

Abstract
Multidrug resistance proteins that belong to the ATP-binding cassette family like the human P-glycoprotein (ABCB1 or Pgp) are responsible for many failed cancer and antiviral chemotherapies because these membrane transporters remove the chemotherapeutics from the targeted cells. Understanding the details of the catalytic mechanism of Pgp is therefore critical to the development of inhibitors that might overcome these resistances. In this work, targeted molecular dynamics techniques were used to elucidate catalytically relevant structures of Pgp. Crystal structures of homologues in four different conformations were used as intermediate targets in the dynamics simulations. Transitions from conformations that were wide open to the cytoplasm to transition state conformations that were wide open to the extracellular space were studied. Twenty-six nonredundant transitional protein structures were identified from these targeted molecular dynamics simulations using evolutionary structure analyses. Coupled movement of nucleotide binding domains (NBDs) and transmembrane domains (TMDs) that form the drug binding cavities were observed. Pronounced twisting of the NBDs as they approached each other as well as the quantification of a dramatic opening of the TMDs to the extracellular space as the ATP hydrolysis transition state was reached were observed. Docking interactions of 21 known transport ligands or inhibitors were analyzed with each of the 26 transitional structures. Many of the docking results obtained here were validated by previously published biochemical determinations. As the ATP hydrolysis transition state was approached, drug docking in the extracellular half of the transmembrane domains seemed to be destabilized as transport ligand exit gates opened to the extracellular space.
ABC transporters constitute a family of proteins that conduct important cellular transport functions. Some members of this family catalyze the import of nutrients, while others are responsible for the export of wastes and toxins, especially amphipathic or hydrophobic cytotoxins.1,2 Several members of this family also cause problems in the treatment of cancer and viral infections because of their role in exporting cytotoxic chemotherapeutics administered for the treatment of cancer or viral infection.3−5 One of these problematic transporters is the multidrug resistance P-glycoprotein (Pgp or ABCB1). Pgp is a 1280-residue, single polypeptide that contains two transmembrane domains (TMDs) and two nucleotide binding domains (NBDs) with a TMD1–NBD1–TMD2–NBD2 topology. The two nucleotide binding sites are shared between the large N- and C-terminal nucleotide binding domains. Each TMD consists of six transmembrane helices responsible for the binding and discharge of transported substrates. The binding sites for transport substrates [drug binding sites (DBS)] are formed by interaction of several transmembrane helices.6−11 Some sites appear to be large enough to accommodate more than one drug molecule at a time.12 ATP hydrolysis is stimulated in the presence of drugs that are transported, indicating direct coupling of drug transport and ATP hydrolysis.13−15 ATP hydrolysis most likely occurs via an alternating site mechanism.16 Formation and collapse of the catalytic transition state may be directly coupled to the transport of drug across the membrane.13
Although high-resolution structures of the human Pgp are not yet available, crystal structures for mouse Pgp17 and several bacterial homologues exist.18−20 In the structural model for mouse Pgp, the catalytic glutamates that likely activate the waters used in hydrolysis of ATP21 are separated by >30 Å, indicative of a complete disengagement of the two NBDs. Very similar structures resulted when the mouse Pgp was cocrystallized with two different stereoisomers of cyclic tris-valineselenazole inhibitors bound in the DBS.17 The widely opened NBDs in these structures create a 9 Å opening for access to the DBS on the cytoplasmic side of the membrane that are formed by transmembrane (TM) helices (TM4 with TM6 and TM10 with TM12).17 Structures of the closely related Staphylococcus aureus multidrug transporter (SAV1866) have been obtained with either ADP bound or the nonhydrolyzable AMP-PNP bound to the nucleotide binding sites.18,19 SAV1866 was shown to transport many compounds that are known transport substrates for human Pgp.22 The crystal structures of SAV1866 show fully engaged and dimerized NBDs with catalytic glutamyl residues separated by only 14 Å. The TMDs of these structures are oriented in an “outward-facing” arrangement with a relatively polar cavity exposed to the extracellular space. This orientation has been equated with a “drug release” conformation.18,19 ATP hydrolysis and the release of ADP and Pi may cause a change to an “inward” orientation that reveals high-affinity drug binding sites.19 Four structures of the bacterial lipid flippase, MsbA,20 show dramatic conformational differences that have been equated with different stages of the transport mechanism.20 These include structures with TMDs opened inward with slightly disengaged NBDs [Protein Data Bank (PDB) entry 3B5X], and structures with an opened outward DBS and fully engaged, dimerized NBDs (PDB entries 3B5Z, 3B5Y, and 3B60). One of these structures is noteworthy in having ADP-Vi, a transition state analogue, bound in one of the nucleotide binding sites (PDB entry 3B5Z). In addition, one MsbA structure displayed fully disengaged NBDs with catalytic glutamates separated by more than 65 Å, similar to the mouse Pgp discussed above.
Powerful new computational schemes for a more complete analysis of protein dynamics and ligand binding have recently been reported.23−25 These methods begin with molecular dynamics (MD) simulations from which representative “non-redundant” structures are identified using statistical methods.23−26 These methods were applied here using targeted MD simulations of Pgp with four crystal structures of homologues of Pgp as targets. In this approach, the current position of each atom is steered toward target coordinates derived from the crystal structures via application of a force vector calculated to decrease the distance between present and target positions.27,28 Targeted MD allows sampling of large conformational changes that would normally be inaccessible because of the large energy barriers that likely lie between such conformations and limitations in computational time. The resulting nonredundant intermediate structures derived from these targeted MD calculations were then used in ligand docking calculations to elucidate binding of the ligand to the different structures. This approach combines the conformational information gained from MD with ligand docking and accounts for the dynamic movement of both protein and ligand.
In this paper, homology models of human P-glycoprotein have been applied to targeted MD simulations to move the Pgp structure through conformations that were based on just a few known, catalytically relevant crystal structures. These crystal structures are germane to the dynamics of transport catalysis by Pgp, because the transporter very likely transitions through structures that are similar to these individual states during catalysis. In this study, 26 novel, nonredundant catalytically intermediate conformations were identified using the targeted molecular dynamics techniques. Ligand docking studies over the dynamic trajectory of the 26 catalytically relevant structures were performed, and results were consistent with published biochemical and biophysical findings. The results of these studies are discussed with regard to the molecular mechanisms that underlie the drug binding and drug transport mechanisms of human P-glycoprotein.
Experimental Procedures
Materials
The Visual Molecular Dynamics program suite (VMD)29 was extensively used in this work. MD experiments were performed using NAMD.30 XPLOR-NIH was employed for simulated annealing calculations.31,32 Secondary structures were identified using STRIDE.33 Procheck version 3.5.4 was used to validate the geometry of calculated protein structures.34 Small commodity computing clusters, the High Performance Computing Cluster at SMU, and the Ranger cluster at the Texas Advanced Computing Center were used for calculations.
Modeling Human Pgp with Sav1866
Models for human P-glycoprotein were created using sequence homologies between Sav186618 and the protein sequence of Pgp (Uniprot entry P08183). After the alignment of sequences with clustalW,35 the coordinates of non-hydrogen backbone and Cβ atoms of the structural template Sav1866 (PDB entry 2HYD,18 with ADP bound) that were homologous to Pgp residues were renamed as Pgp residues. Corresponding backbone and side chains were then built using psfgen.29 The linker that connects NBD1 and TMD2 in the human Pgp, residues 632–693, was not included in these models. Lists of distances and topologies (helical or sheet) were compiled with VMD. These lists were employed as distance or torsional restraints for use in simulated annealing using XPLOR-NIH.31,32 A total of 94414 distance and 1475 dihedral restraints were used in these calculations, and several hundred annealing experiments were performed. The Pgp structure that had the fewest distance and dihedral restraint violations was then positioned into a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayer, and water (TIP3 model) and ions (Na+ and Cl–) were added using the solvate and autoionize functions in VMD. ATP, Mg2+, and a catalytic water were substituted for ADP at NBD1, and a Mg2+ ion was added at NBD2 (see Results). The final system contained two protein chains, one ATP, one ADP, two Mg2+ ions, six Na+ ions, 17 Cl– ions, 203 POPC phospholipids, and 24689 water molecules (120016 total atoms). Energy minimizations to eliminate any van der Waals clashes between atoms and poor geometries of residues and extensive equilibrations of the lipid, the water, the protein, and the entire system were performed using NAMD to ensure the integrity of the system before any simulation experiments were performed.
Molecular Dynamics Simulations
MD simulations used NAMD 2.630 and the CHARMM27 force field36 with constant temperature and pressure (NPT ensemble) in a periodic cell using Langevin temperature and pressure control, as well as particle-mesh Ewald electrostatic calculations at 310 K. Targeted MD simulations27,28 were performed using target coordinates derived from mouse Pgp,17 Sav1866,18 and MsbA structures (PDB entries 3B5X and 3B5Z).20 Structures were aligned using STAMP,37 and the coordinates of homologous Cα atoms for each structure then functioned as target coordinates in sequential simulations. Targeted MD simulations were performed in NAMD using tcl scripts that applied forces to Cα atoms that were directed at individual target coordinates. The magnitudes of these forces were calculated to be inversely proportional to the distances separating the targeted Cα atoms and the target coordinates.
Clustering of Dynamic Structures
The structural redundancy inherent in the molecular dynamics simulations was reduced using the methods applied to protein structures introduced by Luthey-Schulten.38 Coordinate files from targeted MD calculations were saved at ∼1 ps intervals. These files were then analyzed for similarity using the QR factorization methods38 built into Multiseq,26 which employed STAMP structural alignments.37 Phylogenetic trees using QR clustering (or in other cases rmsd clustering) were also determined in Multiseq.26 Nonredundant structures were selected using a QH cutoff of 0.238 or an rmsd cutoff of 2 Å.
In Silico Drug Docking Analyses
AutoDock 4.239−41 was used with each of the nonredundant structures identified from the targeted MD clustering experiments. Grids were calculated using a 0.375 Å spacing in a 126 Å3 cube. Two overlapping grids were used in each set of docking experiments. For each ligand, 100 genetic algorithm experiments were performed with each grid with a population size of 300, 3 million evaluations, and 27000 generations. The preferred mode of binding of each ligand was determined using ligand results clustering at rmsd values of ≤2.0 Å. This type of analysis takes advantage of one of the strengths of the technique (the determination of the mode of binding of the ligand to the protein) while minimizing the reliance on a relative weakness of the method (the estimation of binding affinity).40,42,43 The clustering of ligand docking results by rmsd puts each of the 100 independent docks per experiment per ligand into a set of docking results based on ligand geometry and position in space. These cluster sets can have anywhere between 1 and 100 members in them, depending on the results of the experiments. The cluster set with the greatest number of members is considered here to be the preferred docking cluster because it is the most reproducible docking pose of binding of the ligand to the protein. In general, only the preferred docking cluster for each ligand will be discussed.
