

Abstract
This paper describes the realization of digital loop-mediated DNA amplification (dLAMP) in a sample self-digitization (SD) chip. Digital DNA amplification has become an attractive technique to quantify absolute concentrations of DNA in a sample. While digital polymerase chain reaction is still the most widespread implementation, its use in resource—limited settings is impeded by the need for thermal cycling and robust temperature control. In such situations, isothermal protocols that can amplify DNA or RNA without thermal cycling are of great interest. Here, we showed the successful amplification of single DNA molecules in a stationary droplet array using isothermal digital loop-mediated DNA amplification. Unlike most (if not all) existing methods for sample discretization, our design allows for automated, loss-less digitization of sample volumes on-chip. We demonstrated accurate quantification of relative and absolute DNA concentrations with sample volumes of less than 2 μl. We assessed the homogeneity of droplet size during sample self-digitization in our device, and verified that the size variation was small enough such that straightforward counting of LAMP-active droplets sufficed for data analysis. We anticipate that the simplicity and robustness of our SD chip make it attractive as an inexpensive and easy-to-operate device for DNA amplification, for example in point-of-care settings.
Introduction
The amplification of genetic material is often the first step in nucleic acid analysis. The most widely used method to amplify DNA or RNA is polymerase chain reaction1 (PCR) where the sample is usually cycled between two or three temperatures around 60 °C and 95 °C. Thermal cycling, and the need for robust temperature control, however, impedes the further penetration and use of this technique in resource-limited settings. In an effort to simplify thermal cycling, various isothermal nucleic acid amplification methods, such as rolling circle amplification2 (RCA), multiple strand displacement3 (MDA), nucleic acid sequence based amplification4 (NASBA), recombinant polymerase amplification5 (RPA), helicase dependent amplification6 (HDA), or loop-mediated amplification7 (LAMP) have been developed. These reactions proceed at a fixed temperature, which reduces instrument complexity and lowers energy consumption, making them more suitable for point-of-care diagnostics and home-medicine applications.
LAMP is a particularly promising method, because the simultaneous use of 4 to 6 primers makes it highly specific for its target in the presence of non-target DNA8. LAMP is fast; amplification in test tubes requires less than 1 hour and its relative broad temperature working range (60–65 °C) obviates the demand for highly stable heating devices. Owing to these advantages, various LAMP assays have been developed for the diagnosis of diseases relevant to resource limited settings9–12.
Regardless of the amplification reaction used, it is often important to accurately quantify DNA or RNA copy numbers within a sample. In many therapies, the dose of a therapeutic drug is adjusted to the detected level of viral loads in a patient sample. In rare event diagnostics, forensics, and gene-expression analysis, real-time PCR13 is a sensitive technique providing a measure of initial template concentrations. These bulk reactions, however, are susceptible to errors due to primer dimerization and allelic preference in the amplification process that can impede accurate quantification. To this end, digital or limiting dilution DNA amplification has been developed14, 15, which can quantify the absolute number of template copies in the sample more accurately16. Based on Poisson statistics, the total sample can be diluted and distributed into many small compartments such that on average each compartment contains less than one template molecule. DNA amplification is carried out in all sub-volumes and the number of compartments that show amplification equals the number of initial templates in the sample.
To cover a large dynamic range, the number of compartments has to be large, which results in prohibitively costly reagent consumption (several ml in macroscopic systems). Miniaturization of the reaction in microfluidic devices offers the advantages of reduced sample volumes (few μl), increased sensitivity, and control over liquid handling in a small, portable device. Furthermore, DNA amplification can conveniently be combined with downstream analysis of product on chip.
Various methods have been developed to generate such microreactors and perform digital PCR or isothermal DNA amplification on chip. Examples include continuous flow water-in-oil (w/o) droplets17–19, static microchamber arrays such as the SlipChip20, 21, the OpenArray22 or a multiwell array23, or microvalve-based arrays of reaction chambers24–26. MDA has been reported in localized compartments within a gel matrix27. While each of these platforms offers certain advantages, most of them still require additional mechanical action or complex (and expensive) microfabrication to achieve sample digitization. Many of these platforms further require an excess of sample volume for sample digitization, which is unfavourable, if only small sample volumes are available, e.g. a drop of blood from a needle prick. In resource-limited settings, there is still need for the development of simple, inexpensive and sample-conserving devices that provide self-consistent nanoliter compartments with minimal complexity of chip operation.
To this end, we recently developed an inexpensive and simple alternative for generating stationary water-in-oil nanoliter reactors using a device we called sample self-digitization (SD) chip. Droplet formation is based on an inherent fluidic phenomenon, rather than hydraulic valving or mechanical action28, which greatly simplifies chip operation. Due to the interplay between fluidic forces and interfacial tension, an incoming aqueous fluid spontaneously divides itself into an array of chambers that have been primed with an immiscible fluid (self-digitization). Chip loading can be performed with manual or automated syringe pumps or external air pressure, and chip fabrication does not require glass etching or complex multilayer fabrication. Our platform is robust; once digitized, the chip can be carried over to various instruments without loss of digitization. Furthermore, our design allows us to self-digitize the total sample volume without any sample loss, provided that the number of compartments available on chip is sufficient. This is especially useful in cases of limited sample availability.
