

Abstract
Although formation of urothelial carcinoma of the bladder (UCB) requires multiple steps and proceeds along divergent pathways, the underlying genetic and molecular determinants for each step and pathway remain undefined. By developing transgenic mice expressing single or combinatorial genetic alterations in urothelium, we demonstrated here that overcoming oncogene-induced compensatory tumor barriers was critical for urothelial tumor initiation. Constitutively active Ha-ras (Ras*) elicited urothelial hyperplasia that was persistent and did not progress to tumors over a 10 months period. This resistance to tumorigenesis coincided with increased expression of p53 and all pRb family proteins. Expression of a Simian virus 40 T antigen (SV40T), which disables p53 and pRb family proteins, in urothelial cells expressing Ras* triggered early-onset, rapidly-growing and high-grade papillary UCB that strongly resembled the human counterpart (pTaG3). Urothelial cells expressing both Ras* and SV40T had defective G1/S checkpoint, elevated Ras-GTPase and hyperactivated AKT-mTOR signaling. Inhibition of the AKT-mTOR pathway with rapamycin significantly reduced the size of high-grade papillary UCB but hyperactivated mitogen-activated protein kinase (MAPK). Inhibition of AKT-mTOR, MAPK and STAT3 altogether resulted in much greater tumor reduction and longer survival than did inhibition of AKT-mTOR pathway alone. Our studies provide the first experimental evidence delineating the combinatorial genetic events required for initiating high-grade papillary UCB, a poorly defined and highly challenging clinical entity. Furthermore, they suggest that targeted therapy using a single agent such as rapamycin may not be highly effective in controlling high-grade UCB and that combination therapy employing inhibitors against multiple targets are more likely to achieve desirable therapeutic outcomes.
Introduction
Urothelial carcinoma of the bladder (UCB) is the fifth most common cancer globally and the costliest cancer to treat on a per case basis (1–3). UCB is not homogenous but consists of subtypes characterized by distinct phenotypes, clinical behaviors and genetic alterations (4–10). At presentation, 70–75% of the UCB are of low pathological grade, papillary appearance and confined to the urothelial layer (e.g. pTaG1/2). While these tumors can be initially removed by transurethral resection, they frequently recur, thus requiring additional surgical procedures (11,12). Low-grade urothelial hyperplasias are believed to be the precursors of these tumors (13,14). Genetically, mutations of the components of the receptor tyrosine kinase (RTK)/Ras pathway are extremely prevalent in these tumors, with fibroblast growth factor receptor 3b mutated in 45–75% (15–19), Ha-ras in 15–40% (10,16), PI-3-kinase in 25% (20) and Raf-1 in ∼7% (21) of the tumors. Because most mutations affecting the different components of this pathway do not coexist in a given tumor (22), there is a strong reason to believe that mutations of this signaling pathway occur in at least 90% of the low-grade papillary UCB. Also prevalent in this UCB subtype is the allelic loss of chromosome 9, occurring in over half of the cases (14,23–25).
The second major subtype of UCB accounts for 20–25% of all the UCB and presents as high-grade invasive tumors (e.g. pT1-4). These tumors often assume a highly aggressive clinical course and, despite radical cystectomy and concurrent chemotherapy, about half of them advance to local and distant metastasis for which the 5-year survival rate is only 36 and 6%, respectively (26–29). Patients with high-grade invasive UCB usually do not have a prior history of low-grade papillary UCB, and these two tumor subtypes therefore do not appear to be a continuum in tumor progression (4,15,16,30). Instead, the majority of the high-grade invasive UCB are believed to derive from flat high-grade carcinoma in situ (CIS) or arise de novo (31). Interestingly, mutations of the RTK/Ras pathway components, which occur in over 90% of the low-grade papillary UCB, are relatively uncommon (<20%) in the high-grade invasive UCB. In striking contrast, >50% of the high-grade invasive UCB are associated with mutations and allelic loss of p53 and aberrant expression of retinoblastoma protein (pRb), events that are rare in low-grade papillary UCB (32–35).
Aside from the two major UCB subtypes, a third, much less well-understood subtype, namely the high-grade papillary UCB (e.g. pTaG3), has been suggested to exist. Although constituting ∼3% of all the UCB, the high-grade papillary UCB presents a major challenge in clinical management (5,36). Associated with a high risk of progression to invasive UCB, these tumors are often managed with surgical resection plus local bacillus Calmette–Guérin immunotherapy. When such therapy fails, radical cystectomy is the only remaining option. To date, the few studies that have analyzed the genetic alterations showed that these tumors harbor a significantly greater number of chromosomal abnormalities than the low-grade papillary UCB but much fewer chromosomal changes than the high-grade invasive UCB (6,37). It remains an open question whether the high-grade papillary UCB arise de novo or outgrow from the flat CIS lesion or have progressed from low-grade papillary UCB.
Genetically engineered mice are excellent tools for dissecting the sequential steps of urothelial tumor formation and progression along the divergent phenotypic and genetic pathways. We showed previously that urothelial expression of a constitutively active Ha-ras in transgenic mice could elicit low-grade papillary UCB that strongly resembled its human counterpart (38). The tumor induction was, however, highly dependent on gene dosage, as only homozygous mice bearing two copies of the mutated Ha-ras transgene but not the heterozygous mice bearing one copy of the same transgene, developed early-onset tumors (before 10 months of age) (38,39). Between 1 and 9 months of age, the heterozygous mice had persistent low-grade urothelial hyperplasias that did not progress nor regress. This raised the possibility that one or more tumor barriers that had been established in response to Ras activation prevented urothelial tumor initiation. In another transgenic model, we showed that low-level expression of a Simian virus 40 large T antigen (SV40T) in urothelium elicited exclusively high-grade lesions that bore strong resemblance to flat CIS in humans (40). Interestingly, these CIS-like lesions were also persistent without regression or progression until the mice reached beyond 10 months of age at which point the CIS gradually evolved into high-grade papillary UCB (41). Therefore, data from both of these two transgenic models suggest that activated Ha-ras or SV40T alone at low gene dosages were insufficient to trigger early-onset UCB and that collaborating genetic events are required to initiate UCB.
In the present study, we explored what specific tumor barriers might be present in the urothelial hyperplasias, precursors to the low-grade papillary UCB, in Ha-ras transgenic mice. We tested whether Ras activation, prevalent in human low-grade papillary UCB, was capable of interacting with deficiencies of p53 and pRb pathways, signatures of high-grade invasive UCB, in urothelial tumor initiation. Additionally, we investigated the key signaling molecules whose activation was associated with urothelial tumor formation. Finally, we evaluated the responses of urothelial tumor cells to therapeutic inhibitors and tested the concept that inhibiting multiple signaling targets had a better therapeutic effect than inhibiting a single target. Results from our studies have major implications on the combinatorial molecular events that are necessary to trigger UCB and the potential and challenges of pharmacological therapy of this disease.
Materials and methods
Single and double transgenic mice
Transgenic mice expressing a constitutively active Ha-ras mutant [codon 61 (Q > L); abbreviated as Ras*] in urothelial cells under the control of a 3.6 kb murine uroplakin II (UPII) promoter were generated previously (38). A heterozygous low-copy line (one copy of UPII-Ras* transgene per diploid genome) that reproducibly developed urothelial hyperplasias up to 10 months of age was chosen for this study. Another heterozygous low-copy transgenic line specifically expressed a Simian virus 40 large T antigen (SV40T) in urothelial cells under the control of the same UPII promoter (UPII-SV40T) and reproducibly developed high-grade CIS throughout the urothelium (40,41). Both lines were originally produced and had since been maintained in an inbred FVB/N background in a specific pathogen-free facility. Harboring both the UPII-Ras* and the UPII-SV40T transgenes, 7- to 8-week-old male and female mice from the two lines were crossbred to produce double transgenic mice. Identification of single and double transgenic offspring was carried out by Southern blotting of NcoI-digested tail genomic DNAs using a probe situated at the 3′-end of the UPII promoter. The probe allowed the detection of restriction fragments representing the UPII-Ras* transgene (1.7 kb), the UPII-SV40T transgene (2.7 kb) and the endogenous UPII gene (1.4 kb). All animal studies were conducted in accordance with government guidelines and under an active protocol approved by the Institutional Animal Care and Use Committee.