Results
Catalytic Conformations of Human P-Glycoprotein
The 3.0 Å resolution, “partially opened outward” Sav1866 structure was used as a structural template in simulated annealing calculations to generate an all-atom homology model of human Pgp. Sav1866 was chosen as a template because of its relatively high resolution and because, having a sequence 53% homologous to that of human Pgp, it can be regarded as a close relative.18 Comparison of the secondary structure topologies of Sav1866 with those of mouse Pgp17 demonstrated highly conserved folds for both transporters. Human Pgp structures were built using distance and torsional restraints obtained from Sav1866 that were then applied to the human Pgp sequence. The best Pgp model produced had less than 0.2% distance restraint violations (159 of 94414), less than 0.5% torsional violations (8 of 1475), and an rmsd of 0.96 Å between Cα atoms of the model and template. An overall G factor score (Procheck34) of 0.03 suggested that individual residues approached ideal geometries. A total of 15 of 1190 residues were found in disallowed regions of Ramachandran plots (1.3%). Prior to MD simulations, ATP, Mg2+, and water were substituted for ADP at NBD1. This added water was observed to engage the catalytic glutamyl residue (E556) in what appeared to be a chemically competent manner during energy minimizations. In subsequent MD trials, this nucleotide·Mg2+·H2O complex was retained in NBD1, indicating that these interactions were relatively stable (data not shown). Similar engagement of catalytic glutamyl 1201 with ATP, Mg2+, and H2O placed at NBD2 was not observed. Only the ADP and Mg2+ were therefore retained at NBD2.
Targeted Molecular Dynamics of Human Pgp
Targeted MD simulations were performed on the human Pgp model in attempts to elucidate possible catalytic transitional states. Targeted MD allows the sampling of large conformational changes that would not be computationally accessible with other techniques. In this work, the Sav1866-derived human Pgp structure was initially pushed toward the “fully opened inward” mouse Pgp conformation (PDB entry 3G60).17 Subsequent simulations sequentially targeted the “partially opened inward” MsbA structure (PDB entry 3B5X),20 the Sav1866 partially opened outward, fully engaged NBDs conformation (PDB entry 2HYD(18)), and finally the MsbA transition state structure with fully engaged NBDs and “fully opened outward” DBS (PDB entry 3B5Z).20 Using the force constants described in Experimental Procedures, the close approach of the human Pgp model to each of the crystal structure targets was achieved with rmsds for targeted Cα atoms and modeled atoms varying between 0.4 and 0.6 Å for each of the four target structures.
Several striking characteristics of the conformational changes that Pgp undergoes as it moves from the fully opened inward to fully opened outward conformations were observed in these simulations. Videos of these conformational changes are presented as Figures S1 and S2 of the Supporting Information. One can clearly see the twisting movement of the NBDs relative to the TMDs when engagement of the NBDs occurs. From the extracellular view of the transmembrane domains, one can also clearly observe a gradual rearrangement of TM helices that orient the TMDs from inward-facing to outward-facing structures. In addition, a dramatic widening of the TMDs into the extracellular space occurs, which is very clearly observed when the fully opened outward transition state is adopted.
Reducing Structural Redundancy in the Targeted Molecular Dynamics Simulations
The analysis of protein structures using multidimensional QR factorization to remove redundancy in the structure set has been pioneered by Luthey-Schulten.26 Such analyses were performed as described in ref (23) using the N-terminal and C-terminal halves of Pgp obtained from the targeted MD simulations to identify a small set of structures that represent the entire simulation trajectory. Using these techniques to cluster similar structures allows the creation of structurally based phylogenic trees that aid in choosing nonredundant samples. Two such factorizations performed on the TMD1–NBD1 and TMD2–NBD2 halves of Pgp over the complete targeted MD trajectory were performed to identify “unique” or “nonredundant” conformations of the transporter. A ΔQH cutoff of 0.2, where QH is a measure of structural similarity,26 resulted in 16 representative nonredundant structures for each of the two analyses of the Pgp-targeted MD trajectory. A total of 26 unique nonredundant conformations for the complete Pgp structure were therefore obtained from these simulations (six structures were common to each of the N- and C-terminal QR clusterings). Figure 1 shows the results of these factorizations performed on the NBD1–TMD1 and NBD2–TMD2 motifs over the complete trajectory. Similar results were obtained from rmsd clustering with a 2.0 Å cutoff (not shown). The 26 nonredundant conformations identified via QR factorization were used in all subsequent analyses of the conformational changes and in ligand docking studies presented here.
Figure 1.

Phylogenetic tree representations for human Pgp structures derived from target molecular dynamics trajectories. QR factorizations on the N-terminal half (left) and C-terminal half (right) of the human P-glycoprotein targeted molecular dynamics trajectory were performed as described in Experimental Procedures. A ΔQH cutoff of 0.2 (solid vertical lines) resulted in the selection of 16 representative structures in each half of the protein. Selected representative trajectory structure numbers are shown on the inside of each tree. The locations of the four targets (fully opened inward, partially opened inward, partially opened outward, and fully opened outward) are also indicated with text. The trajectory frame numbering was sequential from fully opened inward to fully opened outward conformations. Model numbers are arbitrary other than indicating the sequential order of the structure snapshots.
Figure 2 (■) shows the separation distances of the catalytically important glutamyl residues 556 and 1201 in the two NBDs over the simulated TMD trajectory. These residues are thought to activate the catalytic water of hydrolysis at the active sites and are therefore at a crucial position in the catalytic sites. One can see that as the simulation progresses from wide open inward to partially opened inward (two leftward arrows in the figure), the separation of the catalytic glutamyl residues, although they do move throughout this phase of the trajectory, remains relatively constant at approximately 30 Å. These initial movements of the transporter correspond to a rotation of the two NBD domains relative to each other while the drug binding domain (DBD) undergoes a transition from fully open inward to partially opened inward states. During this initial transition, two residues of the DBD implicated in verapamil binding [F728 and V982 on TM7 and TM12, respectively (Figure 2, ●)]9,12 show a progressive widening from ∼11 to 17 Å. As the simulation progressed from partially opened inward to fully engaged NBDs with partially opened outward DBD (second and third arrows from the left), the degree of “engagement” of the two NBDs as judged by catalytic glutamate separation was reduced fairly smoothly from 34 to 17 Å (■). This smooth closure between residues 556 and 1201 continued through to the final “transition state” targeted structure with a fully opened outward DBD where the minimal separation of 15 Å between these catalytically important residues was observed. The progression over these final transitions was significantly different for the verapamil-associated residues of the DBD. This residue pair underwent a transition to a minimal separation of ∼14 Å in the fully engaged NBD–partially opened outward structure (third arrow from the left) but then opened up significantly by ∼8 Å to a >22 Å separation. This final conformational change represented a dramatic opening of the verapamil-associated residues as the final transition state target was approached and may represent the “export” state of the transporter.
Figure 2.

Movement of catalytically important residues of the human P-glycoprotein from target molecular dynamics simulations. Shown are the distances that separate two glutamyl residues that have been implicated in the catalytic activation of the waters of hydrolysis (residues 556 and 1201) and two residues of the drug binding domain that have been implicated in verapamil binding (residues 728 and 982). The distances that separate these two pairs of residues were calculated for each of the 26 nonredundant Pgp structures over the course of the simulation.
The dramatic opening in the DBD observed for residues 728 (TM7) and 982 (TM12) was also observed when the extracellular ends of the transmembrane helices were examined in a pairwise fashion as shown in Figure 3. This figure presents the movement of homologous α-helices from the N-terminal and C-terminal halves of the drug binding domains on both the extracellular ends of each pair of transmembrane helices (1/7 through 6/12) and the intracellular cytoplasmic ends of these helices (Figure 3, top and bottom panels, respectively). As observed for the NBD catalytic glutamyl residues and verapamil-associated residues in Figure 2, the initial part of the simulation proceeds with gradual movements of the drug binding domain helices, especially for transitions that occur between the wide open to the cytoplasm and partially open to the cytoplasm targets (first two arrows from the left). These movements correspond to the expected opening of the extracellular helices and closing of the intracellular helices as the transporter undergoes a transition from wide opened inward to partially opened inward conformations. During the next transitions between the partially opened inward and fully engaged NBDs–partially opened outward conformations and finally the transition state fully opened outward conformations, the extracellular ends of helices 1/7 and helices 2/8 continued the relatively smooth trend of opening outward all the way through to the final transition state conformation of the simulation.
Figure 3.

Coordinated movements within the drug binding domain of the human P-glycoprotein from target molecular dynamics simulations. The figure presents the distances between the ends of homologous α-helices in each half of the drug binding domain. The top panel represents paired helical movements on the external face of the membrane for helices 1/7, 2/8, 3/9, 4/10, 5/11, and 6/12 as separation distances between residues 72 and 732, 112 and 753, 210 and 853, 217 and 856, 318 and 961, and 328 and 972, respectively. Similarly, in the bottom panel, helical movements at the cytoplasmic end of helical pairs 1/7, 2/8, 3/9, 4/10, 5/11, and 6/12 very near the NBDs were quantitated as the separation distances between residues 51 and 711, 156 and 797, 168 and 811, 256 and 895, 270 and 916, and 368 and 1010. Distances were calculated for each frame using tcl scripts in VMD. The x-axis numbering represents the 26 nonredundant trajectory-derived structures identified in Figure 1, numbered sequentially from the wide opened inward to wide opened outward targets. The double-headed arrows show the structures closest to the crystal structures that were used as targets, i.e., from left to right in the figure, the fully opened inward, mouse Pgp 3G5U(17) targeted structure; the partially opened inward structure from the MsbA 3B5X(20) structure; the opened outward structure from the Sav1866 2HYD(18) targeted structure; and the fully opened outward structure of the MsbA ADP-Vi-bound 3B5Z(20) structure, respectively.
A dramatic opening and widening movement of the extracellular ends of the four remaining helical pairs (3/9, 4/10, 5/11, and 6/12) was observed, however, as the transporter approached the final catalytic transition state structure from the engaged NBD–partially opened outward state (Figure 3, top panel). These latter movements appeared to be very similar to those presented in Figure 2 for the verapamil-associated residues during this phase of the simulation. The largest changes in this phase of the simulation were observed for helices 4 and 10 and helices 5 and 11. The extracellular ends of these helices separated over the entire simulation by 17 and 24 Å, respectively. Similar extents of movement in the direction of closing of the cytoplasmic NBD-associated helical ends of these same helices (20 and 17 Å, respectively) were observed. It can be seen, however, that the movement on the cytoplasmic NBD ends of these helices occurred with a more constant rate (Figure 3, bottom panel), showing much less if any of the dramatic movement apparent in the extracellular parts of the DBD (Figure 3, top panel). Analogous changes in the other helical pairs (helices 3/9 and helices 6/12) were also observed, but the magnitudes of these changes were smaller than that of the changes observed for helices 4/10 and helices 5/11 (not shown).
Correlation with Previously Observed Helical Movements during ATP Hydrolysis
In a series of studies,44−46 Loo, Clarke, and co-workers were able to show that transmembrane helices 11 and 12, which have been implicated in drug binding,6−11 undergo movement during ATP hydrolysis. In a very large mutational cross-linking study designed to investigate helical interactions with TM11, these authors showed that of 350 possible double-cysteine mutations in TM11 and TM1, -3, -4, -5, or -6, cross-links were observed between only TM1 and TM11 and that all five of the observed cross-links formed only if the transporter turned over via ATP hydrolysis.44 In related work involving disulfide bond formation45 or cross-linking with an extended multifunctional maleimide compound,46 similar observations were made for pairs of residues present on TM6 and -12. Most of the cross-links observed in these latter studies also required ATP hydrolysis to form.
