

Abstract
The locus coeruleus (LC) is the major loci of noradrenergic innervation to the forebrain. Due to the extensive central nervous system innervation of the LC noradrenergic system, a reduction in the number of LC neurons could result in significant changes in noradrenergic function in many forebrain regions. LC noradrenergic neurons were lesioned in adult male C57Bl/6 mice with the unilateral administration of 6-hydroxydopamine (6OHDA) (vehicle on the alternate side). Noradrenergic markers were measured 3 weeks later to determine the consequence of LC loss in the forebrain. Direct administration of 6OHDA into the LC results in the specific reduction of noradrenergic neurons in the LC (as measured by electrophysiology, immunoreactivity and in situ hybridization), the lateral tegmental neurons and dopaminergic neurons in the substantia nigra (SN) and ventral tegmental region were unaffected. The loss of LC noradrenergic neurons did not result in compensatory changes in the expression of mRNA for norepinephrine (NE) synthesizing enzymes. The loss of LC noradrenergic neurons is associated with reduced NE tissue concentration and NE transporter (NET) binding sites in the frontal cortex and hippocampus, as well as other forebrain regions such as the amygdala and SN. Adrenoreceptor (AR) binding sites (α1- and α2-AR) were not significantly affected on the 6OHDA-treated side compared to the vehicle-treated side, although there is a reduction of AR binding sites on both the vehicle- and 6OHDA-treated side in specific forebrain regions. These studies indicate that unilateral stereotaxic injection of 6OHDA into mice reduces noradrenergic LC neurons and reduces noradrenergic innervation to many forebrain regions, including the contralateral side.
Keywords: 6-hydroxydopamine, frontal cortex, hippocampus, norepinephrine transporter, tyrosine hydroxylase, dopamine β-hydroxylase, single-unit extracellular electrophysiology
1.1 Introduction
The noradrenergic nervous system is a major neurotransmitter system in the periphery and in the brain where it is involved in many physiological and behavioral processes; therefore, the central noradrenergic nervous system plays a role in many pathologies including depression, epilepsy, Parkinson’s disease (PD), Alzheimer’s disease (AD), and post traumatic stress disorder (Ressler and Nemeroff, 2000; Weinshenker and Szot, 2002; Zarow et al., 2003; Szot et al., 1999, 2006; Fitzgerald, 2010; McMillan et al., 2011; Olson et al., 2011). The locus coeruleus (LC) contains the vast majority of noradrenergic cell bodies in the brain, which sends projections throughout the forebrain. Therefore, a loss of LC noradrenergic neurons can have a profound effect on the function of the brain. A loss of LC noradrenergic neurons is a major neuropathological characteristic of two neurodegenerative disorders, AD and PD (Bondareff et al., 1981; Tomlinson et al., 1981; Marcyniuk et al., 1986; Cash et al., 1987; Chan-Palay and Asan, 1989, 1991; German et al., 1992; Patt and Gerhard, 1993; Bertrand et al., 1997; Szot et al., 2006; Zarow et al., 2003; McMillan et al., 2011). How this neuronal loss impacts the progression of these disorders remains unclear. To understand the role of LC neuronal loss in the pathogenesis of these neurodegenerative disorders and other disorders involving the noradrenergic nervous system, it is important to determine the consequence of LC noradrenergic neuronal loss on noradrenergic markers in the LC and at terminal forebrain regions. The consequences of LC neuronal loss alone have not been adequately defined.
The vast majority of research on LC neuronal loss has focused on the effects of the noradrenergic neurotoxin N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP4). DSP4 consistently results in a temporary reduction in NE tissue concentration in terminal forebrain regions (Ross, 1976; Jonsson et al., 1981; Grzanna et al., 1989; Theron et al., 1993; Wolfman et al., 1994; Hughes and Stanford, 1998; Kask et al., 1997; Harro et al., 1999; Szot et al., 2010), a temporary reduction in NE transporter (NET) binding sites in specific forebrain regions (Cheetham et al., 1996; Szot et al., 2010) and an increase in α2-AR binding sites in specific forebrain regions (Wolfman et al., 1994; Harro et al., 1999; Szot et al., 2010). However, there is data indicating that DSP4 does not result in a loss of LC noradrenergic neurons but does cause these changes in noradrenergic forebrain markers (Booze et al., 1988; Lyons et al., 1989; Robertson et al., 1993; Matsukawa et al., 2003; Szot et al., 2010).
Another neurotoxin, 6-hydroxydopamine (6OHDA), has been shown to reduce LC noradrenergic neurons when administered directly into the lateral ventricles, but dopaminergic neurons in the substantia nigra (SN) and ventral tegmental area (VTA) are reduced as well (Descarries and Saucier, 1972; Coradazzi et al., 2010). Administration of 6OHDA directly into the LC also results in a significant loss of noradrenergic neurons (Harik, 1984; Biancardi et al., 2008), although a comprehensive analysis of the consequence of this loss on noradrenergic terminals has not been performed. The objective of this study is to determine the effect of unilateral 6OHDA in C57Bl/6 mice on LC neuronal loss and changes in forebrain noradrenergic markers 3 weeks after administration. Alterations in noradrenergic markers 3 weeks after 6OHDA administration would indicate long-term changes. Changes in noradrenergic terminals were assessed by: NET, α1- and α2-adrenoreceptor (AR) binding, and NE tissue concentration in specific forebrain regions. To assess the degree of LC neuron loss tyrosine hydroxylase-immunoreactivity (TH-IR), TH and dopamine β-hydroxylase (DBH) mRNA expression were measured in the same animals where catecholamine or binding assays were performed. Electrophysiological characteristics of viable LC neurons were measured in a separate set of animals.
1.2 Experimental Procedures
1.2.1 Animals
Adult male C57Bl/6 mice were purchased from Charles River Laboratories (Wilmington, MA, USA) and housed in standard enriched environment cages in a temperature controlled room with a 12-h light/dark cycle. Food and water were provided ad libitum. The animals were given at least 2 weeks acclimating period to the facility before administration of 6OHDA. All animal procedures were in accordance with the Animal Care Committee at the VA Puget Sound Health Care System, Seattle, WA, Whitman College, Walla Walla, WA and National Institute of Health guidelines. The minimum number of animals were used for these studies and care was taken to minimize any suffering.
1.2.2 Surgery and LC microinjections
Mice were anesthetized with Isoflurane (Baxter Healthcare, Deerfield, IL, USA) and mounted in a Kopf Model 1900 stereotaxic instrument (David Kopf Instruments, Tujunga, CA, USA) with the skull balanced in the medial/lateral and anterior/posterior planes. Carprofen (5 mg/kg) and ceftriaxone (50 mg/kg) were administered to animals before incision to prevent pain and infection. 6OHDA was administered into the right LC, while vehicle (0.2% ascorbic acid/saline) was administered into the left LC at the following coordinates: A/P: −5.4 mm from bregma; M/L: ± 0.9 mm; D/V: −3.25 mm from the surface of the skull, at a rate of 0.13 μl/min over ~8 min. The needle was left in place an additional 4 min after injection and then the needle was slowly withdrawn. Drilled holes were filled with wax and the skin sutured. Animals were allowed to recover in a cage placed halfway on a heating pad for the next 3 days. During this time animals were given 2 additional doses of carprofen for pain relief.
To assess the dose-response effect of 6OHDA in the LC, 6OHDA (7 (n=7), 10 (n=5) and 14 (n=5) μg/μl) was administered unilaterally to mice as described above. Animals were sacrificed 3 weeks later. In addition to these animals, 5 mice that did not receive LC injection (non-surgery) were sacrificed on the same day as surgery animals to examine the noradrenergic markers in naïve animals. All animals had TH mRNA measured in LC noradrenergic neurons to assess the degree of LC neuronal loss; in addition, TH mRNA was measured in dopaminergic neurons of the SN and VTA in mice that demonstrated a loss of LC neurons.
1.2.3: Functional assessment of neuron loss in the LC following unilateral 6OHDA (10 μg/μl) injection
To examine the loss of noradrenergic neurons following 6OHDA and to assess if any of the surviving LC neurons were functional, we implemented intracellular, sharp electrode recordings to assay the number of electrophysiologically viable cells. Twelve C57Bl/6 mice had 6OHDA (10 μg/μl) injected unilaterally into the LC; vehicle was administered in the alternate LC. Of the 12 animals 1 died following surgery and 2 were lost during acute slice preparation. Three additional animals were administered 1,1′-dilinoleyl-3,3,3′,3′-tetramethylindocarbocynanine and 3,3′-dioctadecyloxacarbocyanine (Biotium, Hayward, CA, USA) bilaterally for verification of stereotaxic placement. For in vitro slice electrophysiology, brain slices containing LC were prepared following a modified protocol as previously published (Henderson et al., 1982; Williams et al., 1984). Four to 7 weeks (n=9) after stereotaxic injection, mice were deeply anesthetized with halothane, rapidly decapitated, and the brains were carefully removed and transferred to cold, oxygenated artificial cerebrospinal fluid (aCSF) containing (in mM) 124 NaCl, 3 KCl, 1.25 NaH2PO4, 2 MgSO4, 26 NaCO3, 2 CaCl2 and 10 dextrose. Brains were blocked to contain the LC by making two coronal cuts at approximately the caudal and rostral limits of the pons. Serial sections were cut at 300 μm thickness using a vibroslicer (Electron Microscopy Sciences, Hatfield, PA, USA) and transferred to a holding chamber at room temperature with oxygenated, circulating aCSF. Individual slices were then transferred to a slice interface recording chamber (Scientific Systems Design Inc, Mississauga, Ontario, Canada) perfused with warmed (32–35 °C), oxygenated aCSF; the slice surface was exposed to a warmed, humidified 95% O2/5% CO2 air. Left and right hemispheres were noted and slice orientation was carefully monitored to track the 6OHDA injected right hemisphere and the vehicle control injected left hemisphere.
