5 cluster is thus driven successively through a five-step catalytic cycle (with the intermediate Si states, i = 0 to 4), coupling the isoenergetic one-electron photochemistry occurring at the PS II reaction center with the four-electron redox chemistry of water at the OEC. Many recent studies have shown that the oxidation or electron transfer processes are tightly coupled to proton-transfer reactions (6, 7). Although the S0 through S3 states are stable over a timescale of seconds, the S4 state is highly reactive and has not yet been experimentally characterized in a conclusive manner (3, 8–11). It is almost generally agreed that O-O bond formation occurs at this S4 oxidation-state level. It likely consists of a sequence of events that include deprotonation, O-O bond formation, O2 evolution, rearrangement of the cluster, and substrate water binding (12–15). Capturing S4 state intermediates by following the S3 → (S4) → S0 transition in a time resolved manner is therefore essential for understanding the water-oxidation mechanism.
oxidizes the Mn4CaO5 cluster of the oxygen-evolving complex (OEC) via a tyrosine residue, YZ. The Mn4CaO5 cluster is thus driven successively through a five-step catalytic cycle (with the intermediate Si states, i = 0 to 4), coupling the isoenergetic one-electron photochemistry occurring at the PS II reaction center with the four-electron redox chemistry of water at the OEC. Many recent studies have shown that the oxidation or electron transfer processes are tightly coupled to proton-transfer reactions (6, 7). Although the S0 through S3 states are stable over a timescale of seconds, the S4 state is highly reactive and has not yet been experimentally characterized in a conclusive manner (3, 8–11). It is almost generally agreed that O-O bond formation occurs at this S4 oxidation-state level. It likely consists of a sequence of events that include deprotonation, O-O bond formation, O2 evolution, rearrangement of the cluster, and substrate water binding (12–15). Capturing S4 state intermediates by following the S3 → (S4) → S0 transition in a time resolved manner is therefore essential for understanding the water-oxidation mechanism.
Extensive X-ray diffraction (XRD) studies using synchrotron radiation (SR) (16–21) and various spectroscopic techniques (22–29) have been used over the last several decades to elucidate the structure and mechanism of the OEC in PS II. Among the various methods, XRD is clearly the technique of choice for obtaining an overall detailed structure of PS II and the Mn4CaO5 cluster. However, a critical problem for the XRD studies has been the inherent radiation sensitivity of the redox-active high-valent Mn4CaO5 site in PS II. X-ray spectroscopy studies have demonstrated that radiation damage to the Mn4CaO5 cluster leads to the reduction of Mn(III) and Mn(IV) present in the intact OEC to Mn(II) at an X-ray dose that is two to three orders of magnitude lower than the dose required to produce an observable decay in diffraction quality of PS II crystals (30, 31). From these radiation damage studies it is inferred that in all current XRD studies of PS II the Mn4CaO5 cluster is reduced at levels ranging from approximately 80% (for structures reported in refs. 16–20) to 25% (for the structure reported by Umena et al., ref. 21). Such specific damage occurred despite the fact that all XRD measurements were carried out at 100–150 K. Extended X-ray absorption fine structure measurements show that this radiation damage also increases the Mn-Mn and Mn-ligand distances as compared to the undamaged Mn4CaO5 cluster (30); an indication that not only are the Mn atoms reduced but also that the structure of the cluster is disrupted. It is now generally recognized that for some redox active metallo-proteins it is difficult to obtain intact structures with SR-based XRD even at cryogenic temperatures. It should also be noted that at least for some proteins conformational changes are induced by the cryocooling procedure (32).
Recently, a new approach to protein crystallography was demonstrated at the Linac Coherent Light Source (LCLS) using ultrashort X-ray pulses of high intensity and crystalline samples in the micrometer size range (0.1–10 μm) (33–40). The use of very short (< 70 fs) pulses enables collection of diffraction data at room temperature before the onset of radiation damage (39–42). This probe before destroy method not only addresses the issue of radiation damage but also opens a way to measure time-resolved XRD at room temperature on enzyme systems. Using a variable-delay trigger, such as photoactivation, or a chemical stimulus, it becomes possible to follow chemical and structural dynamics. This method has recently been applied at the LCLS to probe structural changes upon light excitation in microcrystals of photosystem I (PS I) in complex with its electron acceptor ferredoxin (38).
We report an XRD study of PS II microcrystals at room temperature using ultrashort 9 keV X-ray pulses from the LCLS. Our results are a necessary initial step toward a high-resolution structure determination of the intact OEC in its various catalytic states, opening the way for capturing light-induced structural dynamics of PS II during water oxidation.
Results and Discussion
Single-Shot X-ray Diffraction from PS II.
