

Abstract
| Strategy, Management and Health Policy
| ||||
|---|---|---|---|---|
| Venture Capital Enabling Technology | Preclinical Research | Preclinical Development Toxicology, Formulation Drug Delivery, Pharmacokinetics | Clinical Development Phases I–III Regulatory, Quality, Manufacturing | Postmarketing Phase IV |
The hexahistidine-tagged mouse P2X1 receptor (H-mP2X1R), an ATP-gated ion channel receptor, was expressed in a baculovirus system using the pAcHLT-B transfer vector containing a hexahistidine tag. Both widely used denaturing (8M urea) and nondenaturing (such as 1% Triton X-100) solubilization conditions were compared, resulting in about 30% of the P2X1 receptors being solubilized (S1). However, at pH 13 most of the H-mP2X1R from the initially insoluble pellet fraction was solubilized (S2) and remained in the soluble fraction (S3) after dialyzing against a nondenaturing buffer. H-mP2X1Rs were purified sequentially through cobalt and ATP affinity columns. Receptors purified from S3 had higher purity than those from S1 (i.e., ~90% vs. ~75%). Circular dichroism spectra indicated identical protein secondary structures of the receptors from both sources. Autoradiographic data showed that the purified receptors from S3 had higher affinity for 8-azido-ATP-γ-32P than the receptors from S1. The binding of 8-azido-ATP-γ-32P to H-mP2X1R was inhibited by ATP-γ-S, α,β-me-ATP, and PPADS, but not by a nucleoside analog (N6-methyl-2′-deoxy-adenosine). In the presence of 2 mM Ca2+ or Mg2+ the binding was increased, but not when using a partially purified receptor fraction, in which unidentified proteins bound 8-azido-ATP-γ-32P or were phosphorylated at 4°C in the presence of 2 mM Mg2+. These data suggest that the decrease in potency of ATP in the presence of Ca2+ and Mg2+, as observed in functional studies, is not due to a direct effect of the cations on the binding of ATP to the receptor. Both cyanogen bromide and hydroxylamine cleavage further confirmed the peptide structure of the purified H-mP2X1R. Autoradiographic analysis of the cleavage products showed that 8-azido-ATP-γ-32P was crosslinked to the carboxyl side of the extracellular domain of the receptor.
Keywords: ion channels, nucleotides, affinity chromatography, agonist, binding site
INTRODUCTION
P2X receptors are ATP-gated ion channels that are permeable to both monovalent and divalent cations, including Na+, K+, and Ca2+. To date, seven mammalian P2X receptor subunits (P2X1–7) have been cloned. They differ in a number of features, including desensitization, sensitivity to the agonist α,β-me-ATP and the antagonist PPADS, permeability to cations, receptor localization, and physiological functions [reviewed in Ralevic et al., 1998; Soto et al., 1997; North, 1996; North and Surprenant, 2000]. The predicted structure of P2X receptor subunits contains two transmembrane (TM) domains separated by a long extracellular loop. The functional P2X receptor is suggested to consist of a trimer [Nicke et al., 1998] or tetramer [Kim et al., 1997] of subunits. The pore of the ion channel is formed by TMs of subunits assembled as homomers or heteromers. This hypothesis is supported by heterologous expression systems such as P2X1/P2X5 [Torres et al., 1998a], P2X2/P2X3 [Radford et al., 1997], and P2X4/P2X6 [Lê et al., 1998] heteromers. The characterization of receptor subtypes in vivo is complicated by an overlap in distribution and by the lack of selective agonists and antagonists [Jacobson et al., 1998a].
The P2X1 receptor has been cloned from several cells or tissues, including the rat vas deferens [Valera et al., 1994] and the human and mouse urinary bladder [Valera et al., 1995; 1996]. The recombinant receptor is activated by 2MeSATP≥ATP>α,β-meATP≫ADP, and reversibly blocked by PPADS, with an IC50 value of 98 nM at the rat homolog [Jacobson et al., 1998b]. This receptor is believed to be the most significant P2X subtype in vascular smooth muscle [Valera et al., 1994], although the P2X4 subtype has also been detected [Soto et al., 1996]. A P2X1-like receptor also exists in platelets [MacKenzie et al., 1996] and promyelocytic HL60 cells [Buell et al., 1996]. One of the major characteristics of this receptor is its rapid desensitization, independent of the amounts of agonist used [Werner et al., 1996], whereas the rapid desensitization of the P2X3 receptor is concentration-dependent [Evans and Surprenant, 1996; Robertson et al., 1996].
Like other P2X subtypes, little is know about the functional regulation of the P2X1 receptor. The purification of the recombinant receptor is therefore important for in vitro characterization by methods, such as X-ray crystallography and ion channel reconstitution, and to define the ATP-binding sites and the factors that regulate agonist and antagonist binding.
Although P2X receptors were first cloned in 1994 [Valera et al., 1994], purification has only been reported for a truncated form of the extracellular domain [Kim et al., 1997], as a result of the difficulty in achieving high-level expression and solubilization of the recombinant receptor. In the present study, we optimized conditions for expression of the mouse P2X1 receptor subunit tagged with hexahistidine at the N-terminus (H-mP2X1R) and developed a method for the solubilization to allow large quantity purification of recombinant subunits. This methodology is potentially applicable to other P2X receptor subtypes. In the course of this purification study, many questions were raised, which might be challenges in future studies of the functional regulation of this receptor subtype, as well as other P2X receptors.
EXPERIMENTAL PROCEDURES
Materials
Mouse P2X1 cDNA cloned from urinary bladder was kindly provided by Dr. Gary Buell (Sereno, Switzerland) in pBKCMV (EcoR I-Not I) (Stratagene, La Jolla, CA). Baculovirus transfer vector pAcHLT was purchased from Pharmigen (San Diego, CA). Sf9 insect cells and ESF 921 serum-free insect media were obtained from Expression System LLC (Woodland, CA). TALON Metal Affinity Resins were from Clontech (Palo Alto, CA). 8-Azido-ATP-γ-32P was purchased from ICN (Irvine, CA). All other ATP analogs were purchased from Research Biochemicals International (Natick, MA). Nondetergent sulphobetaines (NDSB) 201 were purchased from Calbiochem (San Diego, CA). The protease inhibitors carbobenzoxy-L-leucyl-L-leucyl-L-norvalinal (MG115), carbobenzoxy-L-isoleucyl-γ-t-butyl-L-glutamyl-L-alanyl-L-leucinal (PSI), and lactacystin were obtained from Peptides International (Louisville, KY). Other protease inhibitors N-[N-(L-trans-3-carboxyoxirane-2-carbonyl)-L-leucyl]-agmatine (E64), N-acetyl-leucyl-leucyl-norleucinal (ALLN), 4-(2-aminoethyl) benzylsulfonylfluoride (AEBSF), phenylmethylsulfonyl fluoride (PMSF), leupeptin, and aprotinin, ATP-agarose (attachment through the ribose-hydroxyls with a 22-carbon atom spacer), anti-polyhistidine monoclonal antibody (clone no. HIS-1), and other detergents and chemicals were purchased from Sigma (St. Louis, MO).
