


Abstract
Identification of transcriptional regulatory regions and tracing their internal organization are important for understanding the eukaryotic cell machinery. Cis-regulatory modules (CRMs) of higher eukaryotes are believed to possess a regulatory ‘grammar’, or preferred arrangement of binding sites, that is crucial for proper regulation and thus tends to be evolutionarily conserved. Here, we present a method CORECLUST (COnservative REgulatory CLUster STructure) that predicts CRMs based on a set of positional weight matrices. Given regulatory regions of orthologous and/or co-regulated genes, CORECLUST constructs a CRM model by revealing the conserved rules that describe the relative location of binding sites. The constructed model may be consequently used for the genome-wide prediction of similar CRMs, and thus detection of co-regulated genes, and for the investigation of the regulatory grammar of the system. Compared with related methods, CORECLUST shows better performance at identification of CRMs conferring muscle-specific gene expression in vertebrates and early-developmental CRMs in Drosophila.
INTRODUCTION
The identification of transcriptional regulatory elements is a key point for the understanding of complexity and development of living organisms. The main transcriptional regulation mechanism in a living cell is provided by transcription factors (TFs) that bind the DNA at their binding sites (TFBSs) and thus activate or repress the transcription. The spatial and temporal specificity in gene expression is achieved by interaction of TFs that yield different expression patterns. In higher eukaryotes, TFBSs tend to be rather short (5–15 bp) and degenerate and they are often spread in extensive non-coding regions. So, thousands of potential binding sites could be found just by chance due to the size of the eukaryotic genomes.
Fortunately, TFBSs tend to cluster along the DNA strand and thus to form cis-regulatory modules (CRMs). Moreover, numerous evidence shows that, in higher eukaryotes, binding sites often form so-called composite elements (1), which are groups of sites in a specific arrangement. The process of formation of a regulatory complex by the TFs sets constraints on this arrangement, defining the grammar, or structure, of the CRMs. Identical patterns of composite elements are assumed to have similar functions in regulatory modules of different genes.
The observation of the TFBSs' tendency to cluster inspired numerous computational approaches to the identification of CRMs in eukaryotic genomes. Many of them start from known motifs represented by position weight matrices (PWMs). Early methods scan a query sequence and detect local clustering of sites representing the input motifs (2–4), typically ignoring the regulatory grammar, i.e. sites order and spacing. Other methods use the hidden Markov model (HMM) for CRM prediction (5–7). Major advantages of the HMM approach are the statistically reliable measure for the CRMs occurrence (8) and the possibility to account for the regulatory grammar of CRM (9,10). Moreover, the use of the expectation maximization algorithm allows one to adjust a large set of parameters computationally rather than manually.
An important and widely used source of data for the CRM prediction is the interspecies comparison (3,4,9,11). However, most methods are based on pairwise or multiple sequence alignments and thus they fail when the regulatory regions are not well alignable (12,13). To work around this problem, Hallikas et al. (14) proposed the algorithm called EEL that aligns significant motif occurrences rather than the sequences; thus, this method does not rely on the raw sequence similarity. However, the EEL processes only two sequences at a time. Moreover, it assumes that TFBSs in the conserved CRMs occur strictly in the same order. A different approach was used in the methods looking for the instances of composite elements shared by all sequences in the dataset (15–18). Their basic assumption is that similar CRMs drive similar expression patterns. These methods can be applied both to search for evolutionary conserved CRMs and to characterize regulatory modules shared by co-regulated genes. Thus, they use interspecies similarity without sequence alignment and can handle multiple orthologous sequences.
Here, we further develop this approach combining it with an HMM-based technique, which allows one to account for the CRM structure. The regulatory modules of both orthologous (different species) and co-regulated (same species) genes are assumed to have a similar structure. We extend the notion of the CRM structure, or regulatory grammar; we describe it not only by a combination of the motifs, but also as the motif frequencies, preferences in the binding sites' order and the distance distributions between adjacent sites in a regulatory module. These characteristics could improve the quality of the CRM prediction and elucidate the rules of combinatorial transcriptional regulation.
CORECLUST (COnservative REgulatory CLUster STructure) uses an HMM-based technique to predict regulatory modules given a set of known PWMs. CORECLUST constructs a CRM model by revealing the conserved structure of regulatory modules of orthologous and/or co-regulated genes without using multiple sequence alignment. Then, the obtained model is used to identify similar regulatory modules throughout the genome and thus to predict candidate co-regulated genes. The model itself is also an interesting object for further analysis, as it comprises conserved properties of regulatory modules, such as co-localization of binding sites of certain types and distance preferences for different motif pairs.
The application of the method to two different biological systems, the vertebrate muscle-specific expression system (3) and the Drosophila anterior–posterior (AP) patterning system (19), demonstrates its ability to identify CRMs for a set of system-specific TFs with a quality higher than that of other methods. By applying CORECLUST to the Drosophila patterning system we show that it can successfully predict co-regulated genes. Based on the trained models, we characterize the regulatory grammars for the Drosophila developmental and vertebrate muscle-specific regulatory systems. The most significant observations are supported by the literature data, which demonstrates the ability of CORECLUST to reveal a reasonable regulatory grammar. The software implementing our method can be freely downloaded from http://bioinf.fbb.msu.ru/~anna.
MATERIALS AND METHODS
The algorithm
Here, we outline the algorithm. Technical details are provided in the Supplementary Materials.
At the preliminary stage of the analysis, candidate sites corresponding to target PWMs are identified. This is done using low thresholds and thus with high sensitivity. Then the HMM combines some of the candidate sites in CRMs, discarding the remaining sites.