The X-ray diffraction pattern of PS II isolated from the thermophilic cyanobacterium Thermosynechococcus elongatus, was collected at the Coherent X-ray Imaging (CXI) instrument at the LCLS (43) using the single-shot approach. PS II microcrystals (approximately 10 μm) (Fig. 1A) were injected into the LCLS X-ray beam in a liquid jet based on the electrohydrodynamic spraying of glycerol in vacuum (44) using the cone-jet mode (45). Several thousand diffraction images were collected at random crystal orientations. A single-shot diffraction pattern from a microcrystal is shown in Fig. 1B. The diffraction pattern exhibits Bragg spots up to 5.4 Å resolution (Fig. 1C), not limited by the experimental geometry at CXI or the LCLS source. The Bragg spots are remarkably sharp and small, spreading over only a few pixels. This is likely due to the combination of low crystal mosaicity and the use of pixel-array detector technology with an extremely narrow point-spread function (46). The diffraction intensities varied strongly from shot to shot due to several factors, such as the size of the probed crystal volume, the quality of the microcrystals and orientation, and variations in the intensity of the beam due to the nature of the LCLS X-ray pulses (47).
Fig. 1.

(A) Light microscope image of crystals (average size of 10 μm) of PS II used for the XRD measurements. (B) X-ray diffraction pattern of PS II obtained at CXI using a pulse width of < 50 fs and a flux of 3.4 × 1011 photons/pulse at 9 keV. Resolutions of some highlighted Bragg spots are given in yellow and resolution at edges of the selected area of the detector are indicated by white dashed circles. The background was removed by subtracting the average image of 1,052 misses recorded directly before and after the crystal diffraction. (C) Enlarged view of an area in the top left corner of the diffraction pattern shown in B (marked by a blue box) to show highest resolution spots observed.
The dose limit for the loss of diffraction in cryocooled (100–150 K) protein crystals is commonly considered to be around 2 to 3 × 107 Grays (Gy or J·kg-1) (the so-called Henderson limit, ref. 48, or Garman limit, ref. 49). Conventional crystallography studies using SR on PS II deposited between 1 × 106 and 1 × 107 Gy, corresponding to about 5–40 absorbed photons/unit cell, with the recent 1.9 Å resolution structure (21) being determined while applying a dose at the lower end of this range. In comparison, at CXI, the flux was between 3 and 5 × 1011 photons/shot at 9 keV, and the beam was focused to 1.5 μm (± 30%) full width at half maximum (FWHM) at the interaction region using Kirkpatrick-Baez mirrors (50). The average dose was 2 × 108 Gy, corresponding to about 1,440 absorbed photons/unit cell, which is an order of magnitude higher than the Henderson/Garman limit (48, 49) and about two orders of magnitude higher than the dose used for the previous SR PS II diffraction experiments. The diffraction measurement at these doses, well above the Henderson/Garman limit and at room temperature, is only possible because the short X-ray pulses (< 50 fs) outrun the X-ray induced damage and the Coulomb explosion (42, 51).
Recently Barty et al. discussed the effect of X-ray pulse length on the measured diffraction intensities using PS I microcrystals at the LCLS and with even higher doses (39). They observed that although molecular order is lost in the system over the duration of longer pulses (> 70 fs), it is still possible to obtain interpretable diffraction data. Their results show that most of the diffraction signal was produced in the earlier part of the pulse, whereas the later disordering only increased the background (39). Lomb et al. (40) also observed decreased intensity of Bragg spots for very high doses of 3 × 109 Gy or more in data collected from lysozyme microcrystals using 70 fs and longer pulses at 2 keV (6.2 Å wavelength).
It is clear that the radiation damage mechanisms using the short intense pulses at the LCLS are quite different from those encountered in SR studies. In the SR experiments, (i) radiation damage (both global and specific) was dose dependent but rate independent (in the range of seconds to hours), (ii) radiation damage was very dependent on temperature, and (iii) the dose leading to specific radiation damage (e.g., reduction of the redox-active center, disulfide bond cleavage, decarboxylation, etc.) is about two to three orders of magnitude less than the dose required for global radiation damage (loss of diffraction). In the X-ray free electron laser studies, specific radiation damage and its temperature dependence that arises from radical migration may not be a limiting factor due to the short femtosecond X-ray pulses. This confers advantages for using the LCLS for XRD studies of the Mn4CaO5 cluster in PS II that are hard to match with SR studies.
Electron Density Map of PS II.
The electron density maps of PS II were computed with data obtained in approximately 4 h 45 min, equivalent to nearly 2 million shots. All shots were initially screened for diffraction from crystals, yielding 113,632 diffraction patterns (5.7% of all shots) classified as potential hits (with a hit defined as showing 16 or more strong Bragg spots). Out of these, a subset of 7,269 individual diffraction patterns, equivalent to 0.4% of all shots, were successfully indexed and used for the dataset. From this set of indexed images, a dataset with 98% completeness was obtained to a resolution of 6.5 Å (82% completeness in the highest resolution shell) with a mean I/σ(I) of 16.9 (Table 1). The unit cell dimensions showed a very narrow distribution with a standard deviation of 2.5% for the a-axis and 1% for the b-and c-axes. They are comparable (within 1–2%) with cryo-SR XRD data at 2.9 Å (20), whereas they differ from the 1.9 Å resolution data (21), which were collected from partially dehydrated crystals exhibiting a contraction along the c-axis by about 20 Å (approximately 7%).
