


Abstract
Computationally retrieving biologically relevant cis-regulatory modules (CRMs) is not straightforward. Because of the large number of candidates and the imperfection of the screening methods, many spurious CRMs are detected that are as high scoring as the biologically true ones. Using ChIP-information allows not only to reduce the regions in which the binding sites of the assayed transcription factor (TF) should be located, but also allows restricting the valid CRMs to those that contain the assayed TF (here referred to as applying CRM detection in a query-based mode). In this study, we show that exploiting ChIP-information in a query-based way makes in silico CRM detection a much more feasible endeavor. To be able to handle the large datasets, the query-based setting and other specificities proper to CRM detection on ChIP-Seq based data, we developed a novel powerful CRM detection method ‘CPModule’. By applying it on a well-studied ChIP-Seq data set involved in self-renewal of mouse embryonic stem cells, we demonstrate how our tool can recover combinatorial regulation of five known TFs that are key in the self-renewal of mouse embryonic stem cells. Additionally, we make a number of new predictions on combinatorial regulation of these five key TFs with other TFs documented in TRANSFAC.
INTRODUCTION
In eukaryotes, transcriptional regulation is mediated by the concerted action of different transcription factors (TFs) (1). Searching for cis-acting regulatory modules (CRMs), or combinations of motifs that often co-occur in a set of coregulated sequences, helps in unraveling the mode of combinatorial regulation. CRM detection is customarily being applied on a set of intergenic regions located upstream of coexpressed genes; such genes are for example identified by microarray experiments. Except for de novo methods (2,3), most CRM detection methods rely on a motif screening step. In this step, all sites that match given motifs of TFs, are located in the selected sequences (4,5). Subsequently, a combinatorial search is performed to identify a set of motifs (called a CRM), that occur frequently in the given set of intergenic sequences. Usually, a score is assigned to each of the obtained CRMs that assess their statistical significance in a set of background sequences (4,5). Some methods apply a highly structured definition in which a CRM consists of combinations of motifs that need to occur in a specific order, with specific orientations, and within certain distances (6–8). Although the overrepresentation of a structured CRM in a gene set is likely to be biologically relevant (9,10), the degree to which biologically relevant CRMs are structured is still largely unknown (11). Therefore, most methods rely on less constrained CRM models, hereafter referred to as unstructured CRM detection methods (4,5,12–16),
The combinatorial search underlying CRM detection is highly complex. Adding to this complexity, is the fact that often the regions containing the binding sites are not well delineated [e.g. when starting from a coexpressed gene sets, the intergenic sequences typically range several 1000s of base pairs upstream of the transcription start site (TSS)]. Such long intergenic sequences reduce the signal/noise ratio to such extent that in silico CRM detection becomes ineffective. Hence the search for combinatorial regulation is often limited to the proximal promoter region, whereas at least some of the sites responsible for the observed expression behavior might also be located in regions distant from the TSS of the given genes (such as for instance in enhancers). Nowadays chromatin-immunoprecipitation (ChIP)-based techniques are becoming increasingly popular for the genome-wide identification of TF binding sites (17,18). Such techniques make it feasible to locate, at least for the assayed TF, the approximate binding regions. Using ChIP-bound sequences thus allows largely reducing the regions in which the binding sites of the assayed TF should be located (typically 500 bp instead of thousands of bp) (19,20) and does not restrict the search for CRMs to the proximal promoter region (7,21–23).
In addition to these obvious advantages, using ChIP-information also allows for a query-based search strategy. Instead of searching for all possible CRMs in the input set as is traditionally being done, we can limit our search to those CRMs that contain the ChIP-assayed TF (7,8). Incorporating knowledge of the assayed TF during CRM detection in such a query-based way allows predicting complex CRMs in which the assayed TF is involved. In this work, we studied the extent to which using ChIP-derived information can help in increasing the performance of CRM detection, compared to the application of CRM detection in a traditional non-query-based setting. To this end, we developed a novel powerful CRM detection method ‘CPModule’, an unstructured CRM detection method based on a constraint programming for itemset mining framework (24). Besides handling the specific challenges of CRM detection on ChIP-Seq based data, CPModule can be used in both a query and non-query-based mode. Its exhaustive search strategy allows making an assessment of the total number of valid CRMs that are present in the input set and of the degree to which a CRM of interest gets prioritized among the total number of candidates.
Applying CPModule on a well-studied ChIP-Seq data set involved in self-renewal of mouse embryonic stem cells (25) showed that using a query-based setting is in most cases the only sensible way to perform CRM detection. Besides recovering well described benchmark CRMs, we also make several novel predictions on the combinatorial regulation of the five key regulators, involved in the process of self-renewal, with other TFs documented in TRANSFAC (26).
MATERIALS AND METHODS
Motif screening and filtering (Figure 1A)
Figure 1.

CPModule analysis flow. (A) The input consists of a library of PWMs and a set of sequences. In the first step, prior to the actual CRM detection a screening with public motif databases is performed. Here, we combine standard PWM screening with filtering based on nucleosome occupancy. Motif sites displaying a high nucleosome occupancy are filtered out (indicated as the transparent shapes in A). (B) The second step consists of the actual combinatorial search. Here, we use a constraint programming for itemset mining approach to enumerate all valid motif sets, i.e. combinations of motifs (i) that occur frequently in the input set (frequency constraint). Only valid motif sets will be considered (indicated in regular boxes), while invalid ones will not (indicated in dashed boxes); (ii) of which the motif sites contributing to the motif set occur in each other’s proximity (proximity constraint). Only valid motif sets will be considered (indicated in regular boxes), while invalid ones will not (indicated in dashed boxes); (iii) that are non-redundant (redundancy constraint). The motif sets in the dashed box are redundant with the motif set in the regular box and will not be considered; (iv) that contain a query-motif (query-based constraint), which corresponds in this work to the motif of the ChIP-assayed TF. Valid motif sets are indicated in regular boxes. (C) Valid motif sets or CRMs are finally assigned a P-value that expresses their specificity for the input set.
Motif screening
The motif models used for screening are position weight matrices (PWMs) from TRANSFAC (26). To remove redundancy among the PWMs, each of them was compared to all the other PWMs using the program MotifComparison (27). MotifComparison calculates the Kullback–Leiber distance between matrices; PWMs showing a mutual distance <0.1 were grouped. All PWMs in a group were removed except for one representative one. This resulted in a final list of 516 PWMs. Motif screening was performed with the tool Clover (28). Given a PWM of W nucleotides and a sequence, it calculates a score for each subsequence of length W. A threshold on the score determines whether the subsequence is considered a potential binding site of the motif, also called a hit or a motif site. The default threshold of 6.0 was used to define a stringent screening, resulting in few hits per motif and sequence on average, while a threshold of 3.0 corresponds to a non-stringent screening resulting in many more hits.
Filtering based on nucleosome occupancy
We filtered potential motif sites by removing sites that exceed a given value for the nucleosome occupancy score (NuOS). To calculate the nucleosome occupancy score of a potential motif site, we first assigned a nucleosome occupancy probability to each base pair position, using the prediction model ‘NuPoP’ of Xi et al. 2010 (29). The nucleosome occupancy score was then calculated as the geometric mean of the nucleosome occupancy probabilities at all positions of the potential motif site (30).
To determine the optimal threshold values for the NuOS, we tested the effect of different filtering thresholds on their ability to: (i) reduce the number of motif site predictions per region and per TF, while (ii) not too much compromising the sensitivity of recovering true binding sites (see Supplementary File S1). Based on these tests, predicted motif sites located within a low probability of nucleosome occupancy (<10%) (when using a filtering based on the NuOS) were retained.
CRM detection using constraint programming for itemset mining (Figure 1B)
CPModule uses as input the motif sites located in the input sequences by motif screening. The result of the screening and filtering step is for each motif M and sequence S a set of motif hits  . Motif M has a hit at
. Motif M has a hit at  with
with 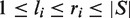 ; here
; here  is an interval between start position
is an interval between start position  and stop position
and stop position  on the sequence S.
on the sequence S.  corresponds to the length of sequence S.
corresponds to the length of sequence S.
The combinatorial search problem of finding a motif set is solved by using the constraint programming (CP) for itemset mining framework (24). The core of CPModule enumerates all possible motif sets, where a motif set  is defined as a subset of all screened motifs. A CRM is a motif set
is defined as a subset of all screened motifs. A CRM is a motif set  that is valid given a set of domain-specific constraints (more details provided below): (i) the frequency constraint, which requires that the motif set occurs in a predefined minimal number of sequences
that is valid given a set of domain-specific constraints (more details provided below): (i) the frequency constraint, which requires that the motif set occurs in a predefined minimal number of sequences  from the input set, (ii) the proximity constraint, which requires that hits of motifs in a set should occur in each other’s proximity. The maximal distance
from the input set, (ii) the proximity constraint, which requires that hits of motifs in a set should occur in each other’s proximity. The maximal distance 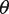 of the region in which hits should co-occur is specified by the user and controls the level of proximity, (iii) the redundancy constraint, which requires that the motif set
of the region in which hits should co-occur is specified by the user and controls the level of proximity, (iii) the redundancy constraint, which requires that the motif set  must be non-redundant with respect to its supersets. Optionally, (iv) a query constraint ensures that a given query motif must be part of the motif set found (Figure 1B).
must be non-redundant with respect to its supersets. Optionally, (iv) a query constraint ensures that a given query motif must be part of the motif set found (Figure 1B).
More formally, a motif set  will only be considered as a potential CRM in a sequence
will only be considered as a potential CRM in a sequence  if and only if its set of hit regions (HR) on that sequence
if and only if its set of hit regions (HR) on that sequence  is not empty, where the set of hit regions (HR) consists of those ranges
is not empty, where the set of hit regions (HR) consists of those ranges  of
of  base pairs in which all selected motifs
base pairs in which all selected motifs  are present, e.g. they have at least one hit with interval
are present, e.g. they have at least one hit with interval  in that range:
in that range:
 |
(1) |
Given a set of sequences S, the subset of sequences in which the set of hit regions is not empty is denoted by  .
.
To find the motif sets that fulfill the above requirements, we use a general and principled approach based on ‘constraint programming’. In constraint programming, problems are modeled as constraint satisfaction problem in terms of constraints on variables. The constraint specification is then solved by a general purpose, yet highly efficient algorithm. Consequently, we need to encode our problem as a constraint satisfaction problem. We do so as follows. Motif sets are represented by Boolean variables: there is a Boolean variable  for every possible motif, indicating whether this motif is part of the motif set
for every possible motif, indicating whether this motif is part of the motif set  . If a certain
. If a certain  , then we say that the motif is in the motif set; otherwise the motif is not in the set. Furthermore, we have a Boolean variable
, then we say that the motif is in the motif set; otherwise the motif is not in the set. Furthermore, we have a Boolean variable  for every genomic sequence, indicating whether the motif set will be considered as a potential CRM in a sequence, i.e. whether
for every genomic sequence, indicating whether the motif set will be considered as a potential CRM in a sequence, i.e. whether  . Lastly, we define a Boolean variable
. Lastly, we define a Boolean variable  for every motif
for every motif  and every sequence
and every sequence  . The variable
. The variable  indicates whether motif
indicates whether motif  is in the proximity of all motifs in motif set
is in the proximity of all motifs in motif set  on sequence
on sequence  .