Construction of Hexahistidine-Tagged Mouse P2X1 Receptor (H-mP2X1R) Transfection Vector and Recombinant H-mP2X1R Virus Production
Mouse P2X1 cDNA in pBKCMV (EcoR I-Not I) was double-digested with Noc I and Not I to release 833-bp of 5′-end Noc I-Noc I fragment (−1 to 832 of open reading frame), and 765-bp of 3′-end Noc I-Not I fragment. The two fragments were sequentially ligated in frame into a pAcHLT-B vector, double-digested with Noc I and Not I. Clones that contained the proper orientation of the 5′-end of the Noc I-Nco I fragment in a pAcHLT-B transfection vector were selected by restriction enzyme mapping. The selected positive clone was further confirmed by sequencing (Biopolymer Laboratory, University of Maryland School of Medicine, Baltimore, MD). Baculovirus containing H-mP2X1R cDNA was obtained by cotransfection of Sf9 cells with H-mP2X1R transfection vectors and linear AcMNPV virus DNAs and purified as described by the manufacturer (InVitrogen, Carlsbad, CA).
Expression of H-mP2X1R in a Baculovirus Insect System
The recombinant virus was amplified and used for large-scale production of H-mP2X1R, either in stationary cultures at a multiplicity of infection (MOI) of 1.5–2.0 × 10−2 pfu/cell or in spinner flask cultures at an MOI of 2.5–5.0 × 10−2 pfu/cell. Sf9 cells expressing H-mP2X1R of 0.5–1.0% of total cell lysate protein were harvested at 90–94 h postinfection and washed twice with PBS. Cell pellets were stored at −80°C.
In Vitro Protease Inhibitor Selection
The inhibition of the degradation of H-mP2X1R was examined in total cell lysates as described previously [McGee et al., 1996]. To a pellet of 1 × 108 cells, 10 ml of 10 mM Tris, pH 7.5, was added. The cell suspension was incubated on ice for 30 min, followed by Polytron homogenization at a setting of 6.5 three times for 10 sec, and sonication at 100 watts three times for 10 sec, both on ice. The homogenate was centrifuged at 1,200 rpm for 10 min at 4°C to remove unbroken cells and nuclei. A cell lysate suspension of 150 μl was transferred into ice-cold Eppendorf tubes containing protease inhibitors in amounts necessary to reach the indicated final concentrations. The mixture was incubated at 37°C for 1, 3, or 6 h, stopped by adding 50 μl of 4X sample buffer, heated at 100°C for 10 min, and stored at −20°C for further analysis.
Solubilization and Purification of H-mP2X1R
After comparing the solubilization methods (Fig. 3), frozen cell pellets of 1 × 109 cells infected with H-mP2X1R baculovirus were resuspended in hypotonic lysis buffer (10 mM Tris, pH 7.5, 2 μg/ml leupeptin, 0.1 mM iodoacetamide, 0.1 mM PMSF, and 5 μg/ml E-64) at 107 cells/ml. The cell suspension was incubated on ice for 30 min, followed by Polytron and sonication as described above. The unbroken cells and nuclei were removed as described. The supernatant was then solubilized by adding detergents to a final concentration of 1% Triton X-100, 0.5% sodium deoxycholate, and 0.2% SDS, or as indicated in each experiment. The mixture was shaken at 4°C for 3 h and centrifuged at 30,000g for 30 min at 4°C. The supernatant (S1) was collected for cobalt affinity chromatography. The pellet was resuspended in 1/10 volume of lysis buffer initially used, and 1 N NaOH was added to a final concentration of 20 mM, followed by sonication to resuspend the pellet. After shaking for 30 min at 4°C, the mixture was centrifuged at 30,000g for 30 min. The resulting supernatant (S2) was dialyzed over a dialysis buffer (10 mM Tris, pH 8.0, 150 mM NaCl, 1 mM EDTA, 1 μg/ml leupeptin, 0.2 mM PMSF, and 0.1% Triton X-100) overnight, and further centrifuged as described to obtain the final supernatant (S3).
Fig. 3.

Solubilization of H-mP2X1R. a: Coomassie Blue staining. b: Immunoblot. Sf9 cells infected with H-mP2X1R baculovirus were harvested at 92 h postinfection. Cells were resuspended in lysate buffer at 1 × 108 cells / 10 ml. After removing cell debris and nuclei, receptors were solubilized with 6 M urea (I), 1% Triton X-100, and 1 M NDSB201 (II), or 1% Triton X-100, 0.5% deoxycholate, and 0.25% SDS (III). The initially insoluble pellets from all preparations were further solubilized with 20 mM NaOH (IV), followed by centrifugation that resulted in S2 and pellet 2 (P2). The S3 and P3 fractions are centrifugation products after dialysis of the S2 fraction against a lysate buffer containing 0.1% Triton X-100 and protease inhibitors (V). Lysate of 1 × 105 cells, or initial supernatant (S1) or pellet (P1) from 1 × 105 cells of cell lysate was loaded. S2 and S3 fractions were 10-fold concentrated. P2 and P3 were 20-fold concentrated. Insert shows S3 (1) and its deglycosylation products (2) in both Coomassie Blue staining (in a) and immunoblotting (in b) analysis.
Cobalt Affinity Chromatography
The solubilized receptors were first adjusted to 250 mM NaCl and 10% glycerol and loaded at 20 ml/h onto a 5 ml cobalt affinity column, prewashed with equilibration buffer (50 mM Tris pH 8.0, 250 mM NaCl, 10% glycerol and 0.5% Triton X-100, and 1 μg/ml leupeptin, 0.2 mM PMSF). The column was washed with 2 column volumes of equilibration buffer, 3 column volumes of equilibration buffer containing 10 mM imidazole, and 5 column volumes of equilibration buffer containing 25 mM imidazole. The column was then equilibrated in equilibration buffer and the H-mP2X1R was eluted with 3 column volumes of elution buffer (50 mM Pipes, pH 5.0, 300 mM NaCl, 10% glycerol, 1 μg/ml E-64 and leupeptin).
ATP-Affinity Chromatography
The receptors eluted from the cobalt affinity column were concentrated to 5 ml and dialyzed against equilibration buffer A (50 mM Tris, pH 7.4, 100 mM NaCl, 5.0 mM MgCl, 1 mM EDTA, 0.02% Triton X-100, 10% glycerol, and 1 μg/ml E64 and leupeptin) overnight and applied to 2 ml ATP-agarose-ribose-hydroxyl affinity column pre-equilibrated with the equilibration buffer A. The flow was stopped and the resins were resuspended in buffer and kept at 4°C for 30 min. The column was opened at a flow rate of 0.5 ml/min, washed with 12 column volumes of the equilibration buffer A, followed by 12 column volumes of equilibration buffer A containing 1 mM AMP, and then eluted with 3 column volumes of the same buffer containing 5 mM ATP. The collected fractions were concentrated by centrifugation through an Amincon Microcon 30 membrane and exchanged with preservation buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 10% glycerol, 0.1% Triton X100, 1 μg/ml E64) and concentrated to 1–2 μg/ml.