Table 1.
Data collection and refinement statistics
| Resolution range (Å) | 85.89–6.56 (6.79–6.56) |
| Space group | P212121 |
| Unit cell | 130.8 (±3.3) 227.8 (±2.7) 308.6 (±3.8) 90 90 90 |
| Total reflections | 873,209 |
| Unique reflections | 17,962 (1,446) |
| Multiplicity | 26 (2.8) |
| Completeness (%) | 98.1 (81.8) |
| Mean I/sigma (I) | 16.9 (4.8) |
| Rwork | 0.366 (0.447) |
| Rfree | 0.385 (0.448) |
| Number of atoms | 50,232 |
| Macromolecules | 41,052 |
| Ligands | 9,180 |
| Protein residues | 5,270 |
| rms (bonds, Å) | 0.007 |
| rms (angles, °) | 1.48 |
| Ramachandran favored (%) | 87 |
| Ramachandran outliers (%) | 2.2 |
| Clashscore | 37.1 |
| Average B-factor | 150.2 |
| Macromolecules | 148.4 |
Numbers in parentheses refer to highest resolution shell except for unit cell, where standard deviation for a, b, c are given in parentheses.
The electron density map of PS II in the dark S1 state was computed as described in the Materials and Methods section. The map at 6.5 Å resolution (Fig. 2) shows that the structure is isomorphous to that published earlier (20). A 2mFo-DFc electron density map was calculated and contoured at 1σ (blue mesh; Fig. 2A). The map clearly shows the presence of 36 trans-membrane helices (TMH) per monomer (shown in yellow in Fig. 2A), five for each of the two central subunits D1 and D2, interlocked in a handshake-like arrangement, typical for a reaction center complex, as well as the six TMH for each of the two chlorophyll-binding proteins CP43 and CP47, located next to D1 and D2, respectively. In addition, electron density for the TMH of all of the 12 small membrane intrinsic subunits, found previously (19, 20), is present in the map, although the density for the TMH of the weakly bound subunit PsbY (not present in the 3.5 and 1.9 Å structures, refs. 18 and 21), located next to the heterodimeric membrane intrinsic cyt b-559 is weaker compared to the density for the other TMHs. In addition to the tubular features attributed to TMHs the map shows extensive features at the lumenal side of the complex, attributed to the large lumenal protrusions of D1, D2, CP43, and CP47 as well as the three extrinsic subunits PsbO, PsbU, and PsbV on the lumenal side of the complex. For example, see the β-barrel-like structure of PsbO indicated in Fig. 2A.
Fig. 2.

Electron density obtained from femtosecond PS II microcrystal diffraction at CXI, 2mFo-DFc electron density maps are shown as blue mesh, omit maps (mFo-DFc) as green (positive) and red (negative) mesh, respectively. (A) Electron density map of PS II computed to a resolution of 6.5 Å, contoured at 1σ, view is along the membrane plane with cytoplasmic side on the top, lumenal side on the bottom. One monomer of PS II is shown as yellow cartoon, cofactors are shown as green sticks. The second monomer in the PS II dimer and the symmetry equivalent dimers in the unit cell are not shown. For better orientation the extrinsic subunit PsbO is labeled. (B) Electron density map of the region around the OEC; view is approximately perpendicular to the membrane plane, from the cytoplasmic side. Protein is shown as cartoon with subunit D1 in yellow and subunit CP43 in magenta. For better orientation the cd-helix and the C terminus of D1 and the ef-loop of CP43 as well as three of the protein ligands (Asp170, Glu333, and Asp342) of the OEC are indicated. The positions of Mn and Ca ions as derived from the 2.9 Å structure (20) are shown as violet and orange spheres, respectively. The electron density map is contoured at 1σ, omit maps at ± 3σ. (C) Electron density in the region around the nonheme Fe (shown as cyan sphere), helices d and e of subunits D1 (yellow) and D2 (orange) are labeled. Electron density map is contoured at 1σ, omit maps at 2.5σ. (D) Electron density around the two central Chl of the reaction center, helices d and cd of subunits D1 (yellow) and D2 (orange) are labeled; Chl are shown in pink with the central Mg2+ ions as gray spheres, 2mFo-DFc, electron density is contoured at 0.8σ, the omit maps at ± 2.5σ.
A positive peak in the refined omit map (mFo-DFc, 3σ green mesh; Fig. 2B) is located at the lumenal side of subunit D1 at the position of the Mn4CaO5 cluster [violet (Mn) and orange (Ca) atoms]. These atoms were omitted from the phasing model; thus the observation of this electron density is an experimental indication that the Mn4CaO5 cluster is present in the sample at the location observed in conventional SR-based XRD (20, 21).