In this work, we used our new SD platform to demonstrate the first on-chip digital loop-mediated DNA amplification (dLAMP). LAMP has recently been performed in microfluidic devices29–31, but so far no on-chip digital format has been reported. We first quantify the reproducibility of droplet formation and show that droplet shrinkage during incubation is negligible in our setup. We address the effect of variation in initial droplet size on the experimental data, and show that in our system droplet-size variation is too small to influence the experimental outcome. Finally, we demonstrate the ability to detect relative changes in template concentration as well as absolute quantification of template numbers in our dLAMP SD chip. Because of its simplicity, we anticipate that our SD chip design will be a useful alternative to existing platforms for the quantification of nucleic acid copy numbers.
Results and discussion
Automated sample self-digitization with reproducible droplet formation
The digital LAMP chip was designed as a series of rectangular cavities to hold the droplets, which were positioned along a smaller rectangular channel used for sample delivery (Figure 1). Based on our previously reported design28, the chip layout was modified for improved droplet stability and retention, automation of the filling process, and operation at elevated temperatures. A reduction of the height ratio between main channel and side cavities to 1/3 reduced the chance of crosstalk between chambers. The depth of the side chamber was extended to 400 μm, which we found to improve droplet retention. Air pressure regulated flow was used to ensure reproducibility and robustness of the automated filling. Evaporation of the aqueous droplets at higher temperatures was minimised by various measures: a) The chambers were arranged in a dense array and embedded between only a thin top and bottom layer of PDMS. b) An additional sacrificial water channel was placed around the array. c) A self-adhesive film was added on top of the chip as a vapour barrier.
Fig. 1.
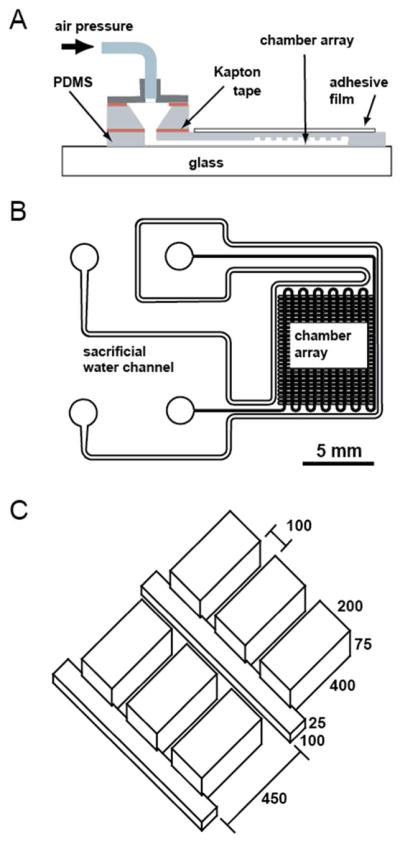
Design of the digital LAMP self-digitization chip. (A) Schematic diagram showing the individual components of a fully assembled chip. The drawing is not to scale. The microfluidic array was embedded in a thin piece of PDMS, which was covered by a sealant film on top and a PDMS coated cover slip on the bottom. Air pressure was delivered via a removable adapter, which was connected to an external pressure source. (B) Layout of the microfluidic network. A dense array of rectangular side chambers was connected to a thin main channel. The whole array was surrounded by a separate water reservoir to saturate the PDMS during incubation at 65 °C. The scale bar represents 5 mm. (C) Geometry of the side chamber array and main channel. All dimensions are in micrometers.
Prior to loading the aqueous phase, the chip was primed with the continuous phase (light mineral oil with 0.025% w/w SPAN-80 surfactant). Fresh LAMP solution was introduced into the inlets and self-digitized into nanoliter-sized droplets inside the side chambers. Figures 2A and 2B show a sequence of images taken during chamber filling and droplet formation. The aqueous phase entered the main channel and slowly displaced the oil from the side chambers. Once the whole sample entered the chip, the tailing oil phase sheared off the fluids at the opening of the side compartments and isolated the nanoliter droplets. The fluidic phenomenon behind this self-digitization process has been discussed in detail in our previous work28.
Fig. 2.

