

Abstract
Factor VII (FVII) consists of an N-terminal gamma-carboxyglutamic acid domain followed by two epidermal growth factor-like (EGF1 and EGF2) domains and the C-terminal protease domain. Activation of FVII results in a two-chain FVIIa molecule consisting of a light chain (Gla-EGF1-EGF2 domains) and a heavy chain (protease domain) held together by a single disulfide bond. During coagulation, the complex of tissue factor (TF, a transmembrane glycoprotein) and FVIIa activates factor IX (FIX) and factor X (FX). FVIIa is structurally “zymogen-like” and when bound to TF, it is more “active enzyme-like.” FIX and FX share structural homology with FVII. Three structural biology aspects of FVIIa/TF are presented in this review. One, regions in soluble TF (sTF) that interact with FVIIa as well as mapping of Ca2+, Mg2+, Na+ and Zn2+ sites in FVIIa and their functions; two, modeled interactive regions of Gla and EGF1 domains of FXa and FIXa with FVIIa/sTF; and three, incompletely formed oxyanion hole in FVIIa/sTF and its induction by substrate/inhibitor. Finally, an overview of the recognition elements in TF pathway inhibitor is provided.
Keywords: Blood Coagulation, Tissue Factor, Factor VIIa, Factors IX and X, Structural Biology, Review
2. Introduction
Human factor VII (FVII) is a vitamin K-dependent trace plasma protein; it is synthesized by hepatocytes and secreted into blood as a single chain molecule of Mr ∼50,000 (1,2). Starting at the N-terminus, FVII contains a gamma-carboxyglutamic acid-rich domain (Gla domain, residues 1-38), a short hydrophobic segment (residues 39-45) that often is considered as part of the Gla domain, two epidermal growth factor (EGF)-like domains (residues 47-84 and 85-131), and the C-terminal serine protease domain (residues 153-406) (3). FVII has negligible activity and participates in the extrinsic pathway of coagulation principally after its activation to FVIIa (4-7). A number of coagulation enzymes including FXa, FIXa, and FVIIa can activate FVII (8-11); however, FXa appears to be the most potent activator of FVII (12). Activation of FVII by each enzyme involves cleavage of a single peptide bond between Arg-c15{152} and Ile-c16{153}, located in the connecting region between the EGF2 and the protease domain. This results in the formation of a two-chain FVIIa molecule consisting of a light chain of 152 amino acids and a heavy chain of 254 amino acids held together by a single disulfide bond (between Cys-135 and Cys-262) (3).
Tissue factor (TF) is a membrane protein with structural homology to the class 2 cytokine receptor family (13). Human TF consists of an N-terminal cytoplasmic domain of 19 residues, a transmembrane domain of 23 residues and a C-terminal ectodomain domain of 219 residues. The ectodomain of TF consists of two fibronectin type-III repeats. The crystal structure of the C-terminal domain pair—soluble tissue factor (sTF)—is known (14-17); the two fibronectin type-III domains are connected end-to-end at an angle of 120 degrees (14-17). Similar to full-length TF, sTF binds FVIIa with high affinity and potentiates its enzymatic activity (18-20).
Binding of TF increases the amidolytic activity of FVIIa several-fold (21-23) by restructuring the active site region. In this regard, Higashi et al. (24) proposed that free FVIIa is structurally more zymogen-like and when bound to TF it is more active enzyme-like. Alanine scanning mutational studies of surface residues in the protease domain of FVIIa support this concept (25). Thus, TF binding induces the concerted structural rearrangements in FVIIa similar to those observed upon the proteolytic activation of trypsinogen to trypsin and related serine proteases (26). Further, these changes occur only when the free NH2-group of the Ile-c16{153} is allowed to make a salt bridge with the carboxylate of Asp-c194{343} of FVIIa (24). TF cannot induce these changes in zymogen FVII in which Arg152-Ile153 bond has not been cleaved (27). Although TF binding to FVIIa increases its activity by several orders of magnitude, it does not appear to do so by forming the oxyanion hole; it is the substrate that induces formation of the oxyanion hole in FVIIa (28). In the absence of TF, the FVIIa regions that play an important role in preventing the transition from the zymogen-like to the active enzyme-like molecule, and how TF binding promotes this transition is an area of active investigation (29-33), which is discussed elsewhere in this issue (34).
Initiation of coagulation begins by exposure of blood to TF in the extravascular space at an injury site and formation of the Ca2+-dependent complex between TF and plasma FVIIa. The Ca2+/FVIIa/TF complex formed on the cell surfaces then activates both FX and FIX leading to thrombin generation and fibrin formation. In this reaction, FVIIa, FX and FIX (see below) anchor to the phospholipid (PL) bilayer through their Gla domains for optimal rates of FXa and FIXa formation. Consequently, in the absence of PL, the rates of activation of FX and FIX by FVIIa/sTF are relatively slow (18-20). TF pathway of coagulation is regulated by tissue factor pathway inhibitor (TFPI), a FXa-dependent inhibitor of the Ca2+/FVIIa/TF complex (35). The translated amino acid sequence of human TFPI (major form) cDNA reveals that it consists of a highly negative N-terminus, three tandemly repeated Kunitz-type domains with intervening linker regions, and a highly positive C-terminus (36). The mechanism of inhibition by TFPI involves its binding first to FXa through domain 2 and then to the TF/FVIIa complex through domain 1 (37, 38). The third Kunitz-type domain has no inhibitory activity and has other biologic functions that will not be discussed here.
Human FX and FIX share structural homology with FVII. FX circulates as a zymogen with a molecular weight of ∼59,000 and consists of a light chain (amino acids 1-139) and a heavy chain (amino acids 143-448) held together by a single disulfide bond between Cys-132 and Cys-c302 (3, 39). Upon activation, a single peptide bond in FX between residues Arg-c15{194} and Ile-c16{195} is cleaved with resultant formation of a serine protease, FXa, and release of a 52-residue activation peptide (3). The NH2 terminus light chain of human FXa contains 11 Gla residues and represents the Gla domain (residues 1-39); the Gla domain is followed by a few aromatic residues (amino acids 40-45), and two EGF-like domains (EGF1 residues 46-84, EGF2 residues 85-128). The heavy chain contains the serine protease domain essential for catalysis and features the active site triad of His-c57{236}, Asp-c102{282}, and Ser-c195{379} (3).
FIX is a single chain protein and contains a Gla domain (residues 1-40), a short hydrophobic segment (residues 41-46), two EGF-like domains [residues 47-84 (EGF1) and 85-127 (EGF2)], an activation peptide region (residues 146-180), and a serine protease module (residues 181-415). Activation of FIX involves proteolytic cleavages at Arg145-Ala146 and Arg-c15{180} and Val-c16{181} bonds with a concomitant release of a 35-residue activation peptide (3, 40). The FIXa thus formed contains a light chain (residues 1-145) and a heavy chain (residues 181-415) held together by a single disulfide bond. The light chain consists of the Gla, EGF1, and EGF2 domains whereas the heavy chain contains the serine protease domain that features the catalytic triad of residues His-c57{221}, Asp-c102{269} and Ser-c195{365} .
In this review, we discuss the structural biology aspects of FVIIa/sTF interface and its interaction with the macromolecular substrates FIX and FX. The functional roles of Ca2+, Mg2+ and Na+, and the inhibitory role of Zn2+ in TF binding and catalysis are described. Unlike other serine proteases of the coagulation cascade, a unique feature of FVIIa bound to TF is the absence of oxyanion hole at the active site and its induction by the substrate. The importance of this mechanism in achieving selectivity of the physiologic substrate by FVIIa/TF is outlined. Lastly, an overview of the important recognition elements in the first and the second domain of TF pathway inhibitor (TFPI) is provided.
3. Molecular Recognition in Tf-Induced Coagulation
3.1. Structures of FVIIa/sTF in Ca2+ and in Ca2+/Mg2+
The formation of FVIIa/TF complex is the first step in the extrinsic pathway of blood coagulation. The crystal structures of human active-site inhibited FVIIa/sTF complex in the presence of Ca2+ only (41, PDB id 1DAN) and in the presence of Ca2+/Mg2+ (28, PDB id 2A2Q) are shown in Figure 1A and Figure 1B, respectively. In the 1DAN structure, the active site is occupied by D-Phe-Phe-Arg-chloromethylketone and the oxyanion hole is fully formed. In the 2A2Q structure, the active site is occupied by p-aminobenzamidine (pAB) and the oxyanion hole is incompletely formed; the significance of this observation is discussed later in section 3.8. In both structures, FVIIa molecule is in an extended form where the protease domain is at the top and the membrane binding Gla domain is at the bottom. In this complex, FVIIa is tightly bound to sTF through salt bridges, hydrogen bonds, ionic contacts and hydrophobic interactions. All domains of FVIIa make strong interactions with sTF. These are outlined in detail earlier (41). The observed interactions between the enzyme FVIIa and the cofactor sTF in the crystal structures (28,41) are consistent with the biochemical and experimental evidence (22,25,29,42-52).
Figure 1.
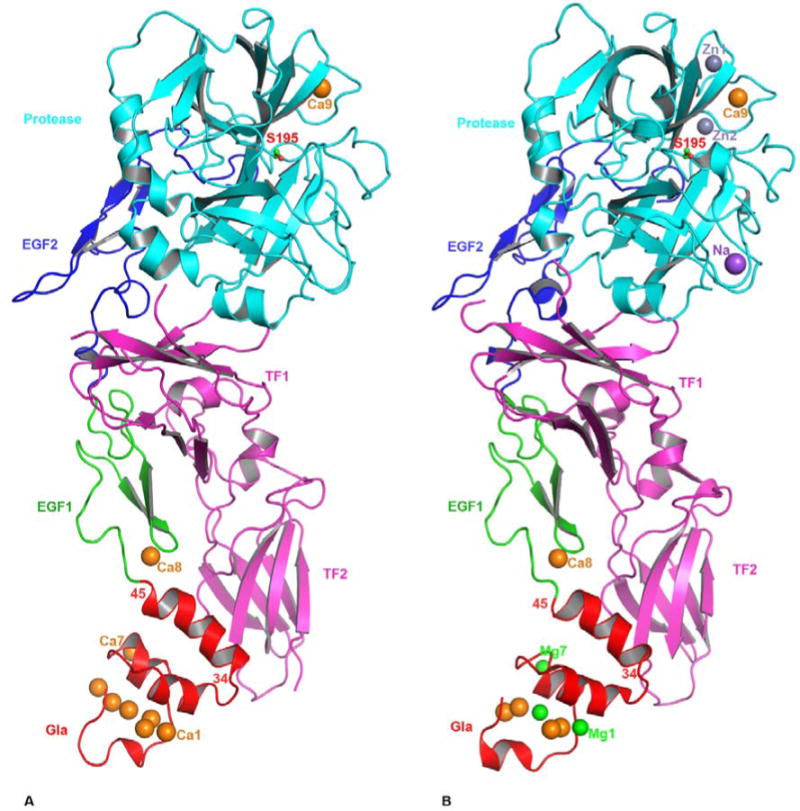
The cartoon representation of FVIIa/sTF complexes. The FVIIa/sTF structure of Banner et al., at 2.0 angstrom resolution (41) (PDB id 1DAN) in the presence of Ca2+ only is shown in A, and that of Bajaj et al., at 1.8 angstrom resolution (28) (PDB id 2A2Q) in the presence of Ca2+ and Mg2+ is shown in B. The locations of Na+ and Zn2+ sites are also shown in the 2A2Q structure. FVIIa consists of Gla (red), EGF1 (green), EGF2 (blue) and protease (cyan) domains, and the sTF (magenta) contains two fibronection type III domains. The different metal ions Ca2+(orange), Mg2+(green), Zn2+ (light blue) and Na+ (purple), bound to different domains of FVIIa are shown as spheres. Three Ca2+ ions in the GLA domain at positions 1, 4 and 7 (53) in the 1DAN structure are replaced by Mg2+ in the 2A2Q structure. The Ca2+ ion bound to the FVIIa EGF1 domain is labeled Ca8 and to the protease domain is labeled Ca9. In the 2A2Q structure Na+ ion is labeled as Na and the two Zn2+ ions labeled Zn1 and Zn2. The position of the active site Ser-195 is shown in ball and stick representation. The position of the C-terminal helix of the Gla domain (residues 34-45) of FVIIa is slightly tilted towards the TF2 domain in the 2A2Q structure resulting in a somewhat more favorable hydrophobic interactions between L13, F31 L39, and F40 of FVIIa with Y156, W158 and V207 of sTF. The residues interacting at the interface regions between FVIIa and sTF have been outlined in detail in an earlier report (41).
