

Bcl-xL localized to mitochondria is necessary and sufficient for apoptotic protection but is unable to restore Ca2+ homeostasis in Bcl-x-KO cells. ER-localized Bcl-xL is required for ER Ca2+ homeostasis but does not affect apoptosis unless Bcl-xL is present in additional cellular compartments.
Abstract
Bcl-2 proteins are major regulators of cellular responses to intrinsic and extrinsic apoptotic stimuli. Among them, overexpression of the antiapoptotic protein Bcl-xL modulates intracellular Ca2+ homeostasis and organelle-specific apoptotic signaling pathways. However, the specific activities of Bcl-xL at mitochondria and the endoplasmic reticulum (ER) have not been fully defined. To further explore this, we generated mouse embryonic fibroblast (MEF) cell lines deficient in Bcl-xL expression (Bcl-x-KO). Deficiency in Bcl-xL expression did not induce compensatory changes in the expression of other Bcl-2 proteins, and Bcl-x-KO MEF cells showed increased sensitivity to various apoptotic stimuli compared with wild-type MEF cells. Targeting Bcl-xL at mitochondria but not at the ER restored apoptosis protection in Bcl-x-KO MEF cells to the degree observed in wild-type MEF cells. However, expression of ER-targeted Bcl-xL but not mitochondrially targeted Bcl-xL was required to restore Ca2+ homeostasis in Bcl-x-KO MEF cells. Of importance, ER-targeted Bcl-xL was able to protect cells against death stimuli in the presence of endogenous Bcl-xL. These data indicate that mitochondrial Bcl-xL can regulate apoptosis independently of ER Bcl-xL and that when localized exclusively at the ER, Bcl-xL impinges on Ca2+ homeostasis but does not affect apoptosis unless Bcl-xL is present in additional cellular compartments.
INTRODUCTION
The Bcl-2 protein family is major regulator of cellular apoptotic signaling (Hardwick and Youle, 2009). Since the founding family member Bcl-2 was shown to protect cells from apoptotic insults (Vaux et al., 1988), >20 Bcl-2–related proteins have been identified possessing either antiapoptotic or proapoptotic function (Hardwick and Youle, 2009). Bcl-2 proteins all share one or more homologous domains designated the Bcl-2 homology (BH) domain. Among antiapoptotic members, Bcl-xL retains the prototypical structural organization of four BH domains (BH1, BH2, BH3, and BH4) and a membrane-targeting hydrophobic carboxyl-terminal region (Boise et al., 1993; Muchmore et al., 1996). One important characteristic of Bcl-2 proteins is that they display differential interactions with each other and with other regulatory proteins in the apoptotic cascade, thus profoundly influence apoptotic signaling (Reed et al., 1998; Gross et al., 1999; Danial and Korsmeyer, 2004; Schwartz and Hockenbery, 2006).
Recent genetic and biochemical studies have revealed a conserved cascade that leads to the disruption of mitochondrial outer membrane (MOM) integrity and the release of apoptosis-inducing factors into the cytosol, subsequently resulting in the destruction of cells (Newmeyer and Ferguson-Miller, 2003; Boyce et al., 2004; Jiang and Wang, 2004). Although antiapoptotic Bcl-2 proteins are generally believed to promote cell survival by preventing MOM disintegration, proapoptotic Bcl-2 family members facilitate MOM permeabilization during apoptosis (Kuwana and Newmeyer, 2003; Chipuk and Green, 2008). Although most studies have focused on how Bcl-2 proteins modulate MOM during apoptosis, there is a large body of evidence indicating that Bcl-2 proteins are also localized and function on the endoplasmic reticulum (ER; Germain and Shore, 2003; Oakes et al., 2006; Pinton and Rizzuto, 2006).