Determination of Purity of the Purified H-mP2X1R
An aliquot (100 μl) of purified H-mP2X1R was separated on a 10% polyacrylamide-SDS gel, and the gel was stained with copper staining solution as described previously [Lee et al., 1987] to visualize the 45 kDa H-mP2X1R band. The band was excised and the receptor was electrophoretically eluted using an Isco little blue tank (Isco Co., Lincoln, NE) according to the method of Takeda and Cone [1984]. The purity of the isolated H-mP2X1R was further confirmed by two-dimensional isoelectric focusing–SDS polyacrylamide electrophoresis, on which a single spot was observed (data not shown). The gel-purified H-mP2X1R was estimated to be >95% pure and served as a standard. The amount of H-mP2X1Rs in each purification fraction was evaluated by immunoblotting and an LKB laser densitometer (Pharmacia Biotech, Piscataway, NJ) scanning, in comparison to a standard curve generated with known amounts (10–200 ng) of H-mP2X1R. The specific content of H-mP2X1R, expressed as mg of H-mP2X1R per mg of total protein, indicated the degree of the purification.
Circular Dichroism
Circular dichroism spectra were recorded at 25°C, in a Jasco J-715 spectropolarimeter, interfaced with a PC and 1 mm quartz cuvettes in 0.1 M sodium phosphate buffer, pH 7.2 [McPhie et al., 1993]. The protein concentration used was 0.1 mg/ml in the presence of 1.0% CHAPS.
Autoradiography of Radioactively-Labeled H-mP2X1R: Inhibition of Binding of 8-Azido-ATP-γ-32P and Phosphorylation
8-Azido-ATP-γ-32P, an ATP analog which has been demonstrated to bind to the P2X1 receptor [Fedan et al., 1986; Giannattasio et al., 1992], was chosen for the autoradiographic study as the azido group on the adenine ring would be involved in UV-induced crosslinking. Conventionally used ATP analogs were used as inhibitors to determine their ability to displace binding, in comparison with the nucleoside analog N6-methyl-2′-deoxy-adenosine as a negative control. About 2 μg of purified H-mP2X1R or 10 μg partially purified H-mP2X1R fraction was used for each incubation. The reaction was carried out in reaction buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% Triton X-100, and 1 μg/ml leupeptin) with a total volume of 20 μl. Some experiments were carried out in the presence of 2 mM MgCl2 or CaCl2. Inhibitors were added to reach the indicated final concentration. Two μl (1 μCi/μl) of 8-azido-ATP-γ-32P (19.7 Ci/mmole) were added to each reaction so that the final 8-azido-ATP-γ-32P concentration was approximately 5 μM. All samples and tubes were pre-ice cooled and samples were added on ice. The reaction mix was incubated at 4°C for 1.5 h. At the end of the incubation a UV irradiation was carried out for 5 min using a Stratagene UV-linker. 7 μl of 4X sample buffer was then added to each tube, which was then heated at 100°C for 10 min. All samples were electrophoresed on 4–20% gradient gels (Owl Separation Systems, Woburn, MA). Gels were stained with GelGode Blue Stain Reagent (Pierce, Rockford, IL), vacuum-dried, and exposed to Kodak X-Omat AR films at −80°C. For cyanogen bromide cleavage, 10 μg of purified H-mP2X1R was used for each reaction. The samples were vacuum-dried after UV crosslinking and subjected to cleavage. All experiments were repeated three times. The crosslinking was also attempted using the same concentration and activity of ATP-γ-33P instead of 8-azido-ATP-γ-32P, with UV irradiation carried out on purified receptor preparations to exclude cross-linking in the absence of the azido group.
To detect protein phosphorylation in partially purified H-mP2X1R preparations under the experimental conditions the incubations were carried out without UV irradiation, using the same concentration and activity of ATP-γ-33P as above. The reaction was stopped by adding 4X SDS sample buffer on ice after 1.5 h incubation and heated as described.
Cyanogen Bromide (CnBr) Cleavage of H-mP2X1R
CnBr cleavage was carried out essentially according to a published procedure [Kaiser and Metzka, 1999]. The purified H-mP2X1R (10 μg) was dissolved in 15 μl of 0.1 N HCl, 8 M urea, and 10 μl of CnBr solution (0.5 g/ ml in acetonitrile) was added. The tube was sealed and the reaction was carried out at RT in the dark for 12–18 h. The reaction was stopped by adding 5 μl of 6X SDS sample buffer. Aliquots of 0.5 μl 10% ammonium hydroxide were added until the yellow color turned blue, and the mixture was heated at 100°C for 10 min.
Hydroxylamine Digestion of H-mP2X1R
The reaction was carried out as described previously [Borstein and Balian, 1977]. The purified H-mP2X1R (10 μg) was resuspended in 20 μl of 2 M hydroxylamine HCl, 2 M guanidine HCl, and 0.2 M potassium carbonate at pH 9.0. The mixture was incubated at 45°C for 4 h and then lyophilized. The dry pellet was resuspended in 2X SDS sample buffer and heated at 100°C for 10 min.
Immunoblotting
Total H-mP2X1R cell lysates and purified H-mP2X1R were electrophoresed on 4–20% gradient gels (Owl Separation Systems) and transferred to nitrocellulose membranes for immunoblotting as described previously [Chen et al., 1997]. The nitrocellulose was incubated overnight in a 1:1,000 dilution of an anti-polyhistidine monoclonal antibody (clone no. HIS1). The blot was developed using a 1:2,000 dilution of goat antimouse IgG conjugated to alkaline phosphatase (Sigma) and 5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium.
Other Methods
H-mP2X1R deglycosylation was performed using N-glycosidase F (New England Biolabs, Beverly, MA) according to the manufacturer. Protein concentration was determined by the protein dye-binding method [Bradford, 1976] using Coomassie Plus protein assay reagent (Pierce).
RESULTS
Expression of H-mP2X1R
The expression of H-mP2X1R was first carried out in six-well plates to determine the optimal MOI by immunoblotting. An MOI of 1.5–2.0 × 10−2 pfu/cell was chosen for infections of 2.8–3.0 × 107 Sf9 cells in 50 ml media in 175 cm2 flasks, with a harvest time of 92–94 h postinfection (pi). Time course studies (Fig. 1) show the accumulation of H-mP2X1R in the cell culture with the chosen MOI. Increasing the MOI to 5.0 × 10−2 pfu/cell could achieve a similar H-mP2X1R expression level in harvested cells at 72 h pi, but with less glycosylated products and ~50% cells lysed (data not shown). Further varying the MOI for infection resulted in less to undetectable H-mP2X1R production (data not shown). Infection with High Five cells, another type of baculovirus insect cell, resulted in better expression but less consistency.
Fig. 1.
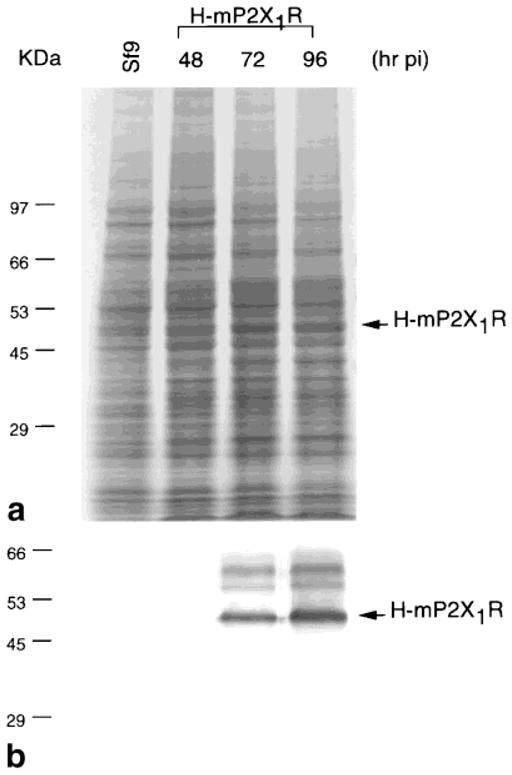
Time course of the expression of H-mP2X1R. a: Coomassie Blue staining. b: Immunoblot with monoclonal anti-polyhistidine antibody, Clone HIS-1. Sf9 cells were infected with 1.5 × 10−2 pfu/cell of H-mP2X1R baculovirus and harvested at the indicated time. Cell lysate of 1 × 105 cells/lane were loaded.