In the 1DAN structure (41), seven Ca2+ ions are bound to the N-terminal Gla domain in a linear fashion. In the 2A2Q structure (28), three of the seven Ca2+ ions at positions 1, 4 and 7 (53) are replaced by Mg2+. In both structures, a single Ca2+ ion is bound to the EGF1 domain as well as to the protease domain. In addition, two putative Zn2+ binding sites and one putative Na+ binding site have been identified in the protease domain of FVIIa in the 2A2Q structure. Further, significant flexibility exists in FVIIa domains in solution (54), which is lost upon binding to sTF. In contrast, sTF undergoes minimal conformational changes upon binding to FVIIa (55). This is consistent with the concept that cofactor TF stabilizes the conformation of enzyme FVIIa in a stable active form (24).
The fold of the Gla domain omega-loop in FVIIa is different in the Ca2+ only structure (41) verses the Ca2+/Mg2+ structure (28). The 2A2Q structure (28) was obtained under concentrations of Mg2+ that were supraphysiologic; however, folding of the omega-loop under 5 mM Ca2+/2.5 mM Mg2+ is similar to the 2A2Q structure, with identical positions for four Ca2+ and three Mg2+ (56, PDB id 3TH2). Further, the folding of the omega-loop under 45 mM Ca2+/5 mM Mg2+ is similar to that reported in the presence of Ca2+ only (41); however, positions 1 and 7 are occupied by Mg2+ (56, PDB id 3TH4). Thus, it would be appear that four Ca2+ and three Mg2+ ions are bound to the Gla domain in the circulating FVII/FVIIa. Moreover, circulating free FVII/FVIIa will need high Ca2+ concentrations to have its position four switched from Mg2+ to Ca2+.
Interestingly, Gla domain of activated protein C (APC) bound to endothelial protein C receptor under 10 mM Ca2+/10 mM Mg2+ has metal ion position four occupied by Ca2+ and only positions 1 and 7 occupied by Mg2+ (56,57; PDB id 3JTC and 1LQV). Similarly, the structure of the ligand bound Gla domain of FIX/FIXa under 5 mM Ca2+/2.5 mM Mg2+ has metal position four occupied by Ca2+ (58); and high concentrations of Ca2+ are needed to substitute Mg2+ at position four by Ca2+ in the ligand free FIX/FIXa molecule (59). Based upon these observations, we propose that vitamin K-dependent clotting and anticlotting proteins circulate in blood with central four Ca2+ ions bound to their Gla domains. The remaining three (or more in FIX and FX) external divalent metal binding sites in each Gla domain are occupied by Mg2+. The conformation of the omega-loop in each circulating vitamin K-dependent protein may not be optimal for binding to PL and is possibly achieved by switching Mg2+ at position four to Ca2+(60). Thus, the metal ion at position four regulates PL-dependent coagulation and anticoagulation reactions. In this context, Mg2+ potentiates PL-dependent physiologic reactions by occupying only the external divalent metal binding sites and none of the central Ca2+ sites (28,56,61,62). One should note that experimental evidence is needed to validate and substantiate this concept.
3.2. Modeled structures of FVIIa/sTF with FXa and with FIXa/FIXalpha
The FVIIa/TF complex activates FIX (63) as well as FX (64), which leads to the activation of prothrombin to thrombin. In a reciprocal fashion, FXa and FIXa activate FVII/TF (2,8,9,12). Although primary sequences of FXa and FIXa are highly homologous, the interaction of FIXa with FVIIa/TF is weaker than FXa (65,66); further, as compared to FXa, FIXa is a poor activator of FVII/TF (12,67). Moreover, the affinities of FX-Gla and FX-EGF1 domains towards FVIIa/TF are stronger than the FIX-Gla and FIX-EGF1 domains (68). To explain these differences, computational models of the ternary complexes of FVIIa/sTF-FXa, FVIIa/sTF-FIXa and FVIIa/sTF-FIXalpha have been proposed (67,69-72). In building these models, biochemical evidence was combined with newly available algorithms for protein-protein docking (73-75). In the absence of experimentally determined structures of FVIIa/sTF-FXa and FVIIa/sTF-FIXa or FVIIa/sTF-FIXalpha, these models serve as reasonable basis for understanding how Gla and EGF1 domains of FIX/IXa and FX/Xa interact with FVIIa/TF.
In 2003, structural models of the FVIIa/sTF-FXa complex were proposed by two independent groups, namely, the Scripps group (70) and the Chapel Hill group (71). Based upon the methodology used to build these complexes, one is referred to as the Scripps static model (Ss model, 70) and the other as the original Chapel Hill solution-equilibrated model (CheA model, 71). Although different approaches were used, the two models have similar overall architecture and comparable residue interactions although with some significant variations. In general, each model was built in three stages using different protein-protein docking algorithms. In stage 1, the Protease-EGF2 domains of FXa were docked onto FVIIa/sTF. In stage 2 and stage 3, EGF1 domain and the Gla domain were docked onto FVIIa/sTF, respectively. At each stage the models were screened and those that corraborated the experimental evidence were selected for the next stage of modeling. The overall architecture of FVIIa/sTF-FXa is shown in Figure 2A. In both models, TF is sandwiched between the extended forms of FVIIa and FXa. Recently, both Ss and CheA model structures (70,71) were subjected to 10 ns all-atom molecular dynamic simulations to obtain solution-equilibrated models (72). The resulting models are referred to as Scripps solution-equilibrated (Se) and current Chapel Hill solution-equilibrated (CheB) models (72). In all models, the Gla domain of FXa interacts with TF domain as well as with Gla domain of FVIIa. The EGF1 domain of FXa interacts only with TF in the Ss and Se models (70,72), whereas it interacts with TF and FVIIa in the CheA and CheB models (71,72). Further, in all models, the EGF2 domain and the serine protease (SP) domain of FXa interact with the serine protease domain of FVIIa and no interaction is noted with TF.
Figure 2.
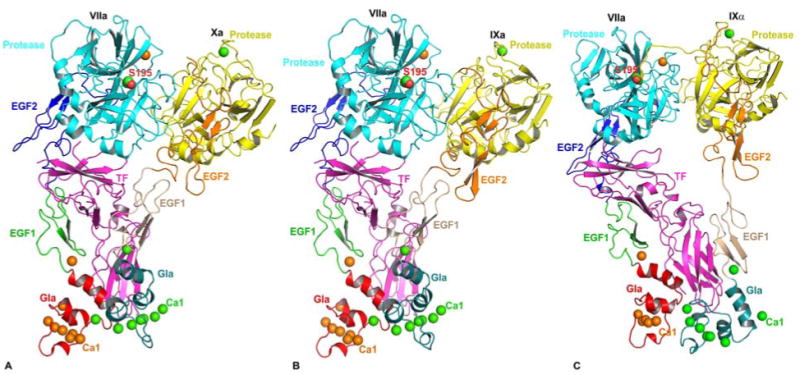
Cartoon representations of modeled structures of the ternary complexes. Structure of the FVIIa/sTF-FXa model is shown in A, structure of the FVIIa/sTF-FIXa model is shown in B, and structure of the FVIIa/sTF-FIXalpha model is shown in C. FIXalpha represents FIX cleaved at Arg145-Ala146 peptide bond, whereas FIXa represents FIX cleaved at Arg145-Ala146 and Arg-c15{180} -Val-c16{181} peptide bonds. The FVIIa/sTF-FXa model (PDB id 1NL8) is from Norledge et al (70) and the energy minimized FVIIa/sTF-FIXa model (67) is based upon 1NL8 as the template. The FVIIa/sTF-FIXalpha model is from Chen et al (69). As in figure 1, FVIIa Gla, EGF1, EGF2, and the protease domains are colored red, green, blue and cyan, respectively. The sTF is shown in magenta. The Gla, EGF1, EGF2 and protease domains of FXa, FIXa and FIXalpha are colored deep teal, salmon, orange, and yellow, respectively. The active site residue (Ser-195) of FVIIa protease domain is shown in the space filling representation. The Ca2+ ions bound to FVIIa are shown as orange spheres, whereas the Ca2+ ions bound to FXa, FIXalpha and FIXa are shown as green spheres. Note that the Gla domains of FVIIa, FXa and FIXa are expected to bind Mg2+ at specific sites under physiologic conditions. These sites have been replaced by Ca2+ during model building.
We used FVIIa/sTF-FXa as a template (1NL8, Ss model) to build the FVIIa-sTF-FIXa model (66). Since the complete structure of FIXa is not available, we modeled the FIXa using the coordinates of FIXa Gla (58), EGF1 (76) and the protease-EGF2 domains (77). The FIXa domains were structurally aligned to the respective domains of FXa in the FVIIa/sTF-FXa complex using the program ‘O’ (78). To remove the steric clashes and the short contacts, the entire FVIIa/sTF-FIXa complex was subjected to minimization using CHARMM (79). The resulting FVIIa/sTF-FIXa model is shown in Figure 2B. The model of FVIIa/sTF-FIXalpha complex (69) is shown in Figure 2C. Similar to FXa, the Gla domain of FIXa/IXalpha in FVIIa/sTF-FIXa or FVIIa/sTF-FIXalpha interacts with the Gla domain of FVIIa and TF, whereas the EGF1 domain interacts with TF only. However, the interactive residues in the FVIIa/sTF-FIXa and the FVIIa/sTF-FIXalpha models are different. Based on these computational models, we outline the possible mode of interactions between FVIIa/sTF and FXa/FIXa/FIXalpha in the next three sections.
3.3. Interactions of FVIIa/sTF with the Gla Domains of FX/Xa and FIX/IXa
The C-terminal region of TF interacts with the Gla domains of FX/Xa (22,80-83) and FIX/IXa (66,67,69). Further, the Gla domains of FX/Xa (68,70-73) and FIX/IXa (66,69) also interact with the Gla domain of FVIIa. The possible modes of interaction between FVIIa/sTF with the Gla domains of FXa (Ss model), FIXa and FXalpha are shown in Figure 3 A, B and C, respectively. Although the structures of FXa and FIXa are similar, the variations in their sequences at the interacting regions with sTF and with the Gla domain of FVIIa make associations different from each other. The residues 9 to 12 (KKGH) in the Gla domain of FXa are basic, whereas residues 10 to 13 in FIXa are hydrophobic and neutral (VQGN). The residues K10 and H12 in FXa make salt bridge with D33 of FVIIa. In comparison, Q11 in FIXa makes weak hydrogen bond with D33 of FVIIa. However, K38 of FVIIa makes a salt bridge with Gla14 in FXa and similarly with Gla15 in FIXa. Overall, the sequence variations in this region make interactions weak for FIXa.
Figure 3.