.
The ‘constraints’ are imposed on these variables as follows:
Proximity constraint
The proximity constraint couples the  variables to the variables representing the motifs. Formally, we define the
variables to the variables representing the motifs. Formally, we define the  variables as follows for every motif on every genomic sequence:
variables as follows for every motif on every genomic sequence:
 |
(2) |
In other words, if in a particular genomic sequence a hit of a particular motif is within a hit region ( ) of the motif set, this motif’s variable for that sequence must be 1. Observe that
) of the motif set, this motif’s variable for that sequence must be 1. Observe that  =1 will hold for all motifs in the motif set
=1 will hold for all motifs in the motif set  , for all sequences that are in
, for all sequences that are in  ; however, there may be additional motifs that have hits in the proximity of regions in
; however, there may be additional motifs that have hits in the proximity of regions in  .
.
Frequency constraint
The constraint that imposes a minimum size on  is formalized as:
is formalized as:  . Here, the
. Here, the  variables are defined in terms of the
variables are defined in terms of the variables, such to ensure that sequences are only counted (
variables, such to ensure that sequences are only counted ( ) if all selected motifs (
) if all selected motifs (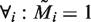 ) occur within each other’s proximity in that sequence (
) occur within each other’s proximity in that sequence ( ):
):
| (3) |
Redundancy constraint
The redundancy constraint requires that we cannot add a motif to a motif set without losing one sequence in its corresponding sequence set  . This can be enforced as follows on the Boolean variables:
. This can be enforced as follows on the Boolean variables:
| (4) |
stating that a motif must be part of the set ( ) if on all selected sequences (
) if on all selected sequences ( ) the motif is within the proximity of the others (
) the motif is within the proximity of the others ( ).
).
Query-based constraint
The query-based constraint requires that each motif set contains at least a given motif. This can be enforced by requiring that the corresponding Boolean variable  satisfies the constraint that
satisfies the constraint that  . Note that the proximity constraint will ensure that only motif sets will be considered with hits close enough to this given motif.
. Note that the proximity constraint will ensure that only motif sets will be considered with hits close enough to this given motif.
This combination of constraints is solved by a constraint programming system by means of a depth first backtracking search. The search strategy alternates between branching, in which a variable is assigned a value from its domain (Boolean value), and propagation, the process of using a constraint to remove values from the domain of variables. The search strategy is similar to strategies that have been used in itemset mining (24). The main difference with traditional itemset mining is the inclusion of proximity constraints and the inclusion of a redundancy constraint for this type of data. The advantage of using an existing CP system (31) is that additional constraints can be added in a modular and straightforward way, preventing the reimplementation of the itemset mining strategy from scratch. For more details on the implementation of the different constraints we refer to ref. (32).
Genome-wide enrichment score calculation and ranking (Figure 1C)
To assess the significance of the found motif sets (CRMs), we calculate for each an enrichment score (P-value) adapting the strategy proposed in Gallo et al. (33). The main modification is that we use a set of background sequences in the calculation of this score. These background sequences are used to estimate the proportion  of sequences in the whole genome that contain the motif set. We compare the number of observed input sequences that contain a particular motif set
of sequences in the whole genome that contain the motif set. We compare the number of observed input sequences that contain a particular motif set  with the number of sequences that is expected to contain this motif set. The latter is estimated by counting the number of background sequences containing the motif set
with the number of sequences that is expected to contain this motif set. The latter is estimated by counting the number of background sequences containing the motif set  . This set is obtained after applying exactly the same screening and filtering strategy on the background sequences as was applied on the input sequences. Based on this estimate of the probability to observe a valid motif set in a random set, we calculate a genome-wide enrichment score (P-value) by means of a cumulative binomial distribution:
. This set is obtained after applying exactly the same screening and filtering strategy on the background sequences as was applied on the input sequences. Based on this estimate of the probability to observe a valid motif set in a random set, we calculate a genome-wide enrichment score (P-value) by means of a cumulative binomial distribution:
 |
(5) |
Where  ;
;  is the set of input sequences;
is the set of input sequences;  is the number of input sequences;
is the number of input sequences;  is the set of background sequences.
is the set of background sequences.
The background sequences were derived by sampling from the mouse genome [Version mm9, NCBI Build 37, UCSC database (34)], a large number of intergenic sequences (2000 background sequences for the synthetic data, 5000 background sequences for each of the ChIP-Seq assays). To exclude the possibility that the composition of the background set would influence the estimated background occurrences of the CRMs, we compiled background sets consisting of either putative promoter sequences, that is sequences located upstream of a gene’s transcription start site, or sets made from background sequences located in putative enhancer regions, that is sequences corresponding to regions bound by the enhancer binding proteins factor CTCF [downloaded from ENCODE (35)]. In our experiments, the composition of the background sets did not influence our final ranking. All results presented in the article use a background set based on proximal promoter regions.
When dealing with ChIP-Seq data, where each sequence is a region around an assayed transcription factor site, we selected the background sequences such that each sequence contains at least one motif site of the assayed TF (which does not overlap with a ChIP-bound region). In this setting, the number of background sequences that qualifies is variable for each data set. To have an equal number of sequences for each background set, we randomly sampled for each data set 5000 sequences from the set of qualifying background sequences (5310 was the maximal number of sequences that could be obtained for the data set with the smallest cognate background set). Note that with this strategy we approximate the P-value calculation in a conservative way as we cannot exclude that the background contains sequences with true sites of the assessed TF that remained unbound under the assessed conditions.
The motif sets are ranked according to their enrichment score [P-value (M), Equation (5)]. Note that with this ranking, for two redundant motif sets that occur in the same sequences, the smaller motif set will never score better than the larger one i.e. if  , with φ(M1,S)=φ(M2,S), motif set M2 will never score worse than M1. This motivates the use of the redundancy constraint during the combinatorial search as it removes from a set of redundant motif sets only the least interesting sets, which would get a very low rank in any case.
, with φ(M1,S)=φ(M2,S), motif set M2 will never score worse than M1. This motivates the use of the redundancy constraint during the combinatorial search as it removes from a set of redundant motif sets only the least interesting sets, which would get a very low rank in any case.
Benchmarking on synthetic data
We first use a synthetic data set to compare CPModule with different CRM detection tools. The synthetic data is retrieved from Xie et al. (36). The data consists of 22 genomic sequences each 1000 base pairs in length. In 20 sequences, sites sampled from the TRANSFAC PWMs of respectively OCT4, SOX2 and FOXD3 were inserted in a region of at most 164 bp (so the CRM encompasses maximally 164 bp). Each PWM was sampled three times per sequence. The last two sequences had no sites inserted.
CPModule was compared with related tools for unstructured CRM detection such as ModuleSearcher, obtained from the author (37) on 6 July 2008; Compo obtained from the author (14) on 14 August 2010; Cister and Cluster-Buster downloaded from the authors website (15,16) on 22 June 2010 and 21 June 2010, respectively; all methods were given the non-redundant motif list described in section ‘Motif screening’.
CPModule was run with a frequency threshold of 60% and a proximity threshold of 165 bp (as this was the maximum distance used when generating the data). Nevertheless, CPModule was shown not to be very sensitive to the exact value of the proximity threshold (see Supplementary File S2). For the other CRM detection tools, we similarly used the best parameter values according to the characteristics of the synthetic data (length of the sequences, the distance between two insertion sites, and the maximum size of CRM) and default values otherwise. Supplementary Table S1 lists the non-default parameters for the used tools.
We evaluated the performance of the different CRM tools using the motif (mCC) and nucleotide correlation coefficients (nCC) (5). At the motif level (mCC), a predicted motif for a sequence is a true positive (TP) if that motif was indeed part of the CRM on that sequence, otherwise it is a false positive (FP). If a motif was not predicted to belong to a CRM on that sequence, although it should have been according to the benchmark, it is counted as a false negative (FN), otherwise as a true negative (TN). As the motif level evaluation does not take the predicted sites into account, a solution is also evaluated at the nucleotide level (nCC): for every nucleotide we verify whether it was predicted to be part of the CRM and whether it should have been predicted or not, again resulting in TP, FP, FN and TN counts. These counts are aggregated over all sequences to obtain the total counts of the predicted CRM. Based on these counts, a correlation coefficient (CC) is defined as follows:
 |
(6) |
The value of this coefficient ranges from −1 to 1. A score of +1 indicates that a prediction corresponds to the correct answer. Random predictions will generally result in CC values close to zero. Ideally, a CRM should score good at both the motif and nucleotide level.
CRM detection on real data
The real data set was derived from genome-wide ChIP data obtained with DNA sequencing (ChIP-Seq) for the KLF4, NANOG, OCT4, SOX2 and STAT3 transcription factors, as described by Chen et al. (25). For each transcription factor, the input set consists of 100 sequences, each corresponding to 500 bp centered around one of the top 100 ChIP-binding peaks of the assayed TF. Binding peaks were taken from the GEO file GSE11431 (38).
Screening is performed using the 516 TRANSFAC PWMs described above (section ‘Motif screening’). However, as a KLF4 PWM was missing in TRANSFAC, we added to our list the PWM described by Whitington et al. (39). Whitington et al. (39) constructed the KLF4 PWM using de novo motif detection on a set of sequences involved in the development of mouse embryonic stem cells, derived from a ChIP-chip experiment by Jiang et al. (40) independent from the one used in this study.
We applied our method using three different screening results: (i) high-stringency screening; (ii) low-stringency screening; (iii) and low-stringency screening in combination with NuOS filtering. Proximity thresholds for CPModule were varied stepwisely as mentioned below (section ‘Results’ section). The frequency threshold was set at 60% unless mentioned otherwise.
RESULTS
CPModule: CRM detection based on constraint programming for itemset mining
Algorithmic design
In contrast to what is often assumed for CRM detection on coexpressed genes, we do not expect that all sequences derived from a ChIP-Seq experiment contain the same CRM. Indeed in the same list of ChIP-bound sequences, different CRMs might be present depending on which other TFs are needed next to the ChIP-assayed one to mediate coregulation of certain subsets of the genes in the list.