Inhibition of H-mP2X1R Degradation In Vitro
H-mP2X1R degradation was inhibited by adding E64, ALLN, MG115, and leupeptin in vitro. Iodoacetamide also inhibited the degradation but to a lesser degree. Other protease inhibitors used did not inhibit the degradation, as H-mP2X1R degradation bands were observed (Fig. 2). Protease inhibitors that inhibited H-mP2X1R degradation in vitro did not reduce the degradation of H-mP2X1R in cell cultures (data not shown).
Fig. 2.

Inhibition of H-mP2X1R Degradation in vitro. Sf9 cells expressing H-mP2X1R receptors were lysed in 10 mM Tris-Cl, pH 7.5, sonicated and spun for 10 min to remove cell debris and nuclei. The supernatants were incubated at 37°C for 6 h with the indicated protease inhibitors. The final concentrations of protease inhibitors were: E64 (50 μg/ml), ALLN (50 μM), PSI (50 μM), MG115 (50 μM), lactacystin (50 μM), leupeptin (10 μg/ml), aprotinin (10 μg/ml), pepstatin A (10 μg/ml), iodoacetamide (100 μM), and PMSF (1 M). After incubation, samples from 1 × 105 cells were subjected to Coomassie Blue Staining (a) and immunoblotting (b) analyses. Control was the supernatant collected after centrifugation. Arrows above the H-mP2X1R band indicate H-mP2X1R with different glycosylation levels.
Solubilization of H-mP2X1R
Solubilization of H-mP2X1R was first compared by using a number of detergents with different combinations. Detergents used were: Triton X-100, deoxycholate, sodium cholate, tergitol NP40, igepal CA-630, digitonin, β-D-octyl glucoside, CHAPS, NDSBs, and urea, all of which resulted in poor solubilization. Examples with relatively better solubilization (~30%) of H-mP2X1R are shown in Figure 3(I, II, and III). However, adding 1 N NaOH to the pellet to a final concentration of 20 mM resulted in >90% H-mP2X1R solubilized from the pellet (S2, Fig. 3: IV) The solubilized receptors stayed in the supernatant after dialyzing over a lysis buffer containing 0.1% Triton X-100 (S3, Fig. 3: V). Glycosylated receptors were mainly solubilized in S2 and S3 fractions. Increasing the pH of the buffer system at the initial solubilization step by using CHES (pH 9.5) or CAPS (pH 11.0) did not improve the solubilization (data not shown).
Purification of H-mP2X1R
Four different HiTrap chelating columns (Ni2+, Zn2+, Cu2+, and Co2+; Pharmacia Biotech) were first tested at a loading pH of 7.5. Both the Zn2+ and the Co2+ columns were relatively selective in binding H-mP2X1R, whereas the Cu2+ column bound 90% of the proteins tightly, and the Ni2+ column bound H-mP2X1R poorly (data not shown). A prechelated Co2+ column, the TALON column (Clontech), was therefore selected for H-mP2X1R purification. Purification of H-mP2X1R with the TALON column resulted in about 15% purity from the S1 fraction. The purity was increased to above 70% after an ATP-affinity column. H-mP2X1R purified from S3 could reach 90% pure. The purification steps and results are shown in Figure 4, and an example of the H-mP2X1R recovery rate from each step is summarized in Table 1.
Fig. 4.
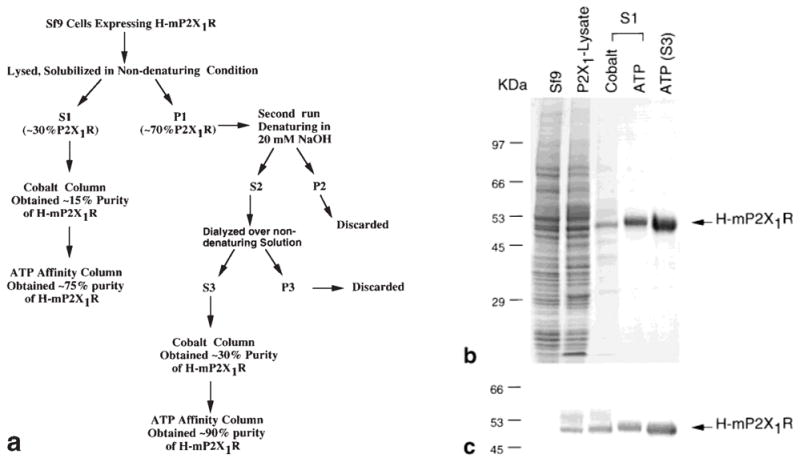
Purification of H-mP2X1R. a: Scheme of H-mP2X1R purification. b: Coomassie Blue staining. c: Immunoblot analysis of purification fractions. Cell lysates of 1 × 105 cells were loaded for control (Sf9 uninfected), and P2X1-lysate (Sf9 cells infected with H-mP2X1R viruses). Samples of 5 μg of fractions from cobalt column (Cobalt) and ATP column (ATP) were loaded. S1 and S3 indicate that receptors were purified from the S1 and S3 fractions. For the immunoblotting analysis, half amounts of samples were loaded for each lane.
TABLE 1.
Purification of H-mP2X1R Expressed in the Baculovirus System
| Cell fraction | Total protein (mg) | P2X/protein1 (mg/mg) | Yield purification | Fold2 (%) |
|---|---|---|---|---|
| Crude extract | 13,000 | 0.0065 | 1.0 | 100.0 |
| S1 | 8500 | 0.0031 | 0.5 | 31.0 |
| Cobalt-sepharose (S1) | 21.0 | 0.1428 | 21.9 | 3.5 |
| ATP-agarose (S1) | 1.5 | 0.7142 | 109.8 | 1.2 |
| S3 | 3500 | 0.0155 | 2.4 | 64.0 |
| Cobalt-spharose (S3) | 15.3 | 0.3269 | 50.3 | 5.9 |
| ATP-agarose (S3) | 2.1 | 0.9047 | 139.2 | 2.2 |
The amount of H-mP2X1R (P2X) in each fraction was determined by immunobloting and the densitometric measurement compared with a standard curve of H-mP2X1R generated with purified standard receptors as described in Experimental Procedures.
Fold purification was determined by the mgH-mP2X1R/mg protein in each fraction divided by the mg h-mP2X1R/mg protein in the crude extract.
Secondary Structure of H-mP2X1R
Circular dichroism (CD) spectroscopy, which is sensitive to the contribution of various secondary structural elements, was used to evaluated the overall conformation of the H-mP2X1R purified from both S1 and S3. The results indicate that there was no difference between the H-mP2X1R purified from both fractions. Proteins from both preparations have minima around 220 nm and maxima around 210 nm (Fig. 5). Analysis of the CD spectrum using the CONTIN program [Provencher and Glockner, 1981] indicates H-mP2X1R with about 8% α-helical (i.e., 10% α-helical in mouse P2X1 receptor) and about 45% β-sheet structures.