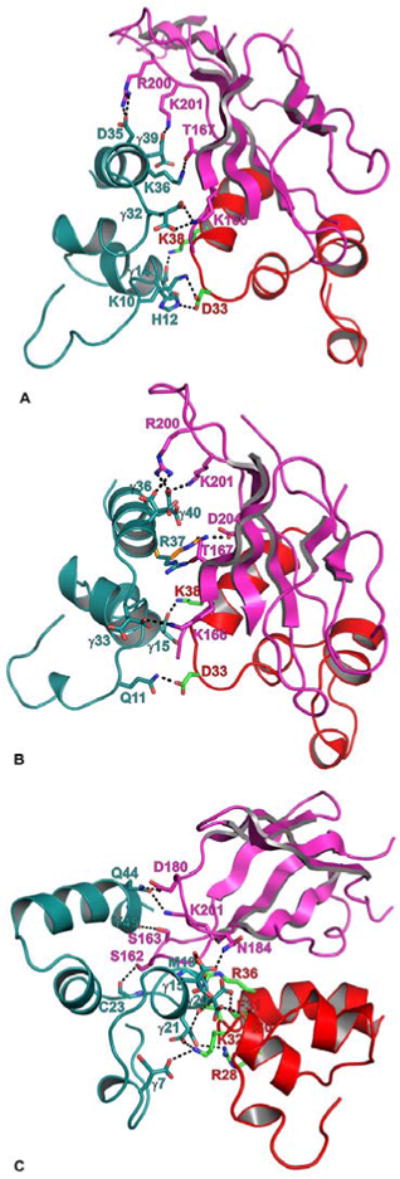
Modeled interactions of FVIIa/sTF with the Gla domains of FXa, FIXaand FIXalpha. The interacting residues of FVIIa-Gla (red), sTF (magenta) and FXa-Gla (deep teal) are shown in stick representation. The carbon atoms are green in FVIIa-Gla, magenta in sTF, and deep teal in the Gla domains of FXa, FIXa and FIXalpha. The N and O atoms are blue and red, respectively. The hydrogen bonds are shown as black dashed lines. A) The modeled interaction of FXa-Gla with sTF and FVIIa-Gla. FXa-Gla:sTF—Gla32:K166, D35:R200, Gla39:K201 and K36:T167; FXa-Gla:FVIIa-Gla—K10:D33, H12:D33 and Gla14:K38. B) The modeled interaction of FIXa-Gla with sTF and FVIIa-Gla. FIXa-Gla:sTF—Gla33:K166, Gla36:R200, R37:D204/T167, and Gla40:R200/K201; FIXa-Gla:VIIa-Gla—Q11:D33 and Gla15:K38. C) The modeled interaction of FIXalpha-Gla with sTF and FVIIa-Gla. FIXalpha-Gla:sTF—Gla15:N184, C23:S162, Q44:D180/201 and Y45:S163; FIXalpha-Gla:FVIIa-Gla—Gla7:K32; M19:R36, Gla20:F31/I30 and Gla21:R28/K32.
The C-terminal helix of the Gla domains in FX/Xa and FIX/IXa interact with the C-terminal domain of TF and make no contact with FVIIa. In the Ss model, FXa residues Gla32, D35, and Gla39 make salt bridges with TF residues K166, R200, and K201, respectively (Figure 3A). In addition, K36 of FXa makes hydrogen bond with T167 of TF. The C-terminal helix of FIXa Gla domain interacts with TF in a similar fashion. In FIXa, corresponding residues Gla33, Gla36, and Gla40 make salt bridges with TF residues K166, R200, and K201, respectively. In addition, R37 of FIXa can make a hydrogen bond with either T167 or D204 of TF (Figure 3B). Mutagenesis studies support the role of TF residues R200, K201, T167 and K165 in its interaction with the Gla domain of FXa/FIXa (80-83). Thus, comparable interactions exist between the C-terminal helix of the Gla domains of FXa or FIXa with TF.
Interactions of the Gla domain of FXa with TF and FVIIa in the CheA/CheB models (71,72) are somewhat different from the Ss/Se models (70,72). The important differences are—a) ionic interactions between K165 of TF and D35 of FXa present in the CheA/CheB models and absent in the Ss/Se models; and b) ionic interactions between R36 of FVIIa and Gla14 of FXa present in the Ss/Se models and absent in the CheA/CheB models. By mutagenesis, residue R36 in Gla domain of FVIIa has also been identified as one of the key residues in substrate recognition of FX (84). However, our recent FVIIa/sTF crystal structure at 1.72 angstrom reveals that R36 in FVIIa is involved in network of hydrogen bonds with TF (56). Further in this structure, the TF region containing residues K165 and K166 are well defined and it appears that only one of these residues side chain can interact with FXa. Moreover, hydrophobic interactions between the Gla domains of FVIIa and FXa are more prominent in the CheA/CheB models as compared to the Ss/Se models. Clearly, experimentally determined structure of FVIIa/sTF-FXa is needed to resolve these discrepancies.
As compared to FIXa, the mode of interaction between the Gla domain of FIXalpha and the FVIIa/sTF is considerably different. The FIXalpha Gla residues that interact with FVIIa/sTF are shown in Figure 3C. These interactions involve possible hydrogen bonds between residues Gla7, Gla15, M19, Gla20, and Gla21 of FIXalpha with R28, I30, F31, K32, and R36 of FVIIa and N184 and S162 of TF. The Gla domain C-terminal helix residues Q44 and Y45 of FIXalpha also interact with residues S163, D180 and K201 of TF via hydrogen bonds. In general, FIXalpha versus FIXa Gla domain interacts strongly with FVIIa/sTF. Again, experimentally determined structures are needed to resolve these anomalies.
3.4. Interactions of FVIIa/sTF with the EGF1 Domains of FX/Xa and FIX/IXa
The experimental evidence indicates that EGF1 domain participates in the complex formation of FVIIa/sTF with FIX (66,85) and FX (66,70,86), and in the reciprocal activation of FVII/TF (67). In FIX/FIXa, side chains of residues E52, N54 and N58 interact with complimentary side chains of K15/N107, N199/E105 and E99 in sTF, respectively (Figure 4). Similarly in the Ss model, side chains of residues E51, S53 and N57 in FX/Xa interact with side chains of K15, E105 and E99 in sTF, respectively. In addition, side chains of T52, Q58 and K60 in FX/Xa interact with side chains of T13, T101 and T197 in sTF, respectively. Carbonyl oxygen of C50 in FXa may also interact with N199 side chain nitrogen of sTF (Figure 4). Thus, based on the Ss model, sTF interacts stronger with EGF1 domain of FX/Xa than of FIX/IXa.
Figure 4.
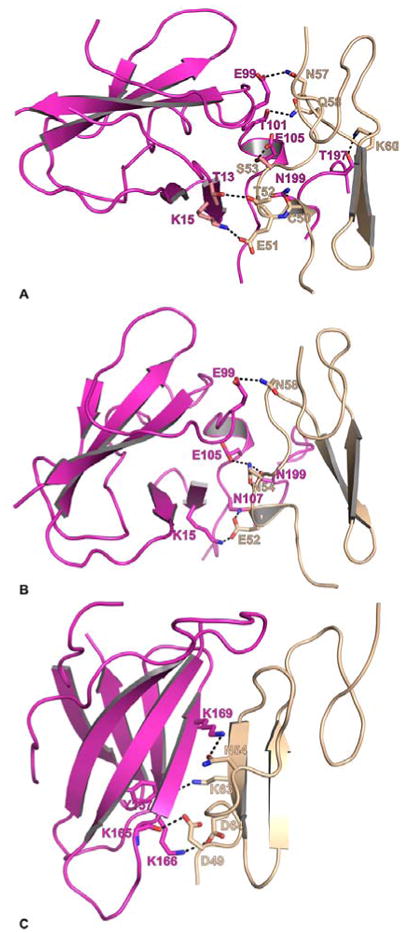
Modeled sTF interactions with the EGF1 domain of FXa, FIXaand FIXalpha. The interacting residues between sTF (magenta) and EGF1 (wheat) are shown in stick representation. The carbon atoms are magenta in sTF, and wheat in FVIIa-EGF1. The N and O atoms are blue and red, respectively. The black dashed lines represent hydrogen bonds between the interacting residues. A) The modeled interaction between FXa-EGF1 domain and sTF. FXa-EGF1:sTFs—C50:N199, E51:K15, T52:T13, S53:E105, N57:E99, Q58:T101 and K60:T197 B) The modeled interaction between FIXa-EGF1 domain and sTF. FIXa-EGF1:sTFs—E52:K15/N107, N54:N199/E105 and N58:E99. C) The modeled interaction between FIXalpha-EGF1 domain and sTF. FIXalpha-EGF1:sTFs—D49:K165, N54:K169, K63:Y157 and D64:K166.
The mode of interactions of EGF1 domain of FX/Xa in the CheA/CheB models is distinct from the Ss/Se models. In the CheA/CheB models, the E51 of FXa interacts with K201 of sTF (72); the residue E51 of FXa interacts with K15/N107 of sTF in the Ss model. None of the other residues of FXa proposed in the Ss/Se models interact with sTF in the CheA/CheB models. In the CheB model, G66 of FXa interacts with E24 of sTF, E77 of FXa interacts with K41/E99 of sTF, and K79 of FXa interacts with E105 of sTF. In addition, E74 of FXa interacts with the K170D of the protease domain of FVIIa. Thus, EGF1 domain of FXa interacts with sTF and FVIIa in the CheA/CheB models.
In the FVIIa/sTF-FIXalpha model, the mode of interaction is different from the FVIIa/sTF-FIXa model. The N54 in FIXalpha interacts with K169 of sTF, whereas N54 in FIXa interacts with N199 of sTF. As compared to FIXa, all other residues in FIXalpha that are proposed to interact with sTF are different (Figures 4B and 4C). In FIXalpha, D49, D64, and K63 interact with K165, K166 and Y157 of sTF, respectively.
3.5. Interactions of FVIIa-protease Domain with the Protease Domains of FXa, FIXa and FIXalpha
The possible mode of interactions between FVIIa protease domain with the protease domains of FXa, FIXa and FIXalpha are shown in Figure 5 A, B and C, respectively. In FVIIa/sTF-FXa Ss model (Figure 5A), the side chains of Q-c20{200}, K-c23{203}, K-c134{318}, K-c186{371}, R-c202{387}, K-c204{389} and D-c205{390} from FXa interact with main and or side chains of L-c153{295}, T-c151{293}, D-c186{334}, E-c154{296}, S-c188A{336}, D-c186{334} and D-c146{289}/R-c147{290} from FVIIa, respectively. In addition, main chain oxygen of V-c160{343} from FXa interact with side chain nitrogen of K-c20{157} from FVIIa. Notably, R-c147{290} in FVIIa is also implicated to be important in FX activation (87).
Figure 5.
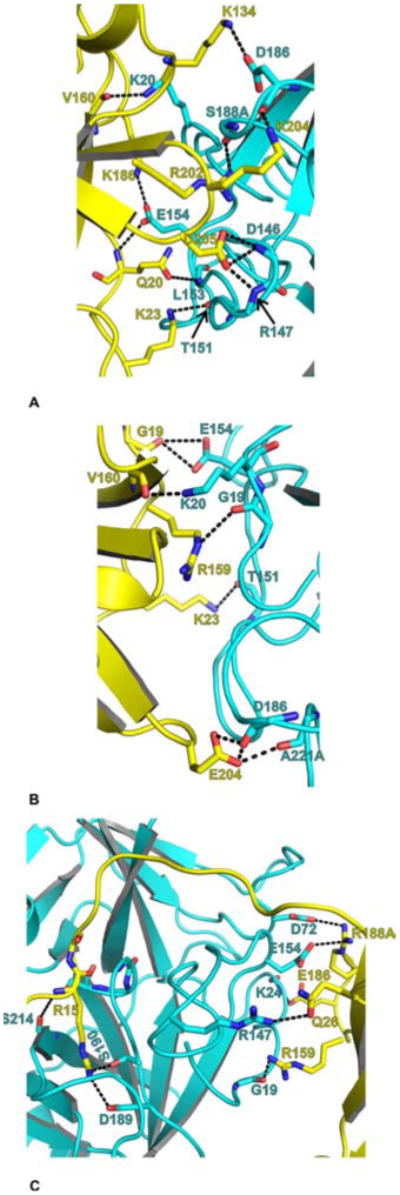
Modeled FVIIa-protease domain interactions with the protease domains of FXa, FIXa and FIXalpha. The interacting residues between FVIIa-protease domain (Cyan) and the protease domains of FXa, FIXa and FIXalpha (yellow) are shown in stick representation. The carbon atoms are cyan in FVIIa, and yellow in FXa, FIXa and FIXalpha. The N and O atoms are blue and red, respectively. The hydrogen bonds are shown as dashed lines. A) The modeled interaction between FXa protease domain and FVIIa protease domain. FXa-protease:FVIIa-protease—Q20:L153/E154; K23:T151; K134:D186; V160:K20; K186:E154; R202:S188A; K204:D186; and D205:D146/R147. B) The modeled interaction between FIXa protease domain and FVIIa protease domain. FIXa-protease:FVIIa-protease—G19:E154; K23:T151; R159:G19; V160:K20 and E204:D186/A221A. C) The modeled interaction between FIXalpha protease domain and FVIIa protease domain. FIXalpha-protease:FVIIa-protease—Q26:R147; R159:G19; E186:K24; R188A:D72/E154. The P1 residue R15 in FIXalpha is shown to interact with D189 and S190 of FVIIa. The typical hydrogen bond observed between the main chain nitrogen of P1 residue (R15 in FIXalpha) and the main chain oxygen of residue S214 in FVIIa is also shown. The chymotrypsin numbering scheme is used to label the residues. The insertion residues are labeled with the number followed by an alphabet A, B, etc.