The HMM
To detect regulatory modules in a DNA sequence we use a HMM (Figure 1). The overall HMM architecture reflects our intuition about the organization of CRMs. A CRM is modeled as a cluster of TFBSs, surrounded by the background sequence. The HMM contains three main types of generative states corresponding to three general types of sequence:
inter-module background sequence; it is modeled as the background and all the candidate sites are ignored there;
sites which are generated in both strands according to the position probability matrices known a priori; and
regions between sites in regulatory modules, i.e. spacers.
Each generative state emits a varying-length sequence of nucleotides. This type of the HMM architecture is known as the generalized hidden Markov model (20) or the ‘HMM with duration’ (21,22). Such models allow for easy use of any predefined length distribution for the emitted states.
Figure 1.

Schematic representation of the HMM. Rectangles represent the emitting states: the BKG (BACKGROUND) state emits background sequence, the S1, S2, etc. states emit, respectively, sites of types S1, S2, etc., and the SPACER:D1 and SPACER:D2 states emit spacer sequences with lengths satisfying distributions D1 and D2, respectively. Ovals represent the silent states: the B (BEGIN) and E (END) states are, respectively, the first and the last states of each HMM path, the CE (CLUSTER ELONGATION) state yields elongation of a site cluster, the NEXT (NEXT SITE) state defines the type of the next site of a cluster. Arrows represent the allowed transitions between the states. The probabilities of the transitions marked by dashed lines are updated during the Baum–Welch training.
Our model characterizes the CRMs by the regulatory structure, that is, by a set of preferences in the site arrangement. First, we take into account preferences in the site ordering by counting the conditional occurrence probability of a site of a certain type given the type of the previous site in a CRM. Second, the spacing between binding sites is considered. For this purpose we introduce several distributions of spacer lengths, the combination of which determines the preferred intersite distances for each motif pair. Currently, two spacer-length distributions are used (Supplementary Figure S2): (i) the geometric distribution, which reflects site clustering without distance specificity, (ii) the exponentially damped sinusoid with a period of 10.5 bp that represents the situation when interacting proteins bind to the same side of the DNA helix. The latter, helical phasing, distribution of intersite distance was observed previously (23–25).
The algorithm stages
The algorithm of CORECLUST comprises two main steps:
training the model on given intergenic sequences of orthologous and/or co-regulated genes; and
applying the trained model to search for regulatory modules with a similar structure.
When the modules are searched in a group of orthologous (or co-regulated) sequences, the algorithm computes the conservation score, which represents the quality and the conservation of TFBS content of the identified CRMs.
Training the HMM parameters
To reveal the regulatory structure from a given set of sequences presumably containing similar CRMs, we train the HMM parameters defining the regulatory structure using the Baum–Welch algorithm (26). The initial values of the parameters are drawn from the uniform distribution.
CRM deciphering and weighting
In the HMM graph that is constructed for a given sequence, each path marks a set of regulatory modules. To optimize the correspondence of the HMM path to the learned regulatory grammar, the posterior-Viterbi decoding algorithm (27) is used. At the first step, the forward–backward algorithm computes the posterior probabilities for each sequence position to be in a given state. Then, the Viterbi algorithm finds the best path (i.e. the path with the maximal posterior probability) through the HMM graph.
The algorithm identifies some (or zero) CRMs in a sequence. Each CRM is scored by the ratio of natural logarithms (base e) of two posterior probabilities: the probability to obtain the nucleotide sub-sequence as generated by the CRM model and the probability to obtain it as generated by the background model. The ratio equals to the ratio of the probabilities of two sub-paths in the HMM graph that emit the CRM and the background sequence, respectively; both sub-paths span from the beginning to the end of the CRM.
Genome-wide search for co-regulated genes
Search for co-regulated genes can be done starting from a group of similarly regulated (i.e. co-regulated or orthologous) genes. Here, we perform the search that starts from one group of orthologous genes. The workflow of the search for co-regulated genes, in outline, consists of the following steps (Figure 2):
Select a gene known to be regulated by TFs of the analyzed system. Define the region supposed to contain the CRMs, for example interval [−20 Kbp, +20 Kbp] relative to the gene start. Take regions situated at the same location relative to the starts of the orthologous genes in all analyzed genomes and train the HMM parameters on this set of sequences.
Apply the trained HMM to sequences surrounding the starts of all known genes in all analyzed genomes to identify and score candidate regulatory modules.
Assign a conservation score to each group of orthologous genes. This score represents the conservation of CRMs identified in the neighborhood of these genes. Sort the groups of orthologs by the conservation score.
Figure 2.

The workflow of the search for co-regulated genes. Horizontal lines denote upstream regions of orthologous genes, the starts of these genes are shown by red arrows. The lowest plot reflects the program output. Blue rectangles represent predicted CRMs; the heights of the rectangles reflect the weights of the CRMs; the red line denotes the threshold for the CRMs weight (see Supplementary Materials for the output examples).
Data
CORECLUST was applied to two biological systems, the vertebrate muscle-specific expression system and the AP patterning system of the Drosophila embryo.
The muscle dataset, initially compiled by Wasserman and Fickett (3), is widely used to assess the quality of the CRM prediction. The dataset including five PWMs (Mef2, Myf, Srf, Tef and Sp1) and 24 sequences from the human, mouse, rat, cow and chicken genomes with the average length of 850 bp, as well as locations of known CRMs conferring muscle-specific gene expression, was obtained from (28).