Reverse transcription–PCR and real-time PCR
The expression of Ras* and/or SV40T in urothelial cells at the messenger RNA level was verified by reverse transcription–PCR. Briefly, urinary bladders freshly dissected out of wild-type, single and double transgenic mice were inverted inside out and their urothelial cells scrapped off using a chemical weighing spatula. After extraction of total RNA using an RNA extraction kit (Invitrogen), reverse transcription and synthesis of the second strand complementary DNA were performed and the double-stranded complementary DNAs were used as a template for PCR. Oligonucleotide primer pairs used were: (i) for Ras* transgene (e.g. rabbit Ras*): forward, 5′-CTAACCAACCCCCTCCTCTC-3′ and reverse, 5′-ATTCGTCCACGAAGTGGTTC-3′; (ii) for control endogenous Ras (e.g. mouse Ras): forward, 5′-CATGTCTACTGGACATCTTA-3′ and reverse, 5′-TCTTGGCTGATGTTTCAATG-3′ and (iii) for SV40T transgene: forward, 5′-GCAGCTAATGGACCTTCTAGG-3′ and reverse, 5′-GCAATTCTGAAGGAAGGTCCT-3′. PCR conditions were 95°C for 3 min for the first cycle; 94°C for 30 s, 55°C for 30 s and 72°C for 30 s for 35 cycles.
The expression levels of growth inhibitors/tumor suppressors (e.g. p16, p19 and p53) in urothelial cells of 3- to 4-week-old UPII-Ras* transgenic mice were assessed using quantitative real-time PCR with a QuantiTect SYBR-Green Kit (Qiagen). Oligonucleotide primer pairs used were (i) for p16: forward, 5′-AGTCCGCTGCAGACAGACTG-3′ and reverse, 5′-CGGGAGAAGGTAGTGGGGTC-3′; (ii) for p19: forward, 5′-CTTGGTCACTGTGAGGATTC-3′ and reverse, 5′-CGGGAGAAGGTAGTGGGGTC-3′ and (iii) for p53: forward, 5′-CACGTACTCTCCTCCCCTCA-3′ and reverse, 5′-ATTTCCTTCCACCCGGATAC-3′. PCR conditions were 95°C for 15 s for the first cycle; 95°C for 15 s, 55°C–58°C for 20 s and 72°C for 30 s for 50 cycles. Amplification of mouse β-actin was carried out in parallel for each sample and used as an internal reference.
Cell cycle distribution
Urinary bladders from 3-week-old mice were inverted inside out and the mucosa was incubated in ice-cold phosphate-buffer saline (PBS) containing 1 mg/ml dispase at 4°C overnight. Bladder mucosa was then gently scraped off, washed with PBS and digested with a 0.2% Trypsin-ethylenediaminetetraacetic acid solution at 37°C for 30 min. After centrifugation, the pellet containing urothelial cells was resuspended in fresh Trypsin-ethylenediaminetetraacetic acid solution and incubated for another 30 min. The digestion mixture was supplemented with fetal bovine serum to a finial concentration of 20%. The urothelial cells were collected by centrifugation at 800g for 5′, washed twice with ice-cold PBS, resuspended in PBS and filtered through a 100 μm pore-size filter. The cells were then fixed with precooled ethanol at 4°C overnight and stained with a solution containing 40 μg/ml propidium iodide and 100 μg/ml RNase. The stained cells were sorted with FACScan (Beckman), and the data were analyzed by ModFit LT 3.2 software (Verity Software House).
Ras activity assay
The Ras activity in urothelial cells derived from the various transgenic strains (n = 8 per strain) was determined using an in vitro Ras activation assay kit (Millipore). Briefly, urothelial cells were treated with a lysis buffer containing 25 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (pH 7.5), 150 mM NaCl, 1% IGEPAL® CA-630 (Sigma), 10 mM MgCl2, 1 mM ethylenediaminetetraacetic acid and 2% glycerol, and the protein concentration of total urothelial proteins was determined using a BCA protein assay kit (Pierce). The urothelial proteins were then incubated with a Raf-1-RBD-GST fusion protein (‘Raf-1-RBD’ represents Ras-binding domain of Raf-1 that specifically binds Ras-GTP; GST represents glutathione transferase). The mixture was then added into a glutathione-coated plate. After washing, an anti-pan-Ras antibody was added, followed by a secondary antibody conjugated with horseradish peroxidase. After additional washing, the reaction was developed by the addition of 3,3′5,5′tetramethylbenzidine (Sigma) and read at 450 nm on a spectrophotometer.
In vivo administration of signaling pathway inhibitors
All the inhibitors used in this study were administered into the mice via intraperitoneal injection and with one dose every 2 days. Treatment commenced immediately after the mice were weaned (3 weeks of age) and lasted for 2 weeks in the case of the single-agent rapamycin treatment or for as long as the mice lived for multiagent treatment to compare survival. All agents were freshly prepared in 50% dimethyl sulfoxide in PBS. Rapamycin (LC Laboratories), a selective mammalian target of rapamycin (mTOR) inhibitor (42), was reconstituted to a final concentration of 5 mg/ml and delivered at a dose of 10 mg/kg body wt per injection. UO126 (LC Laboratories), a phosphorylated mitogen-activated protein kinase (MAPK) inhibitor (43), was reconstituted to a final concentration of 15 mg/ml and delivered at a dose of 30 mg/kg body wt per injection. S3I-201 (Calbiochem), a STAT3 inhibitor (44), was reconstituted to a final concentration of 2.5 mg/ml and delivered at a dose of 5 mg/kg per injection.
Western blotting analysis
Western blotting was carried out routinely using, as starting material, total urothelial proteins prepared in a 20 mM Tris–HCl buffer (pH 7.5) containing 10% sodium dodecyl sulfate, 50 mM NaCl, 5 mM beta-mercaptoethanol and a cocktail of protease inhibitors. Sixty micrograms of proteins per lane were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, electrophoretically transferred onto polyvinylidene fluoride membrane and incubated with primary antibodies. After washing, the membrane was further incubated with peroxidase-conjugated secondary antibodies and then developed using enhanced chemiluminescence (Amersham Biosciences). The primary antibodies employed were: anti-p-AKT (at residue 308), anti-p-AKT (at residue 473), anti-AKT, anti-p-mTOR (at residue 2448), anti-mTOR, anti-p-S6, anti-p-38, anti-p-MAPK, anti-MAPK, anti-p-STAT3 (at residue 705) and anti-STAT3 (Cell Signaling Technology); anti-pan-Ras (Quality Biotech); anti-p16, anti-pRb, anti-p107, anti-p130, anti-p53, anti-CDK4 and anti-CDK6 (Santa Cruz Biotechnology); anti-p21 and anti-proliferating cell nuclear antigen (Abcam). Antibody against beta-actin (Sigma) served as a loading control for all experiments.
Histopathology and immunohistochemistry
Urinary bladders were fixed in PBS-buffered 10% formalin and processed routinely for hematoxylin and eosin staining for histopathological examination. For antibody labeling, deparaffinized sections were microwaved at maximal power setting in citrate buffer (pH 6.0) for 20 min for antigen unmasking, followed by consecutive incubation in solutions containing properly diluted primary and peroxidase-coupled secondary antibodies. The reactions were developed in a solution containing 3,3′-diaminobenzidine tetrahydrochloride and hydrogen peroxide. In addition to the primary antibodies used for western blotting, the following were also used for immunohistochemistry: anti-keratin 14 (Covance), anti-pan Ras (Quality Biotech), anti-SV40T (a gift from Dr Douglas Hanahan of University of California, San Francisco).
Statistical analyses
Student's t-test was used to compare the differences among the experimental groups using Web-based SPSS package. A P-value <0.05 was considered statistically significant. For multigroup comparisons, Mann–Whitney U test was employed using Web-based ‘Statext’ software. Log-rank test was performed for comparison of the survivals of mice treated with different signal pathway inhibitors using an Internet-based ‘R’ statistical software. Values <0.01 were considered statistically significant.
Results
Presence of multiple tumor barriers in Ras*-elicited urothelial hyperplasia
One of the interesting phenomena we observed with our heterozygous UPII-Ras* transgenic mice, which expressed a constitutively active Ha-Ras* oncogene in the urothelium, was the persistence of urothelial hyperplasia. Comprised of conspicuously thickened cell layers with little nuclear atypia and no loss in urothelial polarity (Figure 1A), these hyperplastic lesions persisted but failed to advance to tumors over an extended (e.g. 10 months) period. We hypothesized that such a resistance to tumor initiation had to do with a compensatory induction of tumor defense(s) in response to Ras* activation. To test this, we determined the status of several key growth inhibitors/tumor suppressors whose deficiency has been implicated in human urothelial tumorigenesis (45–49). Using real-time quantitative reverse transcription–PCR, we found the messenger RNAs encoding p16 and p19, two tumor suppressors operative upstream of pRb and p53 pathways, respectively (50), to be significantly upregulated in the hyperplastic urothelial cells of the Ras* transgenic mice, compared with the normal urothelial cells of the wild-type mice (Figure 1B). p53 expression was also elevated, although the difference between the two groups of mice did not reach statistical significance. Western blotting confirmed that there was indeed a marked induction of p16, p19, p53 and p21 (the latter a transcriptional target of p53) in the Ras*-expressing urothelial cells (Figure 1C). In addition, these cells overexpressed pRb and its family members, p107 and p130. These data clearly indicated that the transgenic expression of an active Ras* in urothelial cells elicited multiple lines of tumor defenses involving the p53 signaling pathway and the pRb family proteins. They could explain, at least in part, why the Ras*-induced urothelial hyperplasia failed to progress to tumors over an extended period of time.