Predicted interactions of FVIIa-protease domain with FXa in the Se model are shown in Table 1 (72). Comparative interactions in the CheB are also shown in table 1 (72). Clearly, there are differences between the Se and CheB models. Moreover, only three interactions involving K20, D186, and S188A of FVIIa protease domain and V160, K134 and R202 of FXa protease domain are common between the Ss and the Se model.
Table 1. Predicted interactions of FVIIa protease domain with FXa in the Se and the CheB models (72). M, Main chain; S, Side chain; HI, Hydrophobic interaction; LC, light chain.
| Residues in Fxa, Se Model | Interaction | Residues in VIIa | Interaction | Residues in Fxa, CHeB Model |
|---|---|---|---|---|
| E159 | SS | K20 | MS | Y162 |
| Y185 | HI | V21 | HI | P161 |
| V21 | HI | Y185 | ||
| T185B | SS | E26 | ||
| K186 | Ionic | D72 | ||
| E75 | Ionic | K223 | ||
| I137 | HI | L145 | ||
| M157 | HI | L145 | ||
| Y207 | HI | L145 | ||
| R202 | Ionic | D146 | ||
| E138LC | Ionic | R147 | ||
| E138LC | SS | R147 | ||
| Q20 | SM | L153 | ||
| K186 | Ionic | E154 | SS | Y185 |
| E154 | Ionic | K186 | ||
| E154 | SS | K186 | ||
| D92LC | Ionic | R170C | ||
| D95 | SS | R170C | ||
| K170D | SM | Glu74LC | ||
| K170D | Ionic | D92LC | ||
| K170D | Ionic | D95LC | ||
| K170D | ||||
| K134 | Cation-Pi | Y184 | ||
| K134 | Ionic | D186 | ||
| R202 | SM | S188A | SM | K204 |
In FVIIa/sTF-FIXa complex, G-c19{184}, K-c23{188}, R-c159{327}, V-c160{328} and E-c204{374} from FIXa make hydrogen bonds with E-c154{296}, T-c151{293}, G-c19{156}, K-c20{157} and D-c186{334}/A-c221A{369} from FVIIa, respectively. In the FVIIa/sTF-FIXalpha model, the exosite residues Q-c26{191}, R-c159{327}, E-c186{355} and R-c188A{358} from FIXalpha interact with R-c147{290}, G-c19{156}, R-c147{290} and D-c72{212}/E-c154{296} of FVIIa. There are obvious differences between the FIXa and FIXalpha interactions with FVIIa. In addition to the inherent accuracies of the models, the apparent differences also stem from the fact that FIXa is in the enzyme form and the FIXalpha is in the zymogen form.
From the foregoing, it is clear that there are limitations to the accuracy and reliability of the modeled complexes. Despite these limitations, they provide useful information to design mutagenesis experiments. Experimentally determined structures are needed to precisely define the interactive regions in the ternary complexes. In fact, how protease domains of zymogen FX and FIX interact with FVIIa/sTF should be the primary defining interactions between the FVIIa/sTF and FX/FIX. Further, we know from the KM data (65,66) that FX and FIX interact to FVIIa/TF with equal affinity whereas FXa and FIXa interact differently. Knowing how zymogens FX and FIX interact with FVIIa/TF will answer these lingering questions. One also needs to know, how FXa and FIXa interact with FVII/TF in the reciprocal activation.
3.6. Interface between EGF2/Protease domains of FVIIa with sTF
The protease domain of FVIIa has a fold similar to that of other serine proteases. It consists of two beta-barrel structures with three alpha-helical segments. Based upon the experimentally determined structures (PDB id 1DAN and 2A2Q), the interface between the EGF2 and the protease domains of FVIIa and sTF is shown in Figure 6. Residues from all three alpha-helices of FVIIa protease domain make hydrogen bond with N-terminal domain of TF. These involve residues F-c129F{276}, R-c134{277}, T-c165{307}, Q-c166{308}, D-c167{309}, and R-c230{379} of FVIIa with S39, D44, W45, E91, Y94 and N96 of TF. In addition K85 of EGF2 domain makes hydrogen bond with D61 of TF.
Figure 6.
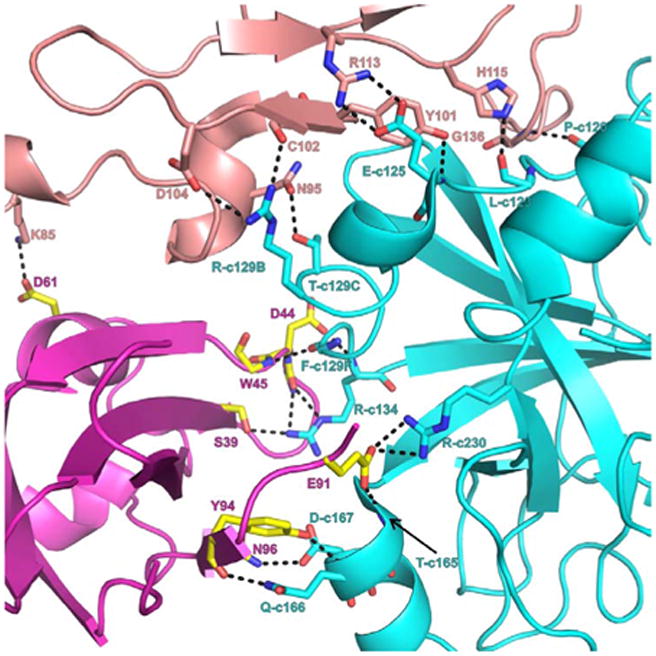
FVIIa-protease, FVIIa-EGF2 and sTF interface interactions. Interactive residues at the interface region of FVIIa protease domain (cyan), EGF2 domain (salmon) and sTF (magenta and yellow) are shown in stick representation. For clarity, either the side chain or main chain atoms that are involved in hydrogen bonding interactions are shown. The hydrogen bonds are represented by black dashed lines. Blue and red represent nitrogen and oxygen atoms, respectively. The residues N95, Y101, C102, D104, R113, H115 and G136 from EGF2 domain interact with P120, L123, E125, R129B and T129C of the protease domain. The residues S39, D44, W45, E91, Y94 and N96 of sTF interact with R134, T165, D167, Q166 and R230 of FVIIa-protease domain. The residue K85 of FVIIa-EGF2 domain makes hydrogen bond with D61 of sTF. Between the FVIIa-EGF2 and sTF, the hydrophobic interactions are more dominant (not shown). The chymotrypsin numbering scheme is used to label the residues.
The loop residues on the either side of the c130 and c160 helical segments of FVIIa interact with TF as well as with the EGF2 domain of FVIIa. The key residues identified to be important in increasing the activity of FVIIa were found in this interface region. The side chain of residue M-c164{306} implicated in allosteric activation of FVIIa is snuggly fitted into the hydrophobic pocket comprised of F76, P92 and Y94 of sTF (28,41). This hydrophobic pocket is lined by sTF residues R74 and N96. Substitution of M-c164{306} by Asp essentially abolishes the TF induced allosteric activation of FVIIa (88). This may be, in part, due to interaction of Asp-c164{306} of FVIIa with side chain of N96 of sTF (88); alternatively Asp-c164{306} could make hydrogen bond with R74. In either case, this could alter the c160 loop in FVIIa and impair its allosteric activation. Moreover, substitution of R74 by Ala in sTF (89) could affect the F76/P92/Y94 hydrophobic pocket and disallow the proper fixation of M-c164{306} in FVIIa. Thus, R74A mutant of sTF appears to have an indirect effect on FVIIa allosteric activation rather than playing a direct role.
The EGF2 domain strongly interacts with the FVIIa-protease domain through the hydrogen bonds and hydrophobic interactions. The residues involved in hydrogen bond formation are shown in Figure 6. EGF2 domain residues C102, D104, N95, Y101, R113, H115, and G136 interact with T-c129C{272}, R-c129B{271}, E-c125{265}, P-c120{260}, and L-c123{263} in the protease domain. In addition, the EGF2 domain stabilizes the FVIIa protease domain by shielding the hydrophobic residues on the surface of the protease domain. In the absence of such contacts, the shielded hydrophobic surface of the protease domain will be exposed to the solvent, which could lead to destabilization of the protease domain. Hence, these two domains are considered to represent a single structural unit.
3.7. Na+-site and Zn2+-sites in the protease domain of FVIIa and their functional roles
In the FVIIa/sTF complex, Na+ ion was located in the region involving c184{332} -c191{340} and c220{368} -c225{374} loops; this region is homologous to the proposed site in FIXa, FXa and APC but not to thrombin (28). Na+ is coordinated to the backbone carbonyl oxygens of Tyr-c184{332}, Ser-c185{333}, His-c224{373}, Thr-c221{370} and two water molecules (Figure 7A). Further, one of the water molecules is hydrogen bonded to Asp-c189{338} . Since there is a three residue insertion in the c183 loop of thrombin (90), Na+ site in thrombin is coordinated to the carbonyl groups of c221 and c224, and four water molecules (91). Binding of Na+ to FVIIa modestly increases its affinity for TF (28). This is consistent with the observations that the Na+-site in FXa and FIXa increases the affinity for FVa and FVIIIa, respectively (92,93). Further, similar to FXa binding to FVa (92) and FIXa binding to FVIIIa (93), binding of FVIIa to TF diminishes the effect of Na+ on its activity (94,95). Notably, zymogen forms of thrombin, FXa, FIXa, and FVIIa each lack Na+-site and is developed during conversion to their respective enzymatic form. Thus, Na+ binding to these enzymes contributes to their structural stability.
Figure 7.
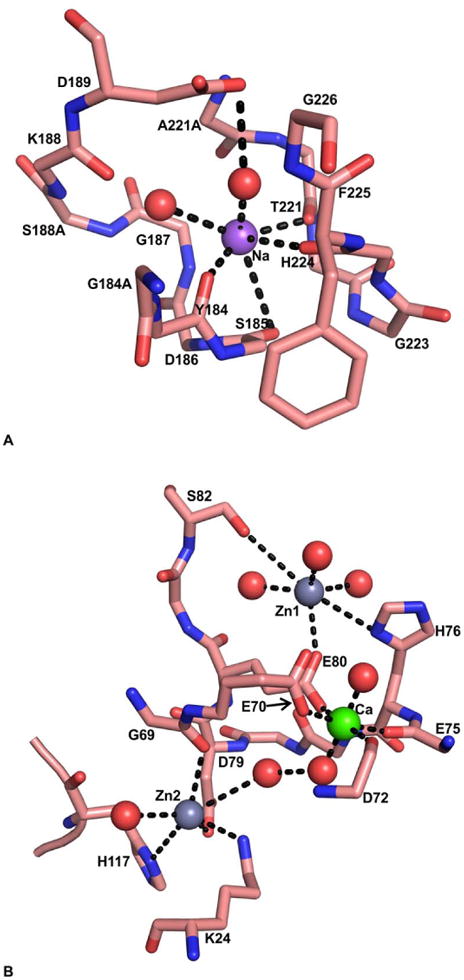
The Ca2+- and putative Na+- and the Zn2+-sites in the protease domain of FVIIa. A) The Na+-site in FVIIa protease domain. The backbone carbonyl oxygens of T221 and H224 from the c220 loop, and Y184 and S185 from the c184 loop serve as ligands for Na+. The side chain of D189 is linked to Na+ through a water molecule. Na+ and water molecules are shown as purple and red spheres, respectively. The Na+ ion coordination to its ligands is shown by dashed lines. B) The Ca2+ site and the Zn2+ sites in FVIIa-protease domain. The residues H76, S82 and E80 for the first Zn2+ (Zn1), and K24, G69, D79, and H117 for the second Zn2+ (Zn2) serve as the protein ligands. The residues E75, D72, E80 and E70 serve as protein ligands for Ca2+. Note that the Zn2+-sites (light gray spheres) are located on each side of the Ca2+-site (green sphere). The coordinated water molecules are shown as red spheres. The black dashed line represents the coordination of metal ions to their respective ligands.