For the AP system, we used PWMs for seven TFs: Bicoid (Bcd), Hunchback (Hb), Caudal (Cad), Kruppel (Kr), Knirps (Kni), Tailles (Tll) and Giant (Gt). All of them were taken from the iDMMPMM database (29). For our analysis we used 12 Drosophila genomes from the FlyBase database (30): D. melanogaster (R5.6), D. ananassae (R1.1), D. erecta (R1.1), D. grimshawi (R1.1), D. mojavensis (R1.1), D. persimilis (R1.1), D. pseudoobscura (R2.1), D. sechellia (R1.1), D. simulans (R1.1), D. virilis (R1.0), D. willistoni (R1.1) and D. yakuba (R1.1). The information on groups of orthologous genes was taken from FlyBase (release FB2008_03) (30). In the genome-wide searches, all annotated genes in all genomes were processed. The search area for a gene was defined as a sequence around the gene start (at most 20 Kbp upstream and 20 Kbp downstream and limited by the adjacent genes). The gene start was defined as the start coordinate of the gene in the FlyBase database. The sense strand of the gene was processed. The CRM was assumed to regulate a gene if it was found in the search area of that gene. The repeat sequences were masked by the RepeatMasker software (http://www.repeatmasker.org).
The expression data for the Drosophila genes were obtained from the DBGP database (31). The GO annotation of genes were taken from the GO database (32); GO statistics was computed by the GOStat program (33).
Evaluation of prediction
The CORECLUST performance was assessed on the muscle dataset of vertebrates and the AP dataset of Drosophila. The quality of predictions on the muscle dataset was evaluated using the benchmarking framework developed by Klepper et al. (28). For a comprehensive assessment of the program's performance, the framework provides six different measures of correspondence of the predicted CRMs to the known ones: correlation coefficient (CC), sensitivity (Sn), specificity (Sp), positive predictive value (PPV), performance coefficient (PC, phi-score) and average site performance (ASP):
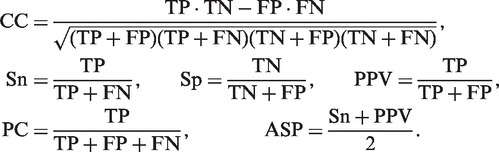 |
Here, TP is the number of nucleotides predicted to be in a CRM and actually belonging to a CRM, TN is the number of nucleotides that do not belong to either predicted or known CRMs, FN is the number of nucleotides that belong to a known CRM but are not predicted as such, and FP is the number of nucleotides that are predicted to be in a CRM, but are not in a known one. Following the procedure in (28), training of the CORECLUST model and CRM search was performed on the entire set of sequences, without dividing the dataset into training and testing parts.
The quality of predictions made for the AP system was assessed on the following dataset: 17 AP genes with known CRMs controlling AP patterning in Drosophila (h, kni, hb, ftz, eve, run, tll, gt, Kr, cad, prd, ems, btd, slp1, bowl, salm and fkh) and PWMs for 7 TFs known to be important in the AP regulation (Bcd, Hb, Cad, Kr, Kni, Tll and Gt). The model training and CRM search were done separately for every gene using all available orthologous sequences. The predictions were made for 40 Kbp sequence fragments ([−20 Kbp, +20 Kbp] relative to the start of each gene). The modules predicted in D. melanogaster sequence fragments were compared with the known ones from the REDFly database (34) (see Supplementary Data); overlapping CRMs were merged.
As a performance measure, we used the CC as it combines all aspects of the prediction quality. The CC was calculated for each gene separately and for the whole gene set. In the former mode, the values of TP, TN, FP and FN were calculated for each fragment separately. In the latter mode, used to assess the overall quality of the predictions for all genes, the values were calculated for all fragments together.
RESULTS
We developed a Java program CORECLUST to search for CRMs in DNA sequences for a set of system-specific TFs. CORECLUST reveals conserved structure (preferred site arrangements) of CRMs of orthologous and/or co-regulated genes and searches for regulatory modules with a similar structure. The program takes as input training sequences presumably containing similar CRMs and a set of user-specified motifs (PWMs). It constructs a model of regulatory modules contained in the training sequences and then uses it to search for similar CRMs in genomic sequences. CORECLUST is available for download at http://bioinf.fbb.msu.ru/~anna.
Testing
We tested the CORECLUST ability to identify CRMs in a set of intergenic regions of co-regulated and orthologous genes on two different biological systems from two distinct clades.
Muscle-specific regulatory modules in vertebrates
We used the benchmarking framework (28) described in the ‘Materials and Methods’ section to assess the CORECLUST performance, which then was compared with the performance of eight published CRM predicting methods, obtained from the benchmarking framework website (http://tare.medisin.ntnu.no/composite/composite.php): Composite Module Analyst (CMA) (16), CisModule (17), ModuleSearcher (18), Stubb (9), MSCAN (2), MCAST (5), Cister (6) and Cluster-Buster (7). All methods except CisModule use known PWMs as input, although CMA and ModuleSearcher can select appropriate matrices from a database, rather than using predefined set of system-specific PWMs. CMA, CisModule and ModuleSearcher look for instances of composite elements shared by all sequences in the dataset. Other methods process each sequence individually. Stubb, MCAST, Cister and Cluster-Buster use a HMM-based approach to search for locally dense clusters of TFBSs, although Stubb can also make use of correlations between binding sites. MSCAN searches for statistically significant clusters of binding sites in a sliding window along the input sequence.
The comparison of the programs' performance (Figure 3) showed that CORECLUST scored better than all other programs for almost all metrics. Losing a little in the sensitivity, CORECLUST scored highest of all for the CC, PC and ASP measures, which capture the over- and underprediction in a single value.
Figure 3.

Comparison of the programs' performance. Notation for the performance measures see in the text.