Since the same CRM must not be present in all sequences, one cannot simply take the intersection of motifs that appear in the sequences. Instead, all possible combinations of motifs have to be considered as candidate CRM, and validated against the data. This makes CRM detection a combinatorial and computational hard problem. Furthermore, it is not known in advance which TFs are part of the CRM hence one would like to consider all TFs having a known motif model. Using a large number of candidate motifs makes the problem more difficult, as 100 candidate motifs means there are 2100 candidate motif sets; the problem is even more severe when using the entire TRANSFAC database, which contains more than 500 motif models. Additionally, ChIP-Seq derived datasets are large in the number of sequences (usually hundreds of peaks are detected for the assayed TF). For each candidate motif set, all these sequences will have to be processed. The fact that each motif can have multiple hits on a single sequence complicates things even further. Despite the fact that several methods for CRM detection have been developed in the past (4), the aforementioned computational issues are still challenging for CRM tools that find complex CRMs consisting of an arbitrary number of TFs.
To deal with these computation issues, we developed CPModule, a CRM detection method based on constraint programming (CP) for itemset mining (24) (for a full description of the method see ‘Materials and Methods’ section). CPModule searches for combinations of motifs (cis-acting regulatory modules) that are sufficiently specific for a given input set. Using a library of PWMs, a set of coregulated sequences is screened and filtered to obtain a list of hits per motif and sequence (Figure 1A). CPModule then uses this input list to enumerate all possible combinations of motifs (motif sets) that meet a set of predefined constraints (Figure 1B). These constraints define what we consider biologically relevant CRMs: first, a CRM should occur in a minimal number of input sequences (frequency constraint, Figure 1B) to be sufficiently specific for the input set, but it does not necessarily have to cover all sequences. Second, we assume that motif sites of a CRM are more likely to reflect true combinatorial regulation when they occur in each other’s proximity on a single sequence than when they are scattered over long genomic distances. Therefore, the motif sites composing a CRM should occur within a maximal genomic distance from each other (proximity constraint, Figure 1B). This is not guaranteed to always be the case, which is compensated by the frequency constraint that does not require all sequences to contain the CRM. Because motifs can have multiple binding sites on the same sequence, we observed that several of the CRMs found were redundant. We consider a found CRM to be redundant to another one if it contains a subset of the motifs of the other CRM and occurs in exactly the same sequences. In this case, the smaller CRM will have a lower statistical score anyway; hence we can discard such CRMs from consideration. To make the search more effective, we will avoid redundant CRMs during search (redundancy constraint, (Figure 1B). Finally, in ChIP-Seq assayed data, one can use the fact that at least the assayed TF should be part of the CRM. For this purpose, we add a query-based option in which a ‘query motif’ can be provided that has to be part of the CRM. Using this knowledge before and during search (query-based constraint, Figure 1B) can make the problem computationally much more feasible.
When using the constraint programming for itemset mining framework with the above constraints, the result is a large collection of CRMs found to fulfill those constraints. We explicitly chose not to statistically evaluate the CRMs during search as this can quickly become computationally intensive, especially when evaluating the specificity of a CRM using a large collection of background sequences. Instead, we calculate an enrichment score (P-value) in a post-processing step and rank the CRMs according to this score (see Figure 1C and ‘Materials and Methods’ section). The added benefit is that our system does not return one CRM as being the most significant CRM, rather it returns an ordered list which a domain expert can choose from.
Benchmarking CPModule
Before applying our method on a real ChIP-Seq data set we compared its performance with that of a number of well performing CRM tools, namely ModuleSearcher (37), Compo (14), Cister (16) and Cluster-Buster (15).
Cister (16) and Cluster-Buster (15) are representative single-sequence tools. They scan each sequence individually, searching for potential CRMs that best match a predefined structure as imposed by model parameters (here a hidden Markov model). In contrast to multiple sequences tools, such as CPModule, they do not explicitly test whether the detected CRMs are specific for the input set as a whole. Nevertheless, these tools are very good at detecting CRMs in individual sequences. Because they treat sequences individually, they are computationally more efficient than multiple sequence tools. However, they cannot take advantage of the fact that sequences are coregulated.
Like CPModule, ModuleSearcher (37) and Compo (14) are multiple sequence tools. Both of them come with their own motif screening tool. ModuleSearcher searches for motif sets by using a genetic algorithm, a heuristic search method that maintains of pool of solutions which are modified to find ever better solutions. This type of search is more ad hoc and gives no guarantees that the best CRMs are found. Compo on the other hand uses techniques from itemset mining, as does CPModule. However, they differ in a number of ways: Compo is a specialized algorithm while CPModule uses a generic constraint-based methodology, allowing to incorporate extra constraints such as the redundancy constraint and the query-based constraint in a principled way. Additionally, while Compo has a strong focus on multi-parameter search and optimization, CPModule does exhaustive search and calculates the significance of a CRM using a large collection of background sequences in a second step.
For benchmarking we used the synthetic data constructed by Xie et al. (36). This data set contains intergenic sequences in which ‘true motif sites’ are inserted. The performance of CRM detection was assessed by comparing the best scoring solution of each algorithm with the known ‘true’ solution. The quality of the solutions was evaluated both at the motif and nucleotide level using respectively a motif correlation coefficient (mCC) and a nCC (5) (see ‘Materials and Methods’ section).
Finding CRMs in multiple long sequences using all 516 TRANSFAC PWMs is a challenging task. Table 1 shows a comparison of the different tools on our synthetic benchmark data. The single sequence tools Cister (16) and Cluster-Buster (15) perform rather poorly (<0.25 for both mCC and nCC). Because they screen sequences individually, they predict CRMs with a large number of motifs that differ from sequence to sequence, resulting in bad scores. ModuleSearcher (41) could not handle the large number of PWMs and repeatedly ran into memory problems, even with 2GB of RAM allocated. When limiting the maximum number of motifs in a CRM to 10 or less, Compo (14) found the solution reported in Table 1. It scores very well at the nucleotide level (0.68), meaning that it finds the binding region on the sequences quite accurately. However, at the motif level it performs rather poorly (0.27); it wrongly identifies the motifs that bind in the identified regions. CPModule scores worse (0.55 versus 0.68) at the nucleotide level than Compo, but scores considerably better (0.57 versus 0.27) at the motif level.
Table 1.
Comparison of CRM prediction algorithms
| Cister | Cluster-Buster | ModuleSearcher | Compo | CPModule | |
|---|---|---|---|---|---|
| mCC | 0.16 | 0.05 | / | 0.27 | 0.57 |
| nCC | 0.23 | 0.23 | / | 0.68 | 0.55 |
The tools were run on the synthetic data set of Xie et al. (36) using a stringent screening with 516 TRANSFAC PWMs. Slash indicates termination by lack of memory.
To allow for a more thorough comparison of different the tools, we reanalyzed the dataset using each time a subset of the 516 motif models. Starting from the PWMs of the three inserted TFs, we gradually increase the number of total PWMs by sampling them from the set of 513 remaining ones. This results in an increasingly larger input in terms of candidate motifs, which contain increasingly more false motif models. This makes the problem increasingly harder. Figure 2 shows the motif and nucleotide-level correlation coefficients (CC) of the different tools on the data. The number of motif models used is shown on the x-axis (for each sample size, 10 different samples are created and all tools are run using the same sample sets). The single-sequence-based tools Cister (16) and Cluster-Buster (15) perform best in the presence of few sampled PWMs and deteriorate as more PWMs are added. With few PWMs their predictions are accurate, especially regarding the binding region of the CRMs (nucleotide level). However, because they treat sequences independently, different false positive motifs are predicted on each sequence separately. This becomes worse as the number of sampled PWMs increases, leading to decreasing scores. For the multiple sequence tools Compo (14), ModuleSearcher (41) and CPModule this problem is less pronounced. The behavior of Compo changes as the number of motifs increases: at the motif level, the score has a decreasing trend, while at the nucleotide level the score increases. Looking at the CRMs found, we observe that Compo finds CRMs with only one true motif and increasingly more false motifs as the number of PWMs increases, explaining the motif-level behavior. Unexpectedly, adding these false motifs seems to contribute rather than to deteriorate the precision at the nucleotide level. The shorter predicted binding regions obtained by adding more motifs seem to coincidentally better approximate the regions in which also the true CRMs are located. The scores of CPModule and ModuleSearcher score well on both the motif- and nucleotide-level, while being less affected by the number of motifs used. However, ModuleSearcher is unable to scale to more than 400 motifs because of memory issues, while this is not a problem for CPModule.
Figure 2.

Performance comparison of CRM detection tools. All CRM detection tools were run on the synthetic data set of Xie et al. (36). Screening was performed with the PWMs used to generate the synthetic data in combination with an additional set of PWMs sampled from TRANSFAC (the number of PWMs added to the true PWMs is indicated on the x-axis). (A) mCC, (B) nCC.
These results show that CPModule is competitive with state-of-the-art tools in detecting CRMs in sets of coregulated sequences. This, in combination with its capability of handling a large number of sequences, prioritizing CRMs by means of ranked lists and the ability to be used in a query-based mode, make it ideally suited for this study and for the analysis of ChIP-Seq data in general.
Assessing the added value of ChIP-based information on detecting CRMs involved in mouse embryonic stem cell
Description of the experimental set up
To show the effect of using ChIP-Seq information on improving the performance of CRM detection, we relied on publicly available ChIP-Seq experiments conducted by Chen et al. (25). The data consist of ChIP-Seq experiments for five key TFs involved in self-renewal of mouse embryonic stem cells, namely KLF4, NANOG, OCT4, SOX2 and STAT3 for which it is known that combinatorial interactions exist amongst at least some of these five TFs. These previously known interactions, corresponding to nine different CRMs were used as a benchmark (Figure 3). Starting from the data of a ChIP-Seq experiment of a single assayed TF, we used CRM detection to discover, in silico, the other TFs with which the assayed TF constitutes a CRM. We then tested to what extent we could recover the previously described benchmark CRMs using either a query-based or non-query-based setting. The non-query-based setting mimics the traditional way in which CRM detection is being performed, that is trying to prioritize a CRM that is enriched in a set of sequences without using any further prior information. In the query-based setting, only CRMs that contain a motif for the assayed TF are searched for. Prior to the CRM detection, we first optimized screening thresholds to reduce the effect of the screening on the success of the CRM detection.
Figure 3.

Known combinatorial regulation of the five assayed TFs. Network representing combinatorial interactions between the five transcription factors (KLF4, SOX2, OCT4, NANOG and STAT3) involved in embryonic stem cell development. Edges indicate that a combinatorial interaction between the indicated TFs exists as reported in literature (with a combinatorial interaction referring to the fact that at least subsets of genes contain binding sites for both TFs in each other’s neighborhood). Dashed lines correspond to the interactions in the benchmark that were missed by CPModule. Solid lines correspond to the interactions in the benchmark that were recovered by CPModule. The thin line indicates that the interaction was detected using CPModule on the ChIP-Seq data set of one TF while the thick line indicates that the interactions was detected by using either ChIP-Seq dataset of the TFs involved in the interaction.