Two putative Zn2+ sites have been identified in the protease domain of FVIIa (28,96). In an earlier report, Zn2+ increased the binding of FVIIa to TF expressed on the bladder carcinoma J82 cells (97). However, in subsequent direct binding studies, it was found that Zn2+ reduces the affinity of FVIIa for sTF (96). Importantly, Zn2+ inhibits the proteolytic activity of FVIIa more potently than FVIIa/TF (96,97). Mutagenesis experiments and modeling studies predicted that the residue His-c76{216} and His-c117{257} are involved in Zn2+ binding to the protease domain of FVIIa (96). The residues His-c76{216}, Glu-c80{220}, Ser-c82{222} and three water molecules provide ligands for the first Zn2+-site and residues His-c117{257}, Lys-c24{161}, Asp-c79{219}, Gly-c69{209} and two water molecules provides ligand for the second Zn2+-site (28). Notably, these interactions are unique for FVIIa. Further, the two Zn2+-sites are located on each side of the protease domain Ca2+-site (Figure 7B). The side chain of residue Glu-c80{220} provides coordination for Zn1 and Ca2+, whereas Zn2 and Ca2+ are interconnected through a network of hydrogen bonds involving two water molecules (Figure 7B). Overlapping Ca2+ and Zn2+ -sites, in part, could provide explanation for experimental observations that the inhibitory effect of Zn2+ is substantially reversed by Ca2+ (96). In this regard, inhibition by Zn2+ is partially overcome by the allosteric activation and stabilization of the FVIIa protease domain induced by binding of TF to residues c164{306} -c167{309} and Ca2+ binding to residues E-c70{210} -E80{220} (96).
3.8. Incompletely formed oxyanion hole in FVIIa/sTF and its induction by substrate/inhibitor
A defining feature of each serine protease, including those contained divergent folds from the chymotrypsin/trypsin, subtilisin, and the carboxypeptidase families, is the presence of an oxyanion-binding site, universally known as the oxyanion hole (98). In the chymotrypsin/trypsin like proteases, Gly-c193 and Ser-c195 backbone amide nitrogens form this binding site. The neculeophilic attack by the hydroxyl group of Ser-c195 on the carbonyl carbon atom of the substrate changes the geometry around this carbon from trigonal planar to tetrahedral. This tetrahedral transition state intermediate is intrinsically unstable because of the negative charge on the peptide carbonyl oxygen atom. However, in serine proteases this charge in the oxyanion hole is stabilized by H-bonds with the amide NH groups of Ser-c195 and Gly-c193 in a site usually referred to as the oxyanion hole. These interactions result in preferential binding of the substrate in the transition state, a necessary requirement for enzyme catalysis.
Although the presence of an oxyanion hole is an essential feature of serine proteases and its geometry is not disrupted by inclusion of benzamidine or pAB at the bottom of S1 site (26,98,99), it is absent in pAB-FVIIa/sTF (PDB id 2A2Q) or the benzamidine-FVIIa/sTF(PDB id 2AER) structures (28). This is shown in Figure 8A. The absence of an oxyanion hole in FVIIa/sTF was initially observed with an amidinophenylurea-based inhibitor (100). However, this was attributed to the unusual nature of the inhibitor, in which the amidino NH groups of the inhibitor induced ∼180 degree rotation of the c192-c193 peptide bond such that the interactions between the NH groups of the inhibitor could occur with the c192 main chain C=O group (100), Thus, a normal conformation of the c192-c193 peptide bond would be changed to the unexpected nonstandard one. Flipped peptide bond between Lys-c192 and Gly-c193 of FVIIa has also been observed by Olivero et al. (101) and by Zbinden et al. (102). In these structures, nitrogen at the para position of benzamidine moiety of the sulfonamide inhibitor (101) or the phenylglycine amide inhibitor (102) makes hydrogen bond with the main chain carbonyl group of Gly-c192 and the hydroxyl group of Ser-c195. Based upon the benzamidine-VIIa/sTF (PDB id 2AER) and the pAB-FVIIa/sTF (PDB id 2A2Q) crystal structures, one can safely conclude that the abnormal conformation of the c192-c193 peptide bond in FVIIa/Stf is not related to the presence of the unusual inhibitors in the active site; instead it is an inherent property of the FVIIa active site.
Figure 8.
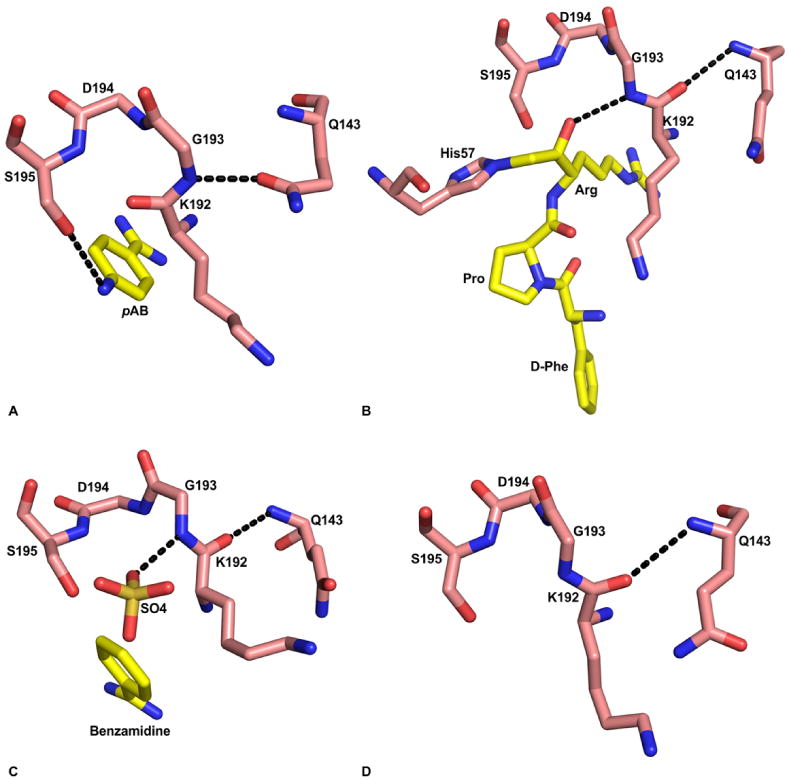
Substrate induced formation of the oxyanion hole in FVIIa protease domain. A) Absence of oxyanion hole in the FVIIa/sTF structure with pAB (PDB id 2A2Q). The hydroxyl group of S195 makes hydrogen bond with the para amino group of pAB. The amide nitrogen of G193 makes hydrogen bond with Odelta of Q143, which prevents formation of the oxyanion hole. The residues 192-195, and Q143 are shown in stick representation. The carbon atoms of protease residues are colored salmon, whereas the carbon atoms in the inhibitor are colored yellow. The ‘N’ and ‘O’ atoms are colored blue and red, respectively. The hydrogen bonds are shown with black dashed lines. B) The fully formed oxyanion hole in FVIIa/sTF with d-FPR (PDB id 2FIR). Note that there is a 180° flip in the peptide bond between K192 and G193 residues as compared to the structure with pAB shown in ‘A’. Now, the carbonyl oxygen of K192 makes hydrogen bond with the backbone amide nitrogen of Q143, and the Odelta of Q143 makes hydrogen bond with R147 (not shown). C) The fully formed oxyanion hole in free FVIIa with benzamidine and sulfate ion in the active site (PDB id 1KLI). The negatively charged sulfate ion in the active site is hydrogen bonded to the amide nitrogen of G193; this interaction induces the formation of oxyanion hole in this structure. D) The K192-G193 peptide bond in standard conformation after removal of the sulfate ion from the FVIIa active site (PDB id 1KLJ). The crystals were obtained under the same condition as that in panel ‘C’; however benzamidine and the sulfate ions were removed by washing the crystal in the buffer lacking benzamidine and sulfate.
Importantly, TF does not increase the activity of FVIIa by inducing the formation of the competent oxyanion hole. It is the occupancy by an oxyanion of the substrate (or substrate analogue) at the active site that provides energy for 180 degree flipping of the c192-c193 peptide bond in FVIIa/sTF crystals. Thus, when the benzamidine-VIIa/sTF crystals were soaked with either EGRck (PDB id 2B8O) or with D-FPRck (PDB id 2FIR), the benzamidine was displaced by the EGR or the DFPR; consequently, the standard conformation of the c192-c193 peptide bond was observed. The D-FPR-FVIIa/sTF active site configuration is shown in Figure 8B. Furthermore, in the benzamidine-FVIIa/sTF crystals the 192-193 conformation is stabilized by the hydrogen bond between Gly-c193 N and the side chain of Gln-c143. In contrast, in the EGR-FVIIa/sTF, d-FPR-FVIIa/sTF, D-FFR-FVIIa/sTF, and the BPTI mutant- FVIIa/sTF (103) crystals, the c192-c193 conformation is stabilized by the hydrogen bond between main chain oxygen of c192 and the main chain N of Q-c143.
The structures (PDB id 1KLI and 1KLJ) of the EGF2/protease domain of FVIIa (104) obtained in the absence of TF support the concept that substrate/inhibitor transition state analog can induce the formation of oxyanion hole in FVIIa. In this structure, the bottom of the S1 site is occupied by benzamidine and top of the S1 site is occupied by a sulfate ion (104). Here, the sulfate anion occupies the position normally occupied by the oxyanion of the carbonyl group of the P1 substrate/inhibitor residue in the transition state. This is shown in Figure 8C. Similar to the negatively charged oxygen in the tetrahedral transition state intermediate during catalysis, oxygen of the sulfate ion in this structure makes a hydrogen bond with amide nitrogen of Gly-c193. Thus, it is quite conceivable that occupancy of sulfate ion in the S1 site of FVIIa induced 180° flip of the c192-c193 peptide bond during crystallization. Moreover, as in the structures of EGR-FVIIa/sTF, D-FPR-FVIIa/sTF, D-FFR-FVIIa/sTF, and mutant BPTI-FVIIa/sTF, carbonyl oxygen of Lys-c192 in the 1KLI structure makes hydrogen bond with the main chain nitrogen of Gln-c143. Upon removal of the sulfate ion by washing the crystal under sulfate free condition, the standard conformation of the oxyanion hole was maintained (Figure 8D). This could be due to stabilization by the hydrogen bond between carbonyl oxygen of c192 and the main chain nitrogen of c143. These observations lend support to a concept that the oxyanion hole in FVIIa is induced by the substrate/inhibitor and not by TF.
The Gla and EGF1 domains of FIX and FX represent the primary recognition determinants in binding to the FVIIa/TF and formation of the ternary complex (66,67,69-71,81-86,105). In the formed ternary complex, the scissile peptide bond sequence in FIX or FX then approaches the active site cleft in FVIIa and induces the formation of the oxyanion hole for efficient proteolysis. This exosite recognition mechanism between the enzyme and the substrate provides unique selectivity in activation of FIX and FX by FVIIa/TF under physiologic conditions in the presence of other plasma proteins.