Early developmental enhancers in Drosophila
Genes of the Drosophila AP system have relatively long CRMs comprising 4500 bp per gene on average. On the other hand, 12 annotated genomes of the Drosophila genus are available in public databases. The large size of the AP CRMs and the number of the available orthologs for each gene allowed us to use upstream regions of genes from just one orthologous group as a training set without overfitting.
The CORECLUST performance was assessed on 17 AP genes as described in the ‘Materials and Methods’ section. The results were compared with those demonstrated by three other publicly available programs: Stubb (9), MOPAT (15) and Cluster-Buster (7). As noted above, Stubb and Cluster-Buster are HMM-based, and Stubb incorporates correlation between binding sites in a module and can take advantage of interspecies comparison. At that, Stubb considers only two sequences at a time and depends on sequence alignment. MOPAT searches for motifs co-occurring in multiple sequences and utilizes both the correlations between binding sites and the comparative information, if provided with a set of orthologous sequences. The test protocol was as follows. All programs were given the same set of PWMs and the same set of genes. MOPAT and CORECLUST were given sequences of all available orthologs for each gene. Stubb was given sequences taken from two species, D. melanogaster and D. virilis, as in the original paper (9) (for the btd gene, the D. mojavensis ortholog was used instead of the D. virilis one because of the absence of the latter). Cluster-Buster was run on sequence fragments from D. melanogaster.
All parameters for Stubb were set to their default values, except the minimum number of motifs in a module. It was set to 1 instead of 3 by default to make it closer to CORECLUST. Cluster-Buster also was run with the default parameters, except for the pseudo-count, which was set to 0.5, as for Stubb. The CRM score threshold was set to 5, as proposed at the Cluster-Buster web-server (http://zlab.bu.edu/cluster-buster/cbust.html). The parameters for the MOPAT program had to be changed to be applicable for our input data. The pseudo-count was set to 0.5; the minimum number of distinct motifs in a cluster (k), to 2; the minimum number of sequence fragments containing instances of a motif cluster (g), to 3. To select the window size (w), we ran MOPAT for four different window sizes: 200, 300, 400 and 500, and selected the one with the highest value of the CC calculated for all genes together (300).
The comparison showed (Table 1) that predictions made by CORECLUST had a higher value of CC than Stubb (P-value <0.05, Wilcoxon signed-rank test), MOPAT (P-value <0.0007, Wilcoxon signed-rank test) and Cluster-Buster (P-value <0.02, Wilcoxon signed-rank test). According to the P-value comparison, Stubb, whose model was quite similar to ours, had the best quality prediction following CORECLUST. At the same time, Stubb scored worse than CORECLUST and Cluster-Buster on the muscle dataset. Interestingly, Cluster-Buster showed rather good results on both the Drosophila and the vertebrate datasets, although it uses no extra information like a regulatory grammar or interspecies comparison.
Table 1.
Comparison of the programs' performance, measured as a Matthews CC
| Gene | CORECLUST | Stubb | MOPAT | Cluster–Buster |
|---|---|---|---|---|
| eve | 0.73 | 0.56 | 0.54 | 0.58 |
| h | 0.69 | 0.17 | 0.26 | 0.49 |
| btd | 0.45 | 0.27 | 0.31 | 0.47 |
| Kr | 0.45 | 0.24 | 0.29 | 0.64 |
| kni | 0.43 | 0.22 | 0.27 | 0.45 |
| gt | 0.41 | 0.48 | 0.27 | 0.40 |
| slp1 | 0.35 | 0.34 | 0.44 | 0.35 |
| hb | 0.32 | 0.33 | 0.17 | 0.22 |
| ftz | 0.31 | 0.36 | 0.32 | 0.27 |
| fkh | 0.31 | 0.28 | 0.27 | −0.02 |
| tll | 0.26 | 0.15 | 0.09 | 0.17 |
| prd | 0.26 | 0.14 | 0.13 | 0.17 |
| salm | 0.23 | 0.07 | −0.01 | 0.17 |
| bowl | 0.20 | 0.10 | −0.01 | 0.17 |
| run | 0.08 | 0.17 | 0.07 | 0.11 |
| ems | −0.02 | 0.15 | −0.01 | −0.02 |
| cad | −0.03 | 0.17 | −0.02 | −0.04 |
| Total | 0.32 | 0.20 | 0.20 | 0.29 |
| Median | 0.31 | 0.22 | 0.26 | 0.22 |
| SD | 0.21 | 0.13 | 0.17 | 0.21 |
| P-value* | <0.05 | <0.0007 | <0.02 |
Total row contains values of CC calculated for the whole gene set (see ‘Materials and Methods’ section). The maximum value in each line is set in bold. *One-tailed P-value for the Wilcoxon signed-rank test.