Fig. 5.

Ultraviolet circular dichroism spectra of H-mP2X1R. CD spectra results were from receptors purified from S1 (a) and S3 (b). Protein concentration of 100 μg/ml was estimated by absorption at A272.
8-Azido-ATP-γ-32P Crosslinking and Inhibition Studies
H-mP2X1Rs purified from S1 (Fig. 6a) and S3 fractions (Fig. 6b) were used for 8-azido-ATP-γ-32P binding and inhibition studies. The results showed no significant difference, except that the receptors from the S3 fraction showed higher affinity. In both cases, the binding of 8-azido-ATP-γ-32P to purified H-mP2X1R was increased in the presence of 2 mM Mg2+ or Ca2+, and inhibited by α,β-me-ATP, ATP-γ-S, and PPADS in a concentration-dependent manner; N6-methyl-2′-deoxy-adenosine, a nucleoside included as a negative control, had no effect (Fig. 6a, b). However, using a partially purified H-mP2X1R fraction for the same experiment, neither Mg2+ nor Ca2+ increased 8-azido-ATP-γ-32P binding to H-mP2X1R. In the presence of 2 mM Mg2+, the γ-phosphate of the 8-azido-ATP-γ-32P was crosslinked to many unidentified proteins (Fig. 6d).
Fig. 6.
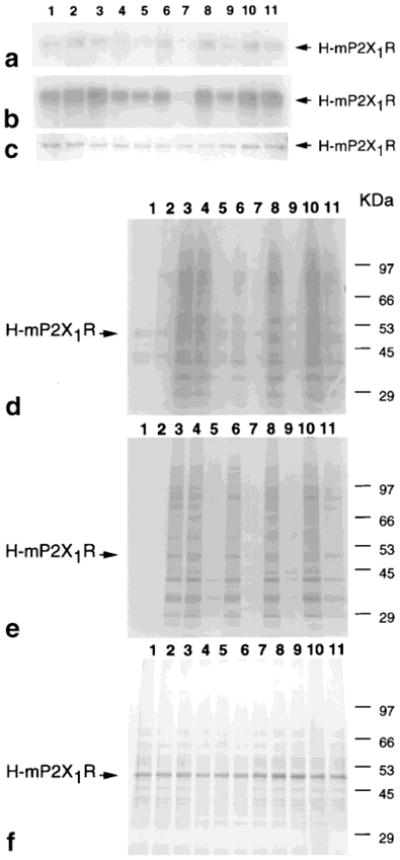
An example of autoradiography of 8-azido-ATP-γ-32P binding to H-mP2X1R and its inhibition. a,b,d: 8-Azido-ATP-γ-32P autoradiographs with purified H-mP2X1R from S1 (a), S3 (b), or with the partially purified H-mP2X1R fraction (d) after 5 min UV crosslinking. e: ATP-γ-33P autoradiograph without UV irradiation using the partially purified H-mP2X1R fraction. c,f: Examples of Coomassie Blue staining gels of a and b (c), and d and e (f). Samples loaded for each lane are proteins with radioactivity marked: ATP alone (1), in the presence of 2 mM Ca2+ (2), or 2 mM Mg2+ (3–11); and in the presence of inhibitory ligands: α,β-me-ATP 10 μM (4) and 300 μM (5), ATP-γ-S 10 μM (6) and 300 μM (7), PPADS 10 μM (8) and 300 μM (9), and N6-methyl-2′-deoxy-adenosine 10 μM (10) and 300 μM (11). Experiments were performed as described in Experimental Procedures. All experiments were repeated three times.
The influence of the UV irradiation time (1, 3, and 5 min) was compared using the same concentration and activities of 8-azido-ATP-γ-32P or ATP-γ-33P for binding to the purified receptors. The results indicate that the irradiation time influenced 8-azido-ATP-γ-32P binding results in a time-dependent manner, with 5 min being optimal. When crosslinking to purified receptors was attempted with ATP-γ-33P, only vague bands were observed under all crosslinking conditions used in the study (data not shown). This indicated that ATP, by itself, did not effectively covalently bind to the receptor.
However, when the incubation experiments were carried out without UV irradiation for 1.5 h at 4°C, using ATP-γ-33P and partially purified H-mP2X1R fractions, many bands appeared. These bands, indicative of protein phosphorylation, disappeared in the presence of the inhibitors (Fig. 6e). Similar results were obtained using 8-azido-ATP-γ-32P (data not shown).
H-mP2X1R Cleavage and Binding Site Detection
Both hydroxylamine·HCl and CnBr digestions confirmed the peptide structure of the purified H-mP2X1R (Fig. 7a,b). Autoradiography, following 8-azido-ATP-γ-32P crosslinking and CnBr digestion, suggested at least one amino acid in the carboxyl terminal of the extracellular domain of H-mP2X1R was crosslinked to 8-azido-ATP-γ-32P (Fig. 7c). The labeled band matched the molecular weight of the expected 13.7 KDa fragment. The same experiment repeated with hydroxylamine·HCl did not succeed, due to the difficulty in electrophoretically separating the digested H-mP2X1R products (data not shown).
Fig. 7.

Confirmation of H-mP2X1R protein by cyanogen bromide and hydroxylamine·HCl cleavage. a: H-mP2X1R peptide map. Mouse P2X1 (399 amino acids) was tagged with 45 amino acids of hexahistidine-tag. Two predicted transmembrane domains and cystine folding domains [Hansen et al., 1997] were boxed. Numbers at the start and end of each domain indicate amino acid positions in the mouse P2X1 peptide. Numbers in parentheses above the peptide indicate CnBr cleavage sites. Numbers in the parentheses under the sequence indicate hydroxylamine·HCl cleavage sites. Double-ended arrows indicate the length of peptides released after cleavage, with molecular weights indicated in Daltons. The triangles indicated GCG program-predicted glycosylation sites. b: Coomassie Blue staining analysis of CnBr and hydroxylamine·HCl cleavage of H-mP2X1R. Samples loaded are: H-mP2X1R without cleavage (lane 1), H-mP2X1R cleaved with CnBr (lane 2), or hydroxylamine HCl (lane 3). c: Autoradiograph of CnBr cleavage of 8-azido-ATP-γ-32P linked H-mP2X1R. The 8-azido-ATP-γ-32P binding and linking was performed as in Experimental Procedures. Lane 4, binding without cleavage; Lane 5, binding with CnBr cleavage. Samples were separated with 15% (15 × 15 cm2) acrylamide gel using Tricine buffer as described previously [Schagger et al., 1987].
DISCUSSION
In the present study we optimized the conditions for the purification of a recombinant ATP-gated ion channel receptor subunit, namely, the H-mP2X1R, expressed in a baculovirus system and initiated the characterization of this receptor. The difficulty in obtaining large quantities of highly purified receptor protein was evident at each step of the purification.
Low MOI has been recommended in many other studies of expression of functional recombinant proteins [Chen et al., 1997; Buters et al., 1995]. Similarly, in our study a low MOI was required for a high level of H-mP2X1R protein expression, which may be due to its being a membrane-bound ion channel protein.