It has been proposed that ER-resident proapoptotic Bcl-2 proteins sense insults to the ER, resulting in death signals being communicated to mitochondria or apoptosis being directly initiated at the ER level (Zong et al., 2003; Rong and Distelhorst, 2008; Wang et al., 2011b). The involvement of the ER in antiapoptotic signaling has been linked to its role as an intracellular Ca2+ store (Rong and Distelhorst, 2008). Apoptosis protection caused by overexpression of antiapoptotic Bcl-2 proteins, including Bcl-2, Bcl-xL, and Mcl-1, can be partially attributed to their ability to modulate ER Ca2+ signals through interactions with the inositol 1,4,5-trisphosphate receptor (InsP3R) Ca2+ release channel (White et al., 2005; Li et al., 2007; Rong et al., 2008, 2009; Eckenrode et al., 2010).
Although these studies have been highly informative, they rely largely on overexpression systems to study antiapoptotic Bcl-2 proteins, which might not reveal the activities of endogenous proteins. More important, there are no studies designed to examine the specific function of Bcl-2 and Bcl-xL at distinct organelles in nonoverexpression systems. To address this, we generated Bcl-xL–deficient mouse embryonic fibroblast (MEF) cell lines to study the effect of organelle-specific Bcl-xL reexpression on apoptosis and ER Ca2+ homeostasis. Bcl-xL–deficient MEF cell lines displayed increased sensitivity to apoptosis and disrupted ER Ca2+ homeostasis. Expressing mitochondrially targeted Bcl-xL in these lines increased apoptotic protection to the levels comparable to wild-type Bcl-xL. In contrast, expression of ER-localized Bcl-xL completely restored ER Ca2+ homeostasis but had no effect on apoptotic protection. When expressed in the wild-type background, however, ER-targeted Bcl-xL did increase apoptotic protection. These data show that mitochondrial localization alone is sufficient to provide protection, but also that mitochondrial localization is an absolute requirement for ER-localized Bcl-xL to be antiapoptotic.
RESULTS
bcl-x gene knockout does not cause compensatory changes in expression of other Bcl-2 proteins
The antiapoptotic Bcl-2 protein Bcl-xL is generated from alternative splicing of the bcl-x gene (Boise et al., 1993). Massive cell death has been found in a variety of tissues in bcl-x–deficient mouse, which dies at embryonic stage, implicating its important role in development (Motoyama et al., 1995). To investigate the function of endogenous Bcl-xL protein in apoptosis regulation, we established MEF cells deficient in Bcl-xL expression (Bcl-x knockout [Bcl-x-KO]). Bcl-x-KO MEF cells were generated from embryos of a pregnant mouse heterozygous for the bcl-x gene (bcl-x+/−). Whereas the expression of Bcl-xL in the established MEF cell lines was readily detected by Western blot analysis, the expression of the other spliced transcript, Bcl-xS, was undetectable, indicating that Bcl-xL is the predominant splicing product in these cells (Figure 1A). Among 10 MEF cell lines developed, cell lines 1, 3, 5, and 6 had higher Bcl-xL expression levels and were considered to express Bcl-xL from both alleles (bcl-x+/+, designated wild-type [WT]). In contrast, no Bcl-xL expression was detected in cell lines 2, 4, and 9, which were designated Bcl-x-KO MEF cells (bcl-x−/−). In cell lines 7, 8, and 10, lower Bcl-xL expression levels were detected, which could be attributed to heterozygous deletion of the bcl-x gene (bcl-x+/−).