Screening and filtering
We started from the top 100 binding peaks identified for each ChIP-Seq-assayed TF (assuming that those represent the most reliable binding sites). It was recently shown that the sites of the assayed TF do not exactly coincide with their binding peaks, but can be located as far as 250 bp from the actual peak (42,43). Therefore, we used a sequence region of 500 bp centered around each of the top 100 peaks as input sequences.
Ideally, the result of the screening should have a high sensitivity, while containing only few false positive motif sites. Using a stringent threshold would bias towards only finding the most ‘conserved motif sites’. However, true sites do not necessarily correspond to the most conserved ones (44,45). Using a low stringency filtering might contrarily result in the inclusion of too many false positives, possibly deteriorating the CRM detection. Therefore, we used in addition to a screening with either a high or a low stringency threshold, also a low stringency screening combined with a filtering based on nucleosome positioning as nucleosome positioning plays a role in determining the accessibility of a site (39). As the information on condition- and tissue-dependent nucleosome occupancy is not readily available, we relied on the NuOS which has also previously been used in the context of motif detection (30) (see ‘Materials and Methods’ section). Motif sites located in regions that show a high NuOS are considered to be transcriptionally inactive (46) and were therefore filtered out.
One advantage of starting from ChIP-Seq based information is that it allows to approximate, at least for the assayed TF, the effect of the screening/filtering on the recovery rate of its binding sites. This effect on recovering the binding sites of the assayed TF can be seen as representative for the effect of the screening/filtering on recovering sites of any other TF. In Figure 4, we display for each input set (sequences corresponding to the 100 binding regions of each assayed TF) the sensitivity in recovering binding sites of the assayed TF after applying different screening/filtering procedures. The sensitivity is expressed as the percentage of the input set in which a motif site of the assayed TF could be detected. A sensitivity of 100% thus corresponds to retrieving at least one motif site for the assayed TF in each of the 100 high scoring binding regions. As can be expected, a stringent screening results in a rather low sensitivity for most of the binding regions of the assayed TFs (sensitivity <50%). Lowering the screening stringency largely increases this sensitivity. At least 80% of the binding peaks for respectively KLF4 (84%), NANOG (80%), OCT4 (98%), SOX2 (95%) and STAT3 (100%) were found to contain a motif site for their respective TFs. However, this increased sensitivity comes at the expense of also predicting many more potentially false positive sites. The number of false positives that result from a screening/filtering procedure is harder to estimate as we have no clue about the identity or location of the true sites. Therefore, we estimated the false discovery rate by the average number of motif sites per TF and per sequence region retained after applying different screening procedures (Figure 4B). The average number of sites per screened TF should be sufficiently low. Note that this average number does not exclude that some TFs can have multiple motif sites in the same sequence while others might have none. Applying a low stringency screening resulted in each of the analyzed data sets in the detection of, on average, four motif sites per TF and per sequence (Figure 4B). This was much lower in case stringent screening was used.
Figure 4.

Effect of different screening/filtering combinations on motif prediction results. (A) Effect of using different screening/filtering combinations on the sensitivity of recovering true sites of the assayed TF. Sensitivity is assessed by the percentage of binding peak regions in which a motif site of the assayed TF could be detected. (B) Effect of using different screening /filtering combinations on the average number of remaining motif sites per sequence and per TF for each of the ChIP-assayed data sets. In each panel, we used a stringent screening, a non-stringent screening without filtering and a non-stringent screening with a filtering based on NuOS (different categories are indicated in the order as mentioned above by bars with increasing gray scales), respectively.
Combining the non-stringent screening with a filtering based on nucleosome occupancy (that is removing sites predicted to be located in nucleosome occupied regions) seems to offer a good trade-off between the sensitivity and the number of false positive sites as is shown in Figure 4. Compared to stringent screening, applying the filtering after a non-stringent screening largely increases the sensitivity for most of the assayed TFs, while still maintaining the number of predicted sites per TF within a reasonable range. In the remainder of the analysis, filtering was applied on the low stringency predicted sites of all TFs except for those of the assayed one. For the assayed TF, filtering was omitted as the ChIP-derived evidence experimentally supports that each ChIP-bound region contains binding sites of that TF.
CRM detection
CPModule was run using for each assayed TF the sequences of the top 100 peak regions, as well as the motif sites identified by the screening and filtering. For the proximity threshold, we started for each data set from 150 bp and step wisely (50 bp at the time) extended this value to maximally 400 bp. The value of the frequency threshold was set to 60%. Predicted CRMs were ranked according to their P-values. The higher the rank of a benchmark CRM (that is a previously known CRM, see Figure 3), the better the algorithm was able to prioritize the CRM amongst the total number of predicted CRMs.
The results obtained by running CPModule after screening with a stringent threshold (for all TFs except the assayed one) (Supplementary Table S2) shows that a stringent screening threshold lowers the sensitivity of retrieving true binding sites to such extent that none of the benchmark CRMs can be retrieved at the predefined frequency threshold of 60%. Only by subsequently lowering the frequency threshold to 50% allowed recovering a benchmark CRM (1 out of the 9). To calculate benchmark recovery we considered all solutions obtained with all possible proximity thresholds irrespective of their rank. Without filtering, the number of binding sites per motif and sequence became so high that the search for CRMs was computationally prohibitive.
The most informative results and highest coverage of benchmark CRMs was obtained using CPModule with non-stringent screening and filtering based on the NuOS. Seven of nine of the previously described CRMs involving KLF4, NANOG, OCT4, SOX2 or STAT3.
Table 2 shows for each of the assayed TFs and for each proximity threshold, the highest ranking recovered benchmark CRM together with its rank amongst the total number of solutions. In addition to their rank we show for each CRM its support as an additional quality criterion, indicating how many of the 100 given peak regions contained the predicted CRMs (the higher this value, the more specific the detected CRM is for the given dataset).
Table 2.
Benchmark CRMs obtained with CPModule in combination with filtering (non-stringent screening with filtering for all TFs except the assayed one)
| ChIP-Seq-assayed TF | CRM | Support (%) | Proximity threshold (bp) | Query-based | Non-query-based | Validation (%) |
|---|---|---|---|---|---|---|
| Rank/Total | Rank/Total | |||||
| KLF4 | KLF4, STAT1 | 66 | 150 | 19/23 | 845/849 | 22.73 |
| KLF4, OCT1 | 60 | 200 | 19/46 | 5562/5694 | 33.33 | |
| KLF4, STAT1, [CEBP] | 61 | 200 | 22/46 | 5635/5694 | 23.33 | |
| KLF4, STAT4, [SMAD] | 61 | 250 | 16/98 | 6160/7029 | 22.95 | |
| KLF4, OCT1 | 60 | 250 | 65/98 | 6994/7029 | 33.33 | |
| KLF4, STAT4, [T3R] | 63 | 300 | 23/183 | 6903/7704 | 22.22 | |
| KLF4, OCT1 | 61 | 300 | 131/183 | 7651/7704 | 32.79 | |
| KLF4, STAT4, STAT1, [CDXA, LEF1] | 60 | 350 | 29/284 | 25 056/26 843 | 23.33 | |
| KLF4, OCT1 | 61 | 350 | 212/284 | 26 771/26 843 | 32.79 | |
| KLF4, STAT4, [SMAD, T3R] | 60 | 400 | 5/468 | 24 930/3 1549 | 21.67 | |
| KLF4, OCT1, [CDXA] | 60 | 400 | 207/468 | 31 220/31549 | 33.33 | |
| NANOG | NANOG, STAT5A_03, STAT5A_04 | 62 | 150 | 1/11 | 5930/5941 | 19.35 |
| NANOG, STAT5A_04, [PU1] | 60 | 200 | 1/39 | 40 171/43 093 | 23.33 | |
| NANOG, STAT5A_03, [PU1] | 61 | 250 | 1/71 | 62 475/64 059 | 21.31 | |
| NANOG, OCT1 | 60 | 250 | 21/71 | 64 006/64 059 | 68.33 | |
| NANOG, OCT1, [FAC1] | 60 | 300 | 1/145 | 66 186/80859 | 70.49 | |
| NANOG, STAT3, [FAC1] | 62 | 300 | 3/145 | 77 724/80 859 | 26.67 | |
| NANOG, STAT5A_04, [PU1, FAC1] | 60 | 350 | 1/406 | 159 818/217 328 | 25.00 | |
| NANOG, OCT1, STAT5A_04, [FAC1] | 60 | 350 | 2/406 | 167 806/217 328 | 66.67 (OCT1); | |
| 26.67 (STAT5A_04) | ||||||
| NANOG, STAT5A_04, [PU1, HNF3, AR] | 60 | 400 | 1/883 | 204 024/299 409 | 23.33 | |
| NANOG, OCT1, STAT6, STAT5A_04, [FAC1] | 60 | 400 | 2/883 | 224 495/299 409 | 70.00 (OCT1); | |
| 26.67 (STAT6) | ||||||
| OCT4 | OCT4, STAT6, [XFD2, FOXJ2, FOXP3] | 63 | 150 | 6/1322 | 30/11966 | 14.75 |
| OCT4, SOX2 | 60 | 150 | 1272/1322 | 10 348/11 966 | 78.33 | |
| OCT4, STAT4, STAT6, [PAX2, PAX4, TITF1] | 62 | 200 | 1/13 141 | 6/111 817 | 16.39 | |
| OCT4, SOX2, [PAX2] | 62 | 200 | 11 740/13 141 | 83 797/111 817 | 79.03 | |
| OCT4, STAT4, STAT6, [PAX4, PAX2, ELF1] | 66 | 250 | 1/29 767 | 23/182 697 | 16.67 | |
| OCT4, SOX2, [CDXA] | 60 | 250 | 28 080/29 767 | 1671 42/182 697 | 81.67 | |
| OCT4, STAT3 | 61 | 300 | 1/73 091 | 7/235 252 | 14.75 | |
| OCT4, SOX2, [CDXA, PAX2] | 60 | 300 | 68 944/73 091 | 217 331/235 252 | 75.00 | |
| OCT4, STAT3, [CDXA] | 60 | 350 | 1/290 997 | 1/859 377 | 12.90 | |
| OCT4, SOX2, [PAX2, FOXP3] | 60 | 350 | 106 443/290 997 | 296 722/859 377 | 73.33 | |
| OCT4, STAT3, STAT5A_03 | 60 | 400 | 1/383 001 | 11/108 0139 | 14.75 | |
| OCT4, SOX2, [PAX2, FOXP3] | 60 | 400 | 150 936/383001 | 449 140/1 080 139 | 73.33 | |
| SOX2 | SOX2, OCT4 | 68 | 150 | 1/6318 | 322/46 471 | 88.24 |
| SOX2, STAT5A_04, [NKX62, AR, HELIOSA] | 60 | 150 | 3/6318 | 840/46 471 | 23.33 | |
| SOX2, STAT5A_04, [GEN_INI2_B, FOXJ2, HNF3ALPHA, CEBP, AR] | 60 | 200 | 2/90 416 | 55/512 702 | 25.00 | |
| SOX2, OCT4, [CDXA, TST1] | 61 | 200 | 4/90 416 | 106/512 702 | 27.87 | |
| SOX2, STAT1, STAT5A_04, [SRY, CAP, NFAT, TEF, AR, CDX, HMGIY, BRCA] | 60 | 250 | 1/168 760 | 55/790 791 | 25.00 | |
| SOX2, OCT4, [CDXA, CDX2] | 62 | 250 | 4/168 760 | 183/79 0791 | 87.10 | |
| SOX2, OCT4, [CDXA, CDX, CEBP] | 60 | 300 | 1/303 533 | 94/1 256 190 | 86.89 | |
| SOX2, STAT, STAT5A_03, [CAP, NFAT, GEN_INI2_B, FOXJ2, CEBP] | 60 | 300 | 2/303 533 | 238/1 256 190 | 21.67 | |
| STAT3 | STAT3, OCT4, [CAP] | 61 | 150 | 36/5649 | 43/6426 | 36.07 |
| STAT3, SOX2, [IRF1] | 61 | 150 | 2651/5649 | 3018/6426 | 31.15 | |
| STAT3, OCT4, [CEBP] | 61 | 200 | 301/32 257 | 312/33 640 | 29.51 | |
| STAT3, SOX2, STAT6, [YY1] | 60 | 200 | 4532/32257 | 4675/33640 | 30.00 | |
| STAT3, OCT4, [CAP, TEF1, YY1, PR] | 60 | 250 | 57/54 549 | 61/56 473 | 31.67 | |
| STAT3, SOX2, STAT6, [SRY, IRF1] | 60 | 250 | 6363/54 549 | 6666/56 473 | 28.33 | |
| STAT3, OCT1, [CAP, FOXM1, YY1, PR] | 60 | 300 | 186/73 378 | 188/74 106 | 33.33 | |
| STAT3, SOX2, STAT6, [XPF1] | 60 | 300 | 8442/73 378 | 8517/74 106 | 28.33 | |
| STAT3, OCT, STAT5A_03, STAT6, [HOXA3, AP2REP, PU1] | 60 | 350 | 6/243 758 | 6/243 979 | 35.00 | |
| STAT3, SOX2, STAT1, STAT4, STAT5A_04, STAT6, [HNF3, YY1] | 60 | 350 | 21 046/243 758 | 21 066/243 979 | 26.67 | |
| STAT3, OCT, STAT5A_03, [AP2REP, PU1, XPF1] | 61 | 400 | 12/308 757 | 12/308 993 | 36.07 | |
| STAT3, SOX2, [HOXA3, AP2REP] | 62 | 400 | 21 375/308 757 | 21 385/308 993 | 29.03 |
In this table, only benchmark CRMs recovered by CPModule are displayed, For reasons of conciseness, we only display for each parameter setting the best ranked versions of each of the benchmark CRMs, for instance, whereas Oct4-Sox2 was found to be the best ranked CRM at a proximity threshold of 150, more combinations of Oct4, Sox2 in combination with other TFs were also detected at this setting of the proximity parameter albeit at lower ranks. These alternative versions with lower rank are not displayed in the table.