Genome-wide search for genes co-regulated with known Drosophila AP genes
CORECLUST can be applied to the genome-wide identification of regulatory modules that are described by the same grammar as the training ones, and thus to detect genes that are co-regulated with a given gene or a set of genes. To test the ability of CORECLUST to predict a set of co-regulated genes starting from a single group of orthologs, we applied CORECLUST to the Drosophila AP patterning system. As the input, the same AP PWMs as in the testing section (Bcd, Hb, Cad, Kr, Kni, Tll and Gt) were used. We performed the search for co-regulated genes for different training genes. For each run, we selected a D. melanogaster gene and trained the HMM on the [−20 Kbp, +20 Kbp] sequence fragments relative to the start of this gene and all its available orthologs from the remaining Drosophila genomes. After the model was trained, it was applied to the regions of all genes in all twelve Drosophila genomes and thus a list of genes that had CRMs similar to the CRMs of the training gene was obtained. Then, the orthologous groups of genes with the best conservation score were selected. Such analysis was performed for 22 AP genes and in all cases developmental genes were overrepresented in the resulting gene lists. For example, for the training gene hairy (h), which is a primary pair-rule gene involved in the establishment of the segments during the developmental stages 4–6, the program predicted 45 co-regulated genes. Six of these genes were predicted as related to the AP patterning system because their search regions overlap with the search regions of well-known AP patterning genes. For a clearer presentation, these genes were removed from the result list. The remaining 39 genes were characterized by strong and conserved predicted CRMs and could be considered as candidates to be co-regulated with h. The analysis of the list using the GOStat program (33) yielded significant overrepresentation of GO-terms (32) related to blastoderm segmentation and to development (Table 2). Moreover, most of the top genes of this list (Figure 4) are well-known genes of the AP patterning process and they do have known CRMs driven by AP TFs. Three genes among the top genes that are not known to be related to the AP system (CG13713, CG5103 and Cyp6v1) are still good candidates to be involved in this system, as they are preceded by regions bound by the AP TFs during the embryogenesis stages 4–6, according to the ChIP-chip data (35).
Table 2.
GO-categories (32) overrepresented in the set of genes predicted to be co-regulated with gene h
| GO term | Npred | Ntotal | P-value |
|---|---|---|---|
| Blastoderm segmentation | 14 | 137 | 2.98E-17 |
| Embrionic pattern specification | 14 | 176 | 5.51E-16 |
| Segmentation | 14 | 181 | 5.51E-16 |
| Periodic partitioning by pair rule gene | 6 | 6 | 2.18E-14 |
| Posterior head segmentation | 7 | 15 | 2.31E-13 |
| Embrionic development | 16 | 532 | 1.78E-12 |
Npred is the number of predicted genes assigned with the GO term, Ntotal is the number of the D. melanogaster genes assigned with the GO term.
Figure 4.

The top genes of the list of genes predicted to be co-regulated with gene h. Genes are sorted by the conservation score value of their predicted CRMs. Expression patterns are presented if available for the developmental stages 4–6 (31).
To assess the quality of the resulting gene lists for all 22 training genes systematically, we compared them with predictions of Cluster-Buster (7). Cluster-Buster was selected for the comparison, as it utilizes neither structure of the regulatory modules, nor their conservation among different species, but still demonstrates rather good performance (see above).
We compiled a list of genes likely belonging to the AP patterning system. These genes are annotated as ‘embryonic pattern specification’ (GO:0009880) in the GO database, and at the same time are expressed at the stages 4–6 of the Drosophila development (31). The list was supplemented by well-known AP genes not found by this procedure. The final positive set contained 115 genes (see Supplementary Data).
As Cluster-Buster does not score genes by potential regulation by the analyzed TFs, we applied two simple measures to select genes with the strongest and most numerous CRMs, as predicted by this program. After the genome-wide search (Cluster-Buster was run with the parameters described in the testing section and on the same set of D. melanogaster sequence fragments as used for the CORECLUST runs), the genes were sorted by:
the maximum weight of the regulatory modules, predicted for a gene; and
the sum of weights of the regulatory modules, predicted for a gene.
Thus, two sorted gene lists for the Cluster-Buster predictions were obtained.
Then for every training gene, hypergeometric tests were applied to estimate the statistical significance of the enrichment between the positive gene set and each of the following three gene lists:
the list of co-regulated genes predicted by CORECLUST which contains m genes;
first m genes from the Cluster-Buster gene list, sorted by the maximum module weight; and
first m genes from the Cluster-Buster gene list, sorted by the sum of the module weights.
The comparison of the lists (Figure 5, Supplementary Table S1) demonstrated that predictions made by CORECLUST better fitted the positive gene set than the Cluster-Buster predictions. On the other hand, the comparison of the lists for different training genes showed that not all of them are equally good, probably because their regulatory regions do not contain enough binding sites to train the CORECLUST model properly.
Figure 5.

Genome-wide prediction of co-regulated genes for different training genes made by CORECLUST and Cluster–Buster (Cbust). The histogram represents the comparison of the hypergeometric P-values of enrichment between the positive gene set and three different gene lists, created by CORECLUST and Cluster–Buster, sorted by the maximum [Cbust(max)] or the sum [Cbust(sum)] of the module weights.
DISCUSSION
Here, we present CORECLUST, a method for prediction of CRMs for a set of known system-specific motifs. CORECLUST constructs a model of CRMs of similarly regulated (i.e. co-regulated or orthologous) genes by identification of conserved grammatical structures of the binding sites. Then, the constructed model may be used for the prediction of CRMs with a similar structure throughout the genome (one eukaryotic genome is processed in 25–60 min on average (for the Drosophila and vertebrate genomes respectively) using one core of Intel Xeon L5520 processor). Testing of CORECLUST on two different biological systems, vertebrate muscle-specific expression system and Drosophila AP patterning system, shows that it may be successfully used for solving the standard problem of CRM detection for a set of system-specific TFs. Application of our program for the identification of co-regulated genes in Drosophila shows that given only one group of orthologous genes as the input, it can predict a considerable number of co-regulated genes.