FIGURE 1:
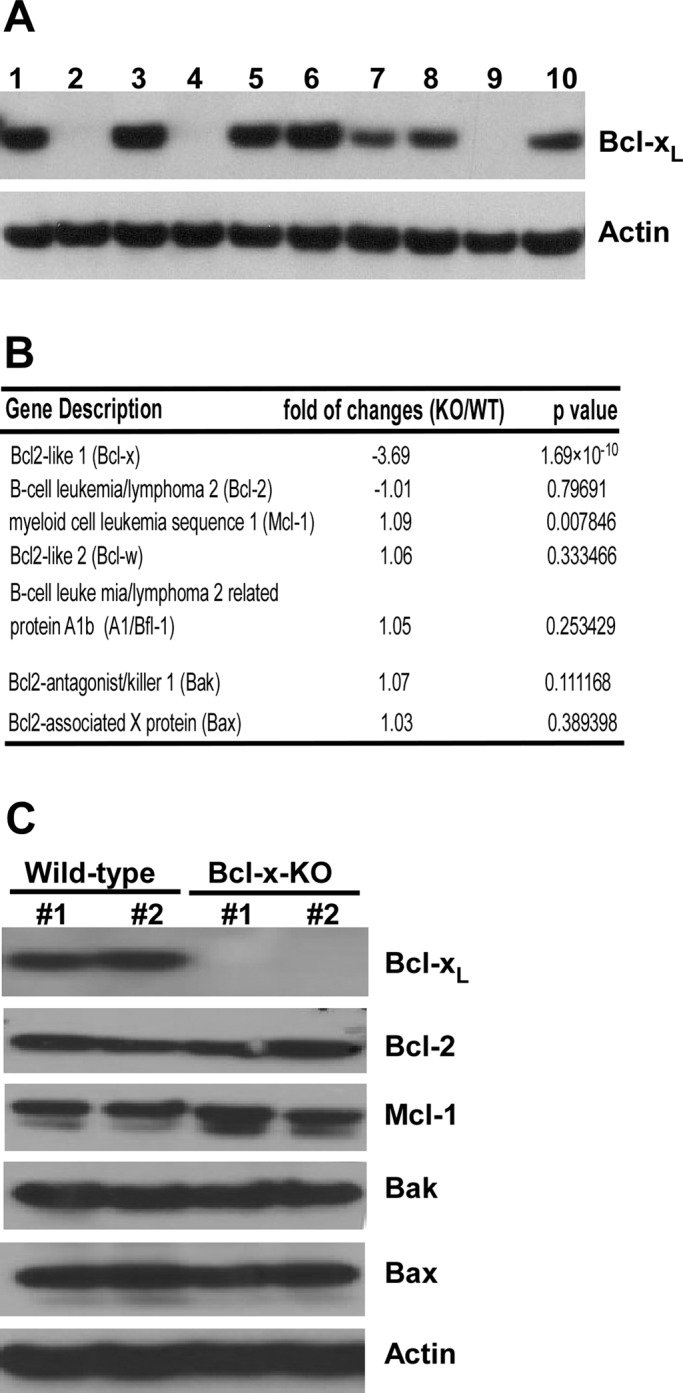
bcl-x deficiency does not cause compensatory changes in expression of other Bcl-2 proteins. (A) Primary embryonic fibroblast cells were isolated from 10 embryos of an 11-d-pregnant bcl-x heterozygous mouse. MEFs were immortalized by expressing SV40 T antigen and cloned. Bcl-xL expression in the established MEF cell lines was detected by Western blot analysis. (B) Changes in mRNA expression of Bcl-2 genes induced by bcl-x deficiency were determined by microarray analysis. Data are represented as fold changes in mean value of mRNA levels in two WT and two Bcl-x-KO cell lines (triplicate RNA samples for each cell line). Negative values indicate down-regulation. (C) Expression of Bcl-2 proteins in the indicated MEF cell lines was determined by Western blot analysis. KO #1, cell line 9; KO #2, cell line 4; WT #1, cell line 3; WT #2, cell line 6.
Decreased expression of antiapoptotic Bcl-2 has been shown to affect the expression of other apoptotic regulators (Rubenstein et al., 2011). We therefore investigated the effect of knocking out the bcl-x gene in MEF cells on the expression of other genes, particularly those of Bcl-2 protein family. Microarray analysis was carried out in two Bcl-x-KO (lines 4 and 9) and two wild-type (lines 3 and 6) MEF cell lines. Gene expression levels were measured and the fold change between Bcl-x-KO and wild-type cells was determined (Figure 1B). In this assay (GeneChip Mouse Gene 1.0 ST Array), analysis of multiple probes on different exons was summarized into an expression value representing all transcripts from the same gene. Because the bcl-x–deficient mouse was generated by homologous recombination, some exons in the targeted bcl-x gene were still intact. By this approach, bcl-x mRNA was still detectable in bcl-x–deficient cells, and bcl-x mRNA level in Bcl-x-KO cells was significantly reduced compared with that in wild-type cells, validating the disruption of intact bcl-x mRNA in Bcl-x-KO cells (Figure 1B). Knocking out the bcl-x gene did not cause significant changes in expression of other Bcl-2 proteins, including antiapoptotic Bcl-2 proteins, proapoptotic multiple-domain Bcl-2 proteins (Figure 1B), and proapoptotic, BH3-only Bcl-2 proteins (Supplemental Table S1). Western blot analysis was performed to further confirm that bcl-x deficiency did not cause significant changes in Bcl-2 protein expression (Figure 1C). These results indicate that alterations in cellular apoptotic signaling in Bcl-x-KO MEF cells are likely attributable to Bcl-xL deficiency, therefore providing an excellent cell system to study the mechanism of endogenous Bcl-xL in apoptosis regulation.
