

Abstract

Pt(II)-catalyzed cycloisomerization of aziridinyl propargylic esters affords 1,2-dihydropyridines with regiodefined installation of substituents. A mild conversion of the 1,2-dihydropyridines to the corresponding substituted pyridines as well as chirality retention from the aziridinyl propargylic ester substrates have been demonstrated.
Nitrogen-containing heterocycles comprise a large percentage of natural products and pharmaceutically important small molecules.1 Methods directed toward the synthesis of azaheterocycles continue to be an important undertaking, and general strategies for the regio- and stereocontrolled introduction of substituents remain a challenge. Dihydropyridines (DHPs) have long been recognized as versatile synthetic intermediates2 that provide ready access to a variety of substituted nitrogen-containing heterocycles such as piperidines (by reduction)3 or to pyridines (by oxidation).4 Historically, 1,4-dihydropyridines (1,4-DHPs) have been the most studied, whereas 1,2-DHPs have received relatively little attention. This has stemmed primarily from the ease with which these compounds can be accessed from pyridinium salts by nucleophilic addition at C(4) and the emergence of Hantzsch-type 1,4-DHPs as important calcium channel blockers.5 The majority of synthetic approaches to 1,2-DHPs has also relied on nucleophilic addition into N-alkyl or N-acylpyridinium salts.6,7 Unfortunately, regioisomeric mixtures of addition products (at the 2, 4, or 6 position) are often obtained if unsymmetrically substituted pyridinium salts, especially those that do not bear a substituent at the C(4) position, are employed.8 More recently, other strategies for the synthesis of 1,2-DHPs have emerged that address some of these limitations.9 Because of the lack of general methods for the regioselective synthesis of highly functionalized 1,2-DHP derivatives, their synthetic and biological potential remains largely unexplored. As such, there is a continued need for the development of more functional-group-tolerant methods that avoid the use of organolithium or Grignard reagents to access 1,2-DHPs as well as general strategies that afford access to bicyclic 1,2-DHPs.
We envisioned that 1,2-DHPs (e.g., 5a, Scheme 1) could be realized from aziridinyl propargylic esters (see 1a) via aPt(II)-catalyzed cascade sequence. Thus, formation of zwitterion 2 from 1a via a 5-exo-dig cyclization of the ester carbonyl functionality onto the alkyne and subsequent rearrangement could afford metallocarbenoid 3. The strained [3.1.0] bicycle 4 could then result from nucleophilic attack of the aziridinyl nitrogen on the metallocarbenoid functionality. At this stage, irreversible valence bond isomerization coupled with the strain-release fragmentation of the aziridine was expected to produce 1,2-DHP 5a.
Scheme 1.
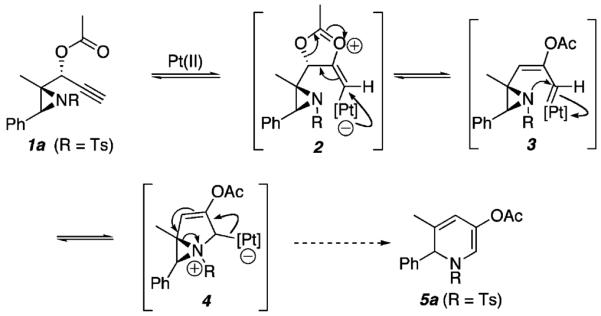
Proposed Formation of 1,2-Dihydropyridines
The expectation that 5a could be formed from 1a is supported by our recent report of a Pt(II)-catalyzed method for the synthesis of acyloxycyclopentenones (e.g., 9, Scheme 2) using epoxide-bearing propargylic ester substrates (6).10 Our earlier investigations on the formation of 9 from 6 support a mechanism involving the intermediacy of 3(2H)-pyran 7 and oxatriene 8. However, unlike 3(2H)-pyrans, which are prone to oxa-6π-electrocyclic ring opening11 and usually exist in equilibrium with the dienone or dienal valence bond isomer, 1,2-DHPs are relatively robust. As such, we hoped to identify optimal conditions for their synthesis from aziridinyl propargylic esters without competing ring-opening reactions.