The dynamics of the interacting pairs of residues implicated in these ATP hydrolysis-dependent cross-linking studies were analyzed in the simulations made here in attempts to further investigate these helical movements. Relatively complex movements of TM1 and -11 and TM6 and -12 were observed as the transporter moved from conformations with the DBD wide open inward to conformations that were wide open to the outside (see Figures S3–S5 of the Supporting Information). These complex movements proceeded in three distinct phases. To start, as the transporter moved from a conformation with DBD wide open to the inside with fully disengaged NBDs (see frame 0 in Figures S3–S5 of the Supporting Information) to a conformation with the DBD partially opened to the inside (frame 12 in the figures), each of the Cα atoms of the cross-linked residue pairs identified in refs (44−46) moved from near minimal separation to near maximal separation. As the simulation progressed from the partially opened inward to partially opened outward conformation (frames 12–22 of Figures S3–S5 of the Supporting Information), movement of the DBD helices to near minimal separation of the identified cross-linked residues of TM1 and -11 and TM6 and -12 occurred. Finally, as the transition state conformation with fully opened outward DBD was reached (frame 26 of the figures), the cross-linked residues once again separated to large and in some cases near maximal values. It is clear that if the transporter ground state in the absence of turnover is at or near the partially opened inward conformation, cross-linking of the paired cysteine residues of TM1 and -11 and TM6 and -12 would be unlikely, because this conformation showed maximal separation of the Cα atoms of the cross-linked residues. It is reasonable to infer that catalytic turnover would likely be required to move the helices into closer contact so that disulfide bond formation could occur. The simulations reported here are consistent with the observations of Loo et al.44−46 about the movements of TM1 and -11 and TM6 and -12.
Mechanistic Insights from the TMD Analyses
Analysis of the structures of the transporter during the TMD simulation phases where the conformation changes from an intracellularly oriented DBD to one with partially and then fully extracellularly oriented DBD reveals several detailed transitions that may serve as testable hypotheses in future studies. Figure S1 of the Supporting Information sequentially shows each of the 26 nonredundant protein structures identified from the targeted MD trajectory as a video animation. An alternate video with color-coded transmembrane helices (Figure S2 of the Supporting Information) has also been included for easier identification of individual substructures. In both videos, concerted movements of the NBDs with the DBD α-helices can be observed. One feature that was very apparent in these simulations was the dramatic widening of the extracellular ends of transmembrane helices 3/9, 4/10, 5/11, and 6/12 (as quantitatively shown in Figures 2 and 3). As Dawson and Locher pointed out previously for the Sav1866 protein,19 helices 2/8 and 3/9 and helices 4/10 and 5/11 of the DBD of the transporter are connected at the interface with the NBDs by short α-helices that are oriented parallel to the membrane and were termed “coupling helices” (CHs) by these authors. The equivalent residues that make up the coupling helices in the human Pgp transporter are 160–167 (CH1), 260–267 (CH2), 801–808 (CH3), and 903–911 (CH4). Because these helices are the connection points between the power-generating NBDs and the structures of the DBD that undergo the energy requiring conformational changes that transport ligands across the membrane, it is reasonable to infer that movement of these coupling helices is critical for the pumping mechanism of the transporter.
Consistent with this inference, in the simulations performed here it appeared that the coupling helices at the NBD interface moved in concert with the nucleotide binding domains as the transporter underwent the transition from the partially opened inward to partially opened outward conformation. This concerted movement continued relatively smoothly as the fully opened outward–transition state conformation was reached. An elucidation of the structures of the NBD that interacted directly with the coupling helices during the targeted MD simulation at this transmission interface was conducted, and the results are listed in Table 1. Consistent with the analyses of the Sav1866 structure conducted in ref (18) and with work that indicated close contact and movement between the CH2–NBD2 and CH4–NBD1 fragments,47 coupling helices CH1 and CH3 each contacted both the NBD1 and NBD2 domains while coupling helices CH2 and CH4 interacted only with the NBD domains of the opposite halves of the transporter [NBD2 and NBD1, respectively (see Table 1)]. From the results presented in Table 1, it can be seen that CH2 and CH4 made many more contacts with the NBDs than did CH1 and CH3 during the simulations. It may be that the CH2 and CH4 contacts are structurally more important in driving the necessary conformational changes in the DBD to effect pumping of substrates than are those contacts made between the NBDs and CH1 and CH3.
Table 1. Close Contacts between NBD Residues and Coupling Helicesa.
| coupling helix | NBD residues in contact with CH residues | NBD | comments |
|---|---|---|---|
| CH1 (residues 160–167) | 371, 372 | NBD1 | residues of NBD1 loop C-terminal to TMD helix 6 |
| 401, 402 | NBD1 | residues of A-loop | |
| 443, 444 | NBD1 | residues C-terminal to Walker A | |
| 1174, 1175 | NBD2 | residues N-terminal to signature sequence | |
| CH2 (residues 260–267) | 1118 | NBD2 | residue of NBD2 loop C-terminal to TMD helix 12 |
| 1077 | NBD2 | Walker A | |
| 1081, 1084, 1086, 1087 | NBD2 | helix following Walker A | |
| 1110, 1114–1117, 1119–1123 | NBD2 | residues at and around Q-loop | |
| 1133, 1135 | NBD2 | loop that returns to CH2 located after Q-loop | |
| 1188, 1189, 1192 | NBD2 | residues just after signature sequence and just before Walker B | |
| 1200 | NBD2 | Walker B | |
| CH3 (residues 801–808) | 1015–1017 | NBD2 | residues on NBD loop at top of helix 12 |
| 1044–1046 | NBD2 | residues at A-loop | |
| 1086–1088 | NBD2 | residues C-terminal to Walker A | |
| 523, 526, 527 | NBD1 | residues of Walker B | |
| CH4 (residues 901–911) | 378 | NBD1 | residue on NBD1 loop at top of helix 6 |
| 438 | NBD1 | helix following Walker A | |
| 441, 443 | NBD1 | loop after the helix following Walker A | |
| 476, 478–480, 491–493 | NBD1 | residues at and around Q-loop | |
| 476, 478–480, 491–493 | NBD1 | residues C-terminal to Q-loop, including the short helix just after Q-loop | |
| 527, 543, 547 | NBD1 | residues before Walker B |
Residues in the nucleotide binding domains (NBDs) found to be within 2.5 Å of coupling helix residues in any of the 26 nonredundant Pgp structures were identified using tcl scripts in VMD.
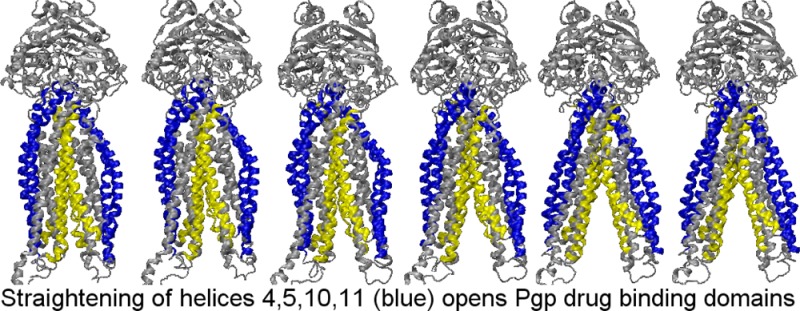
It seemed to be of interest to propose a mechanism for the dramatic movements of the extracellular face of the DBD that occur during the final conformational change to the “transition state–fully opened outward” structure. Figure S6 of the Supporting Information presents a video that allows the visualization of the conformational changes that occurred in the transporter during this transition. Helices 4 and 5 and helices 10 and 11 prior to reaching the catalytic transition state conformation possessed a pronounced bend that produced an arclike shape that was especially pronounced in the fully engaged NBD–partially opened outward structures. This curvature in essence kept the extracellular face of the DBD closed or only partially opened at the inward opened and partially opened outward stages of the simulation. As the catalytic transition state was approached, however, and as CH2 and CH4 moved the final few angstroms in concert with the NBDs, helices 4 and 5 and helices 10 and 11 were positioned in a manner that appeared to allow these helices to adopt much more straightened and more ideal α-helical conformations. This straightening of helices 4 and 5 and helices 10 and 11 resulted in the dramatic widening of the distance between the extracellular ends of these helices (see also third and fourth arrows from the left in Figures 2 and 3). As the catalytic transition state was being approached, the N-terminal ends of CH2 and CH4 were observed to move away from each other by 2.1 Å, while the corresponding C-terminal ends moved together by ∼1.7 Å. This twisting motion of these coupling helices is very likely driven by nucleotide binding domain conformational changes as bound ATP approaches its hydrolytic transition state. This relative motion of coupling helices 2 and 4, although very slight (comprising relative movements of ≤2 Å), is hypothesized here to allow the NBD-located ends of helices 4 and 5 and helices 10 and 11 enough movement to allow the remainder of these helices to straighten out into a more ideal α-helical structure. This associated straightening of helices 4 and 5 and helices 10 and 11 and the consequent coupled movement of helices 3 and 9 (which are connected via very short externally located loops with helices 4 and 10) and helices 6 and 12 (which are connected to helices 5 and 11 by short externally located loops) is hypothesized to move the external face of the drug binding domain in a coordinated manner to its fully opened catalytic transition state conformation. Less relative movement of helices 1/7 and helices 2/8 during this approach to the catalytic transition state is apparent and is consistent with the quantitative data of Figure 3 that show most of the dramatic opening of the drug binding domain to the outside occurring with helices 4/10, 5/11, 3/9, and 6/12. Although these simulations clearly cannot demonstrate the driving force for these rearrangements, it seems evident that the energy input for these movements must originate with rearrangements at the NBDs with nucleotide binding and/or hydrolysis and the consequent movement of the contacted coupling helices.
Drug Docking to Human Pgp
The 26 nonredundant Pgp structures derived from the targeted MD simulations were used in drug docking studies to analyze binding of the ligand to dynamic transitions of human Pgp structures. The ligands analyzed were selected because they are known to bind Pgp, are transport substrates of Pgp, or inhibit Pgp (Table S1 of the Supporting Information). Two molecules, ADP and PPi, served as negative controls for Pgp docking because they do not fit the criteria of good Pgp transport substrates. Two of the Pgp ligands used here are sulfur analogues of the selenium-containing cyclic peptide inhibitors, QZ59-RRR and QZ59-SSS, that were shown previously to crystallize to three closely placed but different binding sites in mouse Pgp.17 The docking of these QZ59 analogues to the fully opened inward conformation of Pgp was closely analyzed here for validation of the human Pgp structure and targeted MD. Figure 4A shows the results of the initial docking to human Pgp. Comparison with the mouse crystal structure (Figure 4B)17 indicates extensive similarities in the location of binding sites for the inhibitors. A clear correlation of both the positioning of the drug within the pockets of the two transporters and the positions of the residues that make up the binding pockets was observed (Figure 4, human Pgp in panel C vs mouse Pgp in panel D). Although the mode of binding to human Pgp and the mouse structure is not identical, there are clearly very similar interactions with homologous residues. Drug binding in the proximity of the partial binding site for QZ59-SSS identified by ref (17) was observed here as a less populated docking cluster (data not shown). Nineteen of the 21 residues identified by ref (17) as interacting with QZ59-SSS were within 3.5 Å of the preferred docking cluster for the QZ59-SSS sulfur analogue in these experiments (Figure 4C,D, bottom). Tryptophan 315 and threonine 837 were >4 Å from the preferred docking cluster for the QZ59-SSS sulfur analogue in the Pgp model. The results from these initial docking experiments strongly support the validity of the structural modeling as well as the drug docking simulations presented here.