Structural aspects of the model
The incorporation of different distributions for the distance between adjacent sites is a novel feature of our algorithm. Nevertheless, CORECLUST can be also run without accounting for the intersite spacing (we shall refer to this option as CORECLUST-FC) or even ignoring distances and correlations between binding sites (CORECLUST-F). The comparison of the quality of the predictions that are made by CORECLUST, CORECLUST-FC and CORECLUST-F on both developmental and muscle datasets shows that, in general, accounting for the structure information increases the sensitivity and slightly decreases the specificity of predictions (Supplementary Figure S4). A possible explanation is that accounting for the regulatory structure reveals weak sites situated in a proper order and at proper distances from each other and from other sites in a CRM. But if we consider each gene of the Drosophila developmental system separately, we see that along with the positive examples, when inclusion of structural aspects allows the algorithm to find proper regulatory modules (Supplementary Figure S5), there are also cases of decreasing overall quality of predictions (Supplementary Table S2), mainly due to the loss in the precision. However, one should take into account that known regulatory modules possibly do not comprise all regulatory regions. Still, the conservation of the site content of the predicted modules is definitely higher for those CRMs, which are predicted with the full CORECLUST model (Supplementary Figure S6), arguing for considering these predictions as more reliable.
There is also a possibility to incorporate other distance distributions in the model, which could yields non-trivial regulatory grammar features. Currently, we use only two distance distributions, geometric and periodic, consistent with the DNA helical pitch. Other possible distributions may utilize, e.g. the nucleosome periodicity. We did not use additional distributions because it could lead to over-fitting of the model on our dataset. The same problem appears with increasing the number of PWMs used in the model, which causes model overfitting and increase in the working time, as the number of the HMM states depends quadratically on the number of the input matrices. In the current version of the program only a relatively small number of PWMs can be used (up to 10; the maximum acceptable number of PWMs may depend on the running parameters and the size of training sequences). A possible way to overcome this limitation is to train only those HMM parameters that are detected to be significant for a given sequence region as in Stubb (9). This could make it possible to use separate CRM regions, rather than large regions around the genes' starts, as training sequences, which could allow one to identify the conserved structure of an individual CRM.
Deciphering the regulatory grammar
By design, CORECLUST identifies a structure of similar regulatory modules, allowing one to detect and analyze the preferred arrangements of binding sites inherent in a particular regulatory system. The structure of training regulatory modules can be obtained directly from the model. For each pair of site types, i and j, the trained model contains the conditional probability to observe a site of type j next to a site of type i. This probability reflects the frequency of occurrence of this site pair in the training sequences, which may mean that the factors binding these sites interact with each other to regulate gene transcription.
According to the parameters of the model trained on the muscle dataset (Supplementary Figure S7), the most probable site pairs are Mef2-Myf [supported by the TransCompel database (36) and (25)], Sp1-Srf [supported by (37,38)] and Sp1-Sp1 [supported by (39)]. Interestingly, according to the model, the intersite distance for the site pair Mef2-Myf is distributed according to the helical phasing distribution, which was also observed in (25). The other observed interactions are also supported by the literature or TransCompel: Tef-Mef2 (40), Myf-Sp1 (36) and Mef2-Sp1 (41).
The analysis of the model, trained on 40 Kb sequence fragments ([−20 Kbp, +20 Kbp] relative to the start of each gene) of 11 well-known developmental genes (h, kni, hb, ftz, eve, run, tll, gt, Kr, cad and prd) and their orthologs (Supplementary Figure S8), shows that Drosophila developmental patterning system is characterized by homotypic TF interactions, which agrees with previous observations (24,42,43). Interestingly, sites in some of these homotypic pairs tend to be codirectional (see Kr–Kr and Hb–Hb pairs on Supplementary Figure S8), and some of them, like Hb–Hb and Bcd-Bcd, are characterized by the periodic distribution of the intersite distances, consistent with the DNA helix step also observed in (23). The analysis of modules predicted near the same developmental genes revealed several interesting distributions of distance between sites in a module (Supplementary Figure S9). For example, for almost all (15 out of 18) observed site pairs 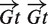 (two Gt binding sites, the arrow shows the direction of a site relative to the gene direction), the distance between sites in a pair is 51–58 bp. The distribution for the site pair
(two Gt binding sites, the arrow shows the direction of a site relative to the gene direction), the distance between sites in a pair is 51–58 bp. The distribution for the site pair 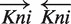 has an unusual peak at distance 135–138 bp, which is rather uncommon and perhaps could indicate that the corresponding TFs interact with packed DNA.
has an unusual peak at distance 135–138 bp, which is rather uncommon and perhaps could indicate that the corresponding TFs interact with packed DNA.
All in one, CORECLUST makes biologically meaningful and useful predictions. It can successfully identify putative CRMs, predict co-regulated genes and decipher common rules of the TF interactions for a regulatory system.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online: Supplementary Tables S1–S2, Supplementary Figures S2–S9, Supplementary Methods, Supplementary Files, and Supplementary References [44–46].
FUNDING
Programs 6 and 17 of the Russian Academy of Sciences; Russian Foundation of Basic Research [grant numbers 09-04-92742, 11-04-02016-a, 10-04-92663-IND_a and 11-04-02051-a]; State Contract of Russian Ministry of Education and Science [grant numbers 07.514.11.4007 and 07.514.11.4005]; Russian Academy of Science Presidium Program on Molecular and Cellular Biology; the Johns Hopkins University Framework for the Future; the Commonwealth Foundation and the SKCCC Center for Personalized Cancer Medicine. Funding for open access charge: Lomonosov Moscow State University.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Mikhail Gelfand and Dmitri Pervouchine for useful discussions and encouragement, and to Dmitry Vinogradov for technical assistance. We thank the BDGP Patterned Gene Expression in Drosophila Development project (NIH R01-GM076655) at Lawrence Berkeley National Laboratory for providing in situ hybridization images used in this study.