Fig. 1.
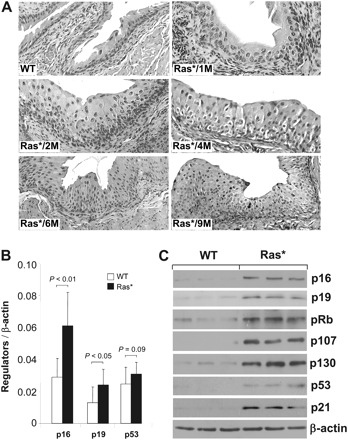
Induction of key tumor suppressors in ‘simple urothelial hyperplasia’ elicited by constitutively active Ha-ras. (A) Hematoxylin and eosin-stained cross sections of bladder urothelia of a 6-month-old wild-type (WT) mouse and five heterozygous UPII/Ha-ras* transgenic mice (1, 2, 4, 6 and 9 months of age) expressing a constitutively active Ras oncogene (Ras*) in the urothelium. Note the persistent urothelial hyperplasia and lack of tumorigenesis irrespective of the age. All panels are ×200. (B and C) Urothelial responses to hyperproliferation. Real-time quantitative PCR (B) and western blotting (C) show marked induction of tumor suppressor genes including those in the p53 pathway (e.g. p19, p53, p21) and those in the pRb pathway (e.g. p16, pRb, p107, p130). Values were means ± SD. N = 8 mice per genotype and per gene analyzed in (B) and N = 3 per genotype in (C).
Rapid induction of high-grade urothelial carcinomas by additionally expressing an SV40 large T antigen in Ras*-expressing urothelial cells
If the tumor suppressors elicited by activated Ras* represented the principal barriers by which the hyperplastic urothelial cells evaded tumors, then disabling these tumor suppressors should in theory overcome these tumor barriers, leading to tumorigenesis. We tested this possibility by expressing an SV40 large T antigen (SV40T) in urothelial cells expressing an activated Ras* via the generation of double transgenic mice. We chose this strategy because SV40T binds to and functionally inactivates p53 as well as the entire pRb family proteins (51), all of which were overexpressed in Ras*-induced urothelial hyperplasia (Figure 1). Crossing the two transgenic lines yielded four major genotypes: (i) wild-type mice, (ii) Ras* single transgenics, (iii) SV40T single transgenics and (iv) Ras*/SV40T double transgenics (Figure 2A and B). While Ras* and SV40T single transgenics were fully viable and had bladders the sizes of those of the wild-type counterparts, the double transgenics had extremely rapid-growing tumors that occupied the entire bladders that were about 20 times heavier than those of the single transgenics (Figure 2C and D). In the single transgenics, obstructive uropathy and hydronephrosis never occurred before 10 months of age; these abnormalities developed in 100% of the double transgenics by 5–7 weeks of age (Figure 2C, lower panel; and data not shown). Because of the obstructive uropathy, all the double transgenics succumbed to death by week 9.
Fig. 2.
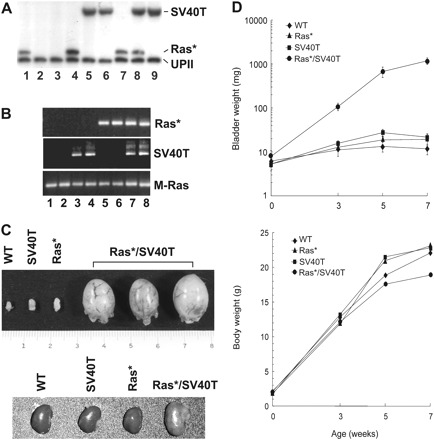
Rapid tumor formation from urothelial cells expressing both Ras* and SV40 large T antigen (SV40T). (A and B) Generation of double transgenic mice expressing both Ras* and SV40T. (A) Genotyping by Southern blotting of a mouse litter showing the generation of (i) single transgenics containing the Ras* transgene (lanes 1, 4 and 7); (ii) single transgenics containing the SV40T transgene (lanes 5, 6 and 9); (iii) double transgenics containing both Ras* and SV40T transgenes (lane 8) and (iv) non-transgenic mice containing neither transgene (lanes 2 and 3). UPII denotes the restrictive digestion fragment from the endogenous mouse UPII gene. (B) Reverse transcription–PCR assay of urothelial expression of Ras* and/or SV40T in the various genotypes as shown in (A), establishing that urothelial cells from non-transgenic mice expressed neither Ras* nor SV40T (lanes 1 and 2); those from single transgenics expressed either SV40T (lanes 3 and 4) or Ras* (lanes 5 and 6) and those from double transgenic expressed both (lanes 7 and 8). (C) Gross anatomy of 5-week-old single and double transgenic mice showing that the bladder sizes of the wild-type (WT) and single transgenic mice were hardly distinguishable and that the bladders of the double transgenics were profoundly enlarged. Lower panel: the corresponding kidneys from these mice, showing the hydronephrosis of a double transgenic mouse. (D) Bladder and body weights of single and double transgenic mice. While the body weights did not significantly differ among the different genotypes, the body weights of the double transgenic mice were about 50-fold higher than those of the WT and single transgenics in 7-week-old groups. Values were means ± SD.
Microscopically, the Ras* single transgenics had normal-appearing urothelium at birth which evolved into simple hyperplasia that persisted throughout the 7 weeks observation period for this cohort (Figure 3A; the hyperplasia could persist for up to 10 months in cohorts of longer observation). The SV40T single transgenics exhibited urothelial dysplasia at birth and then high-grade flat lesions (in distended bladders) that resembled CIS between 3 and 7 weeks of age (Figure 3A). Nuclear irregularity, pleomorphism, giant nuclei, chromatic condensation and mitotic figures, which were absent in the Ras* single transgenics, were common in the SV40T single transgenics. Consistent with the gross anatomy, neither Ras* nor SV40T single transgenics had any microscopic tumor in the bladder by week 7 in this cohort (and by 10 months in cohorts of longer observation). In stark contrast, the Ras*/SV40T double transgenics began with much thickened dysplastic urothelia/CIS at birth, and tumors emerged quickly by as early as week 3. The tumors were of high pathological grade, with large nuclear/cytoplasmic ratio, frequent mitotic figures and conspicuous nucleoli. They had a papillary appearance and remained non-invasive by the time the double transgenics died of obstructive uropathy. Serial sections followed by immunohistochemical staining with an antibody against keratin 14, an epithelial marker specific for basal urothelial cells (52), and antibodies against pan Ras and SV40T did not detect any tumor cell breaching the basement membrane (Figure 3B). These results demonstrated a strong synergistic effect between SV40T and Ras* in initiating high-grade urothelial carcinomas and suggested that inactivation of both the p53 pathway and pRb family proteins plays a crucial role in allowing activated Ras* to overcome the tumor barriers.
Fig. 3.
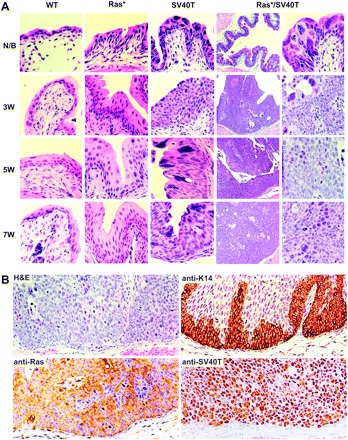
Histopathology. (A) Urinary bladders of WT, Ras* and SV40T single transgenic mice and Ras*/SV40T double transgenic mice of newborn (N/B) and 3, 5 and 7 weeks of age were sectioned and stained with hematoxylin and eosin (H&E). Note the normal urothelial morphology in wild-type (WT) mice of all age groups; simple hyperplasia in Ras* mice; severe dysplasia and CIS-like lesions in SV40T mice and in contrast, the rapid progression from dysplasia/CIS-like lesions in N/B to high-grade papillary tumors in Ras*/SV40T double transgenic mice as early as 3 weeks. Magnification: the left column under Ras*/SV40T, ×50; all other panels, ×200. (B) Examination of the basement membrane zone by H&E and antibody staining. Representative images from serial sections of bladders of 5- to 6-week-old Ras*/SV40T double transgenic mice were stained with H&E or with antibodies against keratin 14, pan-Ras or SV40T. Note the absence of tumor cells beyond the basement membrane.