Figure 4.
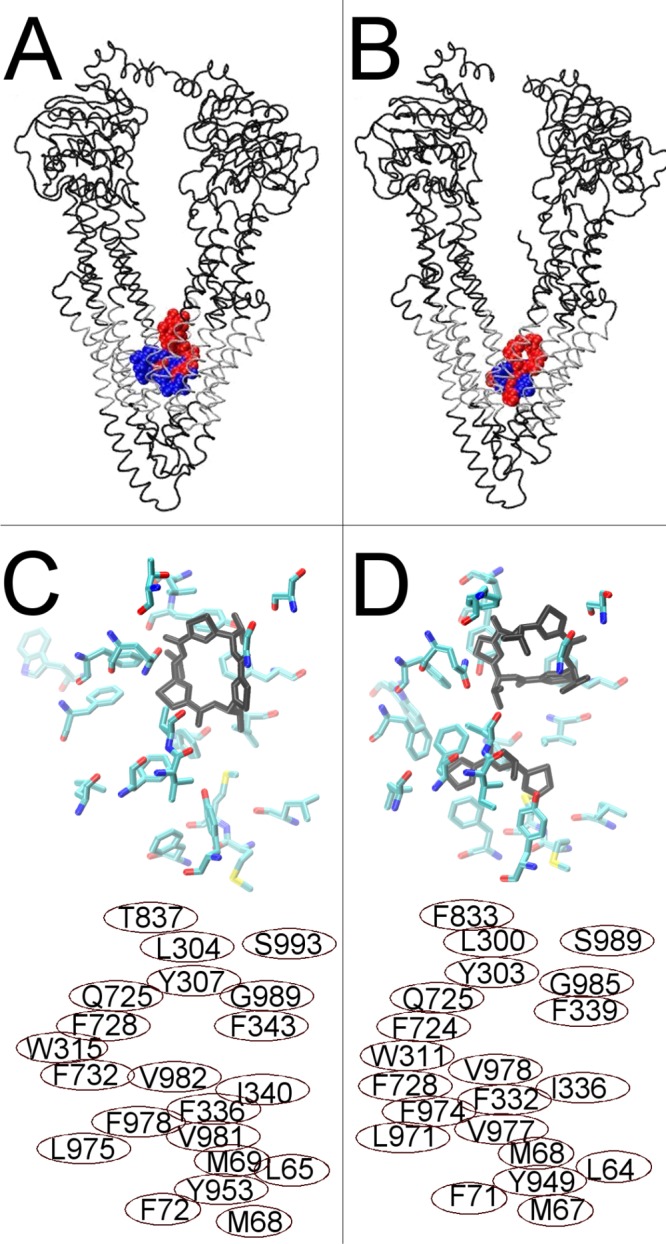
Docking of cyclic inhibitor analogues, QZ59-RRR (blue) and QZ59-SSS (red), to fully opened inward Pgp conformations. (A) Human Pgp showing the preferred docking clusters, the most reproducible drug docking clusters for two simulation frames at the fully opened inward conformation, superposed. (B) Mouse Pgp structures from ref (17) with the QZ59-RRR (blue) and QZ59-SSS (red) cyclic inhibitors bound. Transmembrane regions of both proteins are colored silver. (C) The top portion shows the results for docking of the sulfur analogue of the QZ59-SSS stereoisomer to the fully opened inward conformation of human Pgp. Residues within 3.5 Å of the three most reproducible docking clusters are shown as licorice colored by atom type. The most reproducibly docked QZ59-SSS analogue is shown bound as black licorice. (D) The top portion shows the mouse Pgp structure from ref (17) with QZ59-SSS bound (black). Amino acids identified by Aller et al.17 to be involved in drug binding are shown as licorice colored by atom type. The bottom portions of panels C and D identify the residue numbers of the amino acids shown in the top panel (C, human numbering, and D, mouse numbering). Note that each residue shown in either panel has its homologue shown in the same relative position in the other panel.
Figure 5 shows the docking of the QZ59-RRR analogue to three neighboring conformational states within the dynamic trajectory of Pgp near the fully opened inward conformation (Figure 5A–C, left). For comparison, the mouse Pgp structure in complex with QZ59-RRR is shown in each right-hand panel.17 The positions of side chains in previous and/or subsequent frames are indicated in the figure with thin lines. All 12 of the residues (Figure 5D) that make up the QZ59-RRR “middle” binding site17 are within 3.5 Å of the QZ59-RRR analogue docked into the human fully opened inward conformation, indicating very similar docking of the RRR analogue compared to that seen in the mouse Pgp crystal structure. Comparison of docking to human Pgp in panels A (left), panel B (left) and panel C (left) shows what could represent potential aromatic gates to the drug binding site.67 These results again support the results of docking and simulation reported here as valid representations of the human Pgp transporter.
Figure 5.
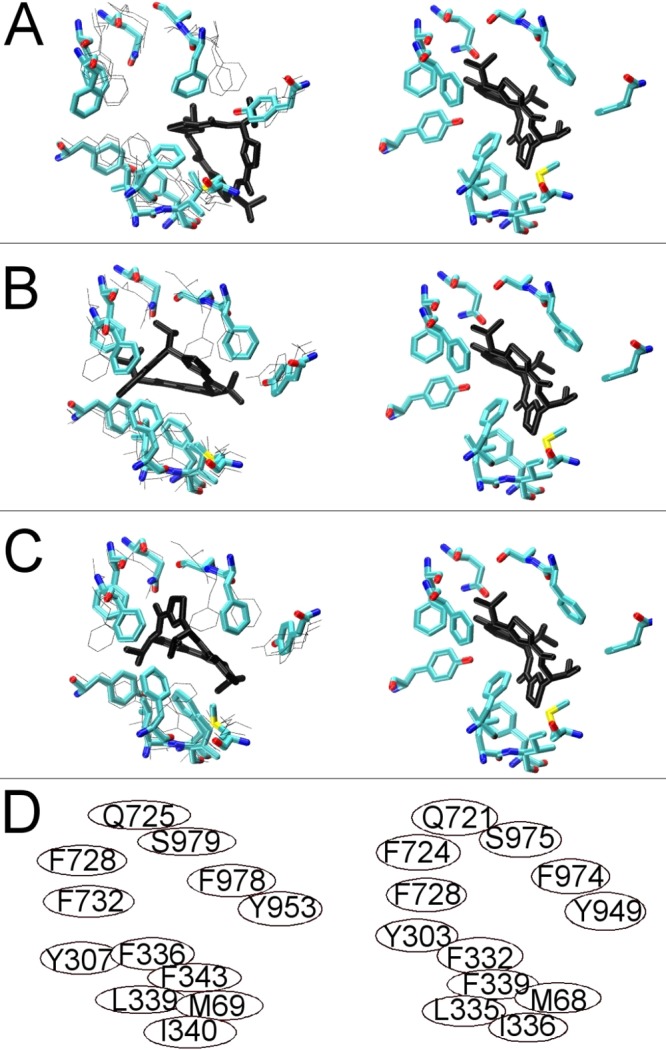
Ligand binding to the drug binding pocket of P-glycoprotein: RRR stereoisomer of the cyclic inhibitor QZ59 binding. The left side of each panel shows the results of docking to three sequential TMD frames bracketing the fully opened inward conformation of the human Pgp model. The right side of each frame shows the crystal structure of mouse Pgp with QZ59_RRR bound. (A) First frame of the cyclic peptide docking to human Pgp as licorice with the positions of side chains in the subsequent two frames (see panels B and C) shown with thin lines. (B and C) Two subsequent frames as licorice with previous and/or subsequent frames shown with thin black lines. Panel D shows the respective positions of the homologous residues that make up the binding pocket for QZ59-RRR as identified by ref (17): left side, human Pgp model; right side, mouse Pgp crystal structure.
Drug–Protein Interaction Dynamics
Different modes of drug interaction throughout the transmembrane regions of the DBS were found in docking experiments performed on the 26 nonredundant Pgp structures using 21 potential transport ligands (see Table S1 of the Supporting Information). It seems noteworthy that the fully opened outward conformations did not show preferential docking in the extracellular half of the transmembrane regions, potentially reflecting structural destabilization of ligand binding to the DBS as Pgp adopts the opened outward conformation (see below). Docking of either PPi or ADP was found exclusively outside of the transmembrane DBS and was limited to a narrow band around the outside of the protein at the cytoplasmic face of the membrane (data not shown). Representative results for docking of daunorubicin to four conformations from the fully opened inward to fully opened outward states are shown in Figure 6. There appear to be several preferred docking sites that are dependent on the conformation of the protein. While both the partially opened inward and partially opened outward conformations preferred daunorubicin docking to the extracellular half of the DBS, the fully opened outward conformation showed only preferred docking in the cytoplasmic half of the DBS. This may reflect structural destabilization of ligand binding to the extracellular half of the DBS as it adopts the opened outward conformation and may be mechanistically important as a “release” state of the transporter.
Figure 6.

Docking interactions of daunorubicin with the drug binding domain of Pgp. The protein backbone is shown as a tube with the transmembrane helices colored silver. Daunorubicin is shown in space-filling representation. Pgp and daunorubicin are shown in the fully open inward, partially opened inward, partially opened outward, and fully opened outward conformations (from left to right, respectively) in side views (top) and in views from the extracellular space (bottom). A similar figure with color-coded helices for ease of identification of individual structures is presented in Figure S7 of the Supporting Information.
Figure 7 summarizes the docking results obtained using all of the ligands tested here by showing the residues of the DBS that were ≤3.5 Å to any of the best docked ligand clusters (colored red). Figure 7A shows the conformational opening of the extracellular parts of Pgp and the exposure of the drug binding surfaces inside the DBS as the conformations progress from fully opened inward to fully opened outward states. Figure 7B depicts Pgp from the cytoplasmic side of the drug binding domains (the nucleotide binding domain portions were removed for the sake of clarity). This row of images demonstrates that the drug binding surfaces of Pgp are exposed to the cytoplasmic face of the membrane and cytoplasm in the fully opened inward conformation and that the drug binding surfaces become progressively less accessible to the cytoplasm as the transporter moves toward the fully opened outward conformation.
Figure 7.