Sample self-digitization in the dLAMP SD chip. (A) Sequential images showing the initial filling of the side-chamber array with aqueous solution. After priming the chip with oil, the aqueous sample entered the main channel and distributed itself into the side compartments, displacing the oil phase in the chambers. (B) Sequence of images showing the self-digitization of aqueous sample in the side chambers. After the whole aqueous phase entered the chip, the tailing oil phase in the main channel isolated individual nanoliter sized droplets in the side chambers. (C) Dependence of the distribution of droplet size in the side chambers on applied pressure. The most uniform distribution with the highest average relative volume fraction (RVF) was obtained for an external pressure of 7 psi. Lower and higher pressures resulted in formation of droplets with more variable volumes and reduced RVF. (D) Size distribution of RVF values for 5000 self-digitized droplets from 16 individual chips. The external pressure was set to 7 psi in all experiments. The average RVF of all droplets was 0.89 ± 0.14. The inset shows the cumulative distribution of RVF values. The numbers correspond to the fraction of droplets with an RVF exceeding 50%, 75% and 90%, respectively. For example, 85% of all droplets filled out more than 75% of the chamber. (E) Reproducibility of sample self- digitization. Shown are the average RVF value and the standard deviation for 15 individual chips, each filled with 7-psi external pressure.
The uniformity of droplet size across the array depended on the applied air pressure as shown in Figure 2C. The volume of the retained droplets was estimated from images taken before chip incubation as described in the experimental section. In our setup, an air pressure between 7–8 psi yielded the most uniform distribution of droplet size, where the majority of droplets showed near complete volume retention, indicated by a relative volume fraction (RVF) near unity. Only a fraction of smaller sizes was found, most of which near the outlet where initial side-chamber filling by the aqueous phase was incomplete. Higher or lower pressures caused the RVF distribution to broaden, with more droplets showing significantly reduced RVF values.
Figure 2D shows a cumulative histogram of 5000 droplets. More than 96% of droplets occupied at least half the chamber volume, while for 56% of all droplets, the RVF exceeded 90%. On average, we found a RVF value of 0.89±0.14. Droplet uniformity was desirable, since it reduced errors in quantification due to difference in initial droplet volume. We will show in a later section that the distribution of droplet sizes did not significantly affect the outcome of our dLAMP analysis.
Figure 2E displays the reproducibility of sample digitization with 7 psi air pressure. For 15 individual chips, the average RVF and its standard deviation were calculated. The average RVF varied within a 10% window, which demonstrates adequate reproducibility in chip performance. Also the standard deviation in droplet size was comparable for all chips, albeit with a somewhat larger chip-to-chip variation.
Droplet stability at elevated temperatures
After filling, the chip was incubated at 65 °C for 70 minutes to carry out isothermal DNA amplification. At elevated temperatures, nanoliter-sized compartments are prone to water evaporation through the PDMS and partitioning of water into the oil phase. This would result in increases in reagent concentration, for example the ionic strength, which might cause inhibition of the reaction. While our chip was carefully designed to minimize sample evaporation, this effect cannot be totally prevented.
We found the amount of sample evaporation during thermal incubation was limited to roughly 10% of the digitized volume (Figure 3A). Chambers located at the periphery of the chip were exposed to more bulk PDMS and showed a slightly higher shrinkage as compared to chambers located in the center of the array (see inlet images of Figure 3A). The overall effect, however, was small and we did not expect it to affect the amplification reaction. If the peripheral evaporation is still a concern, the respective chambers could be ignored and only the innermost chambers could be considered for further analysis, as will be discussed below.
Fig. 3.

Chip performance at elevated temperatures. (A) Effect of incubation on droplet volume. Typical droplet-size distributions are shown before and after incubation at 65 °C for 70 minutes. The sample consisted of a negative control solution without template, hence no amplification was expected to occur. On average the droplets shrank by approximately 10%. As can be seen in the inset, the droplets located at the periphery of the array suffered from slightly increased shrinkage because they were more exposed to the bulk PDMS. Droplets in the center of the array were less affected by shrinkage. (B) Digital LAMP signature observed in our chipA section of the chamber array is shown before and after incubation. The intensity profile corresponds to a line across the centers of several chambers. Loop mediated DNA amplification in some of the chambers caused a sharp increase in their fluorescence. Neighbouring, non LAMP-competent chambers showed no increase in fluorescence, which demonstrated that sample crosstalk between adjacent chambers was negligible.
During incubation at 65 °C, we expect isothermal DNA amplification to proceed in those compartments that contained one or more DNA templates. Positive amplification was evidenced by a large increase in Calcein fluorescence after pyrophosphate is released during the amplification process8. Those compartments that contained no template DNA should only show background fluorescence. We next tested whether we could obtain a digital signature by amplification of a diluted sample of DNA (430-fold dilution of the stock solution). Figure 3B shows images and intensity scans across a section of a chip before and after incubation. As expected, some chambers showed a significant increase in fluorescence, while neighbouring chambers remained dark. Crosstalk between chambers thus appeared not to be an issue: LAMP-competent chambers were well separated by neighbouring chambers that showed only background signal.