Defective G1/S checkpoint and overactivation of Ras-GTPase and AKT-mTOR pathway in Ras*/SV40T double transgenic mice
The cell cycle distribution of urothelial cells from each of the four mouse strains under study was considerably different. At the steady state, the overwhelming majority of the normal urothelial cells from the wild-type mice were quiescent and partitioned in the G0/G1 phase (Figure 4A, upper panel). Expression of either Ras* or SV40T in urothelial cells increased the percentage of S phase cells, albeit only slightly above the normal level. However, expression of both Ras* and SV40T resulted in a dramatic increase in the fraction of S phase cells, which was accompanied by a corresponding rise of S phase-specific cyclin-dependent kinases 4 and 6 (Figure 4A, lower panel). These results suggested that the G1/S checkpoint in Ras*/SV40T-expressing urothelial cells was severely compromised. Urothelial cells expressing SV40T also had lower levels of MDM2, a transcriptional target of p53 and higher levels of E2F1 (Figure 4A, lower panel), a downstream target of pRb, suggesting that p53 and pRb were both functionally disabled by SV40T. Aneuploid cells, which were absent in normal urothelium, were present in Ras* single transgenics and even more so in SV40T single transgenic mice and Ras*/SV40T double transgenics. In vitro quantification of Ras-GTPase activity showed a nearly 10-fold increase in the Ras* transgenics over the wild-type control (Figure 4B). A 3-fold increase of Ras-GTPase activity was also detected in the SV40T single transgenics. This is consistent with earlier observations that SV40T can activate the Ras-GTPase (53–56) and is potentially the basis of why, in the double transgenics, SV40T and Ras* were additive in activating the Ras-GTPase (Figure 4B, right column). Western blotting and immunohistochemistry (Figure 4C and D) demonstrated that the Ras*-expressing urothelial cells had a marked activation of MAPK (as evidenced by the significantly increased level of phosphorylated MAPK) but only a slight activation of the AKT and its downstream effectors mTOR and S6 (as evidenced by the levels of their phosphorylated versions). SV40T-expressing urothelial cells, on the other hand, had only slight activation of MAPK but a strong activation of the AKT pathway components (Figure 4C). Finally, urothelial cells expressing both oncogenes had activation in MAPK as well as AKT pathways, again suggesting a collaborative effect.
Fig. 4.
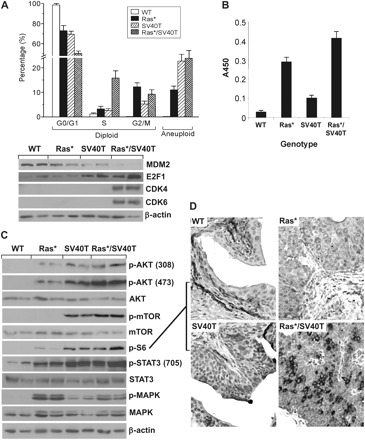
Altered cell cycle and signaling pathways. (A) Urothelial cells from the transgenic mice (6-week old; n = 3) were subject to fluorescein-activated cell sorting (upper panel) or western blotting using anti-CDK4 or anti-CDK6 antibodies (lower panel). Note the marked increase of S phase diploid and aneuploid cells in Ras*/SV40T double transgenic mice. (B) Ras-GTPase assay. In vitro assay of Ras-GTPase was carried out using total urothelial proteins extracted from the transgenic strains (all 6 weeks of age). Values were means ± SD. Note that the Ras activity was considerably higher in Ras* and Ras*/SV40T mice (n = 8 per genotype). (C and D) Western blotting and immunohistochemical detection (anti-p-S6) of signaling pathway components in urothelial cells from the transgenic strains (all 6 weeks of age). Note that the MAPK pathway activation was primarily associated with Ras* activation, whereas AKT-mTOR pathway activation was highly activated in SV40T mice and even more so in Ras*/SV40T mice. All panels in D are ×200.
Inhibition of AKT-mTOR signaling by rapamycin reduced tumor size but hyperactivated the MAPK pathway
To further examine the role of AKT-mTOR pathway activation in urothelial tumorigenesis and to determine whether inhibition of this pathway was sufficient to control high-grade urothelial carcinomas, we treated the double transgenics with rapamycin, an mTOR inhibitor (42,57). Three-week-old Ras*/SV40T double transgenics were randomized into two groups (eight mice per group), one group receiving solvent (50% dimethyl sulfoxide) only and another group receiving the solvent containing 5 mg/ml rapamycin, both administered intraperitoneally every 2 days. Two weeks later, the mice in both groups were killed and their bladder tumors procured. A significant reduction of the tumor size in the treated group as reflected in reduced bladder weights was observed (Figure 5A). Rapamycin-treated mice had bladder weights of about 0.1 g compared with 0.6 g in untreated mice (Figure 5A, lower panel). This corresponded well with a significant reduction in the number of tumor cells histologically (Figure 5B). No significant reduction in the body weights between the two groups was noted (Figure 5A, lower panel), suggesting that at the dosage and time frame employed, rapamycin had minimum systemic toxicity. Despite the reduction in bladder weights, considerable number of tumor cells remained microscopically in the treated mice (Figure 5B). Rapamycin also did not reduce the pathological grade of the tumor cells (Figure 5B).
Fig. 5.
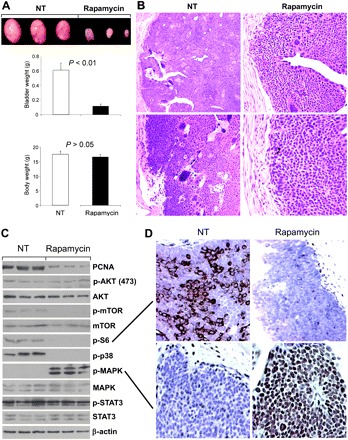
Effects of AKT-mTOR pathway inhibition by rapamycin. (A) Three-week-old Ras*/SV40T double transgenic mice were treated with solvent only (NT; n = 8)) or that containing rapamycin (n = 8) via intraperitoneal delivery every 2 days for a period of 2 weeks. Values were means ± SD. Note the dramatic reduction in bladder (tumor) weights in the rapamycin-treated group. (B) Hematoxylin and eosin-stained bladder cross sections of untreated (NT) and rapamycin-treated double transgenic mice showing the reduced tumor size and tumor cell number in mice receiving rapamycin. Magnification: left panels, ×50; right panels, ×100. (C and D) Responses of signaling effectors to rapamycin treatment in Ras*/SV40T transgenic mice. (C) Western blotting of urothelial proteins extracted from untreated (n = 3) and rapamycin-treated (n = 3) mice. Note the marked inhibition of mTOR and its downstream effector S6 kinase, the inhibition of p38, the unchanged STAT3 and the significantly elevated phosphorylated MAPK. (D) Immunohistochemical staining of untreated and rapamycin-treated mice using antibodies against phosphorylated S6 and MAPK showing the inhibition of the former and marked induction of the latter. Magnification: ×200.
Molecularly, rapamycin treatment was effective, as expected, in decreasing the levels of phosphorylated mTOR and its downstream effector, S6 (Figure 5C). It did not affect the upstream acting phosphorylated AKT, suggesting that the rapamycin-mediated effects on AKT may be context- and tumor cell type dependent (58–62). Rapamycin, however, was not confined to inhibiting mTOR, as it completely inhibited phosphorylated p38 as well. This is in agreement with data from other studies involving rapamycin treatment (63,64). Whether inhibition of S6 led to the inhibition of p38 phosphorylation, as previously suggested (64), remains to be seen. Nevertheless, phosphorylated STAT3 was unaffected by the treatment. Surprisingly, rapamycin-treated bladder tumors had a marked increase of phosphorylated MAPK compared with solvent-treated controls (Figure 5C and D). This suggested that AKT-mTOR pathway inhibition by rapamycin could lead to a hyperactivation of another signaling pathway and that, as a single agent, rapamycin could be ineffective in long-term treatment of certain subtypes of bladder tumors.