Microelectrodes were made from borosilicate glass pulled on a horizontal puller (Sutter Instruments, Novato, CA, USA) and filled with 4M potassium acetate (30 – 60 MΩ). These microelectrodes were used to estimate the number of viable neurons in the vehicle- versus 6OHDA-treated LC. Viable LC neurons exhibited a combination of voltage changes (minimum −15mV shift from baseline), increase in input resistance (in response to a 400 nA current injection) and spontaneous or evoked action potential firing. All electrophysiological data were recorded using an Axon Instruments Multiclamp 700A amplifier (Molecular Devices, Sunnyvale, CA, USA) with a bridge circuit to compensate for electrode resistance, digitized using a Digidata 1440 (Molecular Devices) and stored on a personal computer system using pClamp Software (Molecular Devices). Data were plotted and statistically analyzed using Excel Software (Microsoft Excel for Mac 2011, version 14.1.2). All values are reported as a mean ± SEM.
1.2.4 Experiment 1: Effect of unilateral 6OHDA (10 μg/μl) on NE synthesizing enzymes in surviving LC neurons and forebrain catecholamine levels
Twenty C57Bl/6 mice had 6OHDA (10 μg/μl) injected unilaterally into the LC; vehicle was administered in the alternate LC. Animals were sacrificed 3 weeks later and brains removed, the hindbrain portion containing the LC was dissected free and frozen on dry ice to assess LC neuronal loss. The following forebrain regions were dissected free and separated into vehicle- and 6OHDA-treated side for catecholamine analysis: frontal cortex (FC), septum/bed nucleus of the stria terminalis (Sep/BNST), SN/VTA, HP, striatum (Str) and amygdala (Amy). One set of 10 animals had the FC, Sep/BNST and SN/VTA dissected free; while the other 10 animals had HP, Str and Amy dissected free. In addition, 10 non-surgery mice (5/5) were sacrificed at the same time, hindbrain and forebrain regions were processed as described for animals above. The hindbrain portion from all animals containing the LC was cut on a cryostat at 16 μm onto Superfrost Plus slides (Fisher Scientific, Pittsburgh, PA, USA) in three sets consisting of alternating sections and stored at −80°C. Slides containing the LC had TH and DBH mRNA in situ hybridization (ISH) and TH-immunohistochemistry (IHC) performed to assess neuronal loss in the LC. The lateral tegmental nuclei (nucleus solitary tract (NTS) and A1/A2 nuclei) were also cut in addition to the LC, and TH and DBH mRNA ISH was performed to assess neuronal loss. Slides were initially apposed to Biomax MR film (Eastman Kodak Co, Rochester, NY, USA) for 3 days for TH mRNA and 1 day for DBH mRNA in the LC, and 1 week for TH and DBH mRNA in the NTS and A1/A2 region. Slides containing the LC region were then coated with NTB2 Nuclear Track Emulsion (undiluted) (Eastman Kodak Co, Rochester, NY, USA) after exposure to film and exposed for 1 week for TH mRNA and 4 days for DBH mRNA at −20°C.
1.2.5 Experiment 2: Effect of unilateral 6OHDA (10 μg/μl) on NET, α1- and α2-adrenoreceptor binding in forebrain regions
Eighteen C57Bl/6 mice had 6OHDA (10 μg/μl) injected unilaterally into the LC; vehicle was administered in the alternate LC. Animals were sacrificed 3 weeks later, brains removed and frozen on dry ice. In addition, 6 non-surgery mice were sacrificed concomitantly with 6OHDA mice. Whole brains were cut on a cryostat at 16 μm onto Superfrost Plus slides, alternating sections on three sets of slides and stored at −80°C. Slides containing forebrain regions had NET, α1- and α2-AR binding performed, while slides containing the LC had TH and DBH mRNA ISH and TH-IHC performed. The lateral tegmental nuclei (NTS and A1/A2 nuclei) were also cut in addition to the LC, and TH and DBH mRNA ISH was performed to assess neuronal loss. Slides were apposed to Biomax MR film as described above.
1.2.6 TH-IHC
TH-IR was performed in the LC of Experiment 1 and 2 animals to assess the number of noradrenergic neurons as previously described with some minor modifications (McMillan et al., 2011). The slides were air-dried and fixed 10 min in 4% paraformaldehyde in 0.1M phosphate buffer (PBS pH 7.4) at 4°C, then rinsed and treated with 2:1 ethanol: glacial acetic acid for 5 minutes. Slides were permeabilized with 0.5% Triton X-100 for 30 min and treated with 0.6% H2O2 in PBS for 1 h. Slides were blocked with 10% goat serum, 1% BSA and 0.03% Triton X-100 in PBS for 30 min. The sections were incubated overnight at 4°C with rabbit polyclonal anti-TH (AB152, Millipore, Temecula, CA, USA) at 1:1000. Slides were blocked again briefly for 10 min, and then incubated for 1 h in biotinylated goat anti-rabbit or anti-rat IgG (VectorLabs, Inc., Burlingame, CA, USA). The Vectastain ABC kit (VectorLabs) and 0.5 mg diaminobenzidine/ml PBS + 0.03% hydrogen peroxide were used to visualize TH labeled cells. The primary and secondary antibodies along with the horse radish peroxidase conjugated avidin/biotin complex were diluted in 1% goat serum, 1% bovine serum albumin and 0.03% Triton X-100, and slides were washed 4 times for 3 min between each step. Negative controls with secondary antibody alone did not immunostain tissue sections (data not shown).
The number of TH-IR cell bodies in the LC were counted in 3 consecutive atlas matched sections on the vehicle- and 6OHDA-treated sides and bilaterally in non-surgery animals, and expressed as the average ± SEM of TH-IR neurons unilaterally. Photomicrographs were taken with a digital camera and imported into Adobe Photoshop. To optimize visualization of staining, photomicrographs were modified, when necessary, by adjusting brightness and contrast.
1.2.7 TH and DBH mRNA ISH
TH and DBH ISH was performed in the LC of Experiment 1 and 2 animals to assess the number of noradrenergic neurons as previously described (Szot et al., 1997, 2006). The TH oligonucleotide probe was a 48 base probe complementary to nucleotides 1351–1398 of the rat TH mRNA sequence (Grima et al., 1985). The DBH oligonucleotide probe consisted of two oligonucleotides complementary to nucleotides 454–505 and 1414–1465 of the rat sequence (McMahon et al., 1990). The oligonucleotide probes were 3′ end-labeled with 33P-dATP (PerkinElmer) using terminal deoxyribonucleotidyl transferase (Invitrogen, Piscataway, NJ, USA). The TH probe contained a range from 0.25 × 106 cpm/50 μl to 1.4 × 106 cpm/50 μl for the different experiments and was washed as described in detail in previously published work with the oligonucleotide (Szot et al., 1997). The DBH probe contained a range from 0.37 × 106 cpm/50 μl to 2.7 × 106 cpm/50 μl for the different experiments and was washed as described in detail in previously published work with the oligonucleotide (Szot et al., 2010).
Films were developed by standard procedures as previously described (Szot et al., 1997). TH mRNA expression in the LC was quantitated as optical density (OD) using the MicroComputer Imaging Device system (MCID) (InterFocus Imaging Ltd., Cambridge, England) in 3 consecutive sections atlas matched in animals on the vehicle- and 6OHDA-treated sides and bilaterally in non-surgery animals. Since multiple ISH experiments were performed, to combine the results for each experimental group the data were converted to percent control of the TH mRNA expression in the LC of non-surgery animals and expressed as the average percent change from non-surgery ± SEM. Only animals that exhibited greater than ~25% neuronal loss on the 6OHDA-treated side compared to vehicle-treated side were used for further analysis.
Emulsion coated slides were developed by standard procedures as previously described (Szot et al., 1997). Quantitation of TH and DBH mRNA expression was similar to that performed and described in previous publication (Szot et al., 2006, 2010) using the MCID system. The number of positive labeled neurons that achieved labeling threefold higher than background was counted unilaterally in animals on the vehicle- and 6OHDA-treated side and bilaterally in non-surgery animals on 3–5 consecutive sections that were atlas matched. Data for the number of positive labeled neurons were expressed as the average percent change from non-surgery ± SEM. The density of TH and DBH mRNA expression/neuron was performed by measuring the amount of silver grains over cell bodies of labeled neurons that were threefold higher than background under 20X dark-field illumination using MCID and data for grains/neuron were expressed as the average percent change from non-surgery ± SEM. All labeled neurons that were counted as positively labeled for each oligonucleotide probe were also quantitated for the amount of TH and DBH mRNA expression/neuron.
1.2.8 Receptor Binding
3H-Nisoxetine (80.0 Ci/mmol: American Radiolabeled Chemicals, St. Louis, MO, USA) was used to quantitate NET binding sites, 3H-RX821002 (55.0 Ci/mmol: PerkinElmer) was used to quantitate α2-AR binding sites and 3H-prazosin (85.0 Ci/mmol: PerkinElmer) was used to quantitate α1-AR binding sites. 3H-Nisoxetine binding was performed as previously described (Weinshenker et al., 2002; Szot et al., 2006, 2010). Briefly, slides were thawed as described above, then 600 μl/slide of incubation buffer (~3 nM 3H-nisoxetine in 50mM Tris buffer with 300 mM NaCl and 5 mM KCl, pH 7.7) was placed over the tissue. Non-specific binding was defined in the presence of 1μM mazindol. Slides were incubated for 2 h at room temperature and then washed twice for 2 min in ice-cold 50 mM Tris buffer, pH 7.4, dipped in ice-cold distilled water to remove salts and then dried rapidly under a stream of cool air. 3H-RX821002 binding was performed as described previously (Szot et al., 2006, 2010). Briefly, slides were thawed as described above, and then 600 μl/slide of incubation buffer (~2 nM 3H-RX821002 in 50 mM NaPO4 buffer, pH 7.4) was placed over the tissue. Non-specific binding was defined in the presence of 10 μM rauwolscine. Slides were incubated for 45 min at room temperature and then washed for 2 min in ice-cold 50 mM NaPO4 buffer, pH 7.4, dipped in ice-cold distilled water and dried as described above for 3H-nisoxetine. 3H-Prazosin was performed as described previously (Sanders et al., 2006; Szot et al., 2006, 2010). Briefly, slides were thawed as described above for 3H-nisoxetine and then 600 μl/slide of incubation buffer (~0.2 nM 3H-prazosin in 50 mM Tris buffer, 1 mM EDTA, pH 7.4) was placed over the tissue. Non-specific binding was defined in the presence of 10 μM phentolamine. Slides were incubated for 40 min at room temperature, washed and dried as described above for 3H-nisoxetine. All slides were apposed to Biomax MR film (Eastman Kodak Co) for 2 months.