Views of the drug binding surfaces of Pgp during catalytic transitions. Amino acid residues observed to approach Pgp within 3.5 Å are shown with van der Waals surfaces colored red. Secondary structures of the protein are colored black (extramembranous residues) or silver (transmembrane residues). (A) Extracellular views of Pgp with a viewpoint given by the tip of the arrow in the left image. (B) Views of the drug binding domains from inside of the cytosolic portion of Pgp with a viewpoint given by the tip of the arrow in the left image (nucleotide binding domains have been removed for the sake of clarity). Images of the fully opened inward, partially opened inward, partially opened outward, and fully opened outward conformations are presented from the targeted MD docking trajectory from left to right, respectively. A similar figure with color-coded helices for ease of identification of individual structures is presented in Figure S9 of the Supporting Information.
In analyses designed to determine whether inhibitors of Pgp interacted with the transporter differently than transport substrates, the close contacts between protein and docked ligands of the preferred docking clusters on all 26 nonredundant Pgp structures of the inhibitors and transport substrates listed in Table S1 of the Supporting Information were compared. Of the 46 ≤3.5 Å contacts between ligands and residues of the drug binding domains, one inhibitor (biricodar) was found to contact a residue on TM6 (F335) that was not within 3.5 Å of any other docked inhibitor or substrate. Several different substrates and inhibitors, however, contacted the neighboring residues L332 and F336 of TM6, making it unlikely that residue 335 was of special interest in establishing Pgp inhibition. (A text file containing a table listing Pgp residues within 3.5 Å of any docked inhibitor or transport substrate from these studies can be downloaded from the Supporting Information.) Of the 39 ≤3.0 Å contacts between ligands and Pgp residues of the drug binding domains, no inhibitor was found to contact a residue that was not also contacted by one or more transport substrates. These results suggest that no differences between the inhibitor or modulator binding to Pgp and the normal transport substrate binding to Pgp could be detected using the ligand docking techniques described here.
Discussion
Model Structures of Human Pgp
Homology modeling using a simulated annealing approach with the 3.0 Å partially opened outward Sav1866 structure produced a human Pgp structural model with very few restraint violations and excellent overall geometries. The Sav1866 crystal structure shows ADP bound to NBD1 and -2. Substitution of the ADP at NBD1 in the human Pgp model with ATP, Mg2+, and H2O at NBD1 resulted in stable binding and an orientation that engaged the catalytic glutamyl 556 in a manner that is consistent with known general base ATP hydrolysis mechanisms. Very strong evidence has recently been presented that favors this type of catalytic mechanism for ABC transporters (see the high-resolution transition state crystal structures of the maltose ABC transporter from Oldham and Chen48 and Senior’s recent mini-review21). Similar substitutions of ATP, Mg2+, and H2O at NBD2 did not result in the engagement of the corresponding catalytic glutamyl 1201, so ADP was retained at NBD2 in all subsequent simulations. Comparable results were observed in MD simulations of the BtuCD vitamin B12 transporter where only one of two ATP molecules was observed to fully engage with residues of the nucleotide binding sites of the protein.49 Although the observations that the catalytic glutamyl residue in NBD1 engaged the bound H2O·ATP·Mg ion complex in these simulations while the NBD2 glutamyl remained disengaged from bound substrate are consistent with an alternating sites mechanism for hydrolysis,16 a more systematic study of the phenomena seems to be warranted.
Testing the stability of homology models with MD simulations has been noted to be critical to the validation of such models.50 Indeed, MD simulations of a homology model based on early crystal structures demonstrated instabilities that helped point out problems with these structures.50,51 To investigate the stability of the Pgp model created here, we performed NPT dynamics for ∼28 ns. During these simulations, the secondary structure of Pgp did not significantly change and nucleotides and Mg2+ remained stably bound. These results suggest that this Pgp model was relatively stable and had no obvious energetic problems. Since 2007, when corrected versions of the MsbA structures became available, several homology models for Pgp have been reported.50,52−63 Validations similar to ours were obtained from these models, providing additional support for the dynamic transitions between protein conformations that were investigated here.
Targeted MD of Human Pgp
Several authors have suggested catalytic mechanisms for Pgp that involve the opened inward, nucleotide-free conformations that bind drug and nucleotide, converting the NBDs to fully engaged structures that then allow relatively large conformational changes that reorient the TMDs from inward-facing to outward-facing structures.3,13,18,19,64−69 There is debate about whether the wide opened to the cytoplasm conformations observed in some studies17,20 are stringently required for transport activity. Recent studies suggest that Pgp may not need to open fully to remain active.70,71 Despite these controversies, a progression of structures from the “fully opened inward–disengaged NBDs” of mouse Pgp17 to the “partially opened inward–disengaged NBDs” MsbA structure20 to the “partially opened outward–fully engaged NBDs” of Sav186618 and finally the ADP-Vi transition state MsbA structure showing “fully opened outward TMDs and engaged NBDs”20 may approximate conformational transitions required for catalysis. The experiments presented here attempt to investigate the possible transitions between these known conformations by using a targeted MD approach. Results presented here show that the human Pgp could be moved very close to each of the four targeted structures. This simulation of conformational dynamics reveals interesting subtleties in the catalytic mechanism. The simulations revealed that the helices that form the drug binding sites of Pgp undergo large movements in relative position, on both the inside and the outside of the cell membrane. The largest movements of drug binding site helices were observed for the pairs of helices 4/10 and helices 5/11. A rather abrupt widening of the drug binding sites to the outside of the cell was observed as the ADP-Vi transition state was approached. Twisting of the NBDs as they fully engage appeared to be coupled to the helical movements in the TMDs that seem to open the drug efflux sites.
Drug Docking to Dynamic Human Pgp
To analyze binding of the drug and ligand to Pgp over the course of the catalytic conformational transitions simulated here, a reduction of the structural complexity of the dynamic trajectories was required. Luthey-Schulten26 and McCammon and colleagues23−25 have developed methods for reducing the amount of conformational information of a MD simulation to a small set of nonredundant structures that can be used individually in drug docking experiments. The application of these methods to the targeted MD simulations of Pgp presented here resulted in the identification of 26 nonredundant transitional structures that have allowed the analysis of ligand docking over the entire dynamic transition from the fully opened inward to fully opened outward states.
Drug docking routines have been shown to be very accurate in predicting the mode of binding of small molecules to proteins while not necessarily accurately predicting binding energies.40,42,43 In rigorous studies,40 binding energies for known ligand–protein complexes had standard errors of ∼2.5 kcal/mol (∼3 orders of magnitude for a typical Kd value). The results discussed here will therefore emphasize the location and geometrical interactions between the ligand and protein and will not stress calculated affinities. Ligand clustering at rmsd values of ≤2.0 Å was used to classify docked ligands. This type of clustering analysis reveals docking configurations that are the most reproducible as those clusters with the greatest number of members. It is assumed that these preferred clusters represent the most favorably bound ligand configuration detected. It should be noted that several of the ligands analyzed had more than one well-populated ligand docking cluster, so it should not be assumed that the docking experiments presented here suggest that each ligand has only one possible docking orientation.
It is worth mentioning that the programs used here for ligand docking allow for fully flexible ligand conformations, which in combination with docking analyses performed on the 26 nonredundant Pgp conformations results in docking analyses that fully encompass both the dynamics of ligand binding and the dynamic conformational changes in the protein throughout the targeted MD simulation. Docking of the drug to all 26 nonredundant Pgp structures using 21 compounds known to be transported by or to inhibit transport by Pgp was performed here. In addition, docking of two compounds, ADP and PPi, was performed as a control for nontransported ligands. To validate the structural modeling, the targeted molecular dynamics, and the docking techniques, we closely analyzed docking of two compounds in the fully opened inward conformations, because crystallographic information was available for the mouse homologue.17 It should be noted that these experiments should not be considered typical “redocking” experiments in which a cocrystallized ligand is removed from a protein crystal structure and then docked back into the protein without any intervening dynamic movement of the target protein. All 26 of the protein structures used here were derived from very long molecular dynamics simulations at 310 K on human Pgp without any ligand bound to the drug binding domains. Despite the differences between the docking results presented here and the cocrystallization studies described in ref (17), docking of both the SSS and RRR analogues of the QZ59 inhibitors (Figures 4 and 5) closely reproduced the binding interactions seen in ref (17). More than 90% of the residues identified at the QZ59-SSS inhibitor binding sites in the mouse model were observed within 3.5 Å of the preferred SSS analogue binding cluster in the human Pgp, and 100% were found to be identical for the RRR analogue docking.
Potentially interesting results were obtained for the docking of the QZ59-RRR analogue to three neighboring conformational states close to the fully opened inward conformation (Figure 5). The figure shows what could represent the QZ59 analogue entering the binding pocket as the side chain of phenylalanine 978 (top of the binding site) moves out of the way, allowing access to the homologous pocket shown in the mouse structure.17 This movement is reminiscent of the aromatic gates that control access of ligands to substrate binding sites and active sites as discussed for acetylcholinesterase and membrane transport channels.72,73 In Figure 5C, docking of the QZ59S-RRR analogue to Pgp appears to be very similar to that observed in the mouse transporter. These results support the hypothesis that the docking and simulation results reported here are valid representations of the human Pgp transporter and extend to the postulation of putative aromatic gating structures in the drug binding sites of Pgp.
Preferred docking clusters for 21 ligands to the 26 nonredundant conformations of Pgp were observed throughout the transmembrane regions of the DBS for the fully opened inward to the partially opened outward conformations. In the Pgp conformations that approached the fully opened outward ADP-Vi transition state, ligand binding in the extracellular half of the TMDs was not favored, with preferred docking found in the cytoplasmic half of the membrane regions. This may be mechanistically important, because the destabilization of ligand binding in the extracellular half of the drug binding site, coupled with denied access to ligand binding on the cytoplasmic side, would effectively force release of the ligand to the extracellular space. It should be noted that the docking experiments performed here do not require connected pathways from one ligand cluster to another and in this sense do not reflect actual pathways that a ligand might travel through the transporter. It will be of interest to follow the molecular dynamics of a single ligand through the entire conformational trajectory to directly test this mechanism.
Figure 7 shows the drug binding sites of Pgp by highlighting any side chain within 3.5 Å of a preferred drug binding cluster that was found in these studies. One can observe that the drug binding surfaces are closed off from the extracellular space in the fully opened inward conformation, while access to the extracellular space increases with each step toward the fully opened outward conformation until a large portion of the drug binding surface is exposed to the outside of the cell in the fully opened outward conformation. Figure 7 clearly demonstrates large conformational changes that allow access of the drug binding surfaces to change from the cytoplasm toward the extracellular space as the transporter opens outward. Actual pumping of the drug transport substrate may or may not reflect a significant change in affinity for the drug binding surfaces. If no change occurs, transport may be effectively a matter of changed access from inside to outside as the transport mechanism progresses.
Significantly, all of the ligands tested except for ADP and pyrophosphate were found to bind like daunorubicin to the DBD of Pgp with multiple preferred ligand clustering sites found within the transmembrane regions of the DBS. The results obtained for ADP and pyrophosphate were significantly different because neither of these ligands is a good Pgp transport substrate and neither of these compounds docked within the drug binding sites of Pgp. These results further strengthen the docking obtained for the known transport ligands of Pgp. Very large ligands such as cyclosporine A, valspodar, and ivermectin were found preferentially docked to the cytoplasmic half of the transmembrane DBS. This could be due to the fact that targeted MD trajectory was produced with unoccupied, “empty” drug binding sites. It may be worth investigating how much plasticity in structure the DBS demonstrate when large ligands are incorporated in MD simulations.