If PWMs for TFs belonging to the same family are very similar, we also considered those CRMs as true that contained rather than the TF reported in literature another member of the same family (48) (i.e. this was the case for TFs of the STAT and OCT family).
The set of sequences corresponding to the top 100 scoring peak regions of the assayed TF, were screened with a set of 517 TRANSFAC motifs using a non-stringent screening threshold. Filtering was applied on all motif sites except on the ones of the assayed TF. ChIP-Seq-assayed TF: TF from which the top 100 binding peaks were used to perform the analysis. CRM: obtained CRMs that correspond to previously well described CRMs for the assayed TF; [between brackets are indicated other TFs that were predicted to belong to the same CRM, but that have not previously been described to interact with the assayed TF]. Support: the percentage of sequences from the input set in which this CRM occurs (always higher than the frequency threshold). Proximity threshold (bp): the proximity threshold with which the displayed CRM was found. Query-based Rank/Total: the rank this CRM received in the query-based setting/the number of solutions containing the motif for the ChIP-Seq-assayed TF. Non-query-based Rank/Total: the rank this CRM received in all of the solutions/the total number of valid CRMs. Validation: we started from the ChIP-Seq data of one TF and predicted using CRM detection with which other TFs the assayed TF interacts. We verified whether the motif sites contributing to the predicted CRMs fell within the binding peaks of the other ChIP-Seq-assayed TF.
Table 2 reads as follows, for instance, when starting from the ChIP-Seq data of SOX2, we predicted a previously described CRM containing SOX2-OCT4. This retrieved CRM was ranked first amongst the 6318 potential CRMs that contained SOX2 (rank in the query-based mode) and ranked 322 out of the total number of 46 471 possible CRMs in the non-query based mode. SOX2 and OCT4 co-occurred in 68% of the SOX2 ChIP-Seq identified regions (Support) within a distance of 150 bp and the identified sites for OCT4 in the predicted CRM fell within the identified OCT4 ChIP-Seq regions in 88.24% of the cases. As an example of how the same CRM can be detected at different proximity thresholds: the CRM containing KL4-OCT1 was recovered at a proximity constraint of 200, 250, 300 and 350, but with an increasingly lower absolute rank in the non-query-based setting.
With the current screening/filtering all runs could be performed except those for SOX2 with proximity thresholds of 350 bp and 400 bp, respectively. These did not finish after 7days.
For all data sets, at the one but lowest proximity (200 bp) most of the benchmark CRMs were retrieved (6 of the 9 benchmark CRMs). Further increasing the proximity threshold results in the same benchmark CRMs also obtained with a lower threshold, albeit most of the time at a lower rank and/or in combination with other motifs. Increasing the proximity threshold will increase the number of valid CRMs. For NANOG, one additional benchmark CRMs were retrieved at a higher proximity threshold than the one for which the first CRM containing the assayed TF was found. Most of the TFs involved in the benchmark interactions, therefore, have binding sites in a rather close proximity on the genome.
Predicted CRMs were also validated using the available ChIP-Seq experimental information: if a previously described interaction between the analyzed TF and any of the other four benchmark TFs was predicted by CPModule, the ChIP-Seq data of the other TFs were used to experimentally verify whether their predicted sites in the retrieved CRMs coincided with their binding peaks. Predicted sites of TFs for which experimental data was available, overlapped for at least 10% (and often more) with their corresponding binding peaks (Column ‘Validation’ in Table 2). This indicates that most of the benchmark CRMs predicted by CPModule reflect true CRM signals present in the ChIP-Seq data.
The added value of using ChIP-Seq derived information becomes obvious when comparing the rank obtained for each benchmark CRM in the ‘non-query-based’ setting with the one obtained using the ‘query-based’ setting. The first setting mimics a classical CRM detection set up, i.e. when searching for CRMs that are statistically overrepresented in a set of coregulated sequences. The query-based setting differs from the classical setting by only listing CRMs that contain a site for the ChIP-assayed TF. Table 2 shows that in the query-based setting, the rank of the benchmark CRMs is in many cases much better than the rank in the non-query-based setting. This is especially true for KLF4, NANOG, SOX2 and STAT3. For OCT4, the ranks are more similar for both settings, but in the query-based setting much less candidate CRMs are returned, and hence the computation is much more efficient. Our results show that exploiting ChIP-Seq information, by constraining the candidate CRMs to those that only contain the assayed TF, not only leads to a compact result set with in many cases a better ranking of the true CRMs, but more importantly they also show that in the absence of such information, CRM detection becomes almost infeasible.
Novel predicted CRMs involved in embryonic stem cell regulation
Besides the results on the benchmark set, we also displayed for all assayed TFs their top three ranking CRMs predicted by CPModule, for the different proximity thresholds (Supplementary Table S3). Note that those CRMs score in most cases much better than the benchmark CRMs.
Functional analysis (Ingenuity Pathway analysis and literature-based analysis) of the 56 TFs that were involved in the predicted CRMs of respectively KLF4, NANOG, OCT4, SOX2 or STAT3, showed that most the TFs have functions related to development (embryonic development, cellular development, tissue development, organ development, organismal development), cellular growth and proliferation, cancer and tissue morphology all functionalities that could be related to ESC cell growth, death and differentiation (see Supplementary File S3).
For a handful of the predicted CRMs (KLF4-STAT4; OCT4-CDXA; OCT4-PAX2; OCT4-STAT; OCT4-SRY; SOX2-OCT4), it was previously proven that their contributing TFs were involved in combinatorial regulation (see Supplementary Table S3 for a list of references).
For most of the other CRMs we could find indirect literature-based support, suggesting the plausibility of the predicted interactions, for instance for NANOG-FAC1, the putative transcriptional regulator FAC1 has shown to be expressed in embryonic and extra-embryonic tissues of the early mouse conceptus and was shown to be essential for trophoblast differentiation during early mouse development (47), making an interaction between NANOG and FAC1 plausible. Other examples are described in Table 3.
Table 3.
Indirect evidences for the suggested CRMs in Supplementary Table S3
| Suggested CRM | Indirect evidence |
|---|---|
| NANOG-FAC1 | The putative transcriptional regulator FAC1 is expressed in embryonic and extraembryonic tissues of the early mouse conceptus. Study (47) showed that FAC1 is essential for trophoblast differentiation during early mouse development. Thus, there might be an interaction between NANOG and FAC1. |
| OCT4-FOXM1 | Foxm1 has been hypothesized to be one of the candidates to help reprogramming somatic cells into iPSCs (Induced pluripotent stem cells) (49). FOXM1 is a major stimulator of cell proliferation (50), so it might interact with KLF4 in the self-renewal process. |
| SOX2-CDXA | Binding of homeobox domain from CDX1 protein and SOX2 protein was shown to occur in a system of purified components (51). Note that we identified a CRM with CDXA and not with CDX1. However CDXA and CDX1 belong to the same protein family and have very similar motif models. |
| SOX2-BRCA | Roles of BRCA in both homologous recombination and non-homologous end joining DNA repair have been shown (52,53). Such function of BRCA might also play a role during the self-renewal process to repair DNA damage. |
| STAT3-HOXA3 | As HOXA3 is involved in wound repair (54), interaction with STAT3 in the self-renewal process is plausible. |
| STAT3-GATA1 | GATA1 was known to be one of the major transcription factors that stimulated cardiogenesis during development (55–57) even though it is frequently used as a marker for endodermal derivatives during differentiation of pluripotent stem cells (58). Interaction with STAT3 in the self-renewal process is therefore plausible. |
| STAT1-STAT3-STAT6 | Binding of human STAT3 protein and human STAT6 protein has been shown in a 2-hybrid assay (59). STAT1 and STAT3 can form heterodimers (60,61). Note however that with STAT motif models it is difficult to make the distinction between the different STAT members. |
Indirect evidences derived from literature searches which give indications on possible interactions between the indicated TFs found in the predicted CRMs.