Improved survival via multi-pathway inhibition in mice bearing rapid-growing urothelial carcinomas
The initial results from the rapamycin experiment prompted us to examine whether inhibition of multiple signaling targets that were activated in the Ras*/SV40T double transgenics would be more efficacious than inhibiting a single target. Because rapamycin led to a marked compensatory induction of MAPK signaling and because the STAT3 activity was not detectably inhibited by rapamycin, we added to the single-agent regimen a specific inhibitor for phosphorylated MAPK [UO126, abbreviated as MAPK-i; (43)] and a specific inhibitor for STAT3 [S3I-201, abbreviated as STAT3-i; (44)]. Ras*/SV40T double transgenics were randomized into three groups (eight mice per group) to receive (i) solvent (50% dimethyl sulfoxide) only; (ii) solvent containing (5 mg/ml) of rapamycin and (iii) solvent containing 5 mg/ml of rapamycin, 15 mg/ml of MAPK-i and 2.5 mg/ml of STAT3-i (Figure 6A). Intraperitoneal delivery of the agents (every 2 days) commenced at 3 weeks of age and continued for 4 weeks after which all animals were killed and their bladders weighed. The bladders of mice receiving the triple agents were significantly smaller and weighed less than those of the single-agent-treated mice (P < 0.01) (Figure 6A). Another cohort comprising the three treatment groups (n = 20 per group) was then followed to compare the time of survival (Figure 6B). The untreated mice only survived 63 days compared with the rapamycin-treated mice, which survived 77 days. In contrast, the triple-agent-treated mice survived up to 119 days, an improvement of 42 days over the single-agent group and 56 days (e.g. almost doubling of survival time) over the untreated group (Figure 6B). The combination therapy, however, did not eradicate the bladder tumors, as all the treated mice eventually succumbed to obstructive uropathy. Western blotting and immunohistochemistry showed that all the three intended targets, e.g. phosphorylated mTOR, MAPK and STAT3, were effectively inhibited (Figure 6C and D). The failure to completely eliminate the tumors in our mice even with the three-agent regimen might therefore have to do the compensatory action of other signaling pathway(s). It might also be due to the fact that the treatment might have not begun soon enough, e.g. at 3 weeks of age when the tumors were already full blown.
Fig. 6.
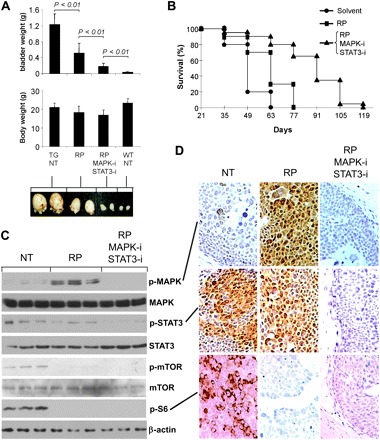
Inhibitory effects of combined inhibitors for mTOR (rapamycin, RP), phosphorylated MAPK (MAPK-i) and phosphorylated STAT3 (STAT3-i). (A) Three-week-old Ras*/SV40T double transgenic mice (n = 8 per group) received solvent only (TG, NT), rapamycin only (RP) or all three inhibitors (RP, MAPK-i and STAT3-i) for 4 weeks before they were killed. Note the three inhibitor combination achieved significantly greater reduction in tumor size than the single-agent treatment. Values were means ± SD. P-values were calculated using Mann–Whitney U test. (B) Another cohort of 3-week-old Ras*/SV40T double transgenic mice (n = 20 per group) received solvent only, rapamycin only (RP) or all three inhibitors continuously until the mice died of obstructive uropathy. Longer survival of the mice receiving the three inhibitors than those receiving only one inhibitor was observed. The log-rank P-values were 0.016 between RP group and solvent group and 1.8 × 10−5 between the three inhibitor group and RP group. (C) Western blotting of urothelial proteins from untreated, rapamycin-treated and three-agent treated mice (n = 3 per group) showing the status of signaling effectors. (D) Immunohistochemical staining verifying the results from western blotting.
Discussion
Genetic events capable of cooperating with activated ras in initiating UCB
Although mutations of the RTK/Ras pathway components affect an overwhelming majority of low-grade papillary UCB in humans (10,15–22), experimental evidence is mounting that these mutations alone are insufficient to initiate UCB (65). In our transgenic mice heterozygous for the mutated Ha-ras transgene, which is expressed in urothelium at a level equivalent to the endogenous wild-type Ras, hyperplasias persist for 10 months (38). Low-grade UCB only emerge between 11 and 28 months and in ∼60% of the mice. Such a long latency and incomplete penetrance suggest that certain cooperating event(s) must be acquired for the mutated Ras to be tumorigenic. We previously found that loss of both p16Ink4a and p19Arf, known to be synergistic with activated Ras and trigger tumors in several non-urothelial tissues, did not accelerate Ras-mediated urothelial tumorigenesis (39). Loss of both p53 alleles in mutant Ras-expressing urothelial cells, however, did shorten tumor latency to 3–7 months, although this cooperative effect occurred in only 30% of the animals (66). Based on data presented here, Ras activation in urothelial cells induces multiple tumor suppressors involving not only the p53 pathway but the entire pRb family (Figure 1). Importantly, these compensatory tumor defenses suppress tumorigenesis from the hyperplastic urothelium. Introduction of SV40T, an oncoprotein that functionally disables p53 and all pRb family proteins(51), into mutant Ras-expressing urothelial cells results in 100% of the mice developing UCB in only 3 weeks (Figures 2 and 3) further supporting this notion. Thus, activated Ha-ras and SV40T-mediated oncogenesis cooperate in urothelial cells and together are sufficient to efficiently convert tumor precursors of the urothelium to UCB.
The dependence of UCB development on cooperation among different oncogenic pathways may not be limited to activated Ras but may apply to other RTK/Ras pathway components. Thus far, expression of a mutated FGFR3b in transgenic mouse urothelium has not resulted in detectable urothelial hyperplasia, let alone UCB (65,67). This is somewhat consistent with our earlier study showing that overexpression of an epidermal growth factor receptor (EGFR) in urothelium only led to urothelial hyperplasia but not UCB (68). Although the presence or absence of urothelial hyperplasia may reflect the level of transgene expression in different transgenic systems, the absence of tumors in FGFR3b and EGFR transgenic mice is consistent. We suggest that, like activated Ras, mutated FGFR3b may require cooperative event(s) to be fully tumorigenic. Because FGFR3b transmits mitogenic signals through Ras (69), it will be of interest to see if the cooperative events for these genes share certain commonalities. Indeed, we showed previously that SV40T was capable of cooperating with overexpressed EGFR in converting SV40T-induced CIS to high-grade papillary UCB (68). Whether SV40T has the same effect on mutated FGFR3b remains unknown and is worthy of exploration.
On a cellular and molecular level, activated Ras and SV40T cooperate in multiple ways to drive urothelial tumorigenesis. First, activated Ras creates significant replicative pressure on urothelial cells, leading to double strand DNA breaks, particularly in common fragile sites (70). Indeed, the number of apurinic and apyrimidinic sites and the level of histone H2A-x, markers of DNA damage, were both elevated in mutant Ras-induced urothelial hyperplasias (Zhou, H.-P. and Wu, X.-R., unpublished results). In response to the DNA damage, urothelial cells increase the synthesis of p53, which plays a principal role in DNA damage repair (71). A new damage/repair balance might be reached that allows urothelial hyperplasias to persist without progression (Figure 1). In the presence of SV40T, however, p53 is functionally impaired (Figure 4A) and as a result, mutant Ras-caused DNA damage cannot be promptly repaired, leading to increased genome instability. Unchecked genome instability is a prerequisite to tumorigenesis (72,73). Second, while mutated Ras is a strong mitogenic signal for urothelial cells, the G1/S restriction point in the Ras-expressing cells is intact via the overexpression of wild-type p53 (Figure 1), which holds urothelial proliferation in check. On the other hand, although SV40T-expressing urothelial cells have a defective G1/S checkpoint, the urothelium remains arrested at a CIS stage due to the absence of a strong mitogenic signal [Figure 3; (41)]. The copresence of a potent mitogenic signal (mutated Ras) and defective cell cycle checkpoint (enabled by SV40T) allow for efficient urothelial tumorigenesis. Third, while mutated Ras is known to upregulate Ras-GTPase, SV40T also has such an effect as shown in Figure 4B and by others previously (55,56). The expression of these two genes is therefore additive, if not synergistic, in raising the Ras-GTPase level in the urothelium. Finally, mutant Ras and SV40T cooperate by activating both MAPK and AKT-mTOR pathways (Figure 4). Mutant Ras alone primarily activated the MAPK pathway, whereas SV40T mainly activated the AKT-mTOR pathway. The latter may be related to the fact that under normal circumstances, p53 upregulates PTEN, a potent upstream inhibitor of PI3K-AKT signaling axis (74,75), and that such an inhibitory effect is compromised by SV40T-mediated inactivation of p53 (Figure 4A). Expression of mutant Ras and SV40T transgenes is therefore necessary to activate both MAPK and AKT-mTOR signaling, consistent with findings in other tumor types (76,77).