It is clear from the analyses of the residues of Pgp that were contacted by ligands in these docking experiments that no apparent differences in the location of binding of transport substrates versus Pgp inhibitors or modulators could be detected using these techniques. These results suggest that there is not a specific “inhibitor binding site” located within the drug binding domain of Pgp and that the mode of inhibition by these compounds, if binding occurs at the locations deduced from these docking studies, may be more simply to compete with transport substrate drugs for the normal pathway(s) of transport through Pgp.
Correlation of Biochemical Data with Dynamic Structural Models
Loo, Clarke, and co-workers were able to show that transmembrane helices 11 and 12 undergo movement relative to helices 1 and 6 during ATP hydrolysis.44−46 In mutational cross-linking studies, these authors showed that double cysteine mutations introduced into TM1 and TM11 formed five different disulfide cross-links, but only if the transporter catalytically turned over via ATP hydrolysis.44 Similar observations of ATP hydrolysis-dependent cross-linking of residues on TM6 and -12 were also made by these investigators.45,46 The simulations presented here showed complex relative movements of transmembrane helices 1 and 11 and helices 6 and 12 that proceeded in three distinct phases (Figures S3–S5 of the Supporting Information). As the transporter moved from a wide open to the inside to partially opened to the inside conformation, distances between cross-linked residues on TM1 and -11 and TM6 and -12 moved from near minimal to near maximal separations. This was followed by movements to near minima as the simulation progressed from the partially opened inward to partially opened outward conformation. Then as the transition state conformation with the fully opened outward DBD was reached, the cross-linked residues on helices 1 and 11 and helices 6 and 12 separated widely again.
Petersen et al.74 in a study of 351 naturally occurring cystines in known protein structures noted that the range of separation of Cα atoms between two half-cystines was 3.4–7.6 Å with averages between 5.3 and 5.8 Å depending on the cystine geometry. The corresponding values for Pgp at its partially opened inward conformation were approximately 3 times these averages, and values at the transition state conformation with the DBD fully opened to the outside were more than twice these averages, making oxidation of cysteines to cystine unlikely in these conformations. The minimal values observed in the simulations for the TM1–TM11 residue pairs were still larger than the range of naturally occurring cystines reported in ref (74), however, with Cα–Cα values for the cross-linked residues ranging from 8.6 to 11.6 Å. Some of this difference might be explained by conformational flexibility of the noncovalently cross-linked helices in these simulations, the fact that two S–H bonds exist in the mutated and reduced transporters (up to ∼1 Å each depending on the orientation), and the need for flexibility in the mutated structures to accommodate the employed oxidant.
Analysis of the geometric orientations of the TM1 and -11 residue pairs suggests that the relative orientation of the interacting residues as also discussed by Loo and Clarke44 may be important in determining whether cystine can form (see Figure S8 of the Supporting Information). In the conformations with Cα–Cα distances at or near observed minima, the wide opened to the inside and partially opened to the outside conformations (first and third panels from the left, respectively, in Figure S8 of the Supporting Information, equivalent to frames 0 and 22, respectively, in Figures S3–S5), the helical surfaces of the interacting residue pairs face each other directly. In the partially opened to the inside conformation (second panel in Figure S8 of the Supporting Information), while the orientation of the helical faces of the interacting pairs would appear to be acceptable for cystine formation, the Cα–Cα distances are close to observed maxima, which may preclude reaction to cystine. In the transition state conformation (fourth panel in Figure S8 of the Supporting Information), distances between reactive residues on TM1 and -11 vary between 12.4 and 14.5 Å and are intermediate between the fully opened inward and partially opened outward minima. A slight reorientation of the reactive helical faces away from each other may have made reaction to disulfide in this latter conformation less probable. Loo et al.44 report that no cross-linking occurred with transporter preparations that were inhibited by ATP-Vi (a transition state analogue).
In summary, the simulations reported here are consistent with the observations of Loo et al.44−46 of the movements of transmembrane helices 1 and 11 and helices 6 and 12. The fact that ATP hydrolysis was required for the cystine formation in these experiments44,45 and enhanced other cross-links46 strongly suggests that the resting ground state of the transporter is not one of the conformations with minimal Cα–Cα distances (conformations with the DBD fully opened to the inside or partially opened to the outside). Catalytic turnover was likely required to bring the reactive residues into closer contact and perhaps to supply required conformational flexibility such that the oxidations to disulfide bonds could proceed. It is a reasonable assumption from refs (44−46) that the transporter ground state without turnover is at or near a conformation with a greater separation of the paired cysteine residues such that cross-linking of these residues in this conformation would be unlikely. Because the transition state conformation of the transporter by definition is quite unlikely to be the ground state, it is tempting to speculate that the remaining conformation reported here with cross-linked residues showing maximal Cα–Cα separation, the partially opened inward conformation, might be similar to the resting conformation of the transporter. It also follows that enough flexibility in the helical backbones is generated during turnover to position the reactive cysteines close enough for disulfide bond formation in conformations where the paired cysteines reach minimal separation (either the conformation with the DBD wide open to the inside or partially opened to the outside). Interestingly, all but one of the ATPase-dependent cross-linked residue pairs examined also showed the dramatic widening as the conformation moved from the partially opened outward to the fully opened outward transition state (Figures S3–S5 of the Supporting Information). The single exception that did not show this effect was the TM6 residue 350–TM12 residue 993 pair. Because these residues are found very near the center of helices 6 and 12, they would not be expected to show an opening as dramatic as those of other residues located close to the exterior. The simulations presented here expand on the demonstrations by Loo, Clarke, and co-workers that helices 1 and 11 and helices 6 and 12 move during ATP hydrolysis by presenting three plausible movements of these helices as catalysis proceeds from transporter conformations fully opened to the cytoplasm to those fully opened to the extracellular space.
Loo and Clarke were also able to identify many residues involved in drug binding using analogues of transport substrates with sulfhydryl reactive functional groups and cysteine or arginine scanning mutagenesis. In their studies, protection of labeling by inclusion of unlabeled substrate was used as the highest criterion for mapping residues of the DBS. Their work demonstrated that S222, L339, A342, and G984 were fully protected by verapamil while I868, F942, and T945 showed less protection.75 All of these seven protected residues were found in this study within 3.5 Å of one or more preferred ligand docking clusters. Similar studies using dibromobimane8,76−78 identified 16 residues that could be protected by transport substrates. All 16 residues were found within 3.5 Å of a preferred cluster. A thio-reactive rhodamine B analogue was used to identify F343,79 and MTS–verapamil labeling at I306 permanently activated ATP hydrolysis.80 Both F343 and I306 were found within 3.5 Å of the preferred ligand clusters elucidated here. F343 in our studies was found to interact very closely with docked ligands (Figure 5). A novel arginine mutagenesis approach for rescuing Pgp folding mutants was used by Loo and Clarke to argue that the bulky arginine side chain, when present in the drug binding site, mimicked the rescue of folding of certain mutations by transport substrates that has been observed.81 Alteration of known substrate affinities was taken as evidence that the A302R, F336R, L339R, G872R, F942R, Q946R, V982R, S993R, and M986R mutations were at drug binding locations. All of these residues were found to be within 3.5 Å of the ligand docking sites identified in our study. Of the 43 residues identified by Loo and Clarke or by Aller et al.17 to be at the drug binding sites of Pgp, 42 of them (all but T837) were found within 3.5 Å of a preferred docking cluster of at least one of the drugs tested. In summary, the docking results presented here are in very good agreement with the biochemical studies that have elucidated the specific residues of the drug binding sites of Pgp.
Mechanistic Implications
Alanine 841, which is found within 3.5 Å of preferred docking clusters, deserves further mention. This residue is present on TM9 and appears in our models to be packed against TM7 during much of the conformational change from the wide opened inward to the partially opened outward conformation (Figure 8). Interestingly, however, as the transporter approaches the opened outward states, A841 moves clear of TM7 as these helices separate widely (Figure 8). One can clearly observe the opening of the drug binding domains in these side views (see also Figures S1 and S2 of the Supporting Information). Whether this represents the opening of a possible transport substrate “exit gate” in analogy to the entrance gate observed in the opened inward conformations will have to be confirmed in future studies. It should be noted that an analogous opening of TM1 and -3 was observed on the opposite side of the transporter during the opened inward to opened outward conformational transitions.
Figure 8.

Possible exit gate in the drug binding domain of Pgp. The transmembrane helices of the drug binding domain of Pgp are shown as silver cartoons, while the extramembranous protein is colored black. Alanine 841 (TM9), V715 (TM7), and G722 (TM7) are shown in space-filling representation. A841 is on the right-hand side of each figure, while V715 and G722 are on the left. V715 is above G722. From left to right are shown the fully opened inward, partially opened inward, partially opened outward, and fully opened outward conformations, respectively. A similar figure with color-coded helices for ease of identification of individual structures is presented in Figure S10 of the Supporting Information.
Loo et al.82 showed in 2003 that different transport substrates differentially changed the cross-linking patterns between residues present in the drug binding domain transmembrane helices. These results were interpreted as evidence of a substrate-induced fit mechanism for the binding of different transport substrates by Pgp. The simulations presented here demonstrated the great plasticity of the drug binding domain structures of Pgp in two different and important ways: the very large movements of the DBD helices observed as the conformation of the transporter changed and the localized, more subtle rearrangements of the side chains of residues surrounding drug binding sites that can be inferred from the three neighboring conformational states in the QZ59-RRR analogue binding site (see Figure 5). This report therefore provides mechanistic support for the proposal that local rearrangements of residues that interact with transport substrates together with more global repositioning of helices between the wide open to the inside to partially opened to the inside conformations of Pgp would be expected to be able to accommodate many different ligand structures in a substrate-induced fit mechanism such as that proposed in ref (82).
Several mechanisms for Pgp catalysis and transport have been postulated.3,67,83−85 Most have in common the hypothesis that ATP binding or hydrolysis promotes the association of NBDs and the adoption of outward-facing DBS, and that release of hydrolysis products promotes the dissociation of NBDs and inward-facing DBS. The results from the targeted MD simulations presented here show some of the details of the progression of the protein conformations from an initial state, where the drug binding domains are opened to the cytoplasm and the nucleotide binding domains are fully disengaged, to a conformation in which the drug binding domains are fully opened to the extracellular space and the nucleotide binding domains are fully engaged. Very large helical movements in the TMD and the relative twisting of the NBDs as they approach each other can be observed. It seems reasonable to conclude that the ADP-Vi transitional state conformation represents a state of the transporter in the catalytic cycle where the drug bound in the DBS would be free to dissociate to the extracellular space. The conformational analyses performed here support this hypothesis because this conformation shows the greatest access of the DBS to the extracellular space. The Sav1866 conformations (with ADP and AMPPNP bound) appear to exist in a more relaxed state where the outside of the cell is still accessible from the DBS, but the extent of opening of the DBS to the extracellular space is smaller. In the simulations presented here, the ADP-bound Sav1866 conformation was taken to be an approximation of a conformation with fully engaged NBDs that followed the partially opened inward conformation with slightly disengaged NBDs but preceded the fully opened outward transitional state. It is very likely that this relaxed opened outward Sav1866 state (or one similar to it) would also follow the transition state as was originally suggested.18,19,65,86 It will be of interest in the future to perform additional targeted molecular dynamics simulations with Pgp as new structures of transporters with other catalytically relevant conformations are discovered.