DISCUSSION
In this work, we developed CPModule, a novel approach for CRM detection with a performance that is competitive to that of other state-of-art tools, while being able to handle larger data sets (such as 100 sequences in combination with a library of 517 PWMs). The advantage of CPModule is that it builds upon a constraint programming for itemset mining framework (24). This approach provides fast search techniques similar to those in itemset mining, while allowing to freely impose additional constraints on the CRMs. These constraints such as the proximity and redundancy constraint can help prioritizing likely CRMs. At this point our system focuses on finding loosely structured CRMs that satisfy multiple constraints, such as a minimum frequency constraint or a constraint on the maximum distance between binding sites. It can easily be extended to find unstructured CRMs under additional constraints. The discovery of structured CRMs, similar to the approaches proposed by Noto and Craven (2006) and by Cartharius et al. (8) in FrameWorker, is outside the scope of the current approach.
The flexible framework also allows us to use CPModule in a query-based setting when dealing with ChIP-Seq derived data, that is, we can search only for CRMs that contain the assayed TF. Because of its enumerative approach, CPModule outputs all valid CRMs as an ordered list rather than returning one CRM as being the most significant CRM. Having an idea of the rank of a true CRM amongst the total number of valid CRMs gives an intuition on the difficulty of computationally retrieving a true CRM in a particular data set. We used this property to assess the contribution of ChIP-Seq data in its ability to prioritize true CRMs. Our results on real datasets showed that in the absence of ChIP-Seq based information, biologically relevant CRM detection is almost infeasible.
The success of CRM detection also depends on the quality of the input data, which is the set of motif sites predicted by screening using motif models such as PWMs. A too dense collection of motif sites (hits) obtained by a non-stringent screening threshold usually results in too many motif combinations, which either make the problem intractable or lower the quality of the outcome; more false positive hits in the screening will also result in the detection of more spurious CRMs. Just increasing the stringency of the screening seems not to be the best option as many true sites, and thus also true CRMs, appear to be missing. Using a lower screening threshold in combination with a filtering procedure based on nucleosome occupancy provided in our case a good trade-off between keeping the number of false positives in a reasonable range and recovering true sites. We applied this filtering to sites of all TFs other than those of the assayed TF. By increasing the recovery rate of sites of the assayed TF, we maximize the chance of finding CRMs and instances of CRMs that contain the assayed TF, as those are the ones we are primarily interested in. Using a more stringent filtering for all other TFs will help reducing the spurious CRMs, but might come at the expense of not being able to detect some of the true interactions between the assayed TF and other TFs in TRANSFAC (which might explain why we couldn’t recover all previously described benchmark CRMs). With more experimental data on condition-dependent nucleosome occupancy and other cell specific features becoming available, filtering will become more reliable and will surely further improve the success of combinatorial CRM detection.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables S1–S3, Supplementary Figures S1–S2 and Supplementary References [62–112].
FUNDING
KULeuven Research Council (GOA/08/011, - SymBioSys -CoE EF/05/007, NATAR C1895-PF/10/10); the agency for Innovation by Science and Technology (SBO-BioFrame); Interuniversity Attraction Poles (P6/25-BioMaGNet); Research Foundation - Flanders (IOK-B9725-G.0329.09); the Human Frontier Science Program (RGY0079/2007C); Ghent University (Multidisciplinary Research Partnership ‘M2N’); Institute for the Promotion and Innovation through Science and Technology in Flanders (IWT-Vlaanderen); A Postdoc grant from the Research Foundation-Flanders. Funding for open access charge: KU Leuven Research Council (SymBioSys, CoE EF/05/007).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Luc De Raedt for the invaluable discussions and his vision on using constraint programming for itemset mining, which enabled the development of the CPModule system. The authors also thank the anonymous reviewers for many useful comments.
REFERENCES
- 1.Davidson EH. Genomic Regulatory Systems: Development and Evolution. San Diego: Academic Press; 2001. [Google Scholar]
- 2.Zhou Q, Wong WH. CisModule: de novo discovery of cis-regulatory modules by hierarchical mixture modeling. Proc. Natl Acad. Sci. USA. 2004;101:12114–12119. doi: 10.1073/pnas.0402858101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gupta M, Liu JS. De novo cis-regulatory module elicitation for eukaryotic genomes. Proc. Natl Acad. Sci. USA. 2005;102:7079–7084. doi: 10.1073/pnas.0408743102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Loo P, Marynen P. Computational methods for the detection of cis-regulatory modules. Brief Bioinform. 2009;10:509–524. doi: 10.1093/bib/bbp025. [DOI] [PubMed] [Google Scholar]
- 5.Klepper K, Sandve GK, Abul O, Johansen J, Drablos F. Assessment of composite motif discovery methods. BMC Bioinformatics. 2008;9:123. doi: 10.1186/1471-2105-9-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Noto K, Craven M. A specialized learner for inferring structured cis-regulatory modules. BMC Bioinformatics. 2006;7:528. doi: 10.1186/1471-2105-7-528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitington T, Frith MC, Johnson J, Bailey TL. Inferring transcription factor complexes from ChIP-seq data. Nucleic Acids Res. 2011;39:e98. doi: 10.1093/nar/gkr341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933–2942. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- 9.Dohr S, Klingenhoff A, Maier H, Hrabe de Angelis M, Werner T, Schneider R. Linking disease-associated genes to regulatory networks via promoter organization. Nucleic Acids Res. 2005;33:864–872. doi: 10.1093/nar/gki230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calva D, Dahdaleh FS, Woodfield G, Weigel RJ, Carr JC, Chinnathambi S, Howe JR. Discovery of SMAD4 promoters, transcription factor binding sites and deletions in juvenile polyposis patients. Nucleic Acids Res. 2011;39:5369–5378. doi: 10.1093/nar/gkr091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kwon AT, Chou AY, Arenillas DJ, Wasserman WW. Validation of skeletal muscle cis-regulatory module predictions reveals nucleotide composition bias in functional enhancers. PLoS Comp. Biol. 2011;7:e1002256. doi: 10.1371/journal.pcbi.1002256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Su J, Teichmann SA, Down TA. Assessing computational methods of cis-regulatory module prediction. PLoS Comp. Biol. 2010;6:e1001020. doi: 10.1371/journal.pcbi.1001020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Loo P, Aerts S, Thienpont B, De Moor B, Moreau Y, Marynen P. ModuleMiner - improved computational detection of cis-regulatory modules: are there different modes of gene regulation in embryonic development and adult tissues? Genome Biol. 2008;9:R66. doi: 10.1186/gb-2008-9-4-r66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sandve GK, Abul O, Drablos F. Compo: composite motif discovery using discrete models. BMC Bioinformatics. 2008;9:527. doi: 10.1186/1471-2105-9-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frith MC, Li MC, Weng Z. Cluster-Buster: finding dense clusters of motifs in DNA sequences. Nucleic Acids Res. 2003;31:3666–3668. doi: 10.1093/nar/gkg540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frith MC, Hansen U, Weng Z. Detection of cis-element clusters in higher eukaryotic DNA. Bioinformatics. 2001;17:878–889. doi: 10.1093/bioinformatics/17.10.878. [DOI] [PubMed] [Google Scholar]
- 17.Buck MJ, Lieb JD. ChIP-chip: considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments. Genomics. 2004;83:349–360. doi: 10.1016/j.ygeno.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 18.Jothi R, Cuddapah S, Barski A, Cui K, Zhao K. Genome-wide identification of in vivo protein-DNA binding sites from ChIP-Seq data. Nucleic Acids Res. 2008;36:5221–5231. doi: 10.1093/nar/gkn488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu ET, Pott S, Huss M. Q&A: ChIP-seq technologies and the study of gene regulation. BMC Biol. 2010;8:56. doi: 10.1186/1741-7007-8-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pepke S, Wold B, Mortazavi A. Computation for ChIP-seq and RNA-seq studies. Nat. Methods. 2009;6:S22–S32. doi: 10.1038/nmeth.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li X, MacArthur S, Bourgon R, Nix D, Pollard D, Iyer V, Hechmer A, Simirenko L, Stapleton M, Luengo Hendriks C, et al. Transcription factors bind thousands of active and inactive regions in the Drosophila blastoderm. PLoS Biol. 2008;6:e27. doi: 10.1371/journal.pbio.0060027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Visel A, Blow M, Li Z, Zhang T, Akiyama J, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van der Meer DL, Degenhardt T, Vaisanen S, de Groot PJ, Heinaniemi M, de Vries SC, Muller M, Carlberg C, Kersten S. Profiling of promoter occupancy by PPARalpha in human hepatoma cells via ChIP-chip analysis. Nucleic Acids Res. 2010;38:2839–2850. doi: 10.1093/nar/gkq012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Raedt L, Guns T, Nijssen S. Proceedings of the 14th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. New York, NY, USA: ACM; 2008. Constraint programming for itemset mining; pp. 204–212. [Google Scholar]