Films were developed by standard procedures (Szot et al., 1997). NET, α2- and α1-AR binding sites were quantitated as optical density (OD) using the MCID system (InterFocus Imaging Ltd., Cambridge, England) in 3 consecutive sections in animals on the vehicle- and 6OHDA-treated sides and bilaterally in non-surgery animals. NET (3H-nisoxetine) binding sites were quantitated (OD) in the following atlas matched regions of vehicle/6OHDA-treated and non-surgery animals: Sep, FC, BNST, paraventricular thalamic nucleus (PVTN), HP, SN, VTA and geniculate (Gen). Specific binding was obtained by taking average OD values minus non-specific OD value from the same region. α2-AR (3H-RX821002) binding sites were quantitated in the following atlas matched regions of vehicle/6OHDA-treated and non-surgery animals: septum, FC, BNST, Str, dorsal thalamic nucleus (DTN), hypothalamus (hypo), HP, Amy, SN, VTA and Gen. Specific binding was obtained as described above for NET. α1-AR (3H-prazosin) binding sites were quantitated in the following atlas matched regions of vehicle/6OHDA-treated and non-surgery animals: Sep, FC, BNST, thalamus (Thal), HP, Amy, SN and VTA. Specific binding was obtained as described above for NET.
Data for NET, α1- and α2-AR binding concentrations were expressed as the average percent change from non-surgery ± SEM. Photomicrographs were taken with a digital camera and imported into Adobe Photoshop; images presented are from the same experiment. To optimize visualization of labeling, all photomicrographs were modified equally, when necessary, by adjusting brightness and contrast.
1.2.9 HPLC measurement of catecholamines
Each unilateral brain region was sonicated in 0.5 ml of 0.1 M perchloric acid. A 100 μl aliquot of the sonicated material was stored at −80°C for protein determination using Pierce BCA™ Protein Assay kit (Thermo Scientific, Rockford, IL, USA). The supernate was collected from centrifugation of the sonicated material at 13,000 g for 15 min and stored at −70°C until catecholamine extraction was performed. Catecholamine levels were assayed for each region together to reduce variability. Catecholamines were extracted by alumina extraction from 100 μl of the sonicated supernate as previously described (Eisenhofer et al., 1986). The eluted catechols were filtered through 0.22 MillexR GV syringe driven filter and detection was performed with the ESA Coulochem II electrochemical detector (conditioning cell set at +350 mV, electrode 1 of analytical cell set at +90 mV, electrode 2 of analytical cell set at −300 mV) ESA, Chelmsford, MA, USA). Phenomonex (Phenomonex, Torrance, CA, USA) reverse phase c18 Gemini column (150X4.6 mm, 3 μC, 110A) and Scientific Software Inc. was to measure levels of NE, dopamine (DA), 3,4-dihydroxyphenylglycol (DHPG), 3,4-dihydroxyphenylalanine (DOPA) and dihydroxyphenylacetic acid (DOPAC) in each unilateral brain region. DOPA is a precursor for both NE and DA; DHPG is a metabolite of NE and DOPAC is a metabolite of DA. Catecholamine values were expressed as ng catecholamine/mg protein ± SEM.
1.2.10 Statistical Analysis
A one-way ANOVA was used to assess statistical difference followed by a post hoc Tukey’s test using the computer program GraphPad Prism (v. 5.0, GraphPad Software Inc.) for all studies except the dose-response effects of 6OHDA and electrophysiological analysis. A two-way ANOVA was used only to compare the effect of different 6OHDA doses in the different brain regions (LC, SN and VTA). Bonferroni tests were used to compare the different groups. Assessment of functional loss of LC neurons by 6OHDA was performed by two-tailed unpaired Student’s t-test. Statistical difference was taken at P values less than 0.05.
Simple linear regressions were performed between NE catecholamine levels and NET binding sites in specific brain region to the number of TH-IR neurons in the LC using GraphPad Prism, with significance at P values less than 0.05.
1.3 Results
1.3.1 6-OHDA dose-response
Direct unilateral LC administration of 6OHDA at 3 different doses (7, 10 and 14 μg/μl) demonstrated a dose-dependent effect; 7 μg/μl 6OHDA had no effect on LC noradrenergic neurons, while the 10 and 14 μg/μl doses significantly reduced TH mRNA expression in the LC compared to vehicle-treated side and non-surgery animals (Figure 1). Expression of TH mRNA in the LC of the vehicle-treated side was similar to TH mRNA expression observed in the LC of non-surgery animals (Figure 1). The effect of the 14 μg/μl dose of 6OHDA on TH mRNA in the LC was greater than the 10 μg/μl dose, but it did not reach statistical difference. Importantly, 6OHDA administration directly into the LC did not affect TH mRNA expression in the dopaminergic neurons of the SN or VTA, despite significant reductions in the LC (Figure 1 middle and lower panel). These data indicate the administration of 6OHDA directly into the LC remains localized to LC noradrenergic neurons. Following 6OHDA injections we observed an acute dose-dependent behavior: the 14 μg/μl dose of 6OHDA elicited a head tilt toward the side administered 6OHDA and some exhibited barrel rotation toward the 6OHDA-treated side. The 10 μg/μl dose of 6OHDA, which also produced a significant loss of LC neurons, usually did not result in head tilt or barrel rotation, and if it did occur, the behavior was milder and shorter in duration than that observed with the 14 μg/μl dose of 6OHDA. Due to the behavior observed consistently with the 14 μg/μl dose of 6OHDA, the remainder of the studies were performed with 6OHDA 10 μg/μl.
Figure 1.
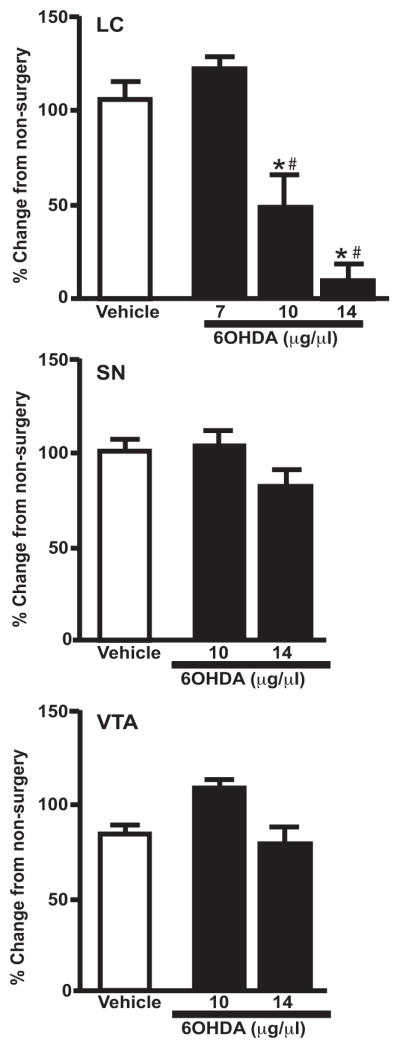
Dose-response effect of unilateral 6OHDA on TH mRNA expression (OD) in the LC, SN and VTA. 6OHDA specifically reduced LC noradrenergic neurons. * Indicates significant difference from non-surgery animals. # Indicates significant difference from vehicle-treated animals.
1.3.2 Functional loss in the LC following unilateral 6OHDA (10 μg/μl) injection
LC neurons were identified by a minimum 15 mV negative shift in potential combined with action potential firing (either spontaneous or induced through positive current injection) and a concomitant increase in electrode tip resistance, estimated by changes in voltage response to current injection. Using anatomical markers, such as the 4th ventricle, the pontine central gray and the mesencephalic root of the trigeminal nerve, we placed our electrodes in the LC and systematically moved our recording sites along the long, dorso-ventral axis of the LC. We recorded from 5–6 sites in each LC (vehicle- and 6OHDA-treated sides, see Figure 2A) and randomly selected which side to initiate recordings. Of the nine animals from which we successfully obtained recordings, we were able to obtain a single 300 μm thick slice containing the majority of the LC from 8 and in the one remaining animal we obtained two slices containing either the rostral or caudal LC. For the latter, we electrophysiologically assayed both slices (as described above) and pooled the data. We recorded from a total of over 140 LC neurons, of which 129 were from the vehicle-injected LC and only 12 were from the 6OHDA-injected LC. An average of 14 cells were observed on the vehicle-treated side compared to an average of one cell on the 6OHDA (see Figure 2B), suggesting a significant (paired, Student’s t-test; p<0.0001) reduction in the number of LC neurons following 6OHDA. LC neurons when observed were commonly in the more ventral aspects of the LC in both hemispheres and only a few cells were observed near the ventricle. This observation was more notable in the 6OHDA-treated side. Of the 12 cells recorded from the 6OHDA-treated side, all cells except for one were observed in the ventral most recording sites, the remaining cell were observed in mid-LC region. These data are consistent with our injection method (drug was injected dorsal to LC) and suggest that neurons in dorsal regions of the LC are likely exposed to more neurotoxin and therefore more susceptible to damage.
Figure 2.
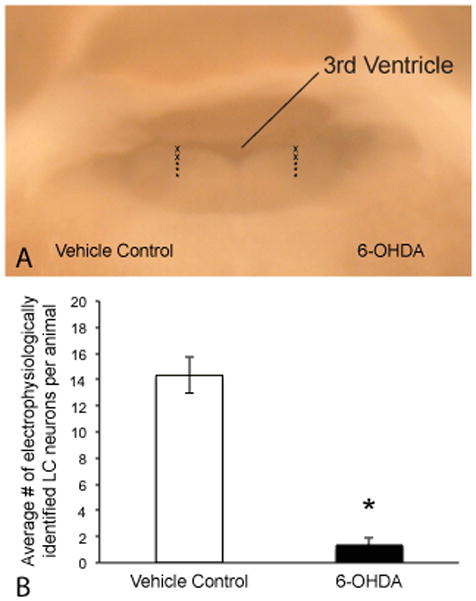
Electrophysiological confirmation of neuron loss following 6-OHDA injection. A) Digital image of 300mm thick acute slice containing the LC (view from caudal most aspect of the LC) just prior to electrophysiological recordings. Image demonstrates the approximate recording sites used to assess viable LC neurons comparing the vehicle-treated side (left) with the 6-OHDA injected side (right). Approximately 5–6 recording tracks were run on each side along the dorsal-ventral axis of the LC. Asterisks (lower three) identify the tracks in which most LC cells were observed (particularly in the 6-OHDA side). B) The average number of viable cells identified by characteristic electrophysiological properties (negative shift in voltage, increased input resistance and action potential firing) were significantly greater in the vehicle-treated side compared to the 6OHDA-treated side (n=9; paired student’s t-test p<0.0001).