The dramatic opening of the external face of the DBD as the transporter moved from the partially opened outward to the fully opened outward conformation of the catalytic transition state observed here is hypothesized to occur by a realignment of coupling helices 2 and 4 caused by movements of the nucleotide binding domains during this catalytic transition. The movement of coupling helices 2 and 4 appeared to allow helices 4 and 5 and helices 10 and 11 to straighten into much more ideal α-helical conformations. The straightening of helices 4 and 5 and helices 10 and 11 is hypothesized to occur as a consequence of the relative twisting of coupling helices 2 and 4. Coordinated movement of helices 3 and 9 and helices 6 and 12 (which are directly connected to the extracellular ends of helices 4 and 10 and helices 5 and 11, respectively) together with the described straightening of helices 4 and 5 and helices 10 and 11 accounted for most of the dramatic opening of the external face of the DBD as the transition state conformation was approached. This opening of the extracellular part of the DBD likely allows dissociation of the bound transport ligand to the extracellular membrane leaflet or the extracellular bulk matrix.
In this work, transitions between well-established ABC transporter conformations have been studied using targeted MD in attempts to assemble a series of conformational changes that reflect structural changes required for catalysis. Over the course of these simulations, drug and inhibitor binding have been investigated and the coordinated movements of individual helices have been correlated with the proposed conformational changes. The detailed movements described here should provide ample hypothetical tests for future experimentation.
Acknowledgments
I thank Pia D. Vogel for valuable discussions and critical reading of the manuscript, Joe Gargiula and Jim Jaeger for their assistance with computer resources, and Justin Ross for help with the High Performance Computing Center at Southern Methodist University. I acknowledge the Texas Advanced Computing Center (TACC) at the University of Texas at Austin for providing some of the computational resources used in this work and the High Performance Computing Center at Southern Methodist University for the majority of the computational resources used here.
Glossary
Abbreviations
- ABCB1
ATP binding cassette transporter, type B1
- DBD
drug binding domain (the domain of the transporter consisting of the 12 transmembrane helices and the intracellular extensions of these helices)
- DBS
drug binding site(s) (residues of the transporter found within the drug binding domain that interact directly with transported ligands)
- MD
molecular dynamics
- NBDs
nucleotide binding domains
- NPT
isothermic–isobaric molecular simulation ensemble in which moles, pressure, and temperature are held constant
- Pgp
P-glycoprotein
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- rmsd
root-mean-square deviation
- TM
transmembrane
- TMD
transmembrane domain.
Supporting Information Available
Figures S1 and S2 present videos showing the 26 sequential nonredundant conformational changes of Pgp that were observed in the targeted molecular dynamics simulations presented here. Figures S3–S5 show the complex helical movements observed previously by Loo, Clarke, and co-workers during ATP hydrolysis for transmembrane helices 1 and 11 and helices 6 and 12. Figure S6 presents video animations of the helical movements within the drug binding domains of Pgp that fully open the extracellular face of the transporter as the catalytic transition state conformation is approached. Figures S7, S9, and S10 are color versions of Figures 6–8, respectively, that have color-coded transmembrane helices that allow identification of specific substructures of Pgp should that be desired. Figure S8 shows changes in the orientation of mutated residues on TM1 and -11 during simulated catalysis that are known to form disulfide cross-links in an ATP hydrolysis-dependent manner. Table S1 presents the list of compounds and their relevant characteristics that were used here in docking studies of the 26 nonredundant Pgp conformations. Table S2 is a text file containing a table of residues of Pgp that come within 3.5 Å of any docked ligand in any of the 26 nonredundant conformations tested here. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This work was supported by Grant 1R15GM094771-01A1 from the National Institute of General Medical Sciences to Pia D. Vogel and John G. Wise.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Holland I. B.; Blight M. A. (1999) ABC-ATPases, adaptable energy generators fuelling transmembrane movement of a variety of molecules in organisms from bacteria to humans. J. Mol. Biol. 293, 381–399. [DOI] [PubMed] [Google Scholar]
- van Veen H. W.; Konings W. N. (1998) Structure and function of multidrug transporters. Adv. Exp. Med. Biol. 456, 145–158. [DOI] [PubMed] [Google Scholar]
- Sauna Z. E.; Ambudkar S. V. (2007) About a switch: How P-glycoprotein (ABCB1) harnesses the energy of ATP binding and hydrolysis to do mechanical work. Mol. Cancer Ther. 6, 13–23. [DOI] [PubMed] [Google Scholar]
- Callaghan R.; Crowley E.; Potter S.; Kerr I. D. (2008) P-Glycoprotein: So many ways to turn it on. J. Clin. Pharmacol. 48, 365–378. [DOI] [PubMed] [Google Scholar]
- Sauna Z. E.; Kim I.; Ambudkar S. V. (2007) Genomics and the mechanism of P-glycoprotein (ABCB1). J. Bioenerg. Biomembr. 39, 481–487. [DOI] [PubMed] [Google Scholar]
- Bruggemann E. P.; Currier S. J.; Gottesman M. M.; Pastan I. (1992) Characterization of the azidopine and vinblastine binding site of P-glycoprotein. J. Biol. Chem. 267, 21020–21026. [PubMed] [Google Scholar]
- Demeule M.; Laplante A.; Murphy G. F.; Wenger R. M.; Béliveau R. (1998) Identification of the cyclosporin-binding site in P-glycoprotein. Biochemistry 37, 18110–18118. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Clarke D. M. (2002) Location of the rhodamine-binding site in the human multidrug resistance P-glycoprotein. J. Biol. Chem. 277, 44332–44338. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Bartlett M. C.; Clarke D. M. (2006) Transmembrane segment 7 of human P-glycoprotein forms part of the drug-binding pocket. Biochem. J. 399, 351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo T. W.; Bartlett M. C.; Clarke D. M. (2006) Transmembrane segment 1 of human P-glycoprotein contributes to the drug-binding pocket. Biochem. J. 396, 537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Collins K. I.; Greenberger L. M. (1995) Functional evidence that transmembrane 12 and the loop between transmembrane 11 and 12 form part of the drug-binding domain in P-glycoprotein encoded by mdr1. J. Biol. Chem. 270, 5441–5448. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Bartlett M. C.; Clarke D. M. (2003) Simultaneous binding of two different drugs in the binding pocket of the human multidrug resistance P-glycoprotein. J. Biol. Chem. 278, 39706–39710. [DOI] [PubMed] [Google Scholar]
- Senior A. E.; al-Shawi M. K.; Urbatsch I. L. (1995) The catalytic cycle of P-glycoprotein. FEBS Lett. 377, 285–289. [DOI] [PubMed] [Google Scholar]
- Senior A. E.; al-Shawi M. K.; Urbatsch I. L. (1995) ATP hydrolysis by multidrug-resistance protein from Chinese hamster ovary cells. J. Bioenerg. Biomembr. 27, 31–36. [DOI] [PubMed] [Google Scholar]
- Urbatsch I. L.; al-Shawi M. K.; Senior A. E. (1994) Characterization of the ATPase activity of purified Chinese hamster P-glycoprotein. Biochemistry 33, 7069–7076. [DOI] [PubMed] [Google Scholar]
- Urbatsch I. L.; Sankaran B.; Bhagat S.; Senior A. E. (1995) Both P-glycoprotein nucleotide-binding sites are catalytically active. J. Biol. Chem. 270, 26956–26961. [DOI] [PubMed] [Google Scholar]
- Aller S. G.; Yu J.; Ward A.; Weng Y.; Chittaboina S.; et al. (2009) Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323, 1718–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson R. J. P.; Locher K. P. (2006) Structure of a bacterial multidrug ABC transporter. Nature 443, 180–185. [DOI] [PubMed] [Google Scholar]
- Dawson R. J. P.; Locher K. P. (2007) Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 581, 935–938. [DOI] [PubMed] [Google Scholar]
- Ward A.; Reyes C. L.; Yu J.; Roth C. B.; Chang G. (2007) Flexibility in the ABC transporter MSBA: Alternating access with a twist. Proc. Natl. Acad. Sci. U.S.A. 104, 19005–19010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior A. E. (2011) Reaction chemistry ABC-style. Proc. Natl. Acad. Sci. U.S.A. 108, 15015–15016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velamakanni S.; Yao Y.; Gutmann D. A. P.; van Veen H. W. (2008) Multidrug transport by the ABC transporter Sav1866 from Staphylococcus aureus. Biochemistry 47, 9300–9308. [DOI] [PubMed] [Google Scholar]
- Amaro R. E.; Baron R.; McCammon J. A. (2008) An improved relaxed complex scheme for receptor flexibility in computer-aided drug design. J. Comput.-Aided Mol. Des. 22, 693–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivetac A.; McCammon J. A. (2011) Molecular recognition in the case of flexible targets. Curr. Pharm. Des. 17, 1663–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J.; Perryman A. L.; Schames J. R.; McCammon J. A. (2002) Computational drug design accommodating receptor flexibility: The relaxed complex scheme. J. Am. Chem. Soc. 124, 5632–5633. [DOI] [PubMed] [Google Scholar]
- Roberts E.; Eargle J.; Wright D.; Luthey-Schulten Z. (2006) Multiseq: Unifying sequence and structure data for evolutionary analysis. BMC Bioinf. 7, 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engels M.; Jacoby E.; Krüger P.; Schlitter J.; Wollmer A. (1992) The T ⇋ R structural transition of insulin; pathways suggested by targeted energy minimization. Protein Eng. 5, 669–677. [DOI] [PubMed] [Google Scholar]
- Schlitter J.; Engels M.; Krüger P. (1994) Targeted molecular dynamics: A new approach for searching pathways of conformational transitions. J. Mol. Graphics 12, 84–89. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. (1996) VMD: Visual molecular dynamics. J. Mol. Graphics 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; et al. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwieters C. D.; Kuszewski J. J.; Tjandra N.; Clore G. M. (2003) The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73. [DOI] [PubMed] [Google Scholar]
- Schwieters C. D.; Kuszewski J. J.; Clore G. M. (2006) Using Xplor-NIH for NMR molecular structure determination. Prog. NMR Spectrosc. 48, 66–74. [Google Scholar]
- Frishman D.; Argos P. (1995) Knowledge-based protein secondary structure assignment. Proteins 23, 566–579. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A.; Rullmannn J. A.; MacArthur M. W.; Kaptein R.; Thornton J. M. (1996) Aqua and Procheck-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486. [DOI] [PubMed] [Google Scholar]