Potential origins of high-grade papillary UCB
High-grade papillary UCB is a clinically important but biologically enigmatic entity whose cell origin and mode of progression remain uncertain (5,36,37). The mutant Ras/SV40T double transgenic mice represent the first experimental model that reproducibly develops this urothelial tumor subtype (Figures 2 and 3). Because SV40T single transgenic mice develop exclusively CIS lesions (Figure 3) and the addition of mutant Ras results in high-grade papillary UCB, these tumors could result from CIS lesions acquiring additional growth advantage by mutational activation and/or overexpression of RTK/Ras components. The development of high-grade papillary tumors by double transgenic mice expressing both SV40T and EGFR supports this scenario (68). Alternatively, high-grade papillary UCB may reflect the progression of urothelial hyperplasias or low-grade papillary UCB upon acquisition of p53- and pRb pathway deficiencies. Regardless of its origin(s), high-grade papillary UCB clearly carries a greater risk of progression than low-grade papillary UCB because of its rapid growth rate, large number of genetic alterations and high level of genome instability (5,36,37). Notably, invasive lesions were not observed in our double transgenic mice despite serial sections and immunohistochemical examination of multiple bladders from our double transgenic mice (Figure 3). It is possible that this lack of detectable invasion results from premature death of the mice due to renal failure. One also cannot completely rule out the possibility that molecular events critical for triggering invasion are absent in the mutant Ras/SV40T mice.
Targeted therapy of UCB: the effectiveness of single agent versus multiple agents
The current mainstay for treating high-grade papillary UCB is local resection followed by intravesicle instillation of bacillus Calmette–Guérin (5,78). If tumors are resistant to this treatment modality and progress to the invasive stage, they have to be treated with radical cystectomy, which nonetheless does not assure cure. The limited treatment options for different subtypes of UCB, in particular high-grade tumors, call for new therapeutic strategies. As the molecular details underlying the development and progression of UCB start to be unraveled, it is more possible than ever to devise targeted therapies for this disease (16). In the present study, we found that inhibition of mTOR, a downstream effector of AKT, by a 2 weeks course of rapamycin significantly reduces the size of high-grade papillary UCB (Figure 5). However, considerable residual tumors exist and consequently the survival of the mice with these tumors did not significantly improve. Molecular analysis revealed a marked induction of activated MAPK in rapamycin-treated tumors (Figure 5), suggesting this as a possible reason for the ineffectiveness of the single-agent therapy. Additionally, compared with the wild-type mouse urothelial cells, those of the transgenic mice particularly the double transgenics had markedly activated STAT3, which was virtually unaffected by rapamycin treatment alone. A combination of three agents was then employed that contained inhibitors-specific mTOR, MAPK and STAT3. This led to significantly greater tumor inhibition and longer survival of the treated mice compared with rapamycin as a single agent (Figure 6). Our data suggest that a single agent targeting one pathway signal could result in a compensatory activation of another signaling pathway, causing drug resistance, and that sustained tumor control might be achievable through the inhibition of several targets in multiple signaling pathways.
We believe that the principle we have demonstrated with mouse models has significant implications on the rational design of targeted therapies for patients with bladder cancer. Mounting evidence indicates that the PI3K-AKT-mTOR pathway is activated in a sizable portion of human bladder cancer, in particular high-grade and late-stage tumors. One recent study found phosphorylated mTOR and S6 in 74% and 55%, respectively, of muscle-invasive tumors (79). Activation of this pathway could be mediated by a range of abnormalities in its components including PTEN (e.g. by deletion, mutation or reduced expression), PI3K (by mutation) and AKT (by mutation or increased phosphorylation), all of which have been found in human bladder cancer (80–82). It is to no great surprise that exposure of cell lines derived from human bladder cancer to rapamycin or other mTOR inhibitors results in inhibition of cell proliferation in vitro and reduced tumor growth in xenograft models (79,83,84). Although much less is known about exactly how MAPK or STAT3 activation contributes to bladder cancer development and progression, evidence is emerging that these two signals are also frequently overexpressed and/or activated in human bladder cancer (85–89). Because of the existence of non-overlapping effects of mTOR, MAPK and STAT3 and the extensive cross talk among the different pathways, it is conceivable that the effective control of human bladder cancer with targeted therapies would also reply on the inhibition of multiple signaling pathways.
Funding
This study was supported in part by a Merit Review Award from the Veterans Administration's Research Service and by a grant-in-aid from the Goldstein Fund for Urological Research of the New York University School of Medicine.
Acknowledgments
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- CIS
carcinoma in situ
- EGFR
epidermal growth factor receptor
- MAPK
mitogen-activated protein kinase
- mTOR
mammalian target of rapamycin
- PBS
phosphate-buffer saline
- RTK
receptor tyrosine kinase
- UCB
urothelial carcinoma of the bladder
References
- 1.American Cancer Society. Cancer Facts and Figures 2011. Atlanta, GA: American Cancer Society; 2011. [Google Scholar]
- 2.Parkin DM, et al. Estimating the world cancer burden: Globocan 2000. Int. J. Cancer. 2001;94:153–156. doi: 10.1002/ijc.1440. [DOI] [PubMed] [Google Scholar]
- 3.Ploeg M, et al. The present and future burden of urinary bladder cancer in the world. World J. Urol. 2009;27:289–293. doi: 10.1007/s00345-009-0383-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McConkey DJ, et al. Molecular genetics of bladder cancer: emerging mechanisms of tumor initiation and progression. Urol. Oncol. 2010;28:429–440. doi: 10.1016/j.urolonc.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goebell PJ, et al. Bladder cancer or bladder cancers? Genetically distinct malignant conditions of the urothelium. Urol. Oncol. 2010;28:409–428. doi: 10.1016/j.urolonc.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 6.Castillo-Martin M, et al. Molecular pathways of urothelial development and bladder tumorigenesis. Urol. Oncol. 2010;28:401–408. doi: 10.1016/j.urolonc.2009.04.019. [DOI] [PubMed] [Google Scholar]
- 7.Wolff EM, et al. Mechanisms of disease: genetic and epigenetic alterations that drive bladder cancer. Nat. Clin. Pract. Urol. 2005;2:502–510. doi: 10.1038/ncpuro0318. [DOI] [PubMed] [Google Scholar]
- 8.Mitra AP, et al. Molecular pathogenesis and diagnostics of bladder cancer. Annu. Rev. Pathol. 2009;4:251–285. doi: 10.1146/annurev.pathol.4.110807.092230. [DOI] [PubMed] [Google Scholar]
- 9.Cohen SM. Comparative pathology of proliferative lesions of the urinary bladder. Toxicol. Pathol. 2002;30:663–671. doi: 10.1080/01926230290166751. [DOI] [PubMed] [Google Scholar]
- 10.Dinney CP, et al. Focus on bladder cancer. Cancer Cell. 2004;6:111–116. doi: 10.1016/j.ccr.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 11.Dalbagni G. The management of superficial bladder cancer. Nat. Clin. Pract. Urol. 2007;4:254–260. doi: 10.1038/ncpuro0784. [DOI] [PubMed] [Google Scholar]
- 12.Grossman HB. Superficial bladder cancer: decreasing the risk of recurrence. Oncology (Huntingt) 1996;10:1617–1624. discussion 1624, 1627–1628. [PubMed] [Google Scholar]