1.3.3. 6OHDA (10 μg/μl) significantly reduces LC noradrenergic neurons but not lateral tegmental neurons
Fourteen of the 20 animals of Experiment 1 and 12 of the 18 animals of Experiment 2 administered unilateral 6OHDA met the requirement of a ~25% loss or greater of LC neurons on the 6OHDA-treated side compared to vehicle-treated side. The number of TH-IR neurons counted in the LC and the OD measurement of TH and DBH mRNA expression in the lateral tegmental areas of the two different experiments were assessed and combined. The number of TH-IR neurons on the vehicle-treated side was not statistically different from the number of TH-IR neurons in the LC of non-surgery (NS) animals (Figure 3A, B photomicrographs of TH-IR labeling). 6OHDA produced a significant reduction in the number of TH-IR neurons in the LC by 56% compared to vehicle-treated side and 58% compared to non-surgery animals (Figure 3A, B). In contrast, noradrenergic neurons in the lateral tegmental area of the NTS (Figure 3C) and A1/A2 (Figure 3D) were unaffected by 6OHDA administration into the LC, indicating the effect of 6OHDA was localized to LC noradrenergic neurons.
Figure 3.

Unilateral LC 6OHDA reduces the number of TH-IR neurons, but not noradrenergic neurons in the lateral tegmental area as determined by TH and DBH ISH. A) Quantitation of TH-IR neurons in the LC of non-surgery animals and vehicle- and 6OHDA-treated animals with B) photomicrograph of TH-IR neurons in the LC of a non-surgery animal and vehicle- and 6OHDA-treated animal. Quantitation of TH and DBH mRNA labeling in the NTS (C) and A1/A2 (D). * Indicates significant difference from non-surgery animals. # Indicates significant difference from vehicle-treated animals.
1.3.4 Unilateral 6OHDA (10 μg/μl) does not result in compensatory changes in TH and DBH mRNA expression in surviving LC neurons
Administration of 6OHDA also significantly reduced the number of TH and DBH mRNA positive labeled neurons in the LC compared to vehicle-treated side and non-surgery animals (Figure 4). The percent loss of LC neurons observed with TH (~65%) and DBH mRNA (~60%) is similar to the percent loss observed with TH-IHC (Figure 3A). Interestingly, the number of DBH mRNA positive labeled neurons on the vehicle-treated side was significantly reduced compared to non-surgery animals (Figure 4).
Figure 4.

Unilateral LC 6OHDA reduces the number of positive labeled TH and DBH mRNA expressing neurons, but not expression/neuron. * Indicates significant difference from non-surgery animals. # Indicates significant difference from vehicle-treated animals.
Administration of 6OHDA and the loss of LC neurons did not affect the amount of TH and DBH mRNA expression/neuron compared to vehicle-treated side (Figure 4). However, the amount of TH mRNA expression/neuron on the 6OHDA-treated side was significantly reduced compared to non-surgery animals (Figure 4). In addition, DBH mRNA expression/neuron on the vehicle- and 6OHDA-treated sides was significantly reduced compared to non-surgery animals (Figure 4). These data indicate that surviving LC noradrenergic neurons on the 6OHDA-treated side do not demonstrate compensation for the loss of neurons.
1.3.5 Unilateral 6OHDA (10 μg/μl) reduces NE concentration in specific forebrain regions
Administration of 6OHDA unilaterally significantly reduced catecholamine concentrations in specific forebrain regions (Table 1). NE tissue concentration on the 6OHDA-treated side was significantly reduced compared to the vehicle-treated side and non-surgery animal in the FC, HP, Amy and SN/VTA (Figure 5A upper panel). DHPG, the metabolite of NE, was significantly reduced on the 6OHDA-treated side compared to vehicle-treated side and non-surgery animals in the FC, Sep/BNST, HP and Amy (Figure 5A bottom panel), closely reflecting NE concentration. Interestingly, NE and DHPG tissue concentration on the vehicle-treated side were also significantly reduced compared to non-surgery animals in the FC and Amy (Figure 5A).
Table 1.
Catecholamine levels expressed as ng per catecholamine/mg protein in specific brain regions of non-surgery (NS) and on the vehicle- and 6OHDA-treated side of animals.
| Region | Group | DHPG | NE | DOPA | DA | DOPAC |
|---|---|---|---|---|---|---|
|
| ||||||
| FC | ||||||
| NS | 0.35 ± 0.01 | 6.9 ± 0.2 | 0.04 ± 0.003 | 4.8 ± 1.6 | 1.7 ± 0.3 | |
| Vehicle | 0.26 ± 0.01# | 5.4 ± 0.4# | 0.04 ± 0.005 | 5.8 ± 4.4 | 1.4 ± 0.6 | |
| 6OHDA | 0.12 ± 0.02*# | 2.1 ± 0.4*# | 0.04 ± 0.008 | 5.9 ± 3.8 | 1.5 ± 0.6 | |
|
| ||||||
| BNST/Sep | ||||||
| NS | 0.41 ± 0.04 | 9.1 ± 0.9 | 0.13 ± 0.007 | 76.7 ± 8.0 | 13.7 ± 1.2 | |
| Vehicle | 0.40 ± 0.03 | 10.7 ± 1.7 | 0.14 ± 0.008 | 45.5 ± 8.1# | 9.3 ± 1.4 | |
| 6OHDA | 0.25 ± 0.04*# | 6.6 ± 1.5 | 0.12 ± 0.008 | 75.9 ± 6.6 | 12.1 ± 1.2 | |
|
| ||||||
| HP | ||||||
| NS | 0.32 ± 0.04 | 6.5 ± 0.4 | 0.04 ± 0.009 | 0.40 ± 0.11 | 0.35 ± 0.09 | |
| Vehicle | 0.27 ± 0.03 | 5.4 ± 0.4 | 0.04 ± 0.004 | 0.25 ± 0.03 | 0.20 ± 0.03 | |
| 6OHDA | 0.12 ± 0.3*# | 2.3 ± 0.5*# | 0.03 ± 0.006 | 0.18 ± 0.04 | 0.19 ± 0.02 | |
|
| ||||||
| Str | ||||||
| NS | 0.28 ± 0.04 | 5.3 ± 1.1 | 0.11 ± 0.02 | 83.2 ± 16.7 | 11.3 ± 2.0 | |
| Vehicle | 0.21 ± 0.04 | 4.3 ± 0.9 | 0.12 ± 0.03 | 61.7 ± 26.1 | 8.6 ± 3.5 | |
| 6OHDA | 0.25 ± 0.08 | 3.7 ± 1.6 | 0.15 ± 0.03 | 54.1 ± 25.9 | 8.2 ± 3.6 | |
|
| ||||||
| Amy | ||||||
| NS | 0.32 ± 0.02 | 6.1 ± 0.4 | 0.03 ± 0.01 | 2.9 ± 0.2 | 0.94 ± 0.07 | |
| Vehicle | 0.23 ± 0.002# | 3.9 ± 0.5# | 0.07 ± 0.03 | 3.5 ± 1.0 | 0.81 ± 0.14 | |
| 6OHDA | 0.08 ± 0.004*# | 1.6 ± 0.5*# | 0.04 ± 0.01 | 3.1 ± 0.2 | 0.74 ± 0.04 | |
|
| ||||||
| SN/VTA | ||||||
| NS | 0.83 ± 0.22 | 14.5 ± 0.7 | 0.27 ± 0.04 | 15.4 ± 2.3 | 4.2 ± 0.27 | |
| Vehicle | 0.42 ± 0.06 | 12.2 ± 1.1 | 0.27 ± 0.11 | 6.1 ± 1.1# | 2.4 ± 0.19# | |
| 6OHDA | 0.40 ± 0.12 | 8.2 ± 1.3*# | 0.22 ± 0.08 | 7.0 ± 1.7# | 2.7 ± 0.19# | |
Indicates significant difference from vehicle-treated side.
Indicates significant difference from non-surgery animals.
Figure 5.

Unilateral LC 6OHDA reduces NE and DHPG in specific forebrain regions. Quantitation (ng/mg protein) of NE concentration (A upper panel) and DHPG concentration (A lower panel) in FC, Sep/BNST, HP, Striatum, Amy and SN/VTA of vehicle- and 6OHDA-treated animals and non-surgery animals. B) Correlation of NE concentration in FC, HP, Amy and SN/VTA to the number of TH-IR neurons in the LC of vehicle- and 6OHDA-treated animals and non-surgery animals. * Indicates significant difference from non-surgery animals. # Indicates significant difference from vehicle-treated animals.
NE tissue concentration in the FC, HP, Amy and SN/VTA of vehicle- and 6OHDA-treated animals that exhibited neuronal loss and non-surgery animals correlate with the number of TH-IR neurons (FC: r2= 0.57; p=0.0001; HP: r2= 0.43; p=0.0001; Amy: r2= 0.55; p<0.0001; SN/VTA: r2= 0.32; p=0.01) (Figure 5B). Therefore, a loss of LC neurons will significantly affect NE concentration in many forebrain regions, including the contralateral side.
DOPA tissue concentration varied in each region and administration of 6OHDA did not significantly affect its concentration in any forebrain region (Table 1). DA and DOPAC tissue concentrations, which represent dopaminergic innervation, in each region also exhibit a large amount of variability but the only region that demonstrated a significant reduction was the SN/VTA (Table 1). In the SN/VTA, DA and DOPAC tissue concentration in the 6OHDA-treated side are not significantly different from vehicle-treated side, but both 6OHDA- and vehicle-treated sides are significantly reduced compared to non-surgery controls (Table 1).