- Thompson J. D.; Higgins D. G.; Gibson T. J. (1994) Clustal W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell A. Jr.; Bashford D.; Bellott M.; Dunbrack R. Jr.; Evanseck J.; et al. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- Russell R. B.; Barton G. J. (1992) Multiple protein sequence alignment from tertiary structure comparison: Assignment of global and residue confidence levels. Proteins 14, 309–323. [DOI] [PubMed] [Google Scholar]
- O’Donoghue P.; Luthey-Schulten Z. (2005) Evolutionary profiles derived from the qr factorization of multiple structural alignments gives an economy of information. J. Mol. Biol. 346, 875–894. [DOI] [PubMed] [Google Scholar]
- Goodsell D. S.; Morris G. M.; Olson A. J. (1996) Automated docking of flexible ligands: Applications of Autodock. J. Mol. Recognit. 9, 1–5. [DOI] [PubMed] [Google Scholar]
- Huey R.; Morris G. M.; Olson A. J.; Goodsell D. S. (2007) A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 28, 1145–1152. [DOI] [PubMed] [Google Scholar]
- Morris G. M.; Goodsell D. S.; Huey R.; Olson A. J. (1996) Distributed automated docking of flexible ligands to proteins: Parallel applications of Autodock 2.4. J. Comput.-Aided Mol. Des. 10, 293–304. [DOI] [PubMed] [Google Scholar]
- Osterberg F.; Morris G. M.; Sanner M. F.; Olson A. J.; Goodsell D. S. (2002) Automated docking to multiple target structures: Incorporation of protein mobility and structural water heterogeneity in Autodock. Proteins 46, 34–40. [DOI] [PubMed] [Google Scholar]
- Rosenfeld R. J.; Goodsell D. S.; Musah R. A.; Morris G. M.; Goodin D. B.; et al. (2003) Automated docking of ligands to an artificial active site: Augmenting crystallographic analysis with computer modeling. J. Comput.-Aided Mol. Des. 17, 525–536. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Bartlett M. C.; Clarke D. M. (2005) ATP hydrolysis promotes interactions between the extracellular ends of transmembrane segments 1 and 11 of human multidrug resistance P-glycoprotein. Biochemistry 44, 10250–10258. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Clarke D. M. (1997) Drug-stimulated ATPase activity of human P-glycoprotein requires movement between transmembrane segments 6 and 12. J. Biol. Chem. 272, 20986–20989. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Clarke D. M. (2001) Cross-linking of human multidrug resistance P-glycoprotein by the substrate, tris(2-maleimidoethyl)amine, is altered by ATP hydrolysis. Evidence for rotation of a transmembrane helix. J. Biol. Chem. 276, 31800–31805. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Bartlett M. C.; Clarke D. M. (2008) Processing mutations disrupt interactions between the nucleotide binding and transmembrane domains of P-glycoprotein and the cystic fibrosis transmembrane conductance regulator (CFTR). J. Biol. Chem. 283, 28190–28197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham M. L.; Chen J. (2011) Snapshots of the maltose transporter during ATP hydrolysis. Proc. Natl. Acad. Sci. U.S.A. 108, 15152–15156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oloo E. O.; Tieleman D. P. (2004) Conformational transitions induced by the binding of MgATP to the vitamin B12 ATP-binding cassette (ABC) transporter BtuCD. J. Biol. Chem. 279, 45013–45019. [DOI] [PubMed] [Google Scholar]
- Ecker G. F.; Stockner T.; Chiba P. (2008) Computational models for prediction of interactions with ABC-transporters. Drug Discovery Today 13, 311–317. [DOI] [PubMed] [Google Scholar]
- Campbell J. D.; Biggin P. C.; Baaden M.; Sansom M. S. P. (2003) Extending the structure of an ABC transporter to atomic resolution: Modeling and simulation studies of MsbA. Biochemistry 42, 3666–3673. [DOI] [PubMed] [Google Scholar]
- Becker J.; Depret G.; Van Bambeke F.; Tulkens P. M.; Prévost M. (2009) Molecular models of human P-glycoprotein in two different catalytic states. BMC Struct. Biol. 9, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Globisch C.; Pajeva I. K.; Wiese M. (2008) Identification of putative binding sites of P-glycoprotein based on its homology model. ChemMedChem 3, 280–295. [DOI] [PubMed] [Google Scholar]
- McDevitt C. A.; Shintre C. A.; Günter Grossmann J.; Pollock N. L.; Prince S. M.; et al. (2008) Structural insights into P-glycoprotein (ABCB1) by small angle X-ray scattering and electron crystallography. FEBS Lett. 582, 2950–2956. [DOI] [PubMed] [Google Scholar]
- O’Mara M. L.; Tieleman D. P. (2007) P-Glycoprotein models of the apo and ATP-bound states based on homology with Sav1866 and MalK. FEBS Lett. 581, 4217–4222. [DOI] [PubMed] [Google Scholar]
- Omote H.; Al-Shawi M. K. (2006) Interaction of transported drugs with the lipid bilayer and P-glycoprotein through a solvation exchange mechanism. Biophys. J. 90, 4046–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajeva I. K.; Globisch C.; Wiese M. (2004) Structure-function relationships of multidrug resistance P-glycoprotein. J. Med. Chem. 47, 2523–2533. [DOI] [PubMed] [Google Scholar]
- Pajeva I. K.; Globisch C.; Wiese M. (2009) Comparison of the inward- and outward-open homology models and ligand binding of human P-glycoprotein. FEBS J. 276, 7016–7026. [DOI] [PubMed] [Google Scholar]
- Ravna A. W.; Sylte I.; Sager G. (2009) Binding site of ABC transporter homology models confirmed by ABCB1 crystal structure. Theor. Biol. Med. Modell. 6, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarcsay A.; Keseru G. M. (2011) Homology modeling and binding site assessment of the human P-glycoprotein. Future Med. Chem. 3, 297–307. [DOI] [PubMed] [Google Scholar]
- Ambudkar S. V.; Kim I.; Xia D.; Sauna Z. E. (2006) The A-loop, a novel conserved aromatic acid subdomain upstream of the Walker A motif in ABC transporters, is critical for ATP binding. FEBS Lett. 580, 1049–1055. [DOI] [PubMed] [Google Scholar]
- Jabeen I.; Wetwitayaklung P.; Klepsch F.; Parveen Z.; Chiba P.; et al. (2011) Probing the stereoselectivity of P-glycoprotein-synthesis, biological activity and ligand docking studies of a set of enantiopure benzopyrano[3,4-b][1,4]oxazines. Chem. Commun. 47, 2586–2588. [DOI] [PubMed] [Google Scholar]
- Bikadi Z.; Hazai I.; Malik D.; Jemnitz K.; Veres Z.; et al. (2011) Predicting P-glycoprotein-mediated drug transport based on support vector machine and three-dimensional crystal structure of P-glycoprotein. PLoS One 6, e25815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tombline G.; Senior A. E. (2005) The occluded nucleotide conformation of P-glycoprotein. J. Bioenerg. Biomembr. 37, 497–500. [DOI] [PubMed] [Google Scholar]
- Dawson R. J. P.; Hollenstein K.; Locher K. P. (2007) Uptake or extrusion: Crystal structures of full ABC transporters suggest a common mechanism. Mol. Microbiol. 65, 250–257. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Bartlett M. C.; Clarke D. M. (2004) The drug-binding pocket of the human multidrug resistance P-glycoprotein is accessible to the aqueous medium. Biochemistry 43, 12081–12089. [DOI] [PubMed] [Google Scholar]
- Rees D. C.; Johnson E.; Lewinson O. (2009) ABC transporters: The power to change. Nat. Rev. Mol. Cell Biol. 10, 218–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Urbatsch I. L.; Senior A. E.; Wilkens S. (2002) Projection structure of P-glycoprotein by electron microscopy. Evidence for a closed conformation of the nucleotide binding domains. J. Biol. Chem. 277, 40125–40131. [DOI] [PubMed] [Google Scholar]
- Lee J.; Urbatsch I. L.; Senior A. E.; Wilkens S. (2008) Nucleotide-induced structural changes in P-glycoprotein observed by electron microscopy. J. Biol. Chem. 283, 5769–5779. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Bartlett M. C.; Clarke D. M. (2010) Human P-glycoprotein is active when the two halves are clamped together in the closed conformation. Biochem. Biophys. Res. Commun. 395, 436–440. [DOI] [PubMed] [Google Scholar]
- Verhalen B.; Wilkens S. (2011) P-Glycoprotein retains drug-stimulated ATPase activity upon covalent linkage of the two nucleotide binding domains at their C-terminal ends. J. Biol. Chem. 286, 10476–10482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.; McCammon J. A. (2010) The gates of ion channels and enzymes. Trends Biochem. Sci. 35, 179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H. X.; Wlodek S. T.; McCammon J. A. (1998) Conformation gating as a mechanism for enzyme specificity. Proc. Natl. Acad. Sci. U.S.A. 95, 9280–9283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen M. T.; Jonson P. H.; Petersen S. B. (1999) Amino acid neighbours and detailed conformational analysis of cysteines in proteins. Protein Eng. 12, 535–548. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Clarke D. M. (2001) Defining the drug-binding site in the human multidrug resistance P-glycoprotein using a methanethiosulfonate analog of verapamil, MTS-verapamil. J. Biol. Chem. 276, 14972–14979. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Clarke D. M. (1997) Identification of residues in the drug-binding site of human P-glycoprotein using a thiol-reactive substrate. J. Biol. Chem. 272, 31945–31948. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Clarke D. M. (1999) Identification of residues in the drug-binding domain of human P-glycoprotein. Analysis of transmembrane segment 11 by cysteine-scanning mutagenesis and inhibition by dibromobimane. J. Biol. Chem. 274, 35388–35392. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Clarke D. M. (2000) Identification of residues within the drug-binding domain of the human multidrug resistance P-glycoprotein by cysteine-scanning mutagenesis and reaction with dibromobimane. J. Biol. Chem. 275, 39272–39278. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Bartlett M. C.; Clarke D. M. (2003) Methanethiosulfonate derivatives of rhodamine and verapamil activate human P-glycoprotein at different sites. J. Biol. Chem. 278, 50136–50141. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Bartlett M. C.; Clarke D. M. (2003) Permanent activation of the human P-glycoprotein by covalent modification of a residue in the drug-binding site. J. Biol. Chem. 278, 20449–20452. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Bartlett M. C.; Clarke D. M. (2009) Identification of residues in the drug translocation pathway of the human multidrug resistance P-glycoprotein by arginine mutagenesis. J. Biol. Chem. 284, 24074–24087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo T. W.; Bartlett M. C.; Clarke D. M. (2003) Substrate-induced conformational changes in the transmembrane segments of human P-glycoprotein. Direct evidence for the substrate-induced fit mechanism for drug binding. J. Biol. Chem. 278, 13603–13606. [DOI] [PubMed] [Google Scholar]
- Higgins C. F.; Linton K. J. (2004) The ATP switch model for ABC transporters. Nat. Struct. Mol. Biol. 11, 918–926. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Clarke D. M. (2008) Mutational analysis of ABC proteins. Arch. Biochem. Biophys. 476, 51–64. [DOI] [PubMed] [Google Scholar]
- McDevitt C. A.; Callaghan R. (2007) How can we best use structural information on P-glycoprotein to design inhibitors?. Pharmacol. Ther. 113, 429–441. [DOI] [PubMed] [Google Scholar]
- Hollenstein K.; Dawson R. J.; Locher K. P. (2007) Structure and mechanism of ABC transporter proteins. Curr. Opin. Struct. Biol. 17, 412–418. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.