3.9. Interactions of FVIIa with Kunitz domain-1 of TFPI and of FXa with Kunitz Domain-2 of TFPI
TF induced coagulation is primarily regulated by TFPI. The N-terminal Kunitz domain-1 of TFPI inhibits FVIIa and the Kunitz domain-2 inhibits FXa. The residues of FVIIa that interact with Kunitz domain-1, and of FXa that interact with Kunitz domain-2 have been detailed earlier (35,103,106). Among others, Kunitz domain-1 residues that are important for interaction with FVIIa appear to be D11, R20, and E46. Residue D11 interacts with R147 and K192 of FVIIa; R20 interacts with D60 of FVIIa, and E46 interacts with K60A/K60C of FVIIa. Similarly, among other residues, Kunitz domain-2 residues that are important for interaction with FXa appear to be Y17, R32, and E46. Residue Y17 fits into a hydrophobic cavity with possible interactions with R143 and Q151 of FXa; R32 interacts with E39 of FXa, and E46 interacts with K62 of FXa. The TF residues that interact with FXa in the FVIIa/sTF-FXa ternary complex appear to be also involved in the FVIIa/sTF-FXa-TFPI quaternary complex (107). The precise interactions between the Kunitz domain-1 and FVIIa protease domain and between the Kunitz domain-2 and FXa protease domain must await until the experimentally determined structures are available.
4. Perspective and Future Directions
At the time of writing this review, we know the experimentally determined structures of only FVIIa with or without sTF. Further all structures are with active site inhibited FVIIa. Thus how FVIIa become more active when bound to sTF remains difficult to define precisely. To obtain insight in to this observable fact, structures of the protease domain of FVIIa plus/minus sTF without occupancy of the active site with an inhibitor are needed. Experimentally determined structure of the zymogen form of FVII also remains to be determined. These are daunting tasks and challenges that need to be undertaken. Further, sodium site and the zinc sites in FVIIa need to be confirmed by anomalous data collection. Crystals of ternary complexes of FVIIa with FIX and FX are to be obtained in order to understand the differences in affinity of VIIa/TF for FIX/IXa and FX/Xa and their interactive sites. Again, this may be a difficult task since FVIIa will cleave FIX and FX when zymogens are used. One way is to use Ser-c195Ala mutant of FVIIa for such studies. Recently, in addition to calcium, magnesium has been implicated in promoting the TF-induced coagulation that has not been addressed structurally. Finally, to understand the molecular regulation in TF-induced coagulation, one needs to obtain crystals of FVIIa/FXa/TFPI/sTF complex and solve its structure.
Acknowledgments
We thank Chang Jun Lee and Lee G. Pedersen of University of North Carolina, Chapel Hill for valuable suggestions and comments.
Abbreviations used are
- F
Factor
- FVIIa
Factor VIIa
- FIXa
Factor IXa
- FIXalpha
Factor IXalpha
- FXa
Factor Xa
- TF
Tissue factor
- sTF
soluble TF
- PL
phospholipid
- TFPI
Tissue factor pathway inhibitor
- BPTI
bovine pancreatic trypsin inhibitor
- Ss model
Scripps static model
- Se model
Scripps solution-equilibrated model
- CheA model
Original Chapel Hill solution-equilibrated model A
- CheB model
Current Chapel Hill solution-equilibrated model B
- pAB
p-aminobenzamidine
- EGRck
Glu-Gly-Arg-chloromethylketone
- D-FPRck
D-Phe-Pro-Arg-chloromethylketone
- D-FFR
D-Phe-Phe-Arg-chloromethylketone
Footnotes
For consistency, the numbering system used is that of bovine pancreatic trypsin inhibitor (BPTI) and chymotrypsin. The chymotrypsin number is preceded by the letter c. Where appropriate, the residue numbers for the proteases as well as for the TFPI are given in curly brackets. When insertions occur, the residue number is followed by the capital letter as A,B,C,D, etc. Enzyme S1, S2, S3 subsite numbering and substrate P1, P2, P3 subsite numbering are according to Schechter and Berger (108) The numbering system used for the metals in the Gla domain is that of Tulinsky, who originally solved the structure of prothrombin fragment 1 (53). It should also be noted that factor IX residues in the Gla and EGF1 domains are comparable to the FX residues with a reduction of one in the numbering system.
References
- 1.Broze GJ, Jr, Majerus PW. Purification and properties of human coagulation factor VII. J Biol Chem. 1980;255:1242–1247. [PubMed] [Google Scholar]
- 2.Bajaj SP, Rapaport SI, Brown SF. Isolation and characterization of human factor VII. Activation of factor VII by factor Xa. J Biol Chem. 1981;256:253–259. [PubMed] [Google Scholar]
- 3.Greenberg DL, Davie EW. The blood coagulation factors—their complementary DNAs, genes, and expression. In: Colman RW, Marder VJ, Clowes AW, George JN, Goldhaber SZ, editors. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. 5th. Chapter 3. New York: Lippincott Williams and Wilkins; 2006. pp. 21–57. [Google Scholar]
- 4.Nemerson Y, Repke D. Tissue factor accelerates the activation of coagulation factor VII: the role of a bifunctional coagulation cofactor. Thromb Res. 1985;40:351–358. doi: 10.1016/0049-3848(85)90270-1. [DOI] [PubMed] [Google Scholar]
- 5.Rao LVM, Rapaport SI. Activation of factor VII bound to tissue factor—A key early step in the tissue factor pathway of blood coagulation. Proc Natl Acad Sci USA. 1988;85:6687–6691. doi: 10.1073/pnas.85.18.6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Williams EB, Krishnaswamy S, Mann KG. Zymogen/enzyme discriminations using peptide chloromethyl ketones. J Biol Chem. 1989;264:7536–7545. [PubMed] [Google Scholar]
- 7.Wildgoose P, Berkner KL, Kisiel W. Synthesis, purification and characterization of an Arg152Glu site-directed mutant of recombinant human blood clotting factor VII. Biochemistry. 1990;29:3413–3420. doi: 10.1021/bi00465a039. [DOI] [PubMed] [Google Scholar]
- 8.Radcliffe R, Nemerson Y. Activation and control of factor VII by activated factor X and thrombin. J Biol Chem. 1975;250:388–395. [PubMed] [Google Scholar]
- 9.Rao LVM, Bajaj SP, Rapaport SI. Activation of factor VII during clotting in vitro. Blood. 1985;65:218–226. [PubMed] [Google Scholar]
- 10.Nagagaki T, Foster DC, Berkner KL, Kisiel W. Initiation of the extrinsic pathway of blood coagulation— Evidence for the tissue factor dependent autoactivation of human coagulation factor VII. Biochemistry. 1991;30:10819–10824. doi: 10.1021/bi00109a001. [DOI] [PubMed] [Google Scholar]
- 11.Neuenschwander PF, Fiore MM, Morrissey JH. Factor VII autoactivation proceeds via interaction of distinct protease-cofactor and zymogen-cofactor complexes. J Biol Chem. 1993;268:21489–21492. [PubMed] [Google Scholar]
- 12.Butenas S, Mann KG. Kinetics of human factor VII activation. Biochemistry. 1996;35:1904–1910. doi: 10.1021/bi951768c. [DOI] [PubMed] [Google Scholar]
- 13.Bazan JF. Structural design and molecular evaluation of a cytokine receptor superfamily. Proc Natl Acad Sci USA. 1990;87:6934–6938. doi: 10.1073/pnas.87.18.6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harlos K, Martin DMA, O'Brien DP, Jones EY, Stuart D, Polikarpov DI, Miller A, Tuddenham EGD, Boys CWG. Crystal structure of the extracellular region of human tissue factor. Nature. 1994;370:662–666. doi: 10.1038/370662a0. [DOI] [PubMed] [Google Scholar]
- 15.Muller YA, Ultsch MH, Kelley RF, de Vos AM. Structure of the extracellular domain of human tissue factor—Location of the factor VIIa binding site. Biochemistry. 33:10864–10870. doi: 10.1021/bi00202a003. [DOI] [PubMed] [Google Scholar]
- 16.Muller YA, Ultsch MH, de Vos AM. The crystal structure of the extracellular domain of human tissue factor refined to 1.7 angstrom resolution. J Mol Biol. 1996;256:144–159. doi: 10.1006/jmbi.1996.0073. [DOI] [PubMed] [Google Scholar]
- 17.Huang M, Syed R, Stura EA, Stone MJ, Stefanko RS, Ruf W, Edgington TS, Wilson IA. The mechanism of an inhibitory antibody on TF-initiated blood coagulation revealed by the crystal structures of human tissue factor, Fab 5G9 and TF.G9 complex. J Mol Biol. 1998;275:873–894. doi: 10.1006/jmbi.1997.1512. [DOI] [PubMed] [Google Scholar]
- 18.Ruf W, Rehemtulla A, Morrissey JH, Edgington TS. Phospholipid-independent and -dependent interactions required for tissue factor receptor and cofactor function. J Biol Chem. 1991;266:2158–2166. [PubMed] [Google Scholar]
- 19.Waxman E, Ross JBA, Laue TM, Guha A, Thiruvikraman SV, Lin TC, Konigsberg WH, Nemerson Y. Tissue factor and its extracellular soluble domain—The relationship between intermolecular association with factor VIIa and enzymatic activity of the complex. Biochemistry. 1992;31:3998–4003. doi: 10.1021/bi00131a015. [DOI] [PubMed] [Google Scholar]
- 20.Neuenschwander PF, Morrissey JH. Deletion of the membrane anchoring region of tissue factor abolishes autoactivation of factor VII but not cofactor function. Analysis of a mutant with a selective deficiency in activity. J Biol Chem. 1992;267:14477–14482. [PubMed] [Google Scholar]
- 21.Butenas S, Lawson JH, Kalafatis M, Mann KG. Cooperative interaction of divalent metal ions, substrate, and tissue factor with factor VIIa. Biochemistry. 1994;33:3449–3456. doi: 10.1021/bi00177a039. [DOI] [PubMed] [Google Scholar]
- 22.Neuenschwander PF, Morrissey JH. Roles of the membrane-interactive regions of factor VIIa and tissue factor. The factor VIIa Gla domain is dispensable for binding to tissue factor but important for activation of factor X. J Biol Chem. 1994;269:8007–8013. [PubMed] [Google Scholar]
- 23.Sabharwal AK, Birktoft JJ, Gorka J, Wildgoose P, Petersen LC, Bajaj SP. High affinity Ca(2+)-binding site in the serine protease domain of human factor VIIa and its role in tissue factor binding and development of catalytic activity. J Biol Chem. 1995;270:15523–15530. doi: 10.1074/jbc.270.26.15523. [DOI] [PubMed] [Google Scholar]
- 24.Higashi S, Matsumoto N, Iwanaga S. Molecular mechanism of tissue factor-mediated acceleration of factor VIIa activity. J Biol Chem. 1996;271:26569–26574. doi: 10.1074/jbc.271.43.26569. [DOI] [PubMed] [Google Scholar]
- 25.Dickinson CD, Kelly CR, Ruf W. Identification of surface residues mediating tissue factor binding and catalytic function of the serine protease factor VIIa. Proc Natl Acad Sci USA. 1996;93:14379–14384. doi: 10.1073/pnas.93.25.14379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huber R, Bode RW. Structural basis of the activation and action of trypsin. Acc Chem Res. 1978;11:114–122. [Google Scholar]