A similar result was obtained for the nonheme Fe near the acceptor quinone molecule, located at the cytoplasmic/stromal side at the interface between the two central subunits D1 and D2. It was possible to observe a difference peak in the mFo-DFc map at the position corresponding to Fe, when Fe was omitted from the initial phasing model (Fig. 2C), confirming the presence of the Fe from the LCLS data. In addition, it is possible to locate the electron density for the Chl molecules bound to subunits D1, D2, CP43, and CP47. As an example, the electron density from the two central Chls of the reaction center of PS II is shown in Fig. 2D. The difference map, computed after omitting the two Chl from the phasing model, revealed positive density at the expected locations (Fig. 2D, green mesh). However, it is not possible to observe the flexible phytyl chains of these Chls at this resolution.
The electron density obtained at the LCLS was compared with SR data by using an electron density map based on the 2.9 Å SR data truncated to a resolution of 6.5 Å to match the limited resolution achieved with LCLS data (Fig. 3 A and B). B-factor sharpening was applied to the LCLS structure factor amplitudes to compensate for the difference in intensity falloff with resolution for comparison with SR data. There are no obvious deviations between these two maps, within the observed resolution. The slight differences observed may be due to data processing, which is different for the partial reflections recorded in LCLS data compared to SR data and to the low signal produced by the crystals at 6.5 Å, leading to low signal-to-noise levels at this resolution. The differences between LCLS and synchrotron density maps are presently not interpretable in terms of data quality or differences in the protein structure. The overall electron density maps show similar helix orientation and structures and density from all the expected subunits in PS II compared to the 2.9 Å structure. The arrangements of the TMHs between the two maps are similar and there are also no deviations visible in the potentially more flexible lumenal regions. This indicates that, at least within the limited resolution achieved, no differences in loop flexibility are encountered when comparing room temperature and cryogenic XRD data despite the fact that only a very limited number of surface residues are engaged in crystal contacts.
Fig. 3.

Comparison of electron density computed from CXI data with SR data truncated to 6.5 Å resolution. Overview of the electron density (blue mesh) for one monomer of PS II computed from the CXI data (A) and from the truncated SR data (B). Protein is shown as cartoon in yellow; 2mFo-DFc electron density is contoured at 1σ; view is along the membrane plane with cytoplasm at top. Electron density in the region of the Mn4CaO5 cluster computed from the CXI data (C) or from the truncated SR data (D), respectively, view direction is approximately 90 deg rotated with respect to Fig. 2B and is along the membrane plane, with cytoplasm at the top, lumen at the bottom. Protein is shown as cartoon in yellow (D1) and magenta (CP43), Mn as violet spheres and Ca as orange sphere. The lumenal end of TMH c as well as helix cd and the C-terminal helix (eC) of D1 and the ef helix of CP43 are labeled. The 2mFo-DFc electron density map is contoured at 1σ (blue mesh); the difference density (mFo-DFc map) is shown at +3σ (green mesh) and at -3σ (red mesh).
Fig. 3 C and D are the respective electron density maps in the region of the OEC. The difference maps (green mesh), calculated in the same way in both cases, occupy equivalent positions, indicating that the position and extent of the cluster in both cases agree at the level of detail given by the study presented here. Therefore, no indication for the dislocation of the Mn4CaO5 cluster is found during the < 50 fs X-ray pulses. This is consistent with the previous observations from other proteins studied by femtosecond-crystallography at LCLS (33, 36, 37, 40, 41). However, more detailed differences at the atomic level or in the chemical nature of the complex cannot be captured at the current resolution.
The highest resolution diffraction peaks observed in the LCLS XRD from PS II microcrystals are at a lower resolution than would be expected from SR experiments with larger crystals. This is not due to the experimental geometry, the photon energy used, or the beam intensity, but likely due to the PS II microcrystals. However, it is not clear whether the quality of the microcrystals is limited at the onset, or whether the PS II crystals, which are known to be mechanically fragile, have a lower quality upon reaching the interaction region due to the delivery process.
Conclusions
We have shown that PS II microcrystals can be used to obtain XRD data at room temperature using the short < 50 fs X-ray pulses at the LCLS. Diffraction spots were observed up to a resolution of 5.4 Å and a complete dataset was obtained to a resolution of 6.5 Å. The electron density map of PS II is similar to that from a SR data set truncated to 6.5 Å. Electron density from the Mn4CaO5 cluster is present, showing that the Mn4CaO5 cluster is not dislocated by the intense X-ray pulses that are several times more intense than those used on SR sources. This prototypical study serves as the basis for future time-resolved measurements and more detailed investigations on the intermediate states of PS II as it demonstrates that the probe before destroy approach is feasible for this system.
Materials and Methods
PS II Sample Preparation.