The degradative bands observed in total cell lysates of Sf9 cells expressing H-mP2X1R and the initially low expression levels led us to evaluate various protease inhibitors as protective additives. E64 [Moriyama et al., 1998], a cysteine protease inhibitor, and ALLN and MG115, two nonspecific proteasome inhibitors, which possess the cysteine protease inhibition functions [Figueiredo-Pereira et al., 1994; Rock et al., 1994], inhibited H-mP2X1R degradation. PSI, another nonspecific proteasome inhibitor, which inhibited cysteine proteases, including calpain and cathepsins [Figueiredo-Pereira et al., 1994; Rock et al., 1994], and lactacystin [Fenteany et al., 1995], a specific proteasome inhibitor, did not inhibit degradation. Interestingly, leupetin, an inhibitor of lysosomal cathepsins [Lukacs et al., 1994], inhibited the degradation, while pepstain, another inhibitor of lysosomal cathepsins [Lukacs et al., 1994], did not inhibit degradation. The commonly used cysteine protease inhibitor iodoacetamide also inhibited the degradation, but to a lesser degree. The serine protease inhibitors AEBSF, PMSF, and aprotinin did not inhibit the degradation. In conclusion, the H-mP2X1R degradation in vitro was mainly catalyzed by a cysteine protease, which was inhibited by E64, ALLN, MG115, and leupeptin.
Traditional nondenaturing and denaturing methods only poorly solubilized the H-mP2X1R. However, upon increasing the pH to 13 by adding NaOH to a concentration of 20 mM, the receptors in the pellet fraction were solubilized. The receptors solubilized under alkaline conditions stayed in the soluble fraction after removal of the insoluble fraction, and even after dialyzing against a buffer that originally did not solubilize the receptors. This result suggested that high pH released H-mP2X1R from some insoluble components, such as cytoskeletal matrix [Strand et al., 1994]. P2X1 has been suggested to associate with actin [Parker, 1998], a cytoskeletal protein of isoelectric point (pI) 5.5 (calculated by GCG program). The calculated pI value of H-mP2X1R (GCG program) is pI 9.5 for H-mP2X1R, pI 10.52 for the N-terminal intracellular peptide, pI 11.48 for the N-terminal intracellular peptide with His-tag, and pI 10.01 for C-terminal intracellular peptide. Other high pH buffers, such as CHES (pH 9.5) and CAPS (pH 11.0), failed to solubilize the receptors, suggesting the involvement of ionic interactions in the aggregation. Basic Lys and Arg residues, which consistently appear in both intracellular domains of P2X subtypes [Soto et al., 1997], possibly contribute to the aggregation. The possibility of high pH causing a disintegration of the receptors was excluded when running a nondenaturing acrylamide gel, in which most of the H-mP2X1R from the S3 fraction remained at the top of the gel.
The purpose of running a nondenaturing acrylamide gel was to investigate whether the glycosylation contributed to the aggregation, as we noticed that highly glycosylated proteins were lost during the purification processes. However, no distinctive bands were observed in a nondenaturing acrylamide gel after S3 was deglycosylated (data not shown). A parallel result from a denaturing gel suggested that the proteins were not degraded (Fig. 3, insert). However, the presence of monomers and different multimers following deglycosylation could explain the observation, since PNGase F treatment only weakened bands around 90 kD position (data not shown) and further, more conclusive experiments are required.
We did not perform crosslinking experiments to determine the multimeric structure of the purified receptors by running a blue native PAGE, since the results are ligand spacer-length dependent, and since a monomer would appear at a different molecular weight because of different glycosylation levels [Nicke et al., 1998].
To examine if the high pH changed the protein structure permanently, we performed CD spectral measurement. The result showed an identical secondary structure of the receptors purified from the S1 and S3 fractions. Together with the 8-azido-ATP-γ-32P crosslinking and inhibition data, the results indicate that high pH did not denature the receptors. Therefore, the S3 fraction seems to be a good choice for purification in order to obtain a large quantity of receptor protein with a high purity.
However, posttranslational modification could be a factor that determines whether H-mP2X1R is present in the soluble or the insoluble fraction. Similar phenomena have been observed with some other cytoskeleton-associated proteins, such as ornithine decarboxylase (ODC), which has a phosphorylated form in the insoluble fraction and a nonphosphorylated form in the soluble fraction [Pomidor et al., 1999]. In our experiment, harvesting infected Sf9 cells 72 h pi provided less total, expressed H-mP2X1R, but a higher concentration of receptors in the S1 fraction (data not shown). This observation suggested that highly posttranslationally modified receptors mainly contribute to the insoluble fraction. Results from the binding study showed that the receptors purified from the S3 fraction had a higher affinity for 8-azido-ATP-γ-32P than receptors from the S1 fraction, which might indicate a differential degree of posttranslational modification of the receptors in the different fractions. The different purity, however, might also have influenced the results. It will be interesting to further determine if posttranslational modification, such as phosphorylation or glycosylation, influences receptor function, including ligand binding. It would also be useful to study differences in glycosylation [Torres et al., 1998b] of this recombinant receptor expressed in mammalian vs. insect cells.
Different approaches have been applied to study ligand affinities at P2X receptor subtypes, such as ion channel functional responses in native tissues and transfected cells [Surprenant et al., 1995], binding affinity studies using membranes of native tissues [Michel et al., 1996] or transfected cells [Michel et al., 1997]. The autoradiographic analysis of the binding experiments proves that P2-ligands bind to the purified receptors. The analysis, however, cannot be used to determine ligand affinity constants or binding inhibition values. Nevertheless, the results suggest that ATP-γ-S has higher affinity at the H-mP2X1R than α,β-me-ATP (compare the inhibition at 300 μM). It has been suggested that raising Ca2+ and Mg2+ might decrease the potency of ATP, possibly by allosteric effects on the receptors [Evans et al., 1996; Li et al., 1997]. Our autoradiographic data indicated that both Ca2+ and Mg2+ increased the 8-azido-ATP-γ-32P binding to purified H-mP2X1R, but not to H-mP2X1R in a partially purified fraction. In this fraction Mg2+ dramatically increased 8-azido-ATP-γ-32P binding to many unidentified proteins, and widespread protein phosphorylation occurred. Our results suggest that the decrease in potency of ATP in the presence of Ca2+ and Mg2+ observed in functional studies is not due to a direct influence on the affinity of ATP at the receptors. It seems that gel autoradiography could be a facile method to determine whether a ligand is specifically bound to the receptor, in correlation with functional responses. A range of functional studies of the purified receptor should now be carried out. In addition, reversible binding to a putative P2 receptor radioligand, e.g., α,β-me-ATP, could be studied.
Since ATP binds to many proteins, CnBr and hydroxylamine·HCl cleavage were applied to further confirm the peptide structure of the purified H-mP2X1Rs. The result from CnBr cleavage combined with an autoradiographic analysis indicated that at least one amino acid at the carboxyl side of extracellular loop [Hansen et al., 1997] is involved in ATP binding. Further cleavage with other proteases might narrow the binding region if combined with electrospray mass spectrometry (C A. Haney, North Carolina State University Mass Spectrometry Facility, personal communication). Furthermore, peptide fragments could be used to study ligand affinity using surface plasmon resonance [Chen et al., 2000], if the binding site could be localized to a single fragment. This procedure would avoid receptor aggregation, which may alter the results when using the whole proteins (unpublished data), and decrease the amount of the purified protein required for a binding kinetics study.