1.3.6 Unilateral 6OHDA (10 μg/μl) reduces NET binding sites, α2-AR binding sites and α1-AR binding sites in specific forebrain regions
Measurement of NET binding sites in forebrain regions provides another means of assessing noradrenergic terminals because NET binding sites are localized exclusively to adrenergic terminals (Iversen et al., 2009). 6OHDA significantly reduced NET binding sites compared to the vehicle-treated side in the FC and HP (~75% reduction) (Fig 6A autoradiographs, B). Interestingly, NET binding sites on the vehicle-treated side in the FC and HP also demonstrated a significant reduction compared to non-surgery animals (~ 20% reduction for FC and ~30% reduction for HP, respectively) (Figure 6A, B). NET binding sites in the FC and HP of vehicle- and 6OHDA-treated animals that exhibited neuronal loss and non-surgery animals correlated strongly with the number of TH-IR neurons (PFC: r2= 0.77; p<0.0001 (Figure 6C); HP: r2= 0.64; p<0.0001). In addition, NET binding sites in the PVTN and Gen on the 6OHDA-treated side were not statistically different to vehicle-treated side, but both sides have significantly reduced NET binding sites compared to non-surgery animals (~40% reduced for PVTN and ~50% reduced for Gen, respectively) (Figure 6B). NET binding sites in the PVNT and Gen of vehicle- and 6OHDA-treated animals that exhibited neuronal loss and non-surgery animals also correlated with the number of TH-IR labeled neurons (PVNT: r2= 0.38; p=0.0015 (Figure 6C); Gen: r2= 0.66; p=0.014). The Sep, BNST, SN and VTA did not demonstrate a significant reduction in NET binding sites for either the vehicle- or 6OHDA-treated side compared to non-surgery animals. However, NET binding sites in the BNST and SN of vehicle- and 6OHDA-treated animals that exhibited neuronal loss and non-surgery animals correlated with the number of TH-IR neurons (BNST: r2= 0.20; p=0.031 (Figure 6C); SN: r2= 0.25; p=0.025). These data indicate the number of LC noradrenergic neurons influences the amount of NET binding sites in many forebrain regions where NET binding sites are localized, including the contralateral side.
Figure 6.

Unilateral LC 6OHDA reduces NET binding sites in specific forebrain regions. A) Photomicrographic images of NET binding at the level of the FC (top images) and HP (bottom images) in non-surgery animals (left panels) and vehicle/6OHDA-treated animal (right panels). Horizontal line on vehicle/6OHDA image denotes midline. B) Quantitation of NET binding sites in forebrain regions of vehicle/6OHDA-treated animals as a percentage of non-surgery animals. C) Correlation of NET binding sites in PFC, PVNT and BNST to the number of TH-IR neurons in the LC of vehicle- and 6OHDA-treated animals and non-surgery animals.
* Indicates significant difference from non-surgery animals. # Indicates significant difference from vehicle-treated animals. FC, frontal cortex; Sep, septum; BNST, bed nucleus stria terminalis; PVNT, paraventricular thalamic nucleus; HP, hippocampus; SN, substantia nigra; VTA, ventral tegmental area; Gen, geniculate.
To assess the consequence of LC loss to noradrenergic receptors in forebrain regions, α1- and α2-AR binding sites were also measured. α2-AR binding sites are localized pre-synaptically on noradrenergic terminals as autoreceptors and post-synaptically on dendrites and axon terminals of other transmitter systems (Aghajanian and VanderMaelen, 1982; L’Heureux et al., 1986; Iversen et al., 2009). Therefore, α2-AR binding sites are observed in many brain regions, although it is not always clear if they are pre- or post-synaptic receptors. Despite the loss of LC noradrenergic neurons due to 6OHDA (Figure 3A and Figure 4) and a reduction in NET binding sites in specific forebrain terminal regions (Figure 6), 6OHDA did not affect α2-AR binding sites in any brain region compared to vehicle-injected side (Figure 7). However, α2-AR binding sites in the HP and SN are equally reduced in both the vehicle- and 6OHDA-treated side compared to non-surgery animals (HP ~10% reduced; SN ~28% reduced) (Figure 7A). It is interesting to note α2-AR binding sites in the FC were not altered in either the vehicle- or 6OHDA-treated side compared to non-surgery animal even though there was a significant loss of NET binding sites on the 6OHDA-treated side (Figure 6).
Figure 7.

Unilateral LC 6OHDA does not alter α2-AR binding sites in forebrain regions compared to vehicle-treated side. A) Quantitation of α2-AR binding sites in forebrain regions of non-surgery animals and vehicle/6OHDA-treated animals as a percentage of non-surgery animals. B) Photomicrographic images of α2-AR binding at the level of the HP in non-surgery animal (left panel) and vehicle/6OHDA-treated animal (right panel). Horizontal line on vehicle/6OHDA image denotes midline. * Indicates significant difference from non-surgery animals. Sep, septum; CTX, cortex; FC, frontal cortex; BNST, bed nucleus of the stria terminalis; DTN, dorsal thalamic nucleus; Str, striatum; Hypo, hypothalamus; HP, hippocampus; Amy, amygdala, SN, substantia nigra; VTA, ventral tegmental nucleus; Gen, geniculate.
α1-AR binding sites were measured to assess noradrenergic receptors localized predominately post-synaptically (Iversen et al., 2009). As with α2-AR binding sites, α1-AR binding sites in all regions examined were not affected by the administration of 6OHDA (Figure 8) compared to vehicle-treated side. However, in the Amy and SN, α1-AR binding sites are equally reduced to a modest but significant degree in both the vehicle and 6OHDA-treated side compared to non-surgery animals (Amy ~14% reduced; SN ~23% reduced) (Figure 8A).
Figure 8.

Unilateral LC 6OHDA does not affect α1-AR binding sites in forebrain regions compared to vehicle-treated side. A) Quantitation of α1-AR binding sites in forebrain regions of vehicle/6OHDA-treated animals as a percentage of non-surgery animals. B) Photomicrographic images of α1-AR binding at the level of the HP in non-surgery animal (left panel) and vehicle/6OHDA-treated animal (right panel). Horizontal line on vehicle/6OHDA image denotes midline. * Indicates significant difference from non-surgery animals.
1.4 Discussion
Administration of 6OHDA directly into the LC unilaterally resulted in a selective reduction in LC noradrenergic neurons; noradrenergic neurons in the lateral tegmental region and dopaminergic neurons in the SN and VTA were unaffected, indicating the specificity of direct administration of 6OHDA to LC noradrenergic neurons. 6OHDA 10 μg/μl resulted in 50–60% loss of LC noradrenergic neurons as determined by TH-IR, TH mRNA and DBH mRNA compared to vehicle-treated side and non-surgery animals. The degree of 6OHDA-induced loss determined by these three methods was similar. LC neuron loss induced by 6OHDA was more extensive with electrophysiological assessment of functional viable LC noradrenergic neurons (~90%) then with TH-IR, TH-mRNA and DBH mRNA. A possible reason for the difference in assessment of LC neuron loss by 6OHDA with electrophysiology versus IHC/ISH is that the electrophysiological measurements were made between 4–7 weeks after 6OHDA, while IR/ISH were made at exactly 3 weeks after stereotaxic administration of 6OHDA; more neurons may have been lost on the 6OHDA-treated side with more time. A more likely explanation is that electrophysiology assessment is detecting the electrical properties of neurons while IR/ISH is looking at what markers are still being expressed in the surviving LC neurons. These data indicate that 6OHDA treatment affects the functional output of LC neurons to a greater degree than indicated by measurement of noradrenergic markers alone.
The loss of LC noradrenergic neurons does not result in a compensatory response of the surviving LC neurons. TH and DBH mRNA expression/neuron is even slightly reduced on the 6OHDA-treated side compared to non-surgery animals. Degeneration of LC noradrenergic neurons is a major neuropathological characteristic of AD and PD (Bondareff et al., 1981; Tomlinson et al., 1981; Marcyniuk et al., 1986; Cash et al., 1987; Chan-Palay and Asan, 1989, 1991; German et al., 1992; Patt and Gerhard, 1993; Bertrand et al., 1997; Szot et al., 2006; Zarow et al., 2003; McMillan et al., 2011). Prior work in our laboratory has shown the loss of LC neurons in these two degenerative disorders results in different response of the surviving LC noradrenergic neurons (Szot et al., 2006; McMillan et al., 2011). In AD, the surviving LC neurons are compensating for the loss by increasing TH mRNA expression/neuron (Szot et al., 2006); while in PD, the surviving neurons exhibit levels of TH mRNA expression/neuron that are comparable to age-matched controls (McMillan et al., 2011). The lack of a compensatory response in TH mRNA expression/neuron in the surviving LC noradrenergic neurons following 6OHDA administration is similar to the response of surviving LC noradrenergic neurons in PD subjects, but not in AD subjects. In contrast, the lack of compensatory response in DBH mRNA expression/neuron in the surviving LC noradrenergic neurons following 6OHDA is in contrast to the response of surviving LC noradrenergic neurons in PD subjects where an increase was observed, but similar to what was observed in AD (McMillan et al., 2011). However, analysis of TH and DBH mRNA in surviving LC neurons in PD subjects should be made with caution. All PD subjects analyzed were on L-DOPA treatment to alleviate motor symptoms (McMillan et al., 2011). It is unclear the consequence of L-DOPA treatment on NE synthesizing enzyme mRNA expression. However, it is possible the lack of compensatory response of DBH mRNA expression/neuron may be the normal response to neuronal loss; L-DOPA treatment in PD subjects may be responsible for the increase in DBH mRNA expression.