REFERENCES
- 1.Kel-Margoulis OV, Kel AE, Reuter I, Deineko IV, Wingender E. TRANSCompel: a database on composite regulatory elements in eukaryotic genes. Nucleic Acids Res. 2002;30:332–334. doi: 10.1093/nar/30.1.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johansson O, Alkema W, Wasserman WW, Lagergren J. Identification of functional clusters of transcription factor binding motifs in genome sequences: the MSCAN algorithm. Bioinformatics. 2003;19(Suppl. 1):i169–i176. doi: 10.1093/bioinformatics/btg1021. [DOI] [PubMed] [Google Scholar]
- 3.Wasserman WW, Fickett JW. Identification of regulatory regions which confer muscle-specific gene expression. J. Mol. Biol. 1998;278:167–181. doi: 10.1006/jmbi.1998.1700. [DOI] [PubMed] [Google Scholar]
- 4.Levy S, Hannenhalli S. Identification of transcription factor binding sites in the human genome sequence. Mamm. Genome. 2002;13:510–514. doi: 10.1007/s00335-002-2175-6. [DOI] [PubMed] [Google Scholar]
- 5.Bailey TL, Noble WS. Searching for statistically significant regulatory modules. Bioinformatics. 2003;19(Suppl. 2):ii16–ii25. doi: 10.1093/bioinformatics/btg1054. [DOI] [PubMed] [Google Scholar]
- 6.Frith MC, Hansen U, Weng Z. Detection of cis-element clusters in higher eukaryotic DNA. Bioinformatics. 2001;17:878–889. doi: 10.1093/bioinformatics/17.10.878. [DOI] [PubMed] [Google Scholar]
- 7.Frith MC, Li MC, Weng Z. Cluster-Buster: finding dense clusters of motifs in DNA sequences. Nucleic Acids Res. 2003;31:3666–3668. doi: 10.1093/nar/gkg540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frith MC, Spouge JL, Hansen U, Weng Z. Statistical significance of clusters of motifs represented by position specific scoring matrices in nucleotide sequences. Nucleic Acids Res. 2002;30:3214–3224. doi: 10.1093/nar/gkf438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sinha S, van Nimwegen E, Siggia ED. A probabilistic method to detect regulatory modules. Bioinformatics. 2003;19(Suppl. 1):i292–i301. doi: 10.1093/bioinformatics/btg1040. [DOI] [PubMed] [Google Scholar]
- 10.Noto K, Craven M. Learning probabilistic models of cis-regulatory modules that represent logical and spatial aspects. Bioinformatics. 2007;23:e156–e162. doi: 10.1093/bioinformatics/btl319. [DOI] [PubMed] [Google Scholar]
- 11.Wong WS, Nielsen R. Finding cis-regulatory modules in Drosophila using phylogenetic hidden Markov models. Bioinformatics. 2007;23:2031–2037. doi: 10.1093/bioinformatics/btm299. [DOI] [PubMed] [Google Scholar]
- 12.Birney E. Evolutionary genomics: come fly with us. Nature. 2007;450:184–185. doi: 10.1038/450184a. [DOI] [PubMed] [Google Scholar]
- 13.Moses AM, Pollard DA, Nix DA, Iyer VN, Li X, Biggin MD, Eisen MB. Large-scale turnover of functional transcription factor binding sites in Drosophila. PLoS Comput. Biol. 2006;2:e130. doi: 10.1371/journal.pcbi.0020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hallikas O, Palin K, Sinjushina N, Rautiainen R, Partanen J, Ukkonen E, Taipale J. Genome-wide prediction of mammalian enhancers based on analysis of transcription-factor binding affinity. Cell. 2006;124:47–59. doi: 10.1016/j.cell.2005.10.042. [DOI] [PubMed] [Google Scholar]
- 15.Hu J, Hu H, Li X. MOPAT: a graph-based method to predict recurrent cis-regulatory modules from known motifs. Nucleic Acids Res. 2008;36:4488–4497. doi: 10.1093/nar/gkn407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kel A, Konovalova T, Waleev T, Cheremushkin E, Kel-Margoulis O, Wingender E. Composite Module Analyst: a fitness-based tool for identification of transcription factor binding site combinations. Bioinformatics. 2006;22:1190–1197. doi: 10.1093/bioinformatics/btl041. [DOI] [PubMed] [Google Scholar]
- 17.Zhou Q, Wong WH. CisModule: de novo discovery of cis-regulatory modules by hierarchical mixture modeling. Proc. Natl. Acad. Sci. USA. 2004;101:12114–12119. doi: 10.1073/pnas.0402858101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aerts S, Van Loo P, Thijs G, Moreau Y, De Moor B. Computational detection of cis-regulatory modules. Bioinformatics. 2003;19(Suppl. 2):ii5–ii14. doi: 10.1093/bioinformatics/btg1052. [DOI] [PubMed] [Google Scholar]
- 19.Rivera-Pomar R, Jckle H. From gradients to stripes in Drosophila embryogenesis: filling in the gaps. Trends Genet. TIG. 1996;12:478–483. doi: 10.1016/0168-9525(96)10044-5. [DOI] [PubMed] [Google Scholar]
- 20.Kulp D, Haussler D, Reese MG, Eeckman FH. A generalized hidden Markov model for the recognition of human genes in DNA. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1996;4:134–142. [PubMed] [Google Scholar]
- 21.Rabiner LR. A tutorial on hidden markov models and selected applications in speech recognition. Proc. IEEE. 1989;77:257–286. [Google Scholar]
- 22.Lukashin AV, Borodovsky M. GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 1998;26:1107–1115. doi: 10.1093/nar/26.4.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Papatsenko D, Goltsev Y, Levine M. Organization of developmental enhancers in the Drosophila embryo. Nucleic Acids Res. 2009;37:5665–5677. doi: 10.1093/nar/gkp619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Makeev VJ, Lifanov AP, Nazina AG, Papatsenko DA. Distance preferences in the arrangement of binding motifs and hierarchical levels in organization of transcription regulatory information. Nucleic Acids Res. 2003;31:6016–6026. doi: 10.1093/nar/gkg799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fickett JW. Coordinate positioning of MEF2 and myogenin binding sites. Gene. 1996;172:GC19–GC32. doi: 10.1016/0378-1119(95)00888-8. [DOI] [PubMed] [Google Scholar]