Droplet size variation does not affect dLAMP analysis
Smaller droplets have a lower probability of containing one or more template molecules; positive events are less likely to occur in smaller droplets and should thus be weighted more in the analysis. To test whether such a procedure would differ from results obtained by straightforward counting of LAMP-active compartments, we compared these two analysis schemes for three representative chips at three different initial template concentrations ci (ci=c0/150 (A), ci=c0/430 (B) and ci=c0/1300 (C)). Table 1 summarizes the results. In the first case, the fraction of LAMP-active chambers, f0, was inferred from simple counting of positive droplets, regardless of initial droplet size. This was compared to an “effective” fraction, feff, which we approximated based on the initial droplet volume. feff was calculated as the sum of the inverse relative volume fractions for all positive droplets. In other words, a droplet that had a RVF of 50%, contributed twice as much as a droplet that occupied the full side chamber. We then compared the ratios of f0 and feff between the three chips that were analysed. We found that for our system, both analysis schemes gave similar results in all parameters. The roughly 10% increase of feff over f0 reflects the 10–15% variation in initial droplet size. From this comparison, we concluded that in our experiments the homogeneity of initial droplet size was good enough to validate the straightforward counting method.
Table 1.
Comparison of dLAMP analysis schemes
| f0 (all droplets) | feff (all droplets) | Finner (inner droplets) | |
|---|---|---|---|
| ci=c0/150 (A) | 0.309 | 0.338 | 0.292 |
| ci=c0/430 (B) | 0.092 | 0.104 | 0.092 |
| ci=c0/1300 (C) | 0.034 | 0.040 | 0.035 |
| fXA/fXB | 3.4 | 3.3 | 3.2 |
| fXA/fXC | 9.0 | 8.5 | 8.3 |
| fXB/fXC | 2.7 | 2.6 | 2.6 |
We also checked whether the preferential shrinkage of the peripheral droplets affected the analysis. We compared the fraction of LAMP-competent chambers for the case when: a) all droplets are considered in the analysis (f0), and b) the peripheral droplets were excluded from the analysis (finner). Recall that the increased shrinkage might increase local solute concentrations, which could affect the amplification process. For all three concentrations considered, the relative fraction of LAMP-competent droplets did change by less than 6%, if only the innermost droplets were analysed. The respective ratios for the different sample concentrations agreed to within 10% of each other. For dLAMP, we therefore considered all droplets in the analysis, thus avoiding unnecessary sample waste due to exclusion of the peripheral droplets.
dLAMP quantifies relative and absolute DNA concentrations
An important application of digital DNA amplification is the quantification of absolute template concentration in a sample. We next analysed the performance of the dLAMP chip with a serial dilution of DNA template in the sample. If on average less than one template is confined in the droplet, then we expect the number of LAMP-competent chambers to scale approximately linearly with DNA concentration. The template consisted of the DNA control sample provided with the LoopAmp® kit. We first performed UV absorption spectroscopy to estimate the template concentration in the DNA stock solution. The amount of DNA was too small to provide a quantifiable absorption at 260 nm. We therefore attempted DNA quantification using an intercalating dye. In this experiment, the target template was compared to a dilution series of λ-phage DNA of known concentration. All samples were incubated with a DNA intercalating dye (Evagreen®, Biotium, Hayward, CA, USA) for 30 minutes. The fluorescence signal of each sample was then quantified on the Typhoon™ imager. Since concentration and length of the λ-phage DNA were known (48502 bp), we obtained at least an accurate estimate of the number of bp present in the template solution. The actual length of the target plasmid was not provided by the manufacturer. As shown in supplemental information Figure S1, the intensity from the unknown sample was comparable to the intensity from solutions of less than 105 λ-phage DNA per μl. Because of the low contrast in this concentration regime, we could not narrow down the concentration further. Assuming the plasmid being comparable in size to a full λ-phage DNA, we thus estimate a concentration between 104 and 105 template copies per μl.
To determine the concentration from dLAMP experiments, we first performed dLAMP on a dilution series of template concentrations ranging from 1/4 to 1/1300 of the stock solution. Each experiment was done at least in triplicate. Supplemental Information Figure S2 shows representative images of several chips after 70 minutes incubation at 65 °C. Figure 4A shows the respective fraction of LAMP-competent chambers, f0. To calculate f0, we first determined the number of initial chambers before incubation, ni. We then counted the number of amplified chambers, na, which were chambers that had at least a three-fold increase in fluorescence over background as quantified from the image taken after incubation. f0 was then calculated as f0=na/ni.
Fig. 4.