- 27.Rand KD, Jørgensen TJ, Olsen OH, Persson E, Jensen ON, Stennicke HR, Andersen MD. Allosteric activation of coagulation factor VIIa visualized by hydrogen exchange. J Biol Chem. 2006;281:23018–23024. doi: 10.1074/jbc.M602968200. [DOI] [PubMed] [Google Scholar]
- 28.Bajaj SP, Schmidt AE, Agah S, Bajaj MS, Padmanabhan K. High resolution structures of p-aminobenzamidine- and benzamidine-VIIa/soluble tissue factor: unpredicted conformation of the 192-193 peptide bond and mapping of Ca2+, Mg2+, Na+, and Zn2+ sites in factor VIIa. J Biol Chem. 2006;281:24873–24888. doi: 10.1074/jbc.M509971200. [DOI] [PubMed] [Google Scholar]
- 29.Ruf W, Dickinson CD. Allosteric regulation of the cofactor-dependent serine protease coagulation factor VIIa. Trends Cardiovasc Med. 1998;8:350–356. doi: 10.1016/s1050-1738(98)00031-0. [DOI] [PubMed] [Google Scholar]
- 30.Pike AW, Brzozowski AM, Roberts SM, Olsen OH, Persson E. Structure of human factor VIIa and its implications for the triggering of blood coagulation. Proc Natl Acad Sci USA. 1999;96:8925–8930. doi: 10.1073/pnas.96.16.8925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kemball-Cook G, Johnson DJ, Tuddeham EG, Harlos K. Crystal structure of active site-inhibited coagulation factor VIIa (des-Gla) J Struct Biol. 1999;127:213–223. doi: 10.1006/jsbi.1999.4158. [DOI] [PubMed] [Google Scholar]
- 32.Petrovan RJ, Ruf W. Residue Met156 contributes to the labile enzyme conformation of coagulation factor VIIa. J Biol Chem. 2001;276:6616–6620. doi: 10.1074/jbc.M004726200. [DOI] [PubMed] [Google Scholar]
- 33.Persson E, Bak H, Østergaard A, Olsen OH. Augmented intrinsic activity of Factor VIIa by replacement of residues 305, 314, 337 and 374: evidence of two unique mutational mechanisms of activity enhancement. Biochem J. 2004;379:497–503. doi: 10.1042/BJ20031596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Persson E, Olsen OH. Allosteric activation of coagulation factor VIIa. Front Biosci. 2011;17:3156–3163. doi: 10.2741/3903. [DOI] [PubMed] [Google Scholar]
- 35.Bajaj MS, Birktoft JJ, Steer SA, Bajaj SP. Structure and biology of tissue factor pathway inhibitor. Thromb Haemost. 2001;86:959–972. [PubMed] [Google Scholar]
- 36.Wun TC, Kretzmer KK, Girard TJ, Miletich JP, Broze GJ., Jr Cloning and characterization of a cDNA coding for the lipoprotein-associated coagulation inhibitor shows that it consists of three tandem Kunitz-type inhibitory domains. J Biol Chem. 1988;263:6001–6004. [PubMed] [Google Scholar]
- 37.Girard TJ, Warren LA, Novotny WF, Likert KM, Brown SG, Miletich JP, Broze GJ., Jr Functional significance of the Kunitz-type inhibitory domains of lipoprotein-associated coagulation inhibitor. Nature. 1989;338:518–520. doi: 10.1038/338518a0. [DOI] [PubMed] [Google Scholar]
- 38.Baugh RJ, Broze GJ, Jr, Krishnaswamy S. Regulation of extrinsic pathway factor Xa formation by tissue factor pathway inhibitor. J Biol Chem. 1998;273:4378–4386. doi: 10.1074/jbc.273.8.4378. [DOI] [PubMed] [Google Scholar]
- 39.Leytus SP, Foster DC, Kurachi K, Davie EW. Gene for human factor X: a blood coagulation factor whose gene organization is essentially identical with that of factor IX and protein C. Biochemistry. 1986;25:5098–5102. doi: 10.1021/bi00366a018. [DOI] [PubMed] [Google Scholar]
- 40.Yoshitake S, Schach BG, Foster DC, Davie EW, Kurachi K. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B) Biochemistry. 1985;24:3736–3750. doi: 10.1021/bi00335a049. [DOI] [PubMed] [Google Scholar]
- 41.Banner DW, D'Arcy A, Chene C, Winkler FK, Guha A, Konigsberg WH, Nemerson Y, Kirchhofer D. The crystal structure of the complex of blood coagulation factor VIIa with soluble tissue factor. Nature. 1996;380:41–46. doi: 10.1038/380041a0. [DOI] [PubMed] [Google Scholar]
- 42.Ruf W, Schullek JR, Stone MJ, Edgington TS. identification of factor VII recognition determinants in both structural modules of the predicted cytokine receptor homology domain. Biochemistry. 1994;33:1565–1572. doi: 10.1021/bi00172a037. [DOI] [PubMed] [Google Scholar]
- 43.Schullek JR, Ruf W, Edgington TS. Key ligand interface residues in tissue factor contribute independently to factor VIIa binding. J Biol Chem. 1994;269:19399–19403. [PubMed] [Google Scholar]
- 44.Gibbs CS, McCurdy SN, Leung LL, Paborsky LR. Identification of the factor VIIa binding site on tissue factor by homologous loop swap and alanine scanning mutagenesis. Biochemistry. 1994;33:14003–14010. doi: 10.1021/bi00251a007. [DOI] [PubMed] [Google Scholar]
- 45.Ruf W, Kelly CR, Schullek JR, Martin DM, Polikarpov I, Boys CW, Tuddenham EG, Edgington TS. Energetic contributions and topographical organization of ligand binding residues of tissue factor. Biochemistry. 1995;34:6310–6315. doi: 10.1021/bi00019a008. [DOI] [PubMed] [Google Scholar]
- 46.Kelley RF, Costas KE, O'Connell MP, Lazarus RA. Analysis of the factor VIIa binding site on human tissue factor: effects of tissue factor mutations on the kinetics and thermodynamics of binding. Biochemistry. 1995;34:10383–10392. doi: 10.1021/bi00033a009. [DOI] [PubMed] [Google Scholar]
- 47.Toomey JR, Smith KJ, Stafford DW. Localization of the human tissue factor recognition determinant of human factor VIIa. J Biol Chem. 1991;266:19198–19202. [PubMed] [Google Scholar]
- 48.Clarke BJ, Ofosu FA, Sridhara S, Bona RD, Rickles FR, Blajchman MA. The first epidermal growth factor domain of human coagulation factor VII is essential for binding with tissue factor. FEBS Lett. 1992;298:206–210. doi: 10.1016/0014-5793(92)80058-o. [DOI] [PubMed] [Google Scholar]
- 49.Kazama Y, Pastuszyn A, Wildgoose P, Hamamoto T, Kisiel W. Isolation and characterization of proteolytic fragments of human factor VIIa which inhibit the tissue factor-enhanced amidolytic activity of factor VIIa. J Biol Chem. 1993;268:16231–16240. [PubMed] [Google Scholar]
- 50.Petersen LC, Schiødt J, Christensen U. Involvement of the hydrophobic stack residues 39-44 of factor VIIa in tissue factor interactions. FEBS Lett. 1994;347:73–79. doi: 10.1016/0014-5793(94)00513-3. [DOI] [PubMed] [Google Scholar]
- 51.O'Brien DP, Kemball-Cook G, Hutchinson D, Martin M, Johnson DJ, Byfield PG, Takamiya O, Tuddenham EG, McVey JH. Surface plasmon resonance studies of the interaction between factor VII and tissue factor. Demonstration of defective tissue factor binding in a variant FVII molecule (FVII-R79Q) Biochemistry. 1994;33:14162–14169. doi: 10.1021/bi00251a027. [DOI] [PubMed] [Google Scholar]
- 52.Persson E. Influence of the gamma-carboxyglutamic acid-rich domain and hydrophobic stack of factor VIIa on tissue factor binding. Haemostasis. 1996;26:31–34. doi: 10.1159/000217237. [DOI] [PubMed] [Google Scholar]
- 53.Soriano-Garcia M, Padmanabhan K, de Vos AM, Tulinsky A. The Ca2+ ion and membrane binding structure of the Gla domain of Ca-prothrombin fragment 1. Biochemistry. 1992;31:2554–2566. doi: 10.1021/bi00124a016. [DOI] [PubMed] [Google Scholar]
- 54.Mosbaek CR, Nolan D, Persson E, Svergun DI, Bukrinsky JT, Vestergaard B. Extensive small-angle X-ray scattering studies of blood coagulation factor VIIa reveal interdomain flexibility. Biochemistry. 2010;49:9739–9745. doi: 10.1021/bi1011207. [DOI] [PubMed] [Google Scholar]
- 55.Minazzo AS, Darlington RC, Ross JB. Loop dynamics of the extracellular domain of human tissue factor and activation of factor VIIa. Biophys J. 2009;96:681–692. doi: 10.1016/j.bpj.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vadivel K, Agah S, Messer AS, Cascio D, Bajaj MS, Krishnaswamy S, Esmon CT, Padmanabhan K, Bajaj SP. Mg2+ is required for optimal folding of the γ-carboxyglutamic acid (Gla) domains of vitamin K-dependent clotting factors at physiological Ca2+ ASH Meeting 2011. abstract number 1172. [Google Scholar]
- 57.Oganesyan V, Oganesyan N, Terzyan S, Qu D, Dauter Z, Esmon NL, Esmon CT. The crystal structure of the endothelial protein C receptor and a bound phospholipid. J Biol Chem. 2002;277:24851–24854. doi: 10.1074/jbc.C200163200. [DOI] [PubMed] [Google Scholar]
- 58.Shikamoto Y, Morita T, Fujimoto Z, Mizuno H. Crystal structure of Mg2+- and Ca2+-bound Gla domain of factor IX complexed with binding protein. J Biol Chem. 2003;278:24090–24094. doi: 10.1074/jbc.M300650200. [DOI] [PubMed] [Google Scholar]
- 59.Agah S, Bajaj SP. Role of magnesium in factor XIa catalyzed activation of factor IX: calcium binding to factor IX under physiologic magnesium. J Thromb Haemost. 2009;7:1426–1428. doi: 10.1111/j.1538-7836.2009.03506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Courcy B, Pedersen LG, Parisel O, Gresh N, Silvi B, Pilmé J, Piquemal JP. Understanding selectivity of hard and soft metal cations within biological systems using the subvalence concept. I. Application to blood coagulation: direct cation-protein electronic effects vs. indirect interactions through water networks. J Chem Theory Comput. 2010;6:1048–1063. doi: 10.1021/ct100089s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sekiya F, Yamashita T, Atoda H, Komiyama Y, Morita T. Regulation of the tertiary structure and function of coagulation factor IX by magnesium (II) ions. J Biol Chem. 1995;270:14325–14331. doi: 10.1074/jbc.270.24.14325. [DOI] [PubMed] [Google Scholar]
- 62.Persson E, Ostergaard A. Mg2+ binding to the Gla domain of factor X influences the interaction with tissue factor. J Thromb Haemost. 2007;5:1977–1978. doi: 10.1111/j.1538-7836.2007.02661.x. [DOI] [PubMed] [Google Scholar]
- 63.Osterud B, Rapaport SI. Activation of factor IX by the reaction product of tissue factor and factor VII: additional pathway for initiating blood coagulation. Proc Natl Acad Sci USA. 1977;74:5260–5264. doi: 10.1073/pnas.74.12.5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nemerson Y. The Reaction between Bovine Brain Tissue Factor and Factors VII and X*. Biochemistry. 1966;5:601–608. doi: 10.1021/bi00866a029. [DOI] [PubMed] [Google Scholar]
- 65.Lu G, Broze GJ, Jr, Krishnaswamy S. Formation of factors IXa and Xa by the extrinsic pathway: differential regulation by tissue factor pathway inhibitor and antithrombin III. J Biol Chem. 2004;279:17241–17249. doi: 10.1074/jbc.M312827200. [DOI] [PubMed] [Google Scholar]
- 66.Ndonwi M, Broze GJ, Jr, Agah A, Schmidt AE, Bajaj SP. Substitution of the Gla domain in factor X with that of protein C impairs its interaction with factor VIIa/tissue factor: lack of comparable effect by similar substitution in factor IX. J Biol Chem. 2007;282:15632–15644. doi: 10.1074/jbc.M701908200. [DOI] [PubMed] [Google Scholar]
- 67.Ndonwi M, Broze GJ, Bajaj SP. The First Epidermal Growth Factor-like Domains of Factor Xa and IXa are Important for the Activation of the Factor VII-Tissue Factor Complex. J Thromb Haemost. 2005;1:112–118. doi: 10.1111/j.1538-7836.2004.01051.x. [DOI] [PubMed] [Google Scholar]