- 25.Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 26.Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–D110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coessens B, Thijs G, Aerts S, Marchal K, De Smet F, Engelen K, Glenisson P, Moreau Y, Mathys J, De Moor B. INCLUSive: a web portal and service registry for microarray and regulatory sequence analysis. Nucleic Acids Res. 2003;31:3468–3470. doi: 10.1093/nar/gkg615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frith MC, Fu Y, Yu L, Chen JF, Hansen U, Weng Z. Detection of functional DNA motifs via statistical over-representation. Nucleic Acids Res. 2004;32:1372–1381. doi: 10.1093/nar/gkh299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xi L, Fondufe-Mittendorf Y, Xia L, Flatow J, Widom J, Wang J-P. Predicting nucleosome positioning using a duration Hidden Markov Model. BMC Bioinformatics. 2010;11:346. doi: 10.1186/1471-2105-11-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramsey SA, Knijnenburg TA, Kennedy KA, Zak DE, Gilchrist M, Gold ES, Johnson CD, Lampano AE, Litvak V, Navarro G, et al. Genome-wide histone acetylation data improve prediction of mammalian transcription factor binding sites. Bioinformatics. 2010;26:2071–2075. doi: 10.1093/bioinformatics/btq405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schulte C, Stuckey PJ. Efficient constraint propagation engines. ACM T Progr. Lang. Sys. 2008;31:43. [Google Scholar]
- 32.Guns T, Sun H, Nijssen S, Marchal K, De Raedt L. Proceedings of IEEE International Conference on Bioinformatics and Biomedicine. Washington, DC, USA: IEEE Computer Society; 2010. Cis-regulatory module detection using constraint programming; pp. 363–368. [Google Scholar]
- 33.Gallo A, De Bie T, Cristianini N. Proceedings of the 11th Conference on Principles and Practice of Knowledge Discovery in Databases. Berlin: Springer; 2007. MINI: mining informative nonredundant itemset; pp. 438–445. [Google Scholar]
- 34.Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A, et al. The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 2011;39:D876–D882. doi: 10.1093/nar/gkq963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Celniker SE, Dillon LA, Gerstein MB, Gunsalus KC, Henikoff S, Karpen GH, Kellis M, Lai EC, Lieb JD, MacAlpine DM, et al. Unlocking the secrets of the genome. Nature. 2009;459:927–930. doi: 10.1038/459927a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie D, Cai J, Chia NY, Ng HH, Zhong S. Cross-species de novo identification of cis-regulatory modules with GibbsModule: application to gene regulation in embryonic stem cells. Genome Res. 2008;18:1325–1335. doi: 10.1101/gr.072769.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aerts S, Van Loo P, Moreau Y, De Moor B. A genetic algorithm for the detection of new cis-regulatory modules in sets of coregulated genes. Bioinformatics. 2004;20:1974–1976. doi: 10.1093/bioinformatics/bth179. [DOI] [PubMed] [Google Scholar]
- 38.Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Marshall KA, et al. NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res. 2009;37:D885–890. doi: 10.1093/nar/gkn764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whitington T, Perkins AC, Bailey TL. High-throughput chromatin information enables accurate tissue-specific prediction of transcription factor binding sites. Nucleic Acids Res. 2009;37:14–25. doi: 10.1093/nar/gkn866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang J, Chan YS, Loh YH, Cai J, Tong GQ, Lim CA, Robson P, Zhong S, Ng HH. A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat. Cell Biol. 2008;10:353–360. doi: 10.1038/ncb1698. [DOI] [PubMed] [Google Scholar]
- 41.Aerts S, Van Loo P, Thijs G, Moreau Y, De Moor B. Computational detection of cis -regulatory modules. Bioinformatics. 2003;19(Suppl. 2):ii5–ii14. doi: 10.1093/bioinformatics/btg1052. [DOI] [PubMed] [Google Scholar]
- 42.Wilbanks EG, Facciotti MT. Evaluation of algorithm performance in ChIP-seq peak detection. PLoS One. 2010;5:e11471. doi: 10.1371/journal.pone.0011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu XS, Brutlag DL, Liu JS. An algorithm for finding protein-DNA binding sites with applications to chromatin-immunoprecipitation microarray experiments. Nat. Biotechnol. 2002;20:835–839. doi: 10.1038/nbt717. [DOI] [PubMed] [Google Scholar]
- 45.Hu J, Li B, Kihara D. Limitations and potentials of current motif discovery algorithms. Nucleic Acids Res. 2005;33:4899–4913. doi: 10.1093/nar/gki791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee CK, Shibata Y, Rao B, Strahl BD, Lieb JD. Evidence for nucleosome depletion at active regulatory regions genome-wide. Nat. Genet. 2004;36:900–905. doi: 10.1038/ng1400. [DOI] [PubMed] [Google Scholar]
- 47.Goller T, Vauti F, Ramasamy S, Arnold HH. Transcriptional regulator BPTF/FAC1 is essential for trophoblast differentiation during early mouse development. Mol. Cell Biol. 2008;28:6819–6827. doi: 10.1128/MCB.01058-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Macintyre G, Bailey J, Haviv I, Kowalczyk A. is-rSNP: a novel technique for in silico regulatory SNP detection. Bioinformatics. 2010;26:i524–i530. doi: 10.1093/bioinformatics/btq378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xie Z, Tan G, Ding M, Dong D, Chen T, Meng X, Huang X, Tan Y. Foxm1 transcription factor is required for maintenance of pluripotency of P19 embryonal carcinoma cells. Nucleic Acids Res. 2010;38:8027–8038. doi: 10.1093/nar/gkq715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang IC, Chen YJ, Hughes D, Petrovic V, Major ML, Park HJ, Tan Y, Ackerson T, Costa RH. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol. Cell Biol. 2005;25:10875–10894. doi: 10.1128/MCB.25.24.10875-10894.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beland M, Pilon N, Houle M, Oh K, Sylvestre JR, Prinos P, Lohnes D. Cdx1 autoregulation is governed by a novel Cdx1-LEF1 transcription complex. Mol. Cell Biol. 2004;24:5028–5038. doi: 10.1128/MCB.24.11.5028-5038.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shafee N, Smith CR, Wei S, Kim Y, Mills GB, Hortobagyi GN, Stanbridge EJ, Lee EY. Cancer stem cells contribute to cisplatin resistance in Brca1/p53-mediated mouse mammary tumors. Cancer Res. 2008;68:3243–3250. doi: 10.1158/0008-5472.CAN-07-5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 54.Mace KA, Restivo TE, Rinn JL, Paquet AC, Chang HY, Young DM, Boudreau NJ. HOXA3 modulates injury-induced mobilization and recruitment of bone marrow-derived cells. Stem Cells. 2009;27:1654–1665. doi: 10.1002/stem.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grepin C, Robitaille L, Antakly T, Nemer M. Inhibition of transcription factor GATA-4 expression blocks in vitro cardiac muscle differentiation. Mol. Cell Biol. 1995;15:4095–4102. doi: 10.1128/mcb.15.8.4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lien CL, Wu C, Mercer B, Webb R, Richardson JA, Olson EN. Control of early cardiac-specific transcription of Nkx2-5 by a GATA-dependent enhancer. Development. 1999;126:75–84. doi: 10.1242/dev.126.1.75. [DOI] [PubMed] [Google Scholar]
- 57.Pikkarainen S, Tokola H, Kerkela R, Ruskoaho H. GATA transcription factors in the developing and adult heart. Cardiovasc. Res. 2004;63:196–207. doi: 10.1016/j.cardiores.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 58.Holtzinger A, Rosenfeld GE, Evans T. Gata4 directs development of cardiac-inducing endoderm from ES cells. Dev. Biol. 2010;337:63–73. doi: 10.1016/j.ydbio.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ravasi T, Suzuki H, Cannistraci CV, Katayama S, Bajic VB, Tan K, Akalin A, Schmeier S, Kanamori-Katayama M, Bertin N, et al. An atlas of combinatorial transcriptional regulation in mouse and man. Cell. 2010;140:744–752. doi: 10.1016/j.cell.2010.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 61.John S, Reeves RB, Lin JX, Child R, Leiden JM, Thompson CB, Leonard WJ. Regulation of cell-type-specific interleukin-2 receptor alpha-chain gene expression: potential role of physical interactions between Elf-1, HMG-I(Y), and NF-kappa B family proteins. Mol. Cell Biol. 1995;15:1786–1796. doi: 10.1128/mcb.15.3.1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Farrar JD, Smith JD, Murphy TL, Murphy KM. Recruitment of Stat4 to the human interferon-alpha/beta receptor requires activated Stat2. J. Biol. Chem. 2000;275:2693–2697. doi: 10.1074/jbc.275.4.2693. [DOI] [PubMed] [Google Scholar]
- 63.Wang C, Zhou GL, Vedantam S, Li P, Field J. Mitochondrial shuttling of CAP1 promotes actin- and cofilin-dependent apoptosis. J. Cell Sci. 2008;121:2913–2920. doi: 10.1242/jcs.023911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kelley CM, Ikeda T, Koipally J, Avitahl N, Wu L, Georgopoulos K, Morgan BA. Helios, a novel dimerization partner of Ikaros expressed in the earliest hematopoietic progenitors. Curr. Biol. 1998;8:508–515. doi: 10.1016/s0960-9822(98)70202-7. [DOI] [PubMed] [Google Scholar]
- 65.Battista S, Pentimalli F, Baldassarre G, Fedele M, Fidanza V, Croce CM, Fusco A. Loss of Hmga1 gene function affects embryonic stem cell lympho-hematopoietic differentiation. FASEB J. 2003;17:1496–1498. doi: 10.1096/fj.02-0977fje. [DOI] [PubMed] [Google Scholar]
- 66.Choi HJ, Geng Y, Cho H, Li S, Giri PK, Felio K, Wang CR. Differential requirements for the Ets transcription factor Elf-1 in the development of NKT cells and NK cells. Blood. 2010;117:1880–1887. doi: 10.1182/blood-2010-09-309468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tang Y, Katuri V, Dillner A, Mishra B, Deng CX, Mishra L. Disruption of transforming growth factor-beta signaling in ELF beta-spectrin-deficient mice. Science. 2003;299:574–577. doi: 10.1126/science.1075994. [DOI] [PubMed] [Google Scholar]
- 68.Beck F, Stringer EJ. The role of Cdx genes in the gut and in axial development. Biochem. Soc. Trans. 2010;38:353–357. doi: 10.1042/BST0380353. [DOI] [PubMed] [Google Scholar]
- 69.Park MJ, Kim HY, Kim K, Cheong J. Homeodomain transcription factor CDX1 is required for the transcriptional induction of PPARgamma in intestinal cell differentiation. FEBS Lett. 2009;583:29–35. doi: 10.1016/j.febslet.2008.11.030. [DOI] [PubMed] [Google Scholar]
- 70.Holdcraft RW, Braun RE. Androgen receptor function is required in Sertoli cells for the terminal differentiation of haploid spermatids. Development. 2004;131:459–467. doi: 10.1242/dev.00957. [DOI] [PubMed] [Google Scholar]