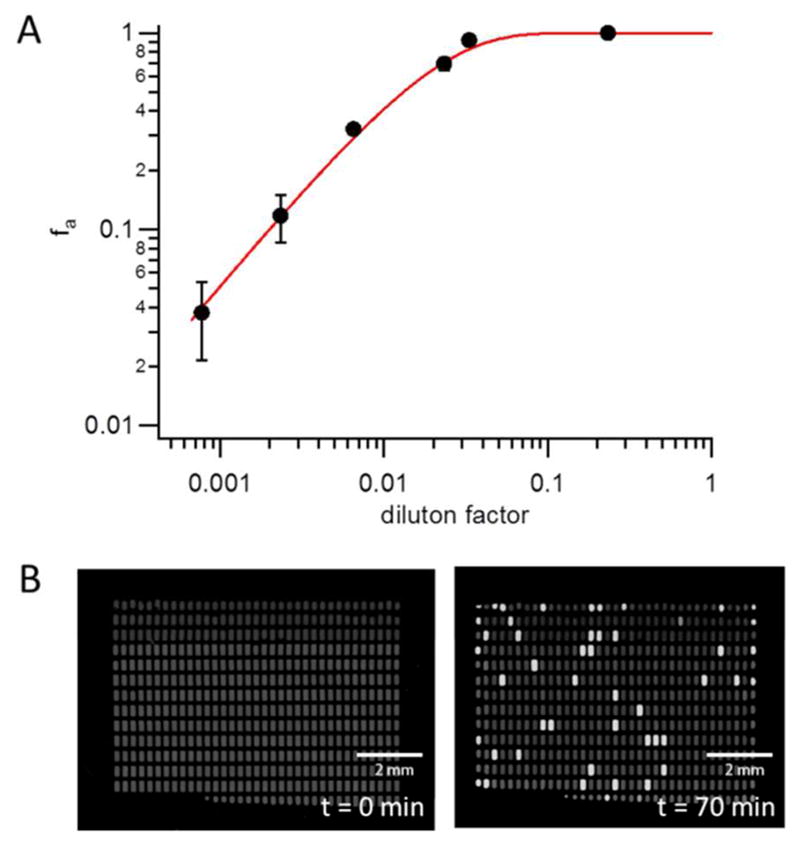
Quantification of relative and absolute changes in DNA concentration. (A) Dilution series of the control DNA sample contained in the LoopAmp® kit. For each sample concentration, at least three different chips were analyzed. The solid line represents the expected fraction of positive events based on a Poisson distribution for a template concentration of 0.99 × 104 molecules per μl in the stock solution. This concentration was determined from the 3 lowest sample concentrations as described in supplemental information, section S3. (B) Absolute quantification of DNA concentration. A 535 well chip is shown before and after incubation at 65 °C for 70 minutes. λ-phage DNA was diluted to a final concentration of 20 copies per μl, for which we expected approximately 12% positive chambers. After incubation we detected 9.8% LAMP-competent droplets in the 535 well array (47 out of 479 initially formed droplets). The difference between expected and measured values for fa may have been caused by pipetting errors accumulated over the 8-fold dilution series.
We observed a linear change in the number of LAMP-competent chambers with DNA concentration for the 3 lowest concentrations analysed. To estimate the concentration of target DNA via dLAMP, we computed a linearized Poisson fit to the three lowest sample concentrations as described in supplemental information, section S3. The fit yielded a concentration of (0.99 ± 0.03) × 104 copies per μl, which is in good agreement with our estimate from the DNA intercalation experiment. This result confirmed that our dLAMP chip was able to accurately reproduce relative changes in DNA concentration in a serial dilution of an unknown sample. Given the inaccuracy in the determination of the original DNA concentration, however, the data did not necessarily demonstrate our ability to determine absolute DNA concentrations with dLAMP. We therefore performed additional dLAMP experiments with a new target template of known concentration.
We chose the full λ-phage DNA template used for the intercalation assay with a set of LAMP primers reported in the literature32. The DNA concentration in the stock solution was measured via UV spectrophotometry to be 465 μg/ml, which corresponded to 8.9 × 109 copies per μl. The sample was then diluted down to 20 copies per μl in the final LAMP solution. For this concentration and a side chamber volume of 6 nanoliter, we expected to observe amplification in about 12% of all chambers to occur. dLAMP was performed for 70 minutes and Figure 4B shows images of a 535 well chip before and after the LAMP reaction. From the initial number of droplets discretized on-chip (479) and the number of LAMP-competent chambers after incubation (47) we computed a relative fraction of 9.8% of positive events. A second experiment in a 351 chamber array yielded a comparable fraction of 10.7% for the same template concentration (see supplemental information, Figure S4). The small difference between expected and measured values for fa can probably be attributed to pipetting errors accumulating over the 8-fold dilution series.
We therefore concluded that our dLAMP chip was capable of correctly determining absolute DNA concentrations as well as relative changes in DNA concentrations. Together with its simple operation, dLAMP in a SD chip provides a convenient platform to perform isothermal, digital DNA quantification with minimum sample consumption.
Conclusions
We demonstrated successful on-chip loop-mediated DNA amplification in a digital format. Driven by the limits of existing platforms to perform digital DNA amplification in a simple-to-use format with minimum sample consumption, we refined our self-digitization concept to allow for isothermal DNA amplification at 65 °C. Our method is simple and robust: once the aqueous phase is pipetted into the chip, the use of a simple pump head and constant air pressure is sufficient to induce sample discretization without the need of further chip manipulation, such as pneumatic valving or mechanical action. Unlike most (if not all) other reported digitization platforms, our design offers complete loss-less sample compartmentalization. This is important in cases where sample availability is limited, such as point-of-care applications.