- 13.Hodges KB, et al. Urothelial dysplasia and other flat lesions of the urinary bladder: clinicopathologic and molecular features. Hum. Pathol. 2010;41:155–162. doi: 10.1016/j.humpath.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 14.van Oers JM, et al. Chromosome 9 deletions are more frequent than FGFR3 mutations in flat urothelial hyperplasias of the bladder. Int. J. Cancer. 2006;119:1212–1215. doi: 10.1002/ijc.21958. [DOI] [PubMed] [Google Scholar]
- 15.Pollard C, et al. Molecular genesis of non-muscle-invasive urothelial carcinoma (NMIUC) Expert Rev. Mol. Med. 2010;12:e10. doi: 10.1017/S1462399410001407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu XR. Urothelial tumorigenesis: a tale of divergent pathways. Nat. Rev. Cancer. 2005;5:713–725. doi: 10.1038/nrc1697. [DOI] [PubMed] [Google Scholar]
- 17.Knowles MA. Role of FGFR3 in urothelial cell carcinoma: biomarker and potential therapeutic target. World J. Urol. 2007;25:581–593. doi: 10.1007/s00345-007-0213-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Rhijn BW, et al. FGFR3 and P53 characterize alternative genetic pathways in the pathogenesis of urothelial cell carcinoma. Cancer Res. 2004;64:1911–1914. doi: 10.1158/0008-5472.can-03-2421. [DOI] [PubMed] [Google Scholar]
- 19.Bakkar AA, et al. FGFR3 and TP53 gene mutations define two distinct pathways in urothelial cell carcinoma of the bladder. Cancer Res. 2003;63:8108–8112. [PubMed] [Google Scholar]
- 20.Platt FM, et al. Spectrum of phosphatidylinositol 3-kinase pathway gene alterations in bladder cancer. Clin. Cancer Res. 2009;15:6008–6017. doi: 10.1158/1078-0432.CCR-09-0898. [DOI] [PubMed] [Google Scholar]
- 21.Boulalas I, et al. Mutational analysis of the BRAF gene in transitional cell carcinoma of the bladder. Int. J. Biol. Markers. 2009;24:17–21. doi: 10.1177/172460080902400103. [DOI] [PubMed] [Google Scholar]
- 22.Jebar AH, et al. FGFR3 and Ras gene mutations are mutually exclusive genetic events in urothelial cell carcinoma. Oncogene. 2005;24:5218–5225. doi: 10.1038/sj.onc.1208705. [DOI] [PubMed] [Google Scholar]
- 23.Sandberg AA, et al. Review of chromosome studies in urological tumors. II. Cytogenetics and molecular genetics of bladder cancer. J. Urol. 1994;151:545–560. doi: 10.1016/s0022-5347(17)35014-0. [DOI] [PubMed] [Google Scholar]
- 24.Aveyard JS, et al. Measurement of relative copy number of CDKN2A/ARF and CDKN2B in bladder cancer by real-time quantitative PCR and multiplex ligation-dependent probe amplification. J. Mol. Diagn. 2004;6:356–365. doi: 10.1016/S1525-1578(10)60532-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cordon-Cardo C, et al. Genetic and molecular markers of urothelial premalignancy and malignancy. Scand. J. Urol. Nephrol. Suppl. 2000;205:82–93. doi: 10.1080/003655900750169338. [DOI] [PubMed] [Google Scholar]
- 26.O'Connor RC, et al. Therapeutic options and treatment of muscle invasive bladder cancer. Expert Rev. Anticancer Ther. 2001;1:511–522. doi: 10.1586/14737140.1.4.511. [DOI] [PubMed] [Google Scholar]
- 27.Lee CT, et al. Treatment of muscle-invasive bladder cancer: is cystectomy enough? Nat. Clin. Pract. Urol. 2007;4:126–127. doi: 10.1038/ncpuro0708. [DOI] [PubMed] [Google Scholar]
- 28.Chang SS, et al. Radical cystectomy for bladder cancer: the case for early intervention. Urol. Clin. North Am. 2005;32:147–155. doi: 10.1016/j.ucl.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 29.Lotan Y, et al. Key concerns about the current state of bladder cancer: a position paper from the Bladder Cancer Think Tank, the Bladder Cancer Advocacy Network, and the Society of Urologic Oncology. Cancer. 2009;115:4096–4103. doi: 10.1002/cncr.24463. [DOI] [PubMed] [Google Scholar]
- 30.Koss LG. Natural history and patterns of invasive cancer of the bladder. Eur. Urol. 1998;33:2–4. doi: 10.1159/000052249. [DOI] [PubMed] [Google Scholar]
- 31.Koss LG. Bladder cancer from a perspective of 40 years. J. Cell. Biochem. Suppl. 1992;16I:23–29. doi: 10.1002/jcb.240501305. [DOI] [PubMed] [Google Scholar]
- 32.Cordon-Cardo C. p53 and Rb: simple interesting correlates or tumor markers of critical predictive nature? J. Clin. Oncol. 2004;22:975–977. doi: 10.1200/JCO.2004.12.994. [DOI] [PubMed] [Google Scholar]
- 33.Grossman HB, et al. p53 and Rb expression predict progression in T1 bladder cancer. Clin. Cancer Res. 1998;4:829–834. [PubMed] [Google Scholar]
- 34.Cordon-Cardo C, et al. Cooperative effects of p53 and pRB alterations in primary superficial bladder tumors. Cancer Res. 1997;57:1217–1221. [PubMed] [Google Scholar]
- 35.Cote RJ, et al. Elevated and absent pRb expression is associated with bladder cancer progression and has cooperative effects with p53. Cancer Res. 1998;58:1090–1094. [PubMed] [Google Scholar]
- 36.Knowles MA. Molecular subtypes of bladder cancer: Jekyll and Hyde or chalk and cheese? Carcinogenesis. 2006;27:361–373. doi: 10.1093/carcin/bgi310. [DOI] [PubMed] [Google Scholar]
- 37.Zhao J, et al. Chromosomal imbalances in noninvasive papillary bladder neoplasms (pTa) Cancer Res. 1999;59:4658–4661. [PubMed] [Google Scholar]
- 38.Zhang ZT, et al. Role of Ha-ras activation in superficial papillary pathway of urothelial tumor formation. Oncogene. 2001;20:1973–1980. doi: 10.1038/sj.onc.1204315. [DOI] [PubMed] [Google Scholar]
- 39.Mo L, et al. Hyperactivation of Ha-ras oncogene, but not Ink4a/Arf deficiency, triggers bladder tumorigenesis. J. Clin. Invest. 2007;117:314–325. doi: 10.1172/JCI30062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang ZT, et al. Urothelium-specific expression of an oncogene in transgenic mice induced the formation of carcinoma in situ and invasive transitional cell carcinoma. Cancer Res. 1999;59:3512–3517. [PubMed] [Google Scholar]
- 41.Cheng J, et al. Allelic loss of p53 gene is associated with genesis and maintenance, but not invasion, of mouse carcinoma in situ of the bladder. Cancer Res. 2003;63:179–185. [PubMed] [Google Scholar]
- 42.Puzio-Kuter AM, et al. Inactivation of p53 and Pten promotes invasive bladder cancer. Genes Dev. 2009;23:675–680. doi: 10.1101/gad.1772909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shankaranarayanan P, et al. Growth factor-antagonized rexinoid apoptosis involves permissive PPARgamma/RXR heterodimers to activate the intrinsic death pathway by NO. Cancer Cell. 2009;16:220–231. doi: 10.1016/j.ccr.2009.07.029. [DOI] [PubMed] [Google Scholar]
- 44.Siddiquee K, et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl Acad. Sci. USA. 2007;104:7391–7396. doi: 10.1073/pnas.0609757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benedict WF, et al. Level of retinoblastoma protein expression correlates with p16 (MTS-1/INK4A/CDKN2) status in bladder cancer. Oncogene. 1999;18:1197–1203. doi: 10.1038/sj.onc.1202452. [DOI] [PubMed] [Google Scholar]
- 46.Chatterjee SJ, et al. Hyperphosphorylation of pRb: a mechanism for RB tumour suppressor pathway inactivation in bladder cancer. J. Pathol. 2004;203:762–770. doi: 10.1002/path.1567. [DOI] [PubMed] [Google Scholar]
- 47.Shariat SF, et al. p53, p21, pRb, and p16 expression predict clinical outcome in cystectomy with bladder cancer. J. Clin. Oncol. 2004;22:1014–1024. doi: 10.1200/JCO.2004.03.118. [DOI] [PubMed] [Google Scholar]
- 48.Rabbani F, et al. Mutation of cell cycle regulators and their impact on superficial bladder cancer. Urol. Clin. North Am. 2000;27:83–102. doi: 10.1016/s0094-0143(05)70237-8. , ix. [DOI] [PubMed] [Google Scholar]
- 49.Wu XR. Biology of urothelial tumorigenesis: insights from genetically engineered mice. Cancer Metastasis Rev. 2009;28:281–290. doi: 10.1007/s10555-009-9189-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sherr CJ. The INK4a/ARF network in tumour suppression. Nat. Rev. Mol. Cell Biol. 2001;2:731–737. doi: 10.1038/35096061. [DOI] [PubMed] [Google Scholar]
- 51.Ahuja D, et al. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene. 2005;24:7729–7745. doi: 10.1038/sj.onc.1209046. [DOI] [PubMed] [Google Scholar]