PS II was isolated from the thermophilic cyanobacterium T. elongatus as in ref. 52. To obtain microcrystals of PS II smaller than 10 μm along the longest dimension, the protocol described previously (52) was changed in the following way. In analogy to the precrystallization procedure, the Chl concentration was set to 0.74 mM, equivalent to 7.4 mg/mL PS II in buffer A [100 mM Pipes (pH 7.0), 5 mM CaCl2] containing 0.03% n-dodecyl-β-D-maltoside (β-DM) and the protein solution was mixed 1∶1 with 10.4% (wt/vol) of the precipitant PEG2000 in buffer A at 4 °C. Crystal formation started within 2–3 h and was completed overnight. Microcrystals were harvested in buffer B [100 mM MES (pH 6.5), 5 mM CaCl2, 0.015% β-DM], containing 10% (wt/vol) PEG2000. Crystals with the desired dimensions (5–10 μm, see Fig. 1A) were separated from larger ones by filtration with Nucleopore track etch membranes (Whatman) with a pore diameter of 8 μm. Microcrystals were transferred stepwise from the harvesting buffer to a final solution containing 10% (wt/vol) PEG2000 and 30% (wt/vol) glycerol in buffer B. The final crystal suspension was adjusted to a Chl concentration of 1 mM in a volume of about 100 μL.
Setup and Data Collection.
Experiments were carried out at the CXI instrument at LCLS. Injection of the samples into the interaction region was achieved by focusing the crystal suspension exiting a silica capillary (100 μm inner diameter) into a < 10 μm jet using an electric potential of 2.1–2.5 kV between the capillary exit and a counter electrode 7 mm away. The crystal suspension flowed at 2.5–3.1 μL/ min using a liquid backing pressure of 15–20 psi. Buffer B with 10% PEG 2000 does not electrospray in vacuum due to freezing at the nozzle exit. Glycerol was added as a cryoprotectant to eliminate freezing and enable formation of a stable cone-jet mode. The combination of glycerol and PEG 2000 in the buffer also contributed to the low flow rate operation and reduced settling of the crystals during the experiment. The X-rays probed the liquid jet 50–100 μm from the exit of the capillary, exposing the crystal suspension to vacuum for fractions of a second.
The CXI instrument (43) at LCLS was operated at an energy of 9 keV with an average intensity of 3–5 × 1011 photons/pulse, a pulse frequency of 120 Hz and a pulse duration of < 50 fs. The beam was focused to a size of about 1.5 μm FWHM at the interaction region using Kirkpatrick-Baez mirrors (50). Forward diffraction was measured using the Cornell-SLAC pixel array detector of the CXI instrument (46) with a pixel size of 110 × 110 μm2 and a total of 2.3 million pixels. The detector metrology was established using Ag behenate, microcrystals of thermolysin, and LCLS-provided optical data. Flux numbers were converted into deposited energy (dose) using the program RADDOSE (53, 54).
Data Processing.
PS II diffraction data was processed with a new software suite (cctbx.xfel) that builds upon components developed previously in the synchrotron context for picking Bragg spots (spotfinder) and autoindexing (labelit) (55, 56), and employs established methods for the integration of Bragg spot intensities by pixel summation (57). Individual reflections were scaled and merged without separately accounting for the partiality fraction of each observation.
The structure was solved by molecular replacement using Phaser (58), searching for two copies of the complete PS II complex [Protein Data Bank (PDB) ID 3bz1; ref. 20) including ligands, but not the central manganese cluster. Because of the low resolution of the data, a single round of tightly restrained refinement was performed in phenix.refine (59). The R-free flags from 3bz1 were preserved for this refinement, with additional reflections flagged to expand the test set to 5% of all reflections. Electron density maps were calculated with B-factor sharpening applied (Bsharp = -75 Å2). The mFo-DFc difference map showed a clear positive peak for the manganese cluster, which was subsequently replaced in the starting model, and the structure re-refined to a final R/R-free of 0.366/0.385 (Table 1). Electron density maps from the PS II SR data (PDB ID 3bz1) truncated to 6.5 Å resolution were generated using the same parameters as those used for the generation of the map for the CXI data but without B-factor sharpening.
ACKNOWLEDGMENTS.