The reduction in LC noradrenergic neurons induced by 6OHDA correlates with a significant loss in NE tissue concentration in the FC and HP, as well as the Amy and SN/VTA. Interestingly, NE tissue concentration on the vehicle-treated side was also significantly reduced compared to non-surgery animals in the FC and Amy. The significant reduction in NE concentration in the FC and HP support previous data that indicates these regions receive sole innervation from the LC (Mason and Fibiger, 1979; Moore and Bloom, 1979; Loughlin et al., 1986a, b). The Amy and SN/VTA have been shown to receive noradrenergic innervation from the LC (Swanson and Hartman, 1975); this data supports an innervation from the LC and indicates a loss of LC neurons could affect the function of these regions. There have been several studies to indicate the ability of LC noradrenergic neurons to modulate midbrain dopaminergic neurons in the SN and VTA, especially when LC noradrenergic neurons are reduced (Antelman and Gaggiula, 1977; Rommelfanger et al., 2007; Guiard et al., 2008; Wang et al., 2010). Noradrenergic innervation to the Amy is involved in normal and pathological fear and anxiety (Aston-Jones et al., 1999; McGaugh, 2004), but this response is attributed to an enhanced noradrenergic activity (Southwick et al., 1999; Strawn et al., 2008). The consequence of LC neuronal loss and reduced NE on the function of Amy neurons is unclear.
The correlation of NE concentration in forebrain regions to the number of LC noradrenergic neurons is also similar to what is observed in PD, but not AD. NE tissue concentration in the cortex, hypothalamus and cerebellum is reduced and the loss is proportional to the loss of neurons in the LC (Gasper and Grey, 1984; Gasper et al., 1991; Kish et al., 1984; Shannak et al., 1994). TH- and DBH-IR are also reduced in the cortex, again comparable to the amount of neuronal loss in the LC (Gasper et al., 1991). While concentration of NE in terminal regions such as the neocortex and HP in postmortem AD subjects is reduced, the reduction in NE tissue concentration does not correspond to the degree of neuronal loss in the LC (Adolfsson et al., 1979; Mann et al., 1981; Tomlinson et al., 1981; Palmer et al., 1987; Hoogendijk et al., 1999). Similar results of a lack of correlation to LC neuronal loss in AD subjects were observed when the synthesizing enzymes for NE were measured in forebrain regions (Cross et al., 1981; Perry et al., 1981; Palmer et al., 1987; Russo-Neustadt et al., 1998).
The reduction in LC neurons induced by 6OHDA also reduces the number of NET binding sites in many forebrain regions. There is a strong correlation of NET binding sites to the number of LC noradrenergic neurons in the FC and HP, again supporting the data that these regions receive sole innervation from the LC (Mason and Fibiger, 1979; Moore and Bloom, 1979; Loughlin et al., 1986a, b). NET binding sites in the PVNT and Gen also correlate to the number of TH-IR neurons, though not as strongly as FC and HP. In addition, NET binding sites in the BNST and SN also correlate to the number of TH-IR neurons in the LC even though these regions do not demonstrate a significant loss of NET binding sites in either the vehicle- or 6OHDA-treated compared to non-surgery animals. The reduction in NET binding sites is probably not a consequence of reduced NE tissue concentration in these regions because NET binding sites in DBH knockout mice, which do not synthesize NE, have NET levels similar to control DBH heterozygote mouse (Weinshenker et al., 2002). Only when DA and NE levels are reduced, as observed in TH knockout mice, or when NET inhibitors are administered are NET binding sites reduced (Weinshenker et al., 2002). DA levels in the FC, HP and Amy are not affected by the administration of 6OHDA. Therefore, the reduced NET binding sites in these forebrain regions reflect a loss of LC terminals. These data indicate the loss of LC noradrenergic neurons has an affect on the amount of NE and the ability of terminal regions to reuptake released NE in many forebrain regions.
The 6OHDA-induced changes observed in forebrain noradrenergic markers are different from the changes observed in noradrenergic markers following DSP4 administration (Szot et al., 2010). DSP4 resulted in a temporary reduction in NE tissue concentration in the FC and HP (as observed with 6OHDA) as well as in the Sep/BNST (not observed with 6OHDA) (Ross, 1976; Jonsson et al., 1981; Grzanna et al., 1989; Theron et al., 1993; Wolfman et al., 1994; Hughes and Stanford, 1998; Kask et al., 1997; Harro et al., 1999; Szot et al., 2010). NET binding sites were temporarily reduced in the FC and HP with DSP4 and 6OHDA, but DSP4 also reduced NET binding sites in the septum (Cheetham et al., 1996; Szot et al., 2010), which was not observed with 6OHDA. The biggest difference between 6OHDA and DSP4 effects is on α2-AR binding sites. DSP4 resulted in an increase in α2-AR binding sites in many forebrain regions (Wolfman et al., 1994; Harro et al., 1999b; Szot et al., 2010), while 6OHDA induced LC neuronal loss did not directly affect α2-AR binding sites compared to vehicle-treated side; however, there was a reduction in α2-AR binding sites in the Amy and SN in both the vehicle- and 6OHDA-treated side compared to non-surgery animals. The effects of 6OHDA on the α2-AR are also in contrast to the changes in α2-AR observed in the DBH knockout mouse where the complete loss of NE results in an increase in α2-AR binding sites in the HP (Sanders et al., 2006). A lack of change in α1-AR binding sites observed in this study is in agreement with other animal models of reduced NE concentration (Sanders et al., 2006; Szot et al., 2010)
Interestingly, α2-binding sites on the 6OHDA-treated side are not significantly altered from the vehicle-treated side in any brain region despite the loss of NET binding sites and NE tissue concentration. A proportion of α2-AR binding sites at terminal regions is considered to be the autoreceptor of the noradrenergic nervous system and is localized to presynaptic noradrenergic terminals. The lack of a change in α2-AR binding sites on the 6OHDA-treated side compared to the vehicle-treated side may be a reflection of compensation of the autoreceptor or postsynaptic receptors at this 3-week time point. Earlier time points may demonstrate a reduction in α2-AR binding sites in specific forebrain regions.
NE and DHPG concentration and binding data generated in this study suggests LC innervation to many forebrain regions is bilateral. Early data has indicated that LC innervation to forebrain regions is mainly unilateral, but that some species exhibit contralateral innervation (Lindvall and Bjorklund, 1974; McBride and Sutin, 1976; Kitt and Brauth, 1986; Inversen et al., 2009). In comparing noradrenergic markers on the vehicle-treated side to animals with non-surgery animals, it appears the mouse is one of these species that exhibits such contralateral innervation. The number of TH-IR and TH mRNA positively labeled neurons on the vehicle-treated side was not different from non-surgery animals, so the direct application of a neurotoxin unilaterally into the LC does not affect the number of LC noradrenergic neurons contralaterally.
1.5 Summary
Administration of 6OHDA unilaterally results in a significant loss of LC noradrenergic neurons 3 weeks later; ventral tegmental regions and midbrain dopaminergic neurons are not affected. The surviving LC noradrenergic neurons do not exhibit compensatory changes in TH and DBH mRNA, synthesizing enzymes for NE. The loss of LC noradrenergic neurons unilaterally is associated with a reduction in NE and NET binding sites on the 6OHDA-treated and vehicle-treated side in many forebrain regions, indicating the loss of LC neurons can reduce noradrenergic innervation to many forebrain regions including the contralateral side. In forebrain regions α1- and α2-AR binding sites are not directly affected on the 6OHDA –treated side. Many noradrenergic markers measured in this study indicate contralateral LC innervation indicating that a unilateral lesion can affect noradrenergic function in both forebrain hemispheres. This suggests that interpretation of results with a unilateral LC lesion cannot be attributed to alteration in noradrenergic markers unilaterally.
Highlights.
6-OHDA reduces LC neurons specifically; No compensation of surviving LC neurons; Reduced NET and NE concentration in specific forebrain regions; No change in adrenoreceptors; evidence of bi-lateral innervation from LC
Acknowledgments
This work was supported by the Department of Veterans Affairs Research and Development Services Northwest Network Mental Illness Research, Education, and Clinical Center (P.S., A.F., M.A.R.), Geriatric Research, Education, and Clinical Center (GRECC) (C.S., C.W.W.), Whitman College Abshire Research Award (S.F., L.K.) and University of Washington Department of Psychiatry and Behavioral Science (S.S.W.).