Scheme 2.
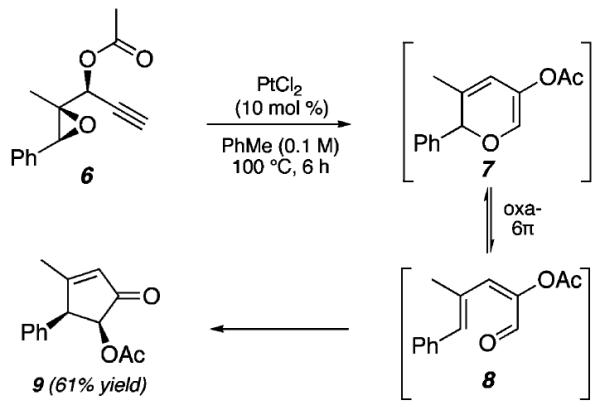
Epoxide Fragmentation/Pentannulation Reaction
To test the viability of our approach to 1,2-DHPs, a judicious choice of protecting group had to be made to overcome the following challenges: (1) oxidation of dihydropyridines in the presence of air;12 (2) the nucleophilicity of the aziridine nitrogen had to be tempered to allow for an initial interaction of the activated Pt(II)-bound alkyne and the propargylic ester (see 1af2) without competing nitrogen addition to the alkyne;13 and (3) the reactivity of the aziridine nitrogen had to be tuned to allow for its addition to the metallocarbenoid functionality of 3 to afford 4 en route to 5a. Importantly, this addition should be rapid relative to a potential vinyl aziridine rearrangement of 3.14
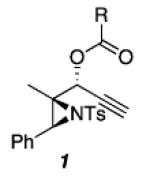
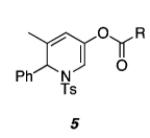
The aziridinyl propargylic ester substrates (see Table 1) were prepared by acylation of the corresponding aziridine propargylic alcohols, which were available from the aziridinyl aldehyde via a highly diastereoselective (>95:5 dr) 1,2-addition15 of the corresponding alkynyllithium or Grignard.16 We were gratified to find that under Pt(II) catalysis (10 mol % of PtCl2, 0.2 M in PhMe, 100 °C, 3 h) a range of aziridinyl propargylic esters afforded the expected 1,2-DHP products in moderate to good yields.
Table 1.
Pt(II)-Catalyzed 1,2-Dihydropyridine Synthesisa
| entry | substrate | product | yield(%)b |
|---|---|---|---|
| 1 |
|
|
|
| a) R = Me | 76 | ||
| b) R = p-CIPh | 72 | ||
| c) R = tBu | 76 | ||
| d) R = Ph | 76 | ||
| 2 |
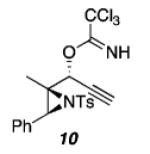
|
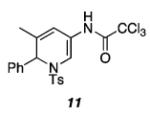
|
56 |
| 3 |
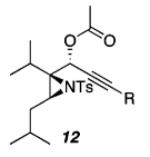
|
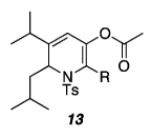
|
|
| a) R = cyclopropyl | 62 | ||
| b) R = Ph | 69 | ||
| c) R = H | 70 | ||
| 4 |
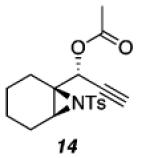
|
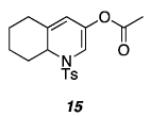
|
76 |
Standard conditions: 10 mol % of PtCl2, 0.2 M in PhMe at 100 °C over 3 h.
Isolated yields after column chromatography are reported.