Digital LAMP signatures reproduced absolute DNA concentrations as well as relative changes with sufficient accuracy within an incubation period of 70 minutes. Droplet shrinkage during sample incubation at elevated temperatures was minimal and did not affect the amplification process. We also addressed the issue of variations in droplet size during chip filling and demonstrated sufficient size homogeneity, with droplet volumes varying around 10–15%. Differences in droplet volume can potentially affect the average number of templates per droplet and the resulting digital signature. We verified that our autonomous chip filling produced droplets with sufficiently homogeneous volumes so that data analysis simplifies to the counting of LAMP-competent chambers, without the need to consider the variation in droplet volume.
Our experimental protocol utilizes standard lab instrumentation for heating and imaging and as such is compatible with most diagnostic settings, as no additional custom-built instrumentation is required. Because of the moderate reaction temperature of 65 °C, chip incubation could also be done in a thermal water bath, which would further simplify chip operation. Finally we note that, besides the increase in Calcein fluorescence, LAMP potentially generates a visual signal from precipitation of the pyrophosphate. Detection could be performed on a simple microscope, which would ultimately obviate the need for fluorescence equipment and reduce assay complexity even further. We thus envision that, in combination with the practical advantages of LAMP, our easy-to-operate chip design and loss-less sample filling method will be an attractive and inexpensive platform for DNA amplification in resource-limited settings.
Experimental
Chemicals and reagents
The LoopAmp® DNA amplification kit and the Calcein fluorescence indicator kit were purchased from SA Scientific (San Antonio, TX, USA). Positive control DNA and with a set of corresponding primers was included in the kit. Light mineral oil, sorbitane monooleate (SPAN-80), bovine serum albumin (BSA), propylene glycol methyl ether acetate (PGMEA) and isopropyl alcohol were obtained from Sigma Aldrich (St. Louis, MO, USA). Polydimethylsiloxane (PDMS, Sylgard 184 kit) was purchased from Dow Corning (Midland, MI, USA).
For the LAMP experiments on the full λ-phage DNA, a set of 6 primers was used as reported previously in the literature32 (forward inner primer (FIP): 5′-CAGCATCCCTTT-CGGCATACCAGGTGGCAAGGGTAATGAGG-3′, backward inner primer (BIP): 5′-GGAGGTTGAAGAACTGCGGCA-GTCGATGGCGTTCGTACTC-3′, forward outer primer (F3): 5′-GAATGCCCGTTCTGCGAG-3′, backward outer primer (B3): 5′-TTCAGTTCCTGTGCGTCG-3′, loop forward primer (LF), 5′-GGCGGCAGAGTCATAAAGCA-3′, and loop backward primer (LB): 5′-GGCAGATCTCCAGCCAGGAACTA-3′. All primers were purchased from IDT (San Diego, CA, USA).
Microfluidic chip fabrication
Microfluidic chips for digital LAMP were replicated in polydimethylsiloxane (PDMS) with standard soft lithography. The network of microfluidic channels and side chambers was designed in AutoCAD (Autodesk, San Rafael, CA, USA) and printed onto a Mylar photomask (Fineline Imaging, Colorado Springs CO, USA). The mask was used to fabricate a two layer SU-8 on-silicon master. For each layer, the following steps were performed: SU-8 photoresist (SU-8 2050, MicroChem, Newton, MA, USA) was spin coated onto a freshly cleaned silicon wafer. After soft baking, wafer and photomask were aligned and exposed to UV in a commercial mask aligner (Newport, Irvine CA, USA). UV exposure led to crosslinking of SU-8 underneath the transparent areas of the photomask. After curing, non-exposed SU-8 was dissolved in PGMEA and the wafer was cleaned with isopropyl alcohol and hard baked for 10 minutes at 155 °C. The height of the positive features in the master was measured in a home-built interferometer33 to be around 75 μm. To avoid sticking of PDMS onto the wafer during replication, the wafer was coated with (tridecafluoro)-1,1,2,2-tetrahydrooctyl) trichlorosilane (Gelest, Morrisville, PA, USA) by gas-phase deposition.
For chip replication, PDMS base and catalyst were mixed in a 10:1 weight ratio as recommended by the manufacturer. The mix was degased for 15–20 minutes and spin coated onto the SU-8 on-silicon master to form a thin film, approximately 300 μm thick. After curing at 70 °C for 3 hours, the PDMS was peeled off the wafer. Access holes were punched into the elastomer with a sharpened 15 gauge punch. The PDMS replica was bonded to a microscope slide coated with a thin layer of cured PDMS via oxygen plasma treatment. The PDMS chip was stored at 115 °C for 2 days, which reverted the surface back to hydrophobic.