We thank Prof. Ken Sauer for continuing scientific discussion; Pavel Afonine for refinement advice; Matthias Broser, Selcan Ceylan, and Alyssa Lampe for support with sample preparation; and Christopher Kenney, Ryan Herbst, Jack Pines, Philip Hart, John Morse, Gunther Haller, and Sven Herrmann for support with the Cornell-SLAC pixel array detector. We thank the staff at LCLS/SLAC for their support. This work was supported by the Director, Office of Science, Office of Basic Energy Sciences (OBES), Division of Chemical Sciences, Geosciences, and Biosciences (CSGB) of the Department of Energy (DOE) under Contract DE-AC02-05CH11231 (J.Y. and V.K.Y.) for structural studies and instrumentation; Director, Office of Science under Contract DE-AC02-05CH11231 and National Institutes of Health (NIH) Grant R01-GM095887 (N.K.S.) for data processing methods; and NIH Grant GM55302 (V.K.Y) for PS II biochemistry. In addition the Deutsche Forschungsgemeinschaft-Cluster of Excellence “UniCat” coordinated by the Technische Universität Berlin (A.Z.), the Solar Fuels Strong Research Environment (Umeå University, J.M.), the Artificial Leaf Project (K&Wallenberg Foundation, J.M.), the Vetenskapsrådet (J.M.), and the Alexander von Humboldt Foundation (J.K.) are acknowledged for supporting this project. The injector work was supported by LCLS (M.J.B., D.W.S.); the Atomic, Molecular and Optical Sciences Program; CSGB Division, OBES, DOE (M.J.B); and through the SLAC Laboratory Directed Research and Development Program (M.J.B., H.L.). Experiments were carried out at the LCLS at SLAC National Accelerator Laboratory operated by Stanford University on behalf of DOE, OBES. Testing of crystals and various parts of the setup were carried out at synchrotron facilities that were provided by the Advanced Light Source (ALS) in Berkeley, Stanford Synchrotron Radiation Lightsource (SSRL) in Stanford, and the Advanced Photon Source in Argonne, funded by DOE OBES. The SSRL Biomedical Technology program is supported by the NIH, the National Center for Research Resources, and the DOE Office of Biological and Environmental Research.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 4FBY).
References
- 1.Wydrzynski T, Satoh K, editors. Photosystem II: The Light-Driven Water:Plastoquinone Oxidoreductase. Dordrecht: Springer; 2005. [Google Scholar]
- 2.Kern J, Renger G. Photosystem II: Structure and mechanism of the water:plastoquinone oxidoreductase. Photosynth Res. 2007;94:183–202. doi: 10.1007/s11120-007-9201-1. [DOI] [PubMed] [Google Scholar]
- 3.Rappaport F, Diner B. Primary photochemistry and energetics leading to the oxidation of the Mn4Ca cluster and to the evolution of molecular oxygen in photosystem II. Coord Chem Rev. 2008;252:259–272. [Google Scholar]
- 4.Messinger J, Noguchi T, Yano J. Photosynthetic O2 evolution. In: Wydrzynski T, Hillier W, editors. Molecular Solar Fuels. Vol 5. London: Royal Society of Chemistry; 2012. pp. 163–207. (RSC Energy and Environment Series). [Google Scholar]
- 5.Nelson N, Ben-Shem A. The complex architecture of oxygenic photosynthesis. Nat Rev Mol Cell Biol. 2004;5:971–982. doi: 10.1038/nrm1525. [DOI] [PubMed] [Google Scholar]
- 6.Tommos C, Babcock GT. Proton and hydrogen currents in photosynthetic water oxidation. Biochim Biophys Acta. 2000;1458:199–219. doi: 10.1016/s0005-2728(00)00069-4. [DOI] [PubMed] [Google Scholar]
- 7.Renger G. Coupling of electron and proton transfer in oxidative water cleavage in photosynthesis. Biochim Biophys Acta. 2004;1655:195–204. doi: 10.1016/j.bbabio.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 8.Razeghifard MR, Pace RJ. EPR kinetic studies of oxygen release in thylakoids and PSII membranes: A kinetic intermediate in the S3 to S0 transition. Biochemistry. 1999;38:1252–1257. doi: 10.1021/bi9811765. [DOI] [PubMed] [Google Scholar]
- 9.Clausen J, Junge W. Search for intermediates of photosynthetic water oxidation. Photosynth Res. 2005;84:339–345. doi: 10.1007/s11120-005-3480-1. [DOI] [PubMed] [Google Scholar]
- 10.Dau H, Haumann M. Eight steps preceding O–O bond formation in oxygenic photosynthesis—A basic reaction cycle of the Photosystem II manganese complex. Biochim Biophys Acta. 2007;1767:472–483. doi: 10.1016/j.bbabio.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 11.Shevela D, Beckmann K, Clausen J, Junge W, Messinger J. Membrane-inlet mass spectrometry reveals a high driving force for oxygen production by photosystem II. Proc Natl Acad Sci USA. 2011;108:3602–3607. doi: 10.1073/pnas.1014249108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Messinger J, Renger G. Photosynthetic water splitting. In: Renger G, editor. Primary Processes of Photosynthesis: Basic Principles and Apparatus. Vol II. Cambridge: Royal Society of Chemistry; 2008. pp. 291–349. [Google Scholar]