As shown in Table 1, acetates, para-chlorobenzoate, pivalate, and benzoate propargylic esters can be employed (entries 1a-d). The permissibility of the nucleophile at the propargylic position is further highlighted by the reaction of trichloroacetimidate 10 (entry 2) to afford 11 in 56% yield. To the best of our knowledge, this constitutes the first report of the use of a trichloroacetimidate in a Pt(II)-catalyzed cycloisomerization reaction.17 In addition, internal alkynes bearing cyclopropyl or phenyl substituents at the alkyne terminus (entries 3a and b, respectively) as well as cyclic substrates (entry 4) readily participate in this transformation to give good isolated yields (62-76%) of the desired 1,2-DHP product.
In an effort to expand the scope of the substrates that participate in this reaction, other substituents at the aziridine nitrogen were explored. The N-acyl substrates (16a-d, Scheme 3) were considered in a preliminary study.16 On the basis of literature precedent, which highlights the ambident nucleophilicity of acylated aziridines,18 we hypothesized that the presumed metallocarbenoid intermediate could lead to 1,2-DHP 17 or amide-incorporated heterocycle 18 products. Although 17 would arise via the initially proposed mechanism in which the aziridine nitrogen engages the metallocarbenoid intermediate (Scheme 1), 18 could be formed through a pathway involving the nucleophilic addition of the N-acyl carbonyl to the metallocarbenoid intermediate.19
Scheme 3.
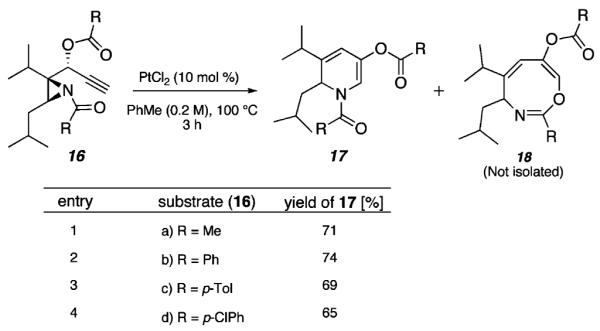
Pt(II)-Catalyzed Cycloisomerization of Acylated Aziridine Substrates
As detailed in Scheme 3, several acylated aziridines were subjected to the standard reaction conditions and yielded the 1,2-DHPs (17a-d) as the major product after column chromatography (65-74% yield). An X-ray crystal structure of 1,2-DHP 17c (Figure 1)20 provides further support for the assigned structure of the 1,2-DHP products that result from the preferred mode of cyclization of the N-acyl aziridine substrates.
Figure 1.
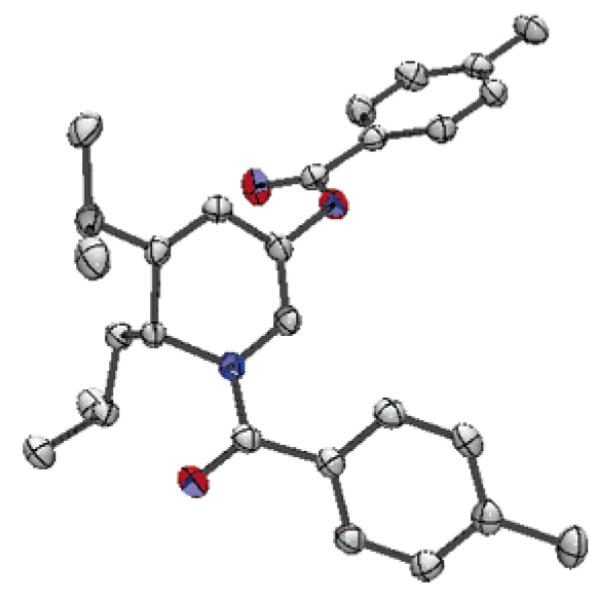
ORTEP structure of 1,2-dihydropyridine 17c with ellipsoids shown at the 50% probability level.