Acknowledgments
We thank Dr. Ivar von Kügelgen (Univ. of Bonn, Germany) for suggesting using 8-azido-ATP-γ-32P for the binding study and for manuscript proofreading.
Abbreviations
- H-mP2X1R
recombinant mouse P2X1 receptor extended with hexahistidine tag
- Sf9
Spodoptera frugiperda
- AcMNPV
Autographa californica neclear polyhedrosis virus
- MOI
multiplicity of infection (the number of virus used per cell)
- pfu
plaque-forming units (measurement of infectivity of a virus)
- E64
N-[N-(L-3-trans-carboxirane-2-carbonyl)-L-leucyl]-agmatine
- ALLN
N-acetyl-leucyl-leucyl-norleucinal
- MG115
carbobennzoxy-L-leucyl-L-leucyl-L-norvalinal
- PSI
carbobenzoxy-L-isoleucyl-γ-t-butyl-L-glutamyl-L-alanyl-L-leucinal
- AEBSF
A-(2-aminoethyl)benzylsulfonylfluoride
- PMSF
phenylmethylsulfonyl fluoride
- NDSBs
non-detergent sulphobetaines
- CHAPS
3-[(3-cholamidopropyl)dimethylammonio]-1-propane-sulfonate
- CHES
2-(N-cyclohexylamino)ethanesulfonic acid
- CAPS
3-(cyclohexylamino)-1-propanesulfonic acid
- GCG
University of Wis-consin Genetics Computer Group
- CD
circular dichroism
- CnBr
cyanogen bromide
- SDS
sodium dodecyl sulfate
References
- Borstein P, Balian G. Cleavage at the Asn-Gly bonds with hydroxylamine. Methods Enzymol. 1977;47:132–145. doi: 10.1016/0076-6879(77)47016-2. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Buell GN, Michel AD, Lewis C, Collo G, Humphrey PP, Surprenant A. P2X1 receptor activation in HL60 cells. Blood. 1996;87:2659–2664. [PubMed] [Google Scholar]
- Buters JTM, Shou M, Hardwick JP, Korzekwa KR, Gonzalez FJ. cDNA-directed expression of human cytochrome P450 CYP1A1 using baculovirus. Drug Metab Dispos. 1995;23:696–701. [PubMed] [Google Scholar]
- Chen L, Buters JTM, Hardwick JP, Tamura S, Penman BW, Gonzalez FJ, Crespi CL. Coexpression of cytochrome P4502A6 and human NADPH-P450 oxidoreductase in the baculovirus system. Drug Metab Dispos. 1997;25:399–405. [PubMed] [Google Scholar]
- Chen L, Broad RM, Sitkovsky MV, Linden J. Affinity comparison of ligands binding to recombinant human A2A adenosine receptors using surface plasmon resonance. Biochem Biophys Res Commun. 2000 (in press) [Google Scholar]
- Evans RJ, Surprenant A. P2X receptors in autonomic and sensory neurons. Semin Neurosci. 1996;8:217–223. [Google Scholar]
- Evans RJ, Lewis C, Virginio C, Lundström K, Buell GN, Surprenant A, North RA. Ionic permeability of, and divalent cation effects on, two ATP-gated cation channels (P2X receptors) expressed in mammalian cells. J Physiol. 1996;497:413–422. doi: 10.1113/jphysiol.1996.sp021777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedan JS, Hogaboom GK, O’Donnell JP. Further comparison of contractions of the smooth muscle of the guinea-pig isolated vas deferens induced by ATP and related analogs. Eur J Pharmacol. 1986;129:279–291. doi: 10.1016/0014-2999(86)90438-3. [DOI] [PubMed] [Google Scholar]
- Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ, Schreiber SL. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science. 1995;268:726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- Figueiredo-Pereira ME, Berg KA, Wilk S. A new inhibitor of the chymotrypsin-like activity of the multicatalytic proteinase complex (20S proteasome) induces accumulation of ubiquitin-protein conjugates in a neuronal cell. J Neurochem. 1994;63:1578–1581. doi: 10.1046/j.1471-4159.1994.63041578.x. [DOI] [PubMed] [Google Scholar]
- Giannattasio B, Powers K, Scarpa A. Characterization of myocardial extracellular ATP receptors by photoaffinity labelling and functional assays. FEBS Lett. 1992;308:327–333. doi: 10.1016/0014-5793(92)81305-6. [DOI] [PubMed] [Google Scholar]
- Hansen MA, Barden JA, Balcar VJ, Keay KA, Bennett MR. Structural motif and characteristics of the extracellular domain of P2X receptors. Biochem Biophys Res Commun. 1997;236:670–675. doi: 10.1006/bbrc.1997.6815. [DOI] [PubMed] [Google Scholar]
- Jacobson KA, Kim Y-C, Camaioni E, van Rhee AM. Structure activity relationships of P2 receptor agonists and antagonists. In: Turner JT, Weisman G, Fedan J, editors. The P2 nucleotide receptors. Clifton, NJ: Humana Press; 1998a. pp. 81–107. (Series: The receptors.) [Google Scholar]
- Jacobson KA, Kim Y-C, Wildman SS, Mohanram A, Harden TK, Boyer JL, King BF, Burnstock G. A pyridoxine cyclic-phosphate and its 6-arylazo-derivative selectively potentiate and antagonize activation of P2X1 receptors. J Med Chem. 1998b;41:2201–2206. doi: 10.1021/jm980183o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser R, Metzka L. Enhancement of cyanogen bromide cleavage yields for methionyl-serine and methionyl-threonine peptide bonds. Anal Biochem. 1999;266:1–8. doi: 10.1006/abio.1998.2945. [DOI] [PubMed] [Google Scholar]
- Kim M, Yoo OJ, Choe S. Molecular assembly of the extracellular domain of P2X2, an ATP-gated ion channel. Biochem Biophys Res Commun. 1997;240:618–622. doi: 10.1006/bbrc.1997.7713. [DOI] [PubMed] [Google Scholar]
- Lê K-T, Babinski K, Séguéla P. Central P2X4 and P2X6 channel subunits coassemble into a novel heterometric ATP receptor. J Neurosci. 1998;18:7152–7159. doi: 10.1523/JNEUROSCI.18-18-07152.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Levin A, Branton D. Copper staining: a five-minute protein stain for sodium dodecyl sulfate-polyacrylamide gels. Anal Biochem. 1987;166:308–312. doi: 10.1016/0003-2697(87)90579-3. [DOI] [PubMed] [Google Scholar]
- Li C, Peoples RW, Weight FF. Mg2+ inhibition of ATP-activated current in rat nodose ganglion neurons: evidence that Mg2+ decreases the agonist affinity of the receptor. J Neurophysiol. 1997;77:3391–3395. doi: 10.1152/jn.1997.77.6.3391. [DOI] [PubMed] [Google Scholar]
- Lukacs GL, Mohamed A, Kartner N, Chang X-B, Riordan JR, Grinstein S. Conformational maturation of CFTR but not its mutant counterpart (ΔF508) occurs in the endoplasmic reticulum and requires ATP. EMBO J. 1994;13:6076–6086. doi: 10.1002/j.1460-2075.1994.tb06954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie AB, Mahaut-Smith MP, Sage SO. Activation of receptor-operated cation channels via P2X1 not P2T purinoceptors in human platelets. J Biol Chem. 1996;271:2879–2881. doi: 10.1074/jbc.271.6.2879. [DOI] [PubMed] [Google Scholar]