Experimental protocol
Prior to the experiment, a set of small troughs matching the inlet and outlet holes was replicated into PDMS and attached to the chip with double-sided tape. Each trough provided a reservoir of approximately 50 μl. The chip was placed under vacuum for 20–30 minutes to remove excess air from the bulk PDMS. 40 μl of light mineral oil supplemented with 0.025% w/w SPAN-80 were placed into the inlet of the main channel to prime the chip. Air pressure was applied to maintain oil flow until the air was driven out of the chip. After priming, the chip was covered with a small piece of adhesive PCR sealant film (Bio-Rad, Hercules, CA, USA) to reduce water evaporation during the incubation.
13 μl of LAMP solution were freshly prepared following the manufacturer’s protocol. The mix was supplemented with 1.2 g/l BSA to stabilise the polymerase during the reaction and 0.6 μl of the Calcein-based fluorescence detection kit. 1.8 – 2μl of LAMP solution were pipetted into the inlet and formed an aqueous plug at the bottom of the trough that was covered with excess oil. External air pressure was applied to move the aqueous plug through the channel network until the whole sample became digitized on-chip. Digitization was visually checked on a microscope (AZ 100, Nikon Instruments, Melville NY, USA). The sacrificial water channel was manually filled with degased water using negative pressure on the outlet. All inlets were covered with at least 20 μl of oil before the chip was incubated at 65 °C for 70 minutes on a Thermocycler fitted with in situ adaptor (Mastercycler, Eppendorf, Westbury, NY, USA).
Chip imaging and quantification
Before and after thermal incubation, all chips were scanned on a variable mode imager (Typhoon FLA9000, GE Healthcare, Pittsburgh, PA, USA). The Calcein fluorescence inside the chambers was excited at 473nm and images were taken through a long pass filter (510LP) with 10 μm pixel resolution and a voltage of 350V applied to the photomultiplier tube. Subsequent image analysis was performed in ImageJ (http://rsbweb.nih.gov) to quantify the number and size distribution of discretized volumes as well as the number of chambers that showed DNA amplification. Further data analysis was done with IGOR Pro (WaveMetrics, Lake Oswego, OR, USA). Although initially the Calcein fluorescence was highly quenched, its residual emission could still be monitored with the Typhoon imager to determine initial droplet size in the chamber. The distribution of initial droplet sizes was quantified in terms of the retained volume fraction (RVF) estimated as the ratio of the droplet area to the area of the side chamber. A threshold equal to 1/3 of the average pixel intensity of the Calcein fluorescence in droplets was used to discriminate against background. Only droplets with a RVF of at least 0.15 were considered for further analysis.
Supplementary Material
Acknowledgments
We gratefully acknowledge support from the NIH (R21RR032399) and NSF (CHE-0844688) for this work.
Footnotes
Electronic Supplementary Information (ESI) available: See DOI: 10.1039/b000000x/
Contributor Information
Luke P. Lee, Email: lplee@berkeley.edu.
Daniel T. Chiu, Email: chiu@chem.washington.edu.
Notes and references
- 1.Mullis KB, Faloona FA. Methods Enzymol. 1987;155:335–50. doi: 10.1016/0076-6879(87)55023-6. [DOI] [PubMed] [Google Scholar]
- 2.Lizardi PM, Huang X, Zhu Z, Bray-Ward P, Thomas DC, Ward DC. Nat Genet. 1998;3:225–32. doi: 10.1038/898. [DOI] [PubMed] [Google Scholar]
- 3.Dean FB, Hosono S, Fang L, Wu X, Faruqi A Fawad, Bray-Ward P, Sun Z, Zong Q, Du Y, Du J, Driscoll M, Song W, Kingsmore SF, Egholm M, Lasken RS. Proc Natl Acad Sci USA. 2002;99(8) doi: 10.1073/pnas.082089499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Compton J. Nature. 1991;350(633):91–92. doi: 10.1038/350091a0. [DOI] [PubMed] [Google Scholar]
- 5.Piepenburg O, Williams CH, Stemple DL, Armes NA. PLoS Biol. 2006;4:1115–1121. doi: 10.1371/journal.pbio.0040204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vincent M, Xu Y, Kong HM. EMBO Rep. 2004;5:795–800. doi: 10.1038/sj.embor.7400200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T. Nucleic Acids Res. 2000;28:e63. doi: 10.1093/nar/28.12.e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tomita N, Yasuyoshi M, Kanda H, Notomi T. Nat Protocols. 2008;3:877–882. doi: 10.1038/nprot.2008.57. [DOI] [PubMed] [Google Scholar]