Under the standard reaction conditions, the Pt(II)-catalyzed transformation of substrates possessing alkyl substitution on the aziridine nitrogen (e.g., benzyl) has led to complex mixtures of products from which none of the desired 1,2-DHPs could be isolated. Presumably, the enhanced basicity of the aziridine lone pair leads to competing nonproductive pathways. This supports our identification of the aziridine protecting group as an important parameter in the design of substrates for this reaction.21
Although ample literature precedent exists for the conversion of 1,2-DHPs to pyridines, the conditions required for these processes often involve the use of strong oxidants or acids.22 We envisioned that our 1,2-DHP products may provide a suitable synthetic starting point to highly substituted pyridines under mild conditions. In a preliminary study, 5b was readily converted in 79% yield to hydroxy pyridine 20 upon treatment with potassium trimethylsilanoate (TMSOK) at room temperature over 3 h (Scheme 4). Access to 20 now sets the stage for further functionalization (e.g., by activation of the hydroxyl group by triflation, followed by organometallic coupling).
Scheme 4.
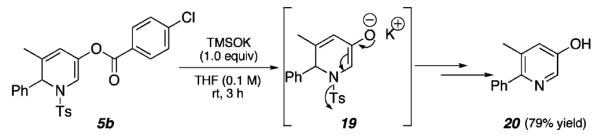
Conversion of 1,2-Dihydropyridines to Pyridines
We next investigated chirality retention in converting the aziridinyl propargylic ester substrates to the 1,2-DHP products. As shown in Scheme 5, enantiopure 1a23 is transformed in 70% yield with only a minor loss of enantiomeric excess to the corresponding highly enantioenriched 1,2-DHP 5a.
Scheme 5.
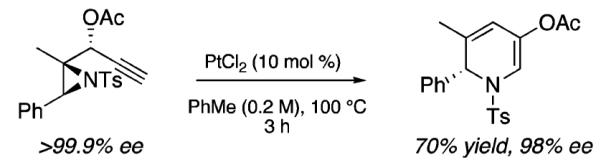
Chirality Retention in the Formation of 1,2-Dihydropyridines
This result attests to the stability of the 1,2-DHP compounds toward epimerization (e.g., via subsequent ring-opening/ring-closing processes) under our reaction conditions. With access to highly enantioenriched 1,2-DHPs now defined, the diastereoselective functionalization of these compounds should provide stereoselective access to a range of important azaheterocycles. These efforts are currently under investigation.
In conclusion, we report a novel Pt(II)-catalyzed cycloisomerization strategy for the synthesis of 1,2-DHPs that achieves the regiodefined installation of substituents using readily prepared aziridinyl propargylic ester substrates. In addition, a mild conversion of the 1,2-DHPs to the corresponding substituted pyridines as well as high levels of chirality retention from the aziridinyl propargylic ester substrates have been demonstrated. Efforts to further expand the scope of these transformations to include other examples of chirality retention and the identification of other modes of cyclization of the aziridinyl propargylic ester substrates are underway in our laboratories.
Supplementary Material
Acknowledgment
The authors are grateful to UC Berkeley, GlaxoSmithKline, Johnson Matthey, and Boehringer-Ingelheim for support of this research and to Ms. Christina Kraml (AccelaPure Corp., Frick Lab Rm 201, Princeton University, Princeton, NJ 08544) for enantiopure samples of 5a. The authors also thank Drs. Fred Hollander and Hye Jin Choi (UC Berkeley) for crystallographic data.
Footnotes
Supporting Information Available: Experimental details and characterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).(a) Cordell AB. The Alkaloids: Chemistry and Biology. Vol. 60. Elsevier Science; San Diego, CA: 2003. [Google Scholar]; (b) Hesse M. Alkaloids: Nature’s Curse or Blessing? WILEY-VCH; Weinheim: 2003. [Google Scholar]
- (2).Lavilla R. J. Chem. Soc., Perkin Trans. 1. 2002:1141–1156. [Google Scholar]
- (3).For an example, see: Charette AB, Grenon M, Lemire A, Pourashraf M, Martel J. J. Am. Chem. Soc. 2001;123:11829–11830. doi: 10.1021/ja017136x.
- (4).Chai LZ, Zhao YK, Sheng QJ, Liu Z-Q. Tetrahedron Lett. 2006;47:9283–9285. [Google Scholar]
- (5).For a review, see: Gaudio AC, Korolkovas A, Takahata Y. J. Pharm. Sci. 1994;83:1110–1115. doi: 10.1002/jps.2600830809.