Endogenous Bcl-xL modulates cellular responses to apoptotic stimuli
To investigate the functional activity of endogenous Bcl-xL in apoptotic signaling, we treated two wild-type and two Bcl-x-KO MEF cells with a range of reagents known to induce apoptosis. Cells were exposed to the topoisomerase inhibitor etoposide, the transcriptional inhibitor actinomycin D, the DNA intercalating agent doxorubicin, the protein kinase inhibitor staurosporine, or the oxidative stress inducer hydrogen peroxide (H2O2). Treatment with all reagents resulted in more cell death in Bcl-x-KO cells than in wild-type cells, except that the protection against actinomycin D in wild-type cells seems to be transient (Figure 2A). Given that Bcl-xL is the major splicing product of the bcl-x gene in MEF cells, higher sensitivity of Bcl-x-KO cells to apoptotic insults suggests that endogenous Bcl-xL is able to protect cells against apoptotic insults.
FIGURE 2:

Endogenous Bcl-xL modulates cellular response to different apoptotic stimuli. (A) Apoptosis was induced by the indicated apoptosis agents: etoposide (5.0 μM), doxorubicin (1.0 μM), actinomycin D (0.2 μg/μl), staurosporine (5.0 nM), or H2O2 (0.4 mM). Bcl-x-KO MEFs were more sensitive to these reagents. Data represent mean ± SD of three independent experiments. (B) Caspase 3/7 activity was measured 12 h after the treatment of etoposide, doxorubicin, or staurosporine, 9 h after treatment with H2O2, or 7 h after actinomycin D treatment, using fluorometric assay. Values are normalized to the values obtained in the untreated control cells. Data are shown as mean ± SD of three independent experiments.
To further confirm that endogenous Bcl-xL is able to inhibit apoptosis, we measured caspase 3/7 activity in both wild-type and Bcl-x-KO MEF cells treated with apoptotic stimuli. As shown in Figure 2B, upon apoptotic insult treatment, caspase 3/7 activity was enhanced in Bcl-x-KO MEFs to higher levels than those in wild-type cells. The increase in caspase 3/7 activity in Bcl-x-KO cells was correlated with the increase in cell death, suggesting that endogenous Bcl-xL protects cells against apoptotic stimuli.
To ensure that enhanced apoptosis observed in Bcl-x-KO MEF cells was due to the lack of Bcl-xL expression, we reexpressed Bcl-xL cDNA in Bcl-x-KO MEF cells (cell line 9) and obtained cells reexpressing different levels of Bcl-xL (Figure 3A). Three cell lines with different Bcl-xL expression levels were selected: cell line H (high), with Bcl-xL expression level higher than the endogenous Bcl-xL level; cell line M (medium), with Bcl-xL expression level comparable to the endogenous level; and cell line L (low), with expression level lower than the endogenous Bcl-xL level. When cells were treated with different apoptotic reagents as described previously (Figure 2A), reexpression of Bcl-xL restored apoptotic resistance of Bcl-x-KO cells (Figure 3B). Of importance, Bcl-xL protected cells against apoptotic stimuli in a dose-dependent manner, providing additional evidence that increased sensitivity to death stimuli in Bcl-x-KO cells is caused by the lack of endogenous Bcl-xL expression. This is further strengthened by the observation that the caspase 3/7 activity detected in the different cell lines after apoptosis induction closely correlated with the cell viability measurements (Figure 3C).