- 26.Durbin R, Eddy SR, Krogh A, Mitchison G. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge, UK: Cambridge University Press; 1998. [Google Scholar]
- 27.Fariselli P, Martelli PL, Casadio R. A new decoding algorithm for hidden Markov models improves the prediction of the topology of all-beta membrane proteins. BMC Bioinf. 2005;6(Suppl. 4):S12. doi: 10.1186/1471-2105-6-S4-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klepper K, Sandve GK, Abul O, Johansen J, Drablos F. Assessment of composite motif discovery methods. BMC Bioinformatics. 2008;9:123–123. doi: 10.1186/1471-2105-9-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kulakovskiy IV, Makeev VJ. Discovery of DNA motifs recognized by transcription factors through integration of different experimental sources. Biophysics. 2010;54:667–674. [Google Scholar]
- 30.Tweedie S, Ashburner M, Falls K, Leyland P, McQuilton P, Marygold S, Millburn G, Osumi-Sutherland D, Schroeder A, Seal R, et al. FlyBase: enhancing Drosophila Gene Ontology annotations. Nucleic Acids Res. 2009;37:D555–D559. doi: 10.1093/nar/gkn788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tomancak P, Berman BP, Beaton A, Weiszmann R, Kwan E, Hartenstein V, Celniker SE, Rubin GM. Global analysis of patterns of gene expression during Drosophila embryogenesis. Genome Biol. 2007;8:R145. doi: 10.1186/gb-2007-8-7-r145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beissbarth T, Speed TP. GOstat: find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics. 2004;20:1464–1465. doi: 10.1093/bioinformatics/bth088. [DOI] [PubMed] [Google Scholar]
- 34.Halfon MS, Gallo SM, Bergman CM. REDfly 2.0: an integrated database of cis-regulatory modules and transcription factor binding sites in Drosophila. Nucleic Acids Res. 2008;36:D594–D598. doi: 10.1093/nar/gkm876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X, MacArthur S, Bourgon R, Nix D, Pollard DA, Iyer VN, Hechmer A, Simirenko L, Stapleton M, Cris LLH, et al. Transcription factors bind thousands of active and inactive regions in the Drosophila blastoderm. PLoS Biol. 2008;6:e27. doi: 10.1371/journal.pbio.0060027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–D110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Biesiada E, Hamamori Y, Kedes L, Sartorelli V. Myogenic basic helix-loop-helix proteins and Sp1 interact as components of a multiprotein transcriptional complex required for activity of the human Cardiac alpha-actin promoter. Mol. Cell. Biol. 1999;19:2577–2584. doi: 10.1128/mcb.19.4.2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Madsen CS, Regan CP, Owens GK. Interaction of CArG elements and a GC-rich repressor element in transcriptional regulation of the smooth muscle myosin heavy chain gene in vascular smooth muscle cells. J. Biol. Chem. 1997;272:29842–29851. doi: 10.1074/jbc.272.47.29842. [DOI] [PubMed] [Google Scholar]
- 39.Anderson GM, Freytag SO. Synergistic activation of a human promoter in vivo by transcription factor Sp1. Mol. Cell. Biol. 1991;11:1935–1943. doi: 10.1128/mcb.11.4.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maeda T, Gupta MP, Stewart AFR. TEF-1 and MEF2 transcription factors interact to regulate muscle-specific promoters. Biochem. Biophys. Res. Commun. 2002;294:791–797. doi: 10.1016/S0006-291X(02)00556-9. [DOI] [PubMed] [Google Scholar]
- 41.Grayson J, Bassel-Duby R, Williams RS. Collaborative interactions between MEF-2 and Sp1 in muscle-specific gene regulation. J. Cell. Biochem. 1998;70:366–375. [PubMed] [Google Scholar]
- 42.Lebrecht D, Foehr M, Smith E, Lopes FJP, Vanario-Alonso CE, Reinitz J, Burz DS, Hanes SD. Bicoid cooperative DNA binding is critical for embryonic patterning in Drosophila. Proc. Natl. Acad. Sci. USA. 2005;102:13176–13181. doi: 10.1073/pnas.0506462102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lifanov AP, Makeev VJ, Nazina AG, Papatsenko DA. Homotypic regulatory clusters in Drosophila. Genome Res. 2003;13:579–588. doi: 10.1101/gr.668403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stark A, Lin MF, Kheradpour P, Pedersen JS, Parts L, Carlson JW, Crosby MA, Rasmussen MD, Roy S, Deoras AN, et al. Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures. Nature. 2007;450:219–232. doi: 10.1038/nature06340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gerstein M, Sonnhammer EL, Chothia C. Volume changes in protein evolution. J. Mol. Biol. 1994;236:1067–1078. doi: 10.1016/0022-2836(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 46.Lawrence C, Altschul S, Boguski M, Liu J, Neuwald A, Wootton J. Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science. 1993;262:208–214. doi: 10.1126/science.8211139. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}