- 52.Kurzrock EA, et al. Label-retaining cells of the bladder: candidate urothelial stem cells. Am. J. Physiol. Renal Physiol. 2008;294:F1415–F1421. doi: 10.1152/ajprenal.00533.2007. [DOI] [PubMed] [Google Scholar]
- 53.Farassati F, et al. Ras signaling influences permissiveness of malignant peripheral nerve sheath tumor cells to oncolytic herpes. Am. J. Pathol. 2008;173:1861–1872. doi: 10.2353/ajpath.2008.080376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grammatikakis N, et al. Simian virus 40 large tumor antigen modulates the Raf signaling pathway. J. Biol. Chem. 2001;276:27840–27845. doi: 10.1074/jbc.M101987200. [DOI] [PubMed] [Google Scholar]
- 55.Lee GH, et al. Detection of activated c-H-ras oncogene in hepatocellular carcinomas developing in transgenic mice harboring albumin promoter-regulated simian virus 40 gene. Carcinogenesis. 1990;11:1145–1148. doi: 10.1093/carcin/11.7.1145. [DOI] [PubMed] [Google Scholar]
- 56.Tack LC, et al. The p53 complex from monkey cells modulates the biochemical activities of simian virus 40 large T antigen. J. Virol. 1989;63:1310–1317. doi: 10.1128/jvi.63.3.1310-1317.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cully M, et al. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat. Rev. Cancer. 2006;6:184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- 58.O'Reilly KE, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu X, et al. PI3K/Akt/mTOR signaling regulates glutamate transporter 1 in astrocytes. Biochem. Biophys. Res. Commun. 2010;393:514–518. doi: 10.1016/j.bbrc.2010.02.038. [DOI] [PubMed] [Google Scholar]
- 60.Podsypanina K, et al. An inhibitor of mTOR reduces neoplasia and normalizes p70/S6 kinase activity in Pten+/- mice. Proc. Natl Acad. Sci. USA. 2001;98:10320–10325. doi: 10.1073/pnas.171060098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zeng Z, et al. Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood. 2007;109:3509–3512. doi: 10.1182/blood-2006-06-030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Anderl S, et al. Therapeutic value of prenatal rapamycin treatment in a mouse brain model of tuberous sclerosis complex. Hum. Mol. Genet. 2011;20:4597–4604. doi: 10.1093/hmg/ddr393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cuenda A, et al. Stress-activated protein kinase-2/p38 and a rapamycin-sensitive pathway are required for C2C12 myogenesis. J. Biol. Chem. 1999;274:4341–4346. doi: 10.1074/jbc.274.7.4341. [DOI] [PubMed] [Google Scholar]
- 64.Oh CD, et al. Immunosuppressant rapamycin inhibits protein kinase C alpha and p38 mitogen-activated protein kinase leading to the inhibition of chondrogenesis. Eur. J. Pharmacol. 2001;427:175–185. doi: 10.1016/s0014-2999(01)01241-9. [DOI] [PubMed] [Google Scholar]
- 65.Ahmad I, et al. K-Ras and {beta}-catenin mutations cooperate with Fgfr3 mutations in mice to promote tumorigenesis in the skin and lung, but not in the bladder. Dis. Model Mech. 2011;4:548–555. doi: 10.1242/dmm.006874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gao J, et al. p53 deficiency provokes urothelial proliferation and synergizes with activated Ha-ras in promoting urothelial tumorigenesis. Oncogene. 2004;23:687–696. doi: 10.1038/sj.onc.1207169. [DOI] [PubMed] [Google Scholar]
- 67.Logie A, et al. Activating mutations of the tyrosine kinase receptor FGFR3 are associated with benign skin tumors in mice and humans. Hum. Mol. Genet. 2005;14:1153–1160. doi: 10.1093/hmg/ddi127. [DOI] [PubMed] [Google Scholar]
- 68.Cheng J, et al. Overexpression of epidermal growth factor receptor in urothelium elicits urothelial hyperplasia and promotes bladder tumor growth. Cancer Res. 2002;62:4157–4163. [PubMed] [Google Scholar]
- 69.Ornitz DM, et al. Fibroblast growth factors. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-3-reviews3005. : reviews3005-reviews3005.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tsantoulis PK, et al. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome-wide study. Oncogene. 2008;27:3256–3264. doi: 10.1038/sj.onc.1210989. [DOI] [PubMed] [Google Scholar]
- 71.Efeyan A, et al. p53: guardian of the genome and policeman of the oncogenes. Cell Cycle. 2007;6:1006–1010. doi: 10.4161/cc.6.9.4211. [DOI] [PubMed] [Google Scholar]
- 72.Negrini S, et al. Genomic instability—an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010;11:220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- 73.Halazonetis TD, et al. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 74.Mayo LD, et al. The PTEN, Mdm2, p53 tumor suppressor-oncoprotein network. Trends Biochem. Sci. 2002;27:462–467. doi: 10.1016/s0968-0004(02)02166-7. [DOI] [PubMed] [Google Scholar]
- 75.Tamguney T, et al. New insights into PTEN. J. Cell Sci. 2007;120:4071–4079. doi: 10.1242/jcs.015230. [DOI] [PubMed] [Google Scholar]
- 76.Robinson JP, et al. Activated BRAF induces gliomas in mice when combined with Ink4a/Arf loss or Akt activation. Oncogene. 2010;29:335–344. doi: 10.1038/onc.2009.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Robinson JP, et al. Activated MEK cooperates with Ink4a/Arf loss or Akt activation to induce gliomas in vivo. Oncogene. 2011;30:1341–1350. doi: 10.1038/onc.2010.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thorstenson A, et al. Diagnostic random bladder biopsies: reflections from a population-based cohort of 538 patients. Scand. J. Urol. Nephrol. 2010;44:11–19. doi: 10.3109/00365590903419020. [DOI] [PubMed] [Google Scholar]
- 79.Hansel DE, et al. Mammalian target of rapamycin (mTOR) regulates cellular proliferation and tumor growth in urothelial carcinoma. Am. J. Pathol. 2010;176:3062–3072. doi: 10.2353/ajpath.2010.090872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Knowles MA, et al. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer Metastasis Rev. 2009;28:305–316. doi: 10.1007/s10555-009-9198-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schultz L, et al. Expression status and prognostic significance of mammalian target of rapamycin pathway members in urothelial carcinoma of urinary bladder after cystectomy. Cancer. 2010;116:5517–5526. doi: 10.1002/cncr.25502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ching CB, et al. Expanding therapeutic targets in bladder cancer: the PI3K/Akt/mTOR pathway. Lab. Invest. 2010;90:1406–1414. doi: 10.1038/labinvest.2010.133. [DOI] [PubMed] [Google Scholar]
- 83.Makhlin I, et al. The mTOR pathway affects proliferation and chemosensitivity of urothelial carcinoma cells and is upregulated in a subset of human bladder cancers. BJU Int. 2011;108:E84–E90. doi: 10.1111/j.1464-410X.2010.09844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chiong E, et al. Effects of mTOR inhibitor everolimus (RAD001) on bladder cancer cells. Clin. Cancer Res. 2011;17:2863–2873. doi: 10.1158/1078-0432.CCR-09-3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Metalli D, et al. The insulin-like growth factor receptor I promotes motility and invasion of bladder cancer cells through Akt- and mitogen-activated protein kinase-dependent activation of paxillin. Am. J. Pathol. 2010;176:2997–3006. doi: 10.2353/ajpath.2010.090904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dangle PP, et al. Ras-MAPK pathway as a therapeutic target in cancer—emphasis on bladder cancer. Recent Pat. Anticancer Drug Discov. 2009;4:125–136. doi: 10.2174/157489209788452812. [DOI] [PubMed] [Google Scholar]
- 87.Kumar B, et al. Differential effects of MAPKs signaling on the growth of invasive bladder cancer cells. Int. J. Oncol. 2009;34:1557–1564. doi: 10.3892/ijo_00000285. [DOI] [PubMed] [Google Scholar]
- 88.Chen CL, et al. Signal transducer and activator of transcription 3 activation is associated with bladder cancer cell growth and survival. Mol. Cancer. 2008;7:78. doi: 10.1186/1476-4598-7-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mitra AP, et al. Generation of a concise gene panel for outcome prediction in urinary bladder cancer. J. Clin. Oncol. 2009;27:3929–3937. doi: 10.1200/JCO.2008.18.5744. [DOI] [PMC free article] [PubMed] [Google Scholar]