- 13.Brudvig GW. Water oxidation chemistry of photosystem II. Philos Trans R Soc Lond B Biol Sci. 2008;363:1211–1218. doi: 10.1098/rstb.2007.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siegbahn PE. Structures and energetics for O2 formation in photosystem II. Acc Chem Res. 2009;42:1871–1880. doi: 10.1021/ar900117k. [DOI] [PubMed] [Google Scholar]
- 15.Renger G. Light induced oxidative water splitting in photosynthesis: Energetics, kinetics and mechanism. J Photochem Photobiol B. 2011;104:35–43. doi: 10.1016/j.jphotobiol.2011.01.023. [DOI] [PubMed] [Google Scholar]
- 16.Zouni A, et al. Crystal structure of photosystem II from Synechococcus elongatus at 3.8 Å resolution. Nature. 2001;409:739–743. doi: 10.1038/35055589. [DOI] [PubMed] [Google Scholar]
- 17.Kamiya N, Shen JR. Crystal structure of oxygen-evolving photosystem II from Thermosynechococcus vulcanus at 3.7-Å resolution. Proc Natl Acad Sci USA. 2003;100:98–103. doi: 10.1073/pnas.0135651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S. Architecture of the photosynthetic oxygen-evolving center. Science. 2004;303:1831–1838. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- 19.Loll B, Kern J, Saenger W, Zouni A, Biesiadka J. Towards complete cofactor arrangement in the 3.0 Å resolution structure of photosystem II. Nature. 2005;438:1040–1044. doi: 10.1038/nature04224. [DOI] [PubMed] [Google Scholar]
- 20.Guskov A, et al. Cyanobacterial photosystem II at 2.9-angstrom resolution and the role of quinones, lipids, channels and chloride. Nat Struct Mol Biol. 2009;16:334–342. doi: 10.1038/nsmb.1559. [DOI] [PubMed] [Google Scholar]
- 21.Umena Y, Kawakami K, Shen JR, Kamiya N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 angstrom. Nature. 2011;473:55–65. doi: 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- 22.Yano J, Yachandra VK. Where water is oxidized to dioxygen: Structure of the photosynthetic Mn4Ca cluster from X-ray spectroscopy. Inorg Chem. 2008;47:1711–1726. doi: 10.1021/ic7016837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haumann M, et al. Photosynthetic O2 formation tracked by time-resolved X-ray experiments. Science. 2005;310:1019–1021. doi: 10.1126/science.1117551. [DOI] [PubMed] [Google Scholar]
- 24.Messinger J, Badger M, Wydrzynski T. Detection of one slowly exchanging substrate water molecule in the S3 state of photosystem II. Proc Natl Acad Sci USA. 1995;92:3209–3213. doi: 10.1073/pnas.92.8.3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Noguchi T. Fourier transform infrared analysis of the photosynthetic oxygen-evolving center. Coord Chem Rev. 2008;252:336–346. [Google Scholar]
- 26.Berthomieu C, Hienerwadel R. Fourier transform infrared (FTIR) spectroscopy. Photosynth Res. 2009;101:157–170. doi: 10.1007/s11120-009-9439-x. [DOI] [PubMed] [Google Scholar]
- 27.Lizasa M, Suzuki H, Noguchi T. Orientations of carboxylate groups coupled to the Mn cluster in the photosynthetic oxygen-evolving center as studied by polarized ATR-FTIR spectroscopy. Biochemistry. 2010;49:3074–3082. doi: 10.1021/bi1002647. [DOI] [PubMed] [Google Scholar]
- 28.Yeagle GJ, Gilchrist ML, McCarrick RM, Britt RD. Multifrequency pulsed electron paramagnetic resonance study of the S2 state of the photosystem II manganese cluster. Inorg Chem. 2008;47:1803–1814. doi: 10.1021/ic701680c. [DOI] [PubMed] [Google Scholar]
- 29.Cox N, et al. Effect of Ca2+/Sr2+ substitution on the electronic structure of the oxygen-evolving complex of photosystem II: A combined multifrequency EPR, 55Mn-ENDOR, and DFT study of the S2 state. J Am Chem Soc. 2011;133:3635–3648. doi: 10.1021/ja110145v. [DOI] [PubMed] [Google Scholar]
- 30.Yano J, et al. X-ray damage to the Mn4Ca complex in single crystals of photosystem II: A case study for metalloprotein crystallography. Proc Natl Acad Sci USA. 2005;102:12047–12052. doi: 10.1073/pnas.0505207102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grabolle M, Haumann M, Muller C, Liebisch P, Dau H. Rapid loss of structural motifs in the manganese complex of oxygenic photosynthesis by X-ray irradiation at 10–300 K. J Biol Chem. 2006;281:4580–4588. doi: 10.1074/jbc.M509724200. [DOI] [PubMed] [Google Scholar]