- 68.Thiec F, Cherel G, Christophe OD. Role of the Gla and first epidermal growth factor-like domains of factor X in the prothrombinase and tissue factor-factor VIIa complexes. J Biol Chem. 2003;278:10393–10399. doi: 10.1074/jbc.M212144200. [DOI] [PubMed] [Google Scholar]
- 69.Chen SW, Pellequer JL, Schved JF, Giansily-Blaizot M. Model of a ternary complex between activated factor VII, tissue factor and factor IX. Thromb Haemost. 2002;88:74–82. [PubMed] [Google Scholar]
- 70.Norledge BV, Petrovan RJ, Ruf W, Olson AJ. The tissue factor/factor VIIa/factor Xa complex: a model built by docking and site-directed mutagenesis. Proteins. 2003;53:640–648. doi: 10.1002/prot.10445. [DOI] [PubMed] [Google Scholar]
- 71.Venkateswarlu D, Duke RE, Perera L, Darden TA, Pedersen LG. An all-atom solution-equilibrated model for human extrinsic blood coagulation complex (sTF-VIIa-Xa): a protein-protein docking and molecular dynamics refinement study. J Thromb Haemost. 2003;1:2577–2588. doi: 10.1111/j.1538-7836.2003.00421.x. [DOI] [PubMed] [Google Scholar]
- 72.Lee CJ, Chandrasekaran V, Wu S, Duke RE, Pedersen LG. Recent estimates of the structure of the factor VIIa (FVIIa)/tissue factor (TF) and factor Xa (FXa) ternary complex. Thromb Res. 2010;125:S7–S10. doi: 10.1016/j.thromres.2010.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mandell JG, Roberts VA, Pique ME, Kotlovyi V, Mitchell JC, Nelson E, Tsigelny I, Ten Eyck LF. Protein Docking Using Continuum Electrostatics and Geometric Fit. Prot Eng. 2001;14:105–113. doi: 10.1093/protein/14.2.105. [DOI] [PubMed] [Google Scholar]
- 74.Duncan B, Olson A. Applications of evolutionary programming for the prediction of protein–protein interactions. Evolutionary programming V: Proceedings of the 5th Annual Conference on Evolutionary Programming; Boston. MIT Press; 1996. [Google Scholar]
- 75.Gabb HA, Jackson RM, Sternberg MJ. Modelling protein docking using shape complementarity, electrostatics and biochemical information. J Mol Biol. 1997;272:106–120. doi: 10.1006/jmbi.1997.1203. [DOI] [PubMed] [Google Scholar]
- 76.Rao Z, Handford P, Mayhew M, Knott V, Brownlee GG, Stuart D. The structure of a Ca2+-binding epidermal growth factor-like domain: its role in protein-protein interactions. Cell. 1995;82:131–141. doi: 10.1016/0092-8674(95)90059-4. [DOI] [PubMed] [Google Scholar]
- 77.Vadivel K, Schmidt A, Bajaj SP. Does protease domain of human factor ixa contain a sodium site? Evidence from the 1.64 angstrom resolution crystal structure. ASH Meeting; 2011; abstract number 2246. [Google Scholar]
- 78.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for the building of protein models in electron density maps and the location of errors in these models. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 79.Brooks BR, Brooks CL, 3rd, Mackerell AD, Jr, Nilsson L, Petrelle RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. CHARMM: the biomolecular simulation program. J Comp Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huang Q, Neuenschwander PF, Rezaie AR, Morrissey JH. Substrate recognition by tissue factor-factor VIIa. Evidence for interaction of residues Lys165 and Lys166 of tissue factor with the 4-carboxyglutamate-rich domain of factor X. J Biol Chem. 1996;271:21752–21757. doi: 10.1074/jbc.271.36.21752. [DOI] [PubMed] [Google Scholar]
- 81.Kirchhofer D, Lipari MT, Moran P, Eigenbrot C, Kelley RF. The tissue factor region that interacts with substrates factor IX and Factor X. Biochemistry. 2000;39:7380–7387. doi: 10.1021/bi000182+. [DOI] [PubMed] [Google Scholar]
- 82.Kirchhofer D, Eigenbrot C, Lipari MT, Moran P, Peek M, Kelley RF. The tissue factor region that interacts with factor Xa in the activation of factor VII. Biochemistry. 2001;40:675–682. doi: 10.1021/bi002013v. [DOI] [PubMed] [Google Scholar]
- 83.Manithody C, Yang L, Rezaie AR. Identification of a basic region on tissue factor that interacts with the first epidermal growth factor-like domain of factor X. Biochemistry. 2007;46:3193–3199. doi: 10.1021/bi6025193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ruf W, Shobe J, Rao SM, Dickinson CD, Olson A, Edgington TS. Importance of factor VIIa Gla-domain residue Arg-36 for recognition of the macromolecular substrate factor X Gla-domain. Biochemistry. 1999;38:1957–1966. doi: 10.1021/bi982254r. [DOI] [PubMed] [Google Scholar]
- 85.Zhong D, Smith KJ, Birktoft JJ, Bajaj SP. First epidermal growth factor-like domain of human blood coagulation factor IX is required for its activation by factor VIIa/tissue factor but not by factor XIa. Proc Natl Acad Sci USA. 1994;91:3574–3578. doi: 10.1073/pnas.91.9.3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhong D, Bajaj MS, Schmidt AE, Bajaj SP. The N-terminal epidermal growth factor-like domain in factor IX and factor X represents an important recognition motif for binding to tissue factor. J Biol Chem. 2002;277:3622–31. doi: 10.1074/jbc.M111202200. [DOI] [PubMed] [Google Scholar]
- 87.Ruf W. Factor VIIa residue Arg290 is required for efficient activation of the macromolecular substrate factor X. Biochemistry. 1994;33:11631–6. doi: 10.1021/bi00204a026. [DOI] [PubMed] [Google Scholar]
- 88.Persson E, Nielsen LS, Olsen OH. Substitution of aspartic acid for methionine-306 in factor VIIa abolishes the allosteric linkage between the active site and the binding interface with tissue factor. Biochemistry. 2001;40:3251–3256. doi: 10.1021/bi001612z. [DOI] [PubMed] [Google Scholar]
- 89.Kittur FS, Manithody C, Morrissey JH, Rezaie AR. The cofactor function of the N-terminal domain of tissue factor. J Biol Chem. 2004;279:39745–39749. doi: 10.1074/jbc.M406628200. [DOI] [PubMed] [Google Scholar]
- 90.Bajaj SP, Birktoft JJ. Human factor IX and factor IXa. Methods in Enzymology. 1993;222:96–128. doi: 10.1016/0076-6879(93)22009-5. [DOI] [PubMed] [Google Scholar]
- 91.Di Cera E, Guinto ER, Vindigni A, Dang QD, Ayala YM, Wuyi M, Tulinsky A. The Na+ binding site of thrombin. J Biol Chem. 1995;270:22089–22092. doi: 10.1074/jbc.270.38.22089. [DOI] [PubMed] [Google Scholar]
- 92.Camire RM. Prothrombinase assembly and S1 site occupation restore the catalytic activity of FXa impaired by mutation at the sodium-binding site. J Biol Chem. 2002;277:37863–37870. doi: 10.1074/jbc.M203692200. [DOI] [PubMed] [Google Scholar]
- 93.Schmidt AE, Stewart JE, Mathur A, Krishnaswamy S, Bajaj SP. Na+ site in blood coagulation factor IXa: effect on catalysis and factor VIIIa binding. J Mol Biol. 2005;350:78–91. doi: 10.1016/j.jmb.2005.04.052. [DOI] [PubMed] [Google Scholar]
- 94.Schmidt AE. Ph D thesis. St. Louis University; 2005. Allostery in Serine proteases. [Google Scholar]
- 95.Gopalakrishna K, Rezaie AR. The influence of sodium ion binding on factor IXa activity. Thromb Haemost. 2006;95:936–941. doi: 10.1160/TH06-03-0156. [DOI] [PubMed] [Google Scholar]
- 96.Petersen LC, Olsen OH, Nielsen LS, Freskgård PO, Persson E. Binding of Zn2+ to a Ca2+ loop allosterically attenuates the activity of factor VIIa and reduces its affinity for tissue factor. Protein Sci. 2000;9:859–866. doi: 10.1110/ps.9.5.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pedersen AH, Lund-Hansen T, Komiyama Y, Petersen LC, Oestergård PB, Kisiel W. Inhibition of recombinant human blood coagulation factor VIIa amidolytic and proteolytic activity by zinc ions. Thromb Haemost. 1991;65:528–534. [PubMed] [Google Scholar]
- 98.Perona JJ, Craik CS. Structural basis of substrate specificity in the serine proteases. Protein Sci. 1995;4:337–360. doi: 10.1002/pro.5560040301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hopfner HP, Lang A, Karcher A, Sichler K, Kopetzki E, Brandstetter H, Huber R, Bode W, Engh RA. Coagulation factor IXa: the relaxed conformation of Tyr99 blocks substrate binding. Structure. 1999;7:989–996. doi: 10.1016/s0969-2126(99)80125-7. [DOI] [PubMed] [Google Scholar]
- 100.Klingler O, Matter H, Schudok M, Bajaj SP, Czech J, Lorenz M, Nestler HP, Schreuder H, Wildgoose P. Design, synthesis, and structure-activity relationship of a new class of amidinophenylurea-based factor VIIa inhibitors. Bioorg Med Chem Lett. 2003;13:1463–1437. doi: 10.1016/s0960-894x(03)00168-9. [DOI] [PubMed] [Google Scholar]
- 101.Olivero AG, Eigenbrot C, Goldsmith R, Robarge K, Artis DR, Flygare J, Rawson T, Sutherlin DP, Kadkhodayan S, Beresini M, Elliott LO, DeGuzman GG, Banner DW, Ultsch M, Marzec U, Hanson SR, Refino C, Bunting S, Kirchhofer D. A selective, slow binding inhibitor of factor VIIa binds to a nonstandard active site conformation and attenuates thrombus formation in vivo. J Biol Chem. 2005;280:9160–9169. doi: 10.1074/jbc.M409068200. [DOI] [PubMed] [Google Scholar]
- 102.Zbinden KG, Obst-Sander U, Hilpert K, Kühne H, Banner DW, Böhm HJ, Stahl M, Ackermann J, Alig L, Weber L, Wessel HP, Riederer MA, Tschopp TB, Lavé T. Selective and orally bioavailable phenylglycine tissue factor/factor VIIa inhibitors. Bioorg Med Chem Lett. 2005;15:5344–5352. doi: 10.1016/j.bmcl.2005.04.079. [DOI] [PubMed] [Google Scholar]
- 103.Zhang E, St Charles R, Tulinsky A. Structure of extracellular tissue factor complexed with factor VIIa inhibited with a BPTI mutant. J Mol Biol. 1999;285:2089–2104. doi: 10.1006/jmbi.1998.2452. [DOI] [PubMed] [Google Scholar]
- 104.Sichler K, Banner DW, D'Arcy A, Hopfner KP, Huber R, Bode W, Kresse GB, Kopetzki E, Brandstetter H. Crystal structures of uninhibited factor VIIa link its cofactor and substrate-assisted activation to specific interactions. J Mol Biol. 2002;322:591–603. doi: 10.1016/s0022-2836(02)00747-7. [DOI] [PubMed] [Google Scholar]
- 105.Krishnaswamy S. Exosite-driven substrate specificity and function in coagulation. J Thromb Haemost. 2005;3:54–67. doi: 10.1111/j.1538-7836.2004.01021.x. [DOI] [PubMed] [Google Scholar]
- 106.Burgering MJ, Orbons LP, van der Doelen A, Mulders J, Theunissen HJ, Grootenhuis PD, Bode W, Huber R, Stubbs MT. The second Kunitz domain of human tissue factor pathway inhibitor: cloning, structure determination and interaction with factor Xa. J Mol Biol. 1997;269:395–407. doi: 10.1006/jmbi.1997.1029. [DOI] [PubMed] [Google Scholar]
- 107.Carlsson K, Freskgård PO, Persson E, Carlsson U, Svensson M. Probing the interface between factor Xa and tissue factor in the quaternary complex tissue factor-factor VIIa-factor Xa-tissue factor pathway inhibitor. Eur J Biochem. 2003;270:2576–2582. doi: 10.1046/j.1432-1033.2003.03625.x. [DOI] [PubMed] [Google Scholar]
- 108.Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]