FIGURE 3:

Reexpression of Bcl-xL in Bcl-x-KO MEFs restores resistance to apoptotic stimuli in a dose-dependent manner. (A) Bcl-xL was reexpressed in Bcl-x-deficient MEFs. Western blot analysis was used to determine Bcl-xL expression levels. (B) Viability of the indicated MEF cells was measured in the presence of the indicated apoptotic stimuli, including etoposide (5.0 μM), doxorubicin (1.0 μM), actinomycin D (0.2 μg/μl), staurosporine (5.0 nM), and H2O2 (0.4 mM). Data represent mean ± SD of three independent experiments. (C) Caspase 3/7 activity was measured under the conditions described in Figure 2B. Values are normalized to the values of untreated cells. Data show mean ± SD of three independent experiments.
Mitochondrially targeted Bcl-xL prevents apoptosis more effectively than ER-targeted Bcl-xL
To investigate the antiapoptotic functions of Bcl-xL localized at different intracellular organelles, we constructed Bcl-xL mutants that either lacked the membrane-targeting sequence or contained ER-specific or mitochondrial-specific membrane targeting sequences. The carboxyl-terminal transmembrane sequence of Bcl-xL was deleted, which specifically confines Bcl-xL to the cytoplasm (Bcl-xL-ΔC). The C-terminal transmembrane region of Bcl-xL was replaced by the cytochrome b5 (cb5) membrane-targeting sequence, which directs Bcl-xL specifically at the ER membrane, or the membrane-targeting sequence of the listerial protein ActA, which specifically targets Bcl-xL at mitochondria (Supplemental Figure S1A). The same targeting sequences were used previously to target both antiapoptotic and proapoptotic Bcl-2 proteins to a specific organelle (Zhu et al., 1996; Scorrano et al., 2003; Zong et al., 2003; Fiebig et al., 2006). To confirm subcellular localization of Bcl-xL mutant proteins, we subcloned cDNAs of the respective Bcl-xL mutants into a plasmid containing green fluorescent protein (EGFP-C1). GFP-tagged Bcl-xL mutant proteins were transiently expressed in Bcl-x-KO and imaged using confocal microscopy. The mitochondrial protein Tom20 served as a mitochondrial maker, and the ER was labeled with calreticulin. Whereas GFP-tagged Bcl-xL-ActA colocalized with Tom20, the staining patterns for GFP-Bcl-xL-ActA and calreticulin were clearly distinct (Supplemental Figures S1B and S1C). In contrast, GFP-Bcl-xL-cb5 was completely targeted at the ER, with its fluorescence colocalizing with that of calreticulin but not Tom20. The subcellular localization of GFP-tagged Bcl-xL proteins was further confirmed in live cells examined by coexpressing fluorescent protein markers of mitochondria and the ER (Supplemental Figure S1D). The pattern of GFP-Bcl-xL-ActA localization entirely matched that of coexpressed mitochondrial–red fluorescent protein (RFP), whereas GFP-tagged Bcl-xL-cb5 completely colocalized with ER-localized DsRed, providing additional evidence of a high degree of differential expression between mitochondria-localized and ER-localized Bcl-xL.
To investigate whether Bcl-xL proteins targeted at specific organelles mediate different apoptotic signaling pathways, we reexpressed the mutant Bcl-xL proteins in Bcl-x-KO MEF cells (cell line 9) at a level comparable to that of endogenous Bcl-xL (Figure 4A). Bcl-x-KO MEF cells reexpressing different Bcl-xL mutants were treated with various death reagents (etoposide, doxorubicin, actinomycin D, staurosporine, and H2O2), and cell viability was measured. Reexpression of Bcl-xL-ActA in Bcl-x-KO MEF cells provided protection against all apoptotic reagents at a level comparable with that of wild-type Bcl-xL (Figure 4B). In contrast, both Bcl-xL lacking membrane-anchoring region (Bcl-xL-ΔC) and ER-localized Bcl-xL (Bcl-xL-cb5) failed to protect cells against any of the death stimuli. These data suggest that mitochondrial localization is an absolute requirement for Bcl-xL to be protective in various apoptotic paradigms. This was further confirmed by the experiments in which caspase 3/7 activity was measured in response to cell death stimuli. The caspase 3/7 activity of Bcl-x-KO MEF cells reexpressing Bcl-xL-cb5 or Bcl-xL-ΔC was higher than that in cells reexpressing Bcl-xL-ActA or wild-type Bcl-xL (Figure 4C). Similar results were obtained from a second set of cell lines (Supplemental Figure S2).