- 32.Fraser JS, et al. Accessing protein conformational ensembles using room-temperature X-ray crystallography. Proc Natl Acad Sci USA. 2011;108:16247–16252. doi: 10.1073/pnas.1111325108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chapman HN, et al. Femtosecond X-ray protein nanocrystallography. Nature. 2011;470:73–77. doi: 10.1038/nature09750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kirian RA, et al. Structure-factor analysis of femtosecond micro-diffraction patterns from protein nanocrystals. Acta Crystallogr A. 2011;67:131–140. doi: 10.1107/S0108767310050981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hunter MS, et al. X-ray diffraction from membrane protein nanocrystals. Biophys J. 2011;100:198–206. doi: 10.1016/j.bpj.2010.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johansson LC, et al. Lipidic phase membrane protein serial femtosecond crystallography. Nat Methods. 2012;9:263–265. doi: 10.1038/nmeth.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koopmann R, et al. In vivo protein crystallization opens new routes in structural biology. Nat Methods. 2012;9:259–262. doi: 10.1038/nmeth.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aquila A, et al. Time-resolved protein nanocrystallography using an X-ray free-electron laser. Opt Express. 2012;20:2706–2716. doi: 10.1364/OE.20.002706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barty A, et al. Self-terminating diffraction gates femtosecond X-ray nanocrystallography measurements. Nat Photonics. 2012;6:35–40. doi: 10.1038/nphoton.2011.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lomb L, et al. Radiation damage in protein serial femtosecond crystallography using an X-ray free-electron laser. Phys Rev B. 2011;84:214111. doi: 10.1103/PhysRevB.84.214111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caleman C, et al. On the feasibility of nanocrystal imaging using intense and ultrashort X-ray pulses. ACS Nano. 2011;5:139–146. doi: 10.1021/nn1020693. [DOI] [PubMed] [Google Scholar]
- 42.Neutze R, Wouts R, van der Spoel D, Weckert E, Hajdu J. Potential for biomolecular imaging with femtosecond X-ray pulses. Nature. 2000;406:752–757. doi: 10.1038/35021099. [DOI] [PubMed] [Google Scholar]
- 43.Boutet S, Williams GJ. The Coherent X-ray Imaging (CXI) instrument at the Linac Coherent Light Source (LCLS) New J Phys. 2010;12:035024. [Google Scholar]
- 44.Ku BK, Kim SS. Electrohydrodynamic spraying characteristics of glycerol solutions in vacuum. J Electrostat. 2003;57:109–128. [Google Scholar]
- 45.Ganan-Calvo AM. Cone-jet analytical extension of Taylor’s electrostatic solution and the asymptotic universal scaling laws in electrospraying. Phys Rev Lett. 1997;79:217–220. [Google Scholar]
- 46.Koerner LJ, Philipp HT, Hromalik MS, Tate MW, Gruner SM. X-ray tests of a pixel array detector for coherent x-ray imaging at the Linac Coherent Light Source. J Instrum. 2009;4:P03001–P03001. [Google Scholar]
- 47.Emma P, et al. First lasing and operation of an angstrom-wavelength free-electron laser. Nat Photonics. 2010;4:641–647. [Google Scholar]
- 48.Henderson R. Cryoprotection of protein crystals against radiation-damage in electron and X-Ray-diffraction. Proc R Soc Lond B Biol Sci. 1990;241:6–8. [Google Scholar]
- 49.Owen RL, Rudino-Pinera E, Garman EF. Experimental determination of the radiation dose limit for cryocooled protein crystals. Proc Natl Acad Sci USA. 2006;103:4912–4917. doi: 10.1073/pnas.0600973103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Siewert F, et al. Ultra-precise characterization of LCLS hard X-ray focusing mirrors by high resolution slope measuring deflectometry. Opt Express. 2012;20:4525–4536. doi: 10.1364/OE.20.004525. [DOI] [PubMed] [Google Scholar]
- 51.Starodub D, et al. Dose, exposure time and resolution in serial X-ray crystallography. J Synchrotron Radiat. 2008;15:62–73. doi: 10.1107/S0909049507048893. [DOI] [PubMed] [Google Scholar]
- 52.Kern J, et al. Purification, characterisation and crystallisation of photosystem II from Thermosynechococcus elongatus cultivated in a new type of photobioreactor. Biochim Biophys Acta. 2005;1706:147–157. doi: 10.1016/j.bbabio.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 53.Paithankar KS, Owen RL, Garman EF. Absorbed dose calculations for macromolecular crystals: Improvements to RADDOSE. J Synchrotron Radiat. 2009;16:152–162. doi: 10.1107/S0909049508040430. [DOI] [PubMed] [Google Scholar]
- 54.Murray JW, Garman EF, Ravelli RBG. X-ray absorption by macromolecular crystals: The effects of wavelength and crystal composition on absorbed dose. J Appl Crystallogr. 2004;37:513–522. [Google Scholar]
- 55.Zhang Z, Sauter NK, van den Bedem H, Snell G, Deacon AM. Automated diffraction image analysis and spot searching for high-throughput crystal screening. J Appl Crystallogr. 2006;39:112–119. [Google Scholar]
- 56.Sauter NK, Grosse-Kunstleve RW, Adams PD. Robust indexing for automatic data collection. J Appl Crystallogr. 2004;37:399–409. doi: 10.1107/S0021889804005874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leslie AG. Integration of macromolecular diffraction data. Acta Crystallogr D. 1999;55:1696–1702. doi: 10.1107/s090744499900846x. [DOI] [PubMed] [Google Scholar]
- 58.Mccoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]