- McGee TP, Cheng HH, Kumagai H, Omura S, Simoni RD. Degradation of 3-hydroxy-3-methylglutaryl-CoA reductase in endoplasmic reticulum membranes is accelerated as a result of increased susceptibility to proteolysis. J Biol Chem. 1996;271:25630–25638. doi: 10.1074/jbc.271.41.25630. [DOI] [PubMed] [Google Scholar]
- McPhie P, Parkison C, Lee BK, Cheng S. Structure of the hormone binding domain of human beta 1 thyroid hormone nuclear receptor: is it an alpha/beta barrel? Biochemistry. 1993;32:7460–7465. doi: 10.1021/bi00080a017. [DOI] [PubMed] [Google Scholar]
- Michel AD, Humphrey PPA. High affinity P2X-purinoceptor binding sites for [35S]-adenosine 5′-O-[3-thiotriphosphate] in rat vas deferens membranes. Br J Pharmacol. 1996;117:63–70. doi: 10.1111/j.1476-5381.1996.tb15155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AD, Miller KJ, Lundström K, Buell GN, Humphrey PPA. Radiolabeling of the rat P2X4 purinoceptor: evidence for allosteric interactions of purinoceptor antagonists and monovalent cations with p2X purinoceptors. Mol Pharmacol. 1997;51:524–532. [PubMed] [Google Scholar]
- Moriyama T, Sather SK, McGee TP, Simoni RD. Degradation of HMG-CoA reductase in vitro. J Biol Chem. 1998;273:22037–22043. doi: 10.1074/jbc.273.34.22037. [DOI] [PubMed] [Google Scholar]
- Nicke A, Bäumert HG, Rettinger J, Eichele A, Lambrecht G, Mutschler E, Schmalzing G. P2X1 and P2X3 receptors form stable trimers: a novel structural motif of ligand-gated ion channels. EMBO J. 1998;17:3016–3028. doi: 10.1093/emboj/17.11.3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RA. P2X receptors: a third major class of ligand-gated ion channels. Ciba Found Symp. 1996;198:91–109. doi: 10.1002/9780470514900.ch5. [DOI] [PubMed] [Google Scholar]
- North RA, Surprenant A. Pharmacology of cloned P2X receptors. Annu Rev Pharmacol Toxicol. 2000;40:563–580. doi: 10.1146/annurev.pharmtox.40.1.563. [DOI] [PubMed] [Google Scholar]
- Parker KE. Modulation of ATP-gated non-selective cation channel (P2X1 receptor) activation and desensitization by the actin cytoskeleton. J Physiol. 1998;510:19–25. doi: 10.1111/j.1469-7793.1998.019bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomidor MM, Cimildoro R, Lazatin B, Zheng P, Gurr JA, Leigh IM, Janne OA, Tuas RS, Hickok NJ. Phosphorylated human keratinocyte ornithine decarboxylase is preferentially associated with insoluble cellular proteins. Mol Biol Cell. 1999;10:4299–4310. doi: 10.1091/mbc.10.12.4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provencher SW, Glockner J. Estimation of globular protein secondary structure from circular dichroism. Biochemistry. 1981;20:33–37. doi: 10.1021/bi00504a006. [DOI] [PubMed] [Google Scholar]
- Radford KM, Virginio C, Surprenant A, North RA, Kawashima E. Baculovirus expression provides direct evidence for heteromeric assembly of P2X2 and P2X3 receptors. J Neurosci. 1997;17:6529–6533. doi: 10.1523/JNEUROSCI.17-17-06529.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev. 1998;50:413–492. [PubMed] [Google Scholar]
- Robertson SJ, Rae MG, Rowan EG, Kennedy C. Characterization of a P2X-purinoceptor in cultured neurons of the rat dorsal root ganglia. Br J Pharmacol. 1996;118:951–956. doi: 10.1111/j.1476-5381.1996.tb15491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg AL. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78:761–771. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- Schagger H, Von Jagow G. Tricine sodium dodecy sulfate polyacrylamide gel elecrophoresis for the separation of proteins in the range of 1 to 100kDA. Anal Biochem. 1987;166:368–397. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- Soto F, Garcia-Guzman M, Gomez-Hernandez JM, Hollmann M, Karschin C, Stühmer P. P2X4: an ATP-activated ionotropic receptor cloned from rat brain. Proc Natl Acad Sci USA. 1996;93:3684–3688. doi: 10.1073/pnas.93.8.3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto F, Garcia-Guzman M, Stuhmer W. Cloned ligand-gated channels activated by extracellular ATP (P2X receptors) J Membr Biol. 1997;160:91–100. doi: 10.1007/s002329900298. [DOI] [PubMed] [Google Scholar]
- Strand D, Raska I, Mechler BM. The drosophila lethal(2)giant larvae tumor suppressor protein is a component of the cytoskeleton. J Cell Biol. 1994;127:1345–1360. doi: 10.1083/jcb.127.5.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surprenant A, Buell GN, North RA. P2X receptors bring new structure to ligand-gated ion channels. Trends Neurosci. 1995;18:224–229. doi: 10.1016/0166-2236(95)93907-f. [DOI] [PubMed] [Google Scholar]
- Takeda A, Cone RE. Two-dimensional peptide mapping by polyacrylamide-gel electrophoresis with limited proteolysis in SDS. Biochem Biopys Res Commun. 1984;122:932–937. doi: 10.1016/0006-291x(84)91180-x. [DOI] [PubMed] [Google Scholar]
- Torres GE, Haines WR, Egan TM, Voigt MM. Co-expression of P2X1 and P2X5 receptor subunits reveals a novel ATP-gated ion channel. Mol Pharmacol. 1998a;54:989–993. doi: 10.1124/mol.54.6.989. [DOI] [PubMed] [Google Scholar]
- Torres GE, Egan TM, Voigt MM. N-Linked glycosylation is essential for the functional expression of the recombinant P2X2 receptor. Biochemistry. 1998b;37:14845–14851. doi: 10.1021/bi981209g. [DOI] [PubMed] [Google Scholar]
- Valera S, Hussy N, Evans RJ, Adami N, North RA, Surprenant A, Buell G. A new class of ligand-gated ion channel defined by P2X receptor for extracellular ATP. Nature (Lond) 1994;371:516–519. doi: 10.1038/371516a0. [DOI] [PubMed] [Google Scholar]
- Valera S, Talbot F, Evans RJ, Gos A, Antonarakis SE, Morris MA, Buell GN. Characterization and chromosomal localization of a human P2X receptor from the urinary bladder. Receptors Channels. 1995;3:283–289. [PubMed] [Google Scholar]
- Valera S, Talbot F, Evans RJ, Gos A, Antonarakis SF, Morris MA, Buell GN. Direct submission of X84896 to Genbank. 1996. [Google Scholar]
- Werner P, Seward EP, Buell GN, North RA. Domains of P2X receptors involved in desensitization. Proc Natl Acad Sci USA. 1996;93:15485–15490. doi: 10.1073/pnas.93.26.15485. [DOI] [PMC free article] [PubMed] [Google Scholar]