FIGURE 4:

Mitochondrially targeted Bcl-xL inhibits apoptosis more efficiently than Bcl-xL targeted at the ER. (A) The indicated Bcl-xL mutants were expressed in Bcl-x-KO MEFs at levels similar to the endogenous Bcl-xL level. Bcl-xL expression was determined by Western blot analysis. (B) Cells expressing different Bcl-xL mutants were treated with 5.0 μM etoposide, 1.0 μM doxorubicin, 0.2 μg/μl actinomycin D, 5.0 nM staurosporine, or 0.4 mM H2O2, and cell viability was measured. The data depict mean ± SD of three independent experiments. (C) Caspase 3/7 activity was measured as described in Figure 2B. Values are normalized to those of untreated cells. All data shown are the means ± SD of experiments independently performed three times.
ER-targeted Bcl-xL modulates ER Ca2+ homeostasis
Previously, we demonstrated that overexpression of Bcl-xL lowers the steady-state [Ca2+] within ER stores ([Ca2+]ER) and affects intracellular Ca2+ ([Ca2+]i) signaling by modulating channel activities of the ER Ca2+ release channel InsP3 receptor (White et al., 2005; Li et al., 2007). Here we investigated whether organelle-targeted Bcl-xL proteins influenced InsP3-dependent [Ca2+]i signaling. To investigate whether organelle-targeted Bcl-xL proteins influenced InsP3-dependent [Ca2+]i signaling, we monitored [Ca2+]i in response to an increase in intracellular [InsP3] evoked by flash photolysis of caged InsP3 (Figure 5A). Maximal InsP3 uncaging and/or receptor activation in these cells was produced by UV pulses of 250-ms duration. This level of InsP3 evoked larger [Ca2+] transients in Bcl-x-KO cells compared with wild-type cells (Figure 5, A and B). Reexpression of Bcl-xL in Bcl-x-KO localized at the ER but not at mitochondria restored InsP3-evoked Ca2+ signal to the level observed in wild-type cells (Figure 5, A and B). These data are consistent with previous studies showing that Bcl-xL overexpression reduces ER Ca2+ store content and thus the magnitude of ER Ca2+ release in response to maximal InsP3 stimulation (Li et al., 2007). In these studies the presence of the type-3 InsP3R isoform was required for Bcl-xL to lower ER Ca2+ store content (Li et al., 2007). Western blot was used to confirm the presence of each InsP3R isoform in both wild-type and Bcl-x-KO MEF cells (Supplemental Figure S3).
FIGURE 5:

ER-localized Bcl-xL but not mitochondrially targeted Bcl-xL modulates ER Ca2+ homeostasis. (A) Representative [Ca2+]i responses in fluo-2AM–loaded cells in response to flash photolysis of caged InsP3. (B) Summary bar graphs demonstrate the amplitude of [Ca2+]i change after InsP3 uncaging shown in A. Data represent the mean ± SEM amplitude of 31–66 cells pooled from at least three independent coverslip (*p < 0.05; ANOVA). (C) The Ca2+ content of the ER was indirectly assessed by monitoring [Ca2+]i measured by fura-2 in response to acute inhibition of ER Ca2+ uptake by CPA (10 μM). Representative traces of the indicated cell lines are shown. (D) Bar graphs summarizing the total released [Ca2+] assessed by measuring the area under the Ca2+ transient evoked by CPA (area under curve [AUC]). Data represent the mean ± SEM of at least 50 cells