Abbreviations
- 6OHDA
6-hydroxydopamine
- AD
Alzheimer’s disease
- Alpha
α
- aCSF
artificial cerebrospinal fluid
- Amy
amygdala
- AR
adrenoreceptors
- CRF
cerebrospinal fluid
- DA
dopamine
- DBH
dopamine β-hydroxylase
- BNST
bed nucleus of the stria terminalis
- DHPG
3,4-dihydroxyphenylglycol
- DOPA
3,4-dihydroxyphenylalanine
- DOPAC
dihydroxyphenylacetic acid
- DSP4
N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine
- DTN
dorsal thalamic nucleus
- FC
frontal cortex
- Gen
geniculate
- HP
hippocampus
- ICV
intracerebroventricular
- ISH
in situ hybridization
- Hypo
hypothalamus
- IR
immunoreactivity
- LC
locus coeruleus
- PVTN
paraventricular thalamic nucleus
- NE
norepinephrine
- NET
norepinephrine transporter
- NTS
nucleus tractus solitaris
- PBS
phosphate buffer
- PD
Parkinson’s disease
- TH
tyrosine hydroxylase
- Thal
thalamus
- Sep
septum
- SN
substantia nigra
- Str
striatum
- VTA
ventral tegmental area
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
1.7 References
- Adolfsson R, Gottfries CG, Roos BE, Winblad B. Changes in the brain catecholamines in patients with dementia of Alzheimer’s type. Br J Psychiatry. 1979;135:216–223. doi: 10.1192/bjp.135.3.216. [DOI] [PubMed] [Google Scholar]
- Aghajanian GK, VanderMaelen CP. α2-Adrenoreceptor-mediated hyperpolarization of locus coeruleus neurons; intracellular studies in vivo. Science. 1982;215:1394–1396. doi: 10.1126/science.6278591. [DOI] [PubMed] [Google Scholar]
- Antelman SM, Caggiula AR. Norepinephrine-dopamine interaction and behavior. Science. 1977;195:646–653. doi: 10.1126/science.841304. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Rajkowski J, Cohen J. Role of locus coeruleus in attention and behavioral flexibility. Biol Pscyiatry. 1999;46:1309–1320. doi: 10.1016/s0006-3223(99)00140-7. [DOI] [PubMed] [Google Scholar]
- Bertrand E, Lechowicz W, Szpak GM, Dynecki J. Qualitative and quantitative analysis of locus coeruleus neurons in Parkinson’s disease. Folia Neuropathol. 1997;35:80–86. [PubMed] [Google Scholar]
- Biancardi V, Bicego K, Almeida MC, Gargaglioni LH. Locus coeruleus noradrenergic neurons and CO2 drive to breathing. Pflugers Arch-Eur J Physiol. 2008;455:1119–1128. doi: 10.1007/s00424-007-0338-8. [DOI] [PubMed] [Google Scholar]
- Bondareff W, Mountjoy CQ, Roth M. Selective loss of neurons of origin of adrenergic projection to cerebral cortex (nucleus locus coeruleus) in senile dementia. Lancet. 1981;1:783–784. doi: 10.1016/s0140-6736(81)92657-x. [DOI] [PubMed] [Google Scholar]
- Booze RM, Hall JA, Cress NM, Miller GD, Davis JN. DESP-4 treatment produces abnormal tyrosine hydroxylase immunoreactive fibers in rat hippocampus. Exp Neural. 1988;101:75–86. doi: 10.1016/0014-4886(88)90066-0. [DOI] [PubMed] [Google Scholar]
- Cash R, Dennis T, L’Heureux R, Raisman R, Javoy-Agid F, Scatton B. Parkinson’s disease and dementia: norepinephrine and dopamine in locus coeruleus. Neurology. 1987;37:42–46. doi: 10.1212/wnl.37.1.42. [DOI] [PubMed] [Google Scholar]
- Chan-Palay V, Asan E. Alterations in catecholamine neurons of the locus coeruleus in senile dementia of the Alzheimer’s type and in Parkinson’s disease with and without dementia and depression. J Comp Neurol. 1989;287:373–392. doi: 10.1002/cne.902870308. [DOI] [PubMed] [Google Scholar]
- Chan-Palay V, Asan E. Alterations in the locus coeruleus in dementias of Alzheimer’s and Parkinson’s disease. Prog Brain Res. 1991;88:625–630. doi: 10.1016/s0079-6123(08)63839-x. [DOI] [PubMed] [Google Scholar]
- Cheetham SC, Viggers JA, Butler SA, Prow MR, Heal DJ. [3H]Nisoxetine- a radioligand for noradrenaline reuptake sites: correlation with inhibition of [3H]noradrenaline uptake and effect of DSP-4 lesioning and antidepressant treatment. Neuropharmacology. 1996;35:63–70. doi: 10.1016/0028-3908(95)00134-4. [DOI] [PubMed] [Google Scholar]
- Coradazzi M, Gulino R, Garozzo S, Leanza G. Selective lesion of the developing central noradrenergic system: short- and long-term effects and reinnervation by noradrenergic-rich tissue grafts. J Neurochem. 2010;114:761–771. doi: 10.1111/j.1471-4159.2010.06800.x. [DOI] [PubMed] [Google Scholar]
- Cross AJ, Crow TJ, Perry EK, Perry RH, Blessed G, Tomlinson BE. Reduced dopamine-beta-hydroxylase activity in Alzheimer’s disease. BMJ (Clin Res Ed) 1981;282:93–94. doi: 10.1136/bmj.282.6258.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descarries L, Saucier G. Disappearance of the locus coeruleus in the rat after intraventricular 6-hydroxydopamine. Brain Res. 1972;37:310–316. doi: 10.1016/0006-8993(72)90676-2. [DOI] [PubMed] [Google Scholar]
- Eisenhofer G, Goldstein DS, Stull R, Kelser HR, Sunderland T, Murphy DL, Koplin IJ. Simultaneous liquid-chromatographic determination of 3,4-dihydroxyphenylglycol, catecholamines, and 3,4-dihydroxyphenylalanine in plasma, and their response to inhibition of monoamine oxidase. Clin Chem. 1986;32:2030–2033. [PubMed] [Google Scholar]
- Fitzgerald PJ. Is elevated norepinephrine an etiological factor in some cases of Alzheimer’s disease? Curr Alzheimer’s Res. 2010;7:506–516. doi: 10.2174/156720510792231775. [DOI] [PubMed] [Google Scholar]
- Gasper P, Duyckaerts C, Alvarez, Javoy-Agid F, Berger B. Alterations of dopaminergic and noradrenergic innervation in motor cortex in Parkinson’s disease. Ann Neurol. 1991;30:365–374. doi: 10.1002/ana.410300308. [DOI] [PubMed] [Google Scholar]
- Gasper P, Gray F. Parkinson’s disease: a neuropathological study of 32 cases. Acta Neuropathol. 1984;64:43–52. doi: 10.1007/BF00695605. [DOI] [PubMed] [Google Scholar]
- German DC, Manaye KF, White CL, Woodward DJ, McIntyre DD, Smith WK, Kalaria RN, Mann DM. Disease-specific patterns of locus coeruleus cell loss. Ann Neurol. 1992;32:667–676. doi: 10.1002/ana.410320510. [DOI] [PubMed] [Google Scholar]
- Grima B, Lamouroux A, Blanot F, Biguet NF, Mallet J. Complete coding sequence of rat tyrosine hydroxylase mRNA. Proc Natl Acad Sci USA. 1985;82:617–621. doi: 10.1073/pnas.82.2.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzanna R, Berger U, Fritschy J-M, Geffard M. Acute action of DSP-4 on central norepinephrine axons: biochemical and immunohistochemical evidence for differential effects. J Histochem Cytochem. 1989;37:1435–1442. doi: 10.1177/37.9.2768812. [DOI] [PubMed] [Google Scholar]
- Guiard BP, Mansari ME, Merali Z, Blier P. Functional interactions between dopamine, serotonin and norepinephrine neurons: an in-vivo electrophysiological study in rats with monoaminergic lesions. Int J Neuropsychopharm. 2008;11:625–639. doi: 10.1017/S1461145707008383. [DOI] [PubMed] [Google Scholar]
- Harik S. Locus ceruleus lesion by local 6-hydroxydopamine infusion causes marked and specific destruction of noradrenergic neurons, long-term depletion of norepinephrine and the enzymes that synthesize it, and enhanced dopaminergic mechanisms in the ipsilateral cerebral cortex. J Neurosci. 1984;4:699–707. doi: 10.1523/JNEUROSCI.04-03-00699.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogendijk WJ, Freenstra MG, Botterblom MH, Gilhuis J, Sommer IE, Kamphorst W, Eikelenboom P, Swaab DF. Increased activity of surviving locus coeruleus neurons in Alzheimer’s disease. Ann Neurol. 1999;45:82–91. doi: 10.1002/1531-8249(199901)45:1<82::aid-art14>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Hughes ZA, Stanford SC. A partial noradrenergic lesion induced by DSP-4 increases extracellular noradrenaline concentration in rat frontal cortex: a microdialysis study in vivo. Psychopharmacology. 1998;136:299–303. doi: 10.1007/s002130050569. [DOI] [PubMed] [Google Scholar]
- Iversen LL, Iversen SD, Bloom FE, Roth RH. Introduction to Neuropsychopharmacology. New York, NY: Oxford University Press; 2009. pp. 150–213. [Google Scholar]
- Jonsson G, Hallman H, Ponzio F, Ross S. DSP4 (N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine)- a useful denervation tool for central and peripheral noradrenaline neurons. Eur J Pharmacol. 1981;72:173–188. doi: 10.1016/0014-2999(81)90272-7. [DOI] [PubMed] [Google Scholar]
- Kask A, Harro J, Tuomainen P, Rago L, Mannisto PT. Overflow of noradrenaline and dopamine in frontal cortex after [N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine] (DSP-4) treatment: in vivo microdialysis study in anaesthetized rats. Naunyn Schmiedeberg’s Arch Pharmacol. 1997;355:267–272. doi: 10.1007/pl00004942. [DOI] [PubMed] [Google Scholar]
- Kish SJ, Shannak KS, Rajput AH, Gilbert JJ, Hornykiewicz O. Cerebellar norepinephrine in patients with Parkinson’s disease and control subjects. Arch Neurol. 1984;41:612–614. doi: 10.1001/archneur.1984.04210080020007. [DOI] [PubMed] [Google Scholar]
- Kitt CA, Brauth SE. Telencephalic projections from midbrain and isthmal cell groups in the pigeon. I. Locus coeruleus and subcoeruleus. J Comp Neurol. 1986;247:69–91. doi: 10.1002/cne.902470105. [DOI] [PubMed] [Google Scholar]
- L’Heureux R, Dennis T, Curet O, Scatton B. Measurement of endogenous noradrenaline release in the cerebral cortex in vivo by transcortical dialysis: effects of drugs affecting noradrenergic transmission. J Neurochem. 1986;46:1794–1801. doi: 10.1111/j.1471-4159.1986.tb08498.x. [DOI] [PubMed] [Google Scholar]
- Lindvall O, Bjorklund A. The organization of the ascending catecholamine neuron systems in the rat brain as revealed by the glyoxylic acid fluorescence method. Acta Physiol Scand Suppl. 1974;412:1–48. [PubMed] [Google Scholar]
- Loughlin SE, Foote SL, Bloom FE. Efferent projections of nucleus locus coeruleus: topographic organization of cells of origin demonstrated by three-dimensional reconstruction. Neuroscience. 1986a;18:291–306. doi: 10.1016/0306-4522(86)90155-7. [DOI] [PubMed] [Google Scholar]