- 9.Lucchi NW, Demas A, Narayanan J, Sumari D, Kabanywanyi A, Kachur SP, Barnwell JW, Udhayakumar V. PLoS ONE. 2010;5(10):e13733. doi: 10.1371/journal.pone.0013733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Kang Z, Gao H, Gao Y, Qin L, Lin H, Yu F, Qi X, Wang X. Virol J. 2011;8:108. doi: 10.1186/1743-422X-8-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang X, Liao M, Jiao P, Luo K, Zhang H, Ren T, Zhang G, Xu C, Xin C, Cao W. J Clin Microbiol. 2010;48(6):2116–2121. doi: 10.1128/JCM.02530-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu R-Y, Zhang K-X, Zhao M-Q, Liu Y-H, Xu Y-Y, Ju C-M, Li B, Chen J-D. J Microbiol Methods. 2009;78(3):339–343. doi: 10.1016/j.mimet.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 13.Heid CA, Stevens J, Livak KJ, Williams PM. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- 14.Vogelstein B, Kinzler KW. Proc Natl Acad Sci USA. 1999;96:9236–9241. doi: 10.1073/pnas.96.16.9236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sykes PJ, Neoh SH, Brisco MJ, Hughes E, Condon J, Morley AA. BioTechniques. 1992;13:444–449. [PubMed] [Google Scholar]
- 16.Sanders R, Huggett JF, Bushell CA, Cowen S, Scott DJ, Foy CA. Anal Chem. 2011;83:6474–6484. doi: 10.1021/ac103230c. [DOI] [PubMed] [Google Scholar]
- 17.Beer NR, Wheeler EK, Lee-Houghton L, Watkins N, Nasarabadi S, Hebert N, Leung P, Arnold DW, Bailey CG, Colston BW. Anal Chem. 2008;80:1854–1858. doi: 10.1021/ac800048k. [DOI] [PubMed] [Google Scholar]
- 18.Schaerli Y, Wootton RC, Robinson T, Stein V, Dunsby C, Neil MA, French PM, DeMello AJ, Abell C, Hollfelder F. Anal Chem. 2009;81:302–306. doi: 10.1021/ac802038c. [DOI] [PubMed] [Google Scholar]
- 19.Mazutis L, Araghi AF, Miller OJ, Baret JC, Frenz L, Janoshazi A, Taly V, Miller BJ, Hutchison JB, Link D, Griffiths AD, Ryckelynck M. Anal Chem. 2009;81:4813–4821. doi: 10.1021/ac900403z. [DOI] [PubMed] [Google Scholar]
- 20.Shen F, Du WB, Kreutz JE, Fok A, Ismagilov RF. Lab Chip. 2010;10:2666–2672. doi: 10.1039/c004521g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen F, Davydova EK, Du W, Kreutz JE, Piepenburg O, Ismagilov RF. Anal Chem. 2011;83(9):3533–3540. doi: 10.1021/ac200247e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Applied Biosystems, Life Technologies. TaqMan OpenArray Digital PCR Plates. https://products.appliedbiosystems.com/ab/en/US/adirect/b?cmd=catNavigate2&catID=607965.
- 23.Matsubara Y, Kerman K, Kobayashi M, Yamamura S, Morita Y, Takamura Y, Tamiya E. Anal Chem. 2004;76(21):6434–6439. doi: 10.1021/ac0497149. [DOI] [PubMed] [Google Scholar]
- 24.Ottesen EA, Hong JW, Quake SR, Leadbetter JR. Science. 2006;314:1464–1467. doi: 10.1126/science.1131370. [DOI] [PubMed] [Google Scholar]
- 25.Heyries KA, Tropini C, VanInsberghe M, Doolin C, Petriv OI, Singhal A, Leung K, Hughesman CB, Hansen CL. Nat Methods. 2011;8:649–651. doi: 10.1038/nmeth.1640. [DOI] [PubMed] [Google Scholar]
- 26.Jensen EC, Bhat BP, Mathies RA. Lab Chip. 2010;10:685–691. doi: 10.1039/b920124f. [DOI] [PubMed] [Google Scholar]
- 27.Michikawa Y, Sugahara K, Suga T, Ohtsuka Y, Ishikawa K, Ishikawa A, Shiomi N, Shiomi T, Iwakawa M, Imai T. Anal Biochem. 2008;383(2):151–158. doi: 10.1016/j.ab.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 28.Cohen DE, Schneider T, Wang M, Chiu DT. Anal Chem. 2010;82(13):5707–5717. doi: 10.1021/ac100713u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fang X, Chen H, Yu S, Jiang X, Kong J. Anal Chem. 2011;83(3):690–695. doi: 10.1021/ac102858j. [DOI] [PubMed] [Google Scholar]
- 30.Hataoka Y, Zhang L, Mori Y, Tomita N, Notomi T, Baba Y. Anal Chem. 2004;76:3689. doi: 10.1021/ac035032u. [DOI] [PubMed] [Google Scholar]
- 31.Lee SY, Lee CN, Mark H, Meldrum DR, Lin CW. Sens Actuators B. 2007;127:598. [Google Scholar]
- 32.Goto M, Honda E, Ogura A, Nomoto A, Hanaki K. Biotechniques. 2009;46(3):167–72. doi: 10.2144/000113072. [DOI] [PubMed] [Google Scholar]
- 33.Yen GS, Fujimoto B, Schneider T, Huynh DTK, Jeffries GDM, Chiu DT. Lab Chip. 2010;11:974–977. doi: 10.1039/c0lc00470g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.