- (6).For a review, see: Stout DM, Meyers AI. Chem. Rev. 1982;82:233–243.
- (7).Comins DL, Hong H, Salvador JM. J. Org. Chem. 1991;56:7197–7199. See also ref 3.
- (8).For examples, see: Sundberg RJ, Hamilton G, Trindle C. J. Org. Chem. 1986;51:3672–3679. Bennasar ML, Roca R, Monerris M, Juan C, Bosch J. Tetrahedron. 2002;58:8099–8106.
- (9).(a) Ichikawa E, Suzuki M, Yabu K, Albert M, Kanai M, Shibasaki M. J. Am. Chem. Soc. 2004;126:11808–11809. doi: 10.1021/ja045966f. [DOI] [PubMed] [Google Scholar]; (b) Brunner B, Stogaitis N, Lautens M. Org. Lett. 2006;8:3473–3476. doi: 10.1021/ol061086p. [DOI] [PubMed] [Google Scholar]
- (10).Pujanauski BG, Prasad BAB, Sarpong R. J. Am. Chem. Soc. 2006;128:6786–6787. doi: 10.1021/ja061549m. [DOI] [PubMed] [Google Scholar]
- (11).For a discussion of pyran chemistry, see: Katrizky AR, Rees CW, editors. Comprehensive Heterocyclic Chemistry. Vol. 3. Pergamon; Oxford: 1984. pp. 737–874.
- (12).Olah GA, Hunadi RJ. J. Org. Chem. 1981;46:715–718. [Google Scholar]
- (13).We have recently described that other nitrogen nucleophiles (e.g., pyridinyl nitrogens) add faster to activated alkynes relative to the propargylic carboxylate. See: Smith CR, Bunnelle EM, Rhodes AJ, Sarpong R. Org. Lett. 2007;9:1169–1171. doi: 10.1021/ol0701971.
- (14).For a recent discussion of vinyl aziridine rearrangements, see: Ohno H. In: Aziridines and Epoxides in Organic Synthesis. Yudin AK, editor. WILEY-VCH; Weinheim: 2006. pp. 37–72.
- (15).A chelation-controlled 1,2-addition initially proposed to explain the high diastereoselectivity has recently been called into question. See: Righi G, Piertrantonio S, Bonini C. Tetrahedron. 2001;57:10039–10046. And: Schomaker JM, Geiser AR, Huang R, Borhan B. J. Am. Chem. Soc. 2007;129:3794–3795. doi: 10.1021/ja068077w.
- (16).See Supporting Information for a detailed description for the synthesis of the substrates.
- (17).PtCl2 has been used to catalyze the Overman rearrangement of trichloroacetimidates. See: Jaunzeme I, Jirgensons A. Synlett. 2005:2984–2986.
- (18).Mente PG, Heine HW, Scharoubim GR. J. Org. Chem. 1968;33:4547–4548. [Google Scholar]
-
(19).
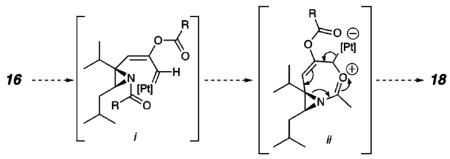 Mechanistically, this could proceed as illustrated below.
Mechanistically, this could proceed as illustrated below.
- (20).Crystallographic data have been deposited in the Cambridge Crystallographic Data Centre (CCDC #637309). Refinement data are provided in the Supporting Information.
- (21).The possibility of reduced stability of N-alkyl 1,2-dihydropyridines relative to N-tosyl or N-acyl 1,2-dihydropyridines under the reaction conditions may also contribute to the lack of success of this reaction.
- (22).For a recent example, see: Lemire A, Grenon M, Pourashraf M, Charette AB. Org. Lett. 2004;6:3517–3520. doi: 10.1021/ol048624n.
- (23).Enantiopure 1a was obtained by preparative chiral column HPLC of the racemate. For more information, see the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.