- Loughlin SE, Foote SL, Grzanna R. Efferent projections of nucleus locus coeruleus noradrenaline neurons: morphologic subpopulations have different efferent’s targets. Neuroscience. 1986b;18:307–319. doi: 10.1016/0306-4522(86)90156-9. [DOI] [PubMed] [Google Scholar]
- Lyons WE, Fritschy JM, Grzanna R. The noradrenergic neurotoxin DSP-4 eliminates the coerulespinal projection but spares projections of the A5 and A7 groups to the ventral horn of the rat spinal cord. J Neurosci. 1989;9:1481–1489. doi: 10.1523/JNEUROSCI.09-05-01481.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann JJ, Stanley M, Neophytides A, deLeon MJ, Ferris SH, Gershon S. Central amine metabolism in Alzheimer’s disease: in vivo relationship to cognitive deficit. Neurobiol Aging. 1981;2:57–60. doi: 10.1016/0197-4580(81)90060-9. [DOI] [PubMed] [Google Scholar]
- Marcyniuk B, Mann DM, Yates PO. Loss of nerve cells from locus coeruleus in Alzheimer’s disease is topographically arranged. Neurosci Lett. 1986;64:247–252. doi: 10.1016/0304-3940(86)90336-8. [DOI] [PubMed] [Google Scholar]
- Mason ST, Fibiger HD. Regional topography within noradrenergic locus coeruleus as revealed by retrograde transport of horseradish peroxidase. J Comp Neurol. 1979;187:703–724. doi: 10.1002/cne.901870405. [DOI] [PubMed] [Google Scholar]
- Matsukawa M, Nakadate K, Ishihara I, Okado N. Synaptic loss following depletion of noradrenaline and/or serotonin in the rat visual cortex: a quantitative electron microscopic study. Neuroscience. 2003;122:627–635. doi: 10.1016/j.neuroscience.2003.08.047. [DOI] [PubMed] [Google Scholar]
- McBride RL, Sutin J. Projections of the locus coeruleus and adjacent pontine tegmentum in the cat. J Comp Neurol. 1976;165:265–84. doi: 10.1002/cne.901650302. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu Rev Neurosci. 2004;27:1–28. doi: 10.1146/annurev.neuro.27.070203.144157. [DOI] [PubMed] [Google Scholar]
- McMahon A, Geertman R, Sabban EL. Rat dopamine β-hydroxylase: molecular cloning and characterization of the cDNA and regulation of the mRNA by reserpine. J Neurosci Res. 1990;25:395–404. doi: 10.1002/jnr.490250317. [DOI] [PubMed] [Google Scholar]
- McMillan PJ, White SS, Franklin A, Greenup JL, Leverenz JB, Raskind MA, Szot P. Differential response of the central noradrenergic nervous system to the loss of locus coerlues neurons in Parkinson’s disease and Alzheimer’s disease. Brain Res. 2011;1373:240–252. doi: 10.1016/j.brainres.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RY, Bloom FE. Central catecholamine neuron system: anatomy and physiology of the norepinephrine and epinephrine systems. Ann Rev Neurosci. 1979;2:113–168. doi: 10.1146/annurev.ne.02.030179.000553. [DOI] [PubMed] [Google Scholar]
- Olson VG, Rockett HR, Reh R, Redila VA, Tran PM, Venkov HA, DeFino MC, Hague C, Peskind ER, Szot P, Raskind MA. The role of norepinephrine in differential response to stress in an animal model of posttraumatic stress disorder. Biol Psychiatry. 2011;70:441–448. doi: 10.1016/j.biopsych.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AM, Wilcock GK, Esiri MM, Francis PT, Bowen DM. Monoaminergic innervation of the frontal and temporal lobes in Alzheimer’s disease. Brain Res. 1987;401:231–238. doi: 10.1016/0006-8993(87)91408-9. [DOI] [PubMed] [Google Scholar]
- Patt S, Gerhard L. A Golgi study of human locus coeruleus in normal brains and in Parkinson’s disease. Neurpoathol Appl Neurobiol. 1993;19:519–523. doi: 10.1111/j.1365-2990.1993.tb00480.x. [DOI] [PubMed] [Google Scholar]
- Perry EK, Blessed G, Tomlinson BE, Perry RH, Crow TJ, Cross AJ, Dockray GJ, Dimaline R, Arregui A. Neurochemical activities in human temporal lobe related to aging and Alzheimer’s-type changes. Neurobiol Aging. 1981;2:251–256. doi: 10.1016/0197-4580(81)90032-4. [DOI] [PubMed] [Google Scholar]
- Ressler KJ, Nemeroff CB. Role of serotonergic and noradrenergic systems in the pathophysiology of depression and anxiety disorders. Depression Anxiety. 2000;12(Suppl 1):2–19. doi: 10.1002/1520-6394(2000)12:1+<2::AID-DA2>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Robertson GB, Fluharty SJ, Zigmond MJ, Sciabassi RJ, Berger TW. Recovery of hippocampal dentate granule cell responsiveness to entorhinal cortical input following norepinephrine depletion. Brain Res. 1993;614:21–28. doi: 10.1016/0006-8993(93)91013-i. [DOI] [PubMed] [Google Scholar]
- Rommelfanger KS, Edwards GL, Freeman KG, Liles LC, Miller GW, Weinshenker D. Norepinephrine loss produces more profound motor deficits than MPTP treatment in mice. Proc Natl Acad Sci. 2007;104:1530–1540. doi: 10.1073/pnas.0702753104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross SB. Long-term effects of N-2-chlorethyl-N-ethyl-2-bromobenzylamine hydrochloride on noradrenergic neurons in the rat brain and heart. Br J Pharmacol. 1976;58:521–527. doi: 10.1111/j.1476-5381.1976.tb08619.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo-Neustadt A, Zomorodian TJ, Cotman CW. Preserved cerebellar tyrosine hydroxylase-immunoreactive neuronal fibers in a behaviorally aggressive subgroup of Alzheimer’s disease patients. Neuroscience. 1998;87:55–61. doi: 10.1016/s0306-4522(98)00134-1. [DOI] [PubMed] [Google Scholar]
- Sanders JD, Szot P, Weinshenker D, Happe HK, Bylund DB, Murrin LC. Analysis of brain adrenergic receptors in dopamine-β-hydroxylase knockout mice. Brain Res. 2006;1109:45–53. doi: 10.1016/j.brainres.2006.06.033. [DOI] [PubMed] [Google Scholar]
- Shannak K, Rajput A, Rozdilsky B, Kish S, Gilbert J, Hornykiewicz O. Noradrenaline, dopamine and serotonin levels and metabolism in the human hypothalamus: observations in Parkinson’s disease and normal subjects. Brain Res. 1994;639:33–41. doi: 10.1016/0006-8993(94)91761-2. [DOI] [PubMed] [Google Scholar]
- Southwick SM, Bremner JD, Rasmusson A, Morgan CA, III, Arnsten A, Charney DS. Role of norepinephrine in the pathophysiology and treatment of posttraumatic stress disorder. Biol Psychiatry. 1999;46:1192–1204. doi: 10.1016/s0006-3223(99)00219-x. [DOI] [PubMed] [Google Scholar]
- Strawn JR, Geracioti TD., Jr Noradrenergic dysfunction and the psychopharmacology of posttraumatic stress disorder. Depress Anxiety. 2008;25:260–271. doi: 10.1002/da.20292. [DOI] [PubMed] [Google Scholar]
- Swanson LW, Hartman BK. The central adrenergic system. An immunofluorescence study of the location of cell bodies and their efferent connections in the rat utilizing dopamine β-hydroxylase as a marker. J Comp Neurol. 1975;163:467–505. doi: 10.1002/cne.901630406. [DOI] [PubMed] [Google Scholar]
- Szot P, Miguelez C, White SS, Franklin A, Sikkema C, Wilkinson CW, Ugedo L, Raskind MA. A comprehensive analysis of the effect of DSP4 on the locus coeruleus noradrenergic system in the rat. Neuroscience. 2010;166:279–291. doi: 10.1016/j.neuroscience.2009.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szot P, Weinshenker D, White SS, Robbins CA, Rust NC, Schwartzkroin PA, Palmiter RD. Norepinephrine-deficient mice have increased susceptibility to seizure-inducing stimuli. J Neurosci. 1999;19:10985–10992. doi: 10.1523/JNEUROSCI.19-24-10985.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szot P, White SS, Greenup JL, Leverenz JB, Peskind ER, Raskind MA. Compensatory changes in the noradrenergic nervous system in the locus coeruleus and hippocampus of postmortem subjects with Alzheimer’s disease and dementia with Lewy bodies. J Neurosci. 2006;26:467–478. doi: 10.1523/JNEUROSCI.4265-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szot P, White SS, Veith RC. Effect of pentylenetetrazol on the expression of tyrosine hydroxylase mRNA and norepinephrine and dopamine transporter mRNA. Mol Brain Res. 1997;44:46–54. doi: 10.1016/s0169-328x(96)00217-3. [DOI] [PubMed] [Google Scholar]
- Theron CN, de Villiers AS, Taljaard JJ. Effects of DSP-4 on monoamine and monoamine metabolites levels and on beta adrenoreceptor binding kinetics in rat brain at different times after administration. Neurochem Res. 1993;18:1321–1327. doi: 10.1007/BF00975054. [DOI] [PubMed] [Google Scholar]
- Tomlinson BE, Irving D, Blessed G. Cell loss in the locus coeruleus in senile dementia of the Alzheimer’s type. J Neurol Sci. 1981;49:419–428. doi: 10.1016/0022-510x(81)90031-9. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhang QJ, Liu J, Ali U, Gui ZH, Hui YP, Chen L, Wu ZH, Li Q. Noradrenergic lesion of the locus coeruleus increases apomorphine-induced circling behavior and the firing activity of substantia nigra pars reticulate neurons in a rat model of Parkinson’s disease. Brain Res. 2010;1310:189–199. doi: 10.1016/j.brainres.2009.10.070. [DOI] [PubMed] [Google Scholar]
- Weinshenker D, Szot P. The role of catecholamines in seizure susceptibility: new results using genetically engineered mice. Pharmacol Therap. 2002;94:213–233. doi: 10.1016/s0163-7258(02)00218-8. [DOI] [PubMed] [Google Scholar]
- Weinshenker D, White SS, Javors MA, Palmiter RD, Szot P. Regulation of norepinephrine transporter abundance by catecholamines and desipramine in vivo. Brain Res. 2002;946:239–246. doi: 10.1016/s0006-8993(02)02889-5. [DOI] [PubMed] [Google Scholar]
- Wolfman C, Abo V, Calvo D, Medina J, Dajas F, Silveiro R. Recovery of central noradrenergic neurons one year after the administration of the neurotoxin DSP4. Neurochem Int. 1994;25:395–400. doi: 10.1016/0197-0186(94)90147-3. [DOI] [PubMed] [Google Scholar]
- Zarow C, Lyness SA, Mortimer JA, Chui HC. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson disease. Arch Neurol. 2003;60:337–341. doi: 10.1001/archneur.60.3.337. [DOI] [PubMed] [Google Scholar]