- 71.Merrill BJ, Gat U, DasGupta R, Fuchs E. Tcf3 and Lef1 regulate lineage differentiation of multipotent stem cells in skin. Genes Dev. 2001;15:1688–1705. doi: 10.1101/gad.891401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Galceran J, Farinas I, Depew MJ, Clevers H, Grosschedl R. Wnt3a-/–like phenotype and limb deficiency in Lef1(-/-)Tcf1(-/-) mice. Genes Dev. 1999;13:709–717. doi: 10.1101/gad.13.6.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bouchard M, Souabni A, Mandler M, Neubuser A, Busslinger M. Nephric lineage specification by Pax2 and Pax8. Genes Dev. 2002;16:2958–2970. doi: 10.1101/gad.240102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Torres M, Gomez-Pardo E, Dressler GR, Gruss P. Pax-2 controls multiple steps of urogenital development. Development. 1995;121:4057–4065. doi: 10.1242/dev.121.12.4057. [DOI] [PubMed] [Google Scholar]
- 75.Kashimada K, Koopman P. Sry: the master switch in mammalian sex determination. Development. 2010;137:3921–3930. doi: 10.1242/dev.048983. [DOI] [PubMed] [Google Scholar]
- 76.Sun L, Ma K, Wang H, Xiao F, Gao Y, Zhang W, Wang K, Gao X, Ip N, Wu Z. JAK1-STAT1-STAT3, a key pathway promoting proliferation and preventing premature differentiation of myoblasts. J. Cell Biol. 2007;179:129–138. doi: 10.1083/jcb.200703184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kang J, DiBenedetto B, Narayan K, Zhao H, Der SD, Chambers CA. STAT5 is required for thymopoiesis in a development stage-specific manner. J. Immunol. 2004;173:2307–2314. doi: 10.4049/jimmunol.173.4.2307. [DOI] [PubMed] [Google Scholar]
- 78.Snow JW, Abraham N, Ma MC, Abbey NW, Herndier B, Goldsmith MA. STAT5 promotes multilineage hematolymphoid development in vivo through effects on early hematopoietic progenitor cells. Blood. 2002;99:95–101. doi: 10.1182/blood.v99.1.95. [DOI] [PubMed] [Google Scholar]
- 79.Wurster AL, Tanaka T, Grusby MJ. The biology of Stat4 and Stat6. Oncogene. 2000;19:2577–2584. doi: 10.1038/sj.onc.1203485. [DOI] [PubMed] [Google Scholar]
- 80.Barak O, Lazzaro MA, Lane WS, Speicher DW, Picketts DJ, Shiekhattar R. Isolation of human NURF: a regulator of engrailed gene expression. EMBO J. 2003;22:6089–6100. doi: 10.1093/emboj/cdg582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jacks T. Tumor suppressor gene mutations in mice. Annu. Rev. Genet. 1996;30:603–636. doi: 10.1146/annurev.genet.30.1.603. [DOI] [PubMed] [Google Scholar]
- 82.Begay V, Smink J, Leutz A. Essential requirement of CCAAT/enhancer binding proteins in embryogenesis. Mol. Cell Biol. 2004;24:9744–9751. doi: 10.1128/MCB.24.22.9744-9751.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Niedernhofer LJ, Essers J, Weeda G, Beverloo B, de Wit J, Muijtjens M, Odijk H, Hoeijmakers JH, Kanaar R. The structure-specific endonuclease Ercc1-Xpf is required for targeted gene replacement in embryonic stem cells. EMBO J. 2001;20:6540–6549. doi: 10.1093/emboj/20.22.6540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wan H, Dingle S, Xu Y, Besnard V, Kaestner KH, Ang SL, Wert S, Stahlman MT, Whitsett JA. Compensatory roles of Foxa1 and Foxa2 during lung morphogenesis. J. Biol. Chem. 2005;280:13809–13816. doi: 10.1074/jbc.M414122200. [DOI] [PubMed] [Google Scholar]
- 85.Tompers DM, Foreman RK, Wang Q, Kumanova M, Labosky PA. Foxd3 is required in the trophoblast progenitor cell lineage of the mouse embryo. Dev. Biol. 2005;285:126–137. doi: 10.1016/j.ydbio.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 86.Ohyama T, Groves AK. Expression of mouse Foxi class genes in early craniofacial development. Dev. Dyn. 2004;231:640–646. doi: 10.1002/dvdy.20160. [DOI] [PubMed] [Google Scholar]
- 87.Granadino B, Arias-de-la-Fuente C, Perez-Sanchez C, Parraga M, Lopez-Fernandez LA, del Mazo J, Rey-Campos J. Fhx (Foxj2) expression is activated during spermatogenesis and very early in embryonic development. Mech. Dev. 2000;97:157–160. doi: 10.1016/s0925-4773(00)00410-x. [DOI] [PubMed] [Google Scholar]
- 88.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 89.Tsai FY, Browne CP, Orkin SH. Knock-in mutation of transcription factor GATA-3 into the GATA-1 locus: partial rescue of GATA-1 loss of function in erythroid cells. Dev. Biol. 1998;196:218–227. doi: 10.1006/dbio.1997.8842. [DOI] [PubMed] [Google Scholar]
- 90.Stecca B, Nait-Oumesmar B, Kelley KA, Voss AK, Thomas T, Lazzarini RA. Gcm1 expression defines three stages of chorio-allantoic interaction during placental development. Mech. Dev. 2002;115:27–34. doi: 10.1016/s0925-4773(02)00095-3. [DOI] [PubMed] [Google Scholar]
- 91.Fijalkowska I, Sharma D, Bult CJ, Danoff SK. Expression of the transcription factor, TFII-I, during post-implantation mouse embryonic development. BMC Res. Notes. 2010;3:203. doi: 10.1186/1756-0500-3-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kameda Y, Nishimaki T, Takeichi M, Chisaka O. Homeobox gene hoxa3 is essential for the formation of the carotid body in the mouse embryos. Dev. Biol. 2002;247:197–209. doi: 10.1006/dbio.2002.0689. [DOI] [PubMed] [Google Scholar]
- 93.Fournier M, Lebert-Ghali CE, Krosl G, Bijl JJ. HOXA4 induces expansion of hematopoietic stem cells in vitro and confers enhancement of pro-B-cells in vivo. Stem Cells Dev. 2011;21:133–142. doi: 10.1089/scd.2011.0259. [DOI] [PubMed] [Google Scholar]
- 94.Kim JI, Li T, Ho IC, Grusby MJ, Glimcher LH. Requirement for the c-Maf transcription factor in crystallin gene regulation and lens development. Proc. Natl Acad. Sci. USA. 1999;96:3781–3785. doi: 10.1073/pnas.96.7.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Han J, Ishii M, Bringas P, Jr, Maas RL, Maxson RE, Jr, Chai Y. Concerted action of Msx1 and Msx2 in regulating cranial neural crest cell differentiation during frontal bone development. Mech. Dev. 2007;124:729–745. doi: 10.1016/j.mod.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chang CP, Neilson JR, Bayle JH, Gestwicki JE, Kuo A, Stankunas K, Graef IA, Crabtree GR. A field of myocardial-endocardial NFAT signaling underlies heart valve morphogenesis. Cell. 2004;118:649–663. doi: 10.1016/j.cell.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 97.Kimura S, Hara Y, Pineau T, Fernandez-Salguero P, Fox CH, Ward JM, Gonzalez FJ. The T/ebp null mouse: thyroid-specific enhancer-binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev. 1996;10:60–69. doi: 10.1101/gad.10.1.60. [DOI] [PubMed] [Google Scholar]
- 98.Henseleit KD, Nelson SB, Kuhlbrodt K, Hennings JC, Ericson J, Sander M. NKX6 transcription factor activity is required for alpha- and beta-cell development in the pancreas. Development. 2005;132:3139–3149. doi: 10.1242/dev.01875. [DOI] [PubMed] [Google Scholar]
- 99.Wang J, Elghazi L, Parker SE, Kizilocak H, Asano M, Sussel L, Sosa-Pineda B. The concerted activities of Pax4 and Nkx2.2 are essential to initiate pancreatic beta-cell differentiation. Dev. Biol. 2004;266:178–189. doi: 10.1016/j.ydbio.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 100.Mansouri A, Chowdhury K, Gruss P. Follicular cells of the thyroid gland require Pax8 gene function. Nat. Genet. 1998;19:87–90. doi: 10.1038/ng0598-87. [DOI] [PubMed] [Google Scholar]
- 101.Selleri L, Depew MJ, Jacobs Y, Chanda SK, Tsang KY, Cheah KS, Rubenstein JL, O'Gorman S, Cleary ML. Requirement for Pbx1 in skeletal patterning and programming chondrocyte proliferation and differentiation. Development. 2001;128:3543–3557. doi: 10.1242/dev.128.18.3543. [DOI] [PubMed] [Google Scholar]
- 102.Shyamala G, Yang X, Cardiff RD, Dale E. Impact of progesterone receptor on cell-fate decisions during mammary gland development. Proc. Natl Acad. Sci. USA. 2000;97:3044–3049. doi: 10.1073/pnas.97.7.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sebastiano V, Dalvai M, Gentile L, Schubart K, Sutter J, Wu GM, Tapia N, Esch D, Ju JY, Hubner K, et al. Oct1 regulates trophoblast development during early mouse embryogenesis. Development. 2010;137:3551–3560. doi: 10.1242/dev.047027. [DOI] [PubMed] [Google Scholar]
- 104.Ryu EJ, Wang JY, Le N, Baloh RH, Gustin JA, Schmidt RE, Milbrandt J. Misexpression of Pou3f1 results in peripheral nerve hypomyelination and axonal loss. J. Neurosci. 2007;27:11552–11559. doi: 10.1523/JNEUROSCI.5497-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Aberg T, Cavender A, Gaikwad JS, Bronckers AL, Wang X, Waltimo-Siren J, Thesleff I, D'Souza RN. Phenotypic changes in dentition of Runx2 homozygote-null mutant mice. J. Histochem. Cytochem. 2004;52:131–139. doi: 10.1177/002215540405200113. [DOI] [PubMed] [Google Scholar]
- 106.Tremblay KD, Dunn NR, Robertson EJ. Mouse embryos lacking Smad1 signals display defects in extra-embryonic tissues and germ cell formation. Development. 2001;128:3609–3621. doi: 10.1242/dev.128.18.3609. [DOI] [PubMed] [Google Scholar]
- 107.James D, Levine AJ, Besser D, Hemmati-Brivanlou A. TGFbeta/activin/nodal signaling is necessary for the maintenance of pluripotency in human embryonic stem cells. Development. 2005;132:1273–1282. doi: 10.1242/dev.01706. [DOI] [PubMed] [Google Scholar]
- 108.Wontakal SN, Guo X, Will B, Shi M, Raha D, Mahajan MC, Weissman S, Snyder M, Steidl U, Zheng D, et al. A large gene network in immature erythroid cells is controlled by the myeloid and B cell transcriptional regulator PU.1. PLoS Genet. 2011;7:e1001392. doi: 10.1371/journal.pgen.1001392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Korinek V, Barker N, Moerer P, van Donselaar E, Huls G, Peters PJ, Clevers H. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat. Genet. 1998;19:379–383. doi: 10.1038/1270. [DOI] [PubMed] [Google Scholar]
- 110.Inukai T, Inaba T, Dang J, Kuribara R, Ozawa K, Miyajima A, Wu W, Look AT, Arinobu Y, Iwasaki H, et al. TEF, an antiapoptotic bZIP transcription factor related to the oncogenic E2A-HLF chimera, inhibits cell growth by down-regulating expression of the common beta chain of cytokine receptors. Blood. 2005;105:4437–4444. doi: 10.1182/blood-2004-08-2976. [DOI] [PubMed] [Google Scholar]
- 111.Wallis K, Sjogren M, van Hogerlinden M, Silberberg G, Fisahn A, Nordstrom K, Larsson L, Westerblad H, Morreale de Escobar G, Shupliakov O, et al. Locomotor deficiencies and aberrant development of subtype-specific GABAergic interneurons caused by an unliganded thyroid hormone receptor alpha1. J. Neurosci. 2008;28:1904–1915. doi: 10.1523/JNEUROSCI.5163-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Affar el B, Gay F, Shi Y, Liu H, Huarte M, Wu S, Collins T, Li E. Essential dosage-dependent functions of the transcription factor yin yang 1 in late embryonic development and cell cycle progression. Mol. Cell Biol. 2006;26:3565–3581. doi: 10.1128/MCB.26.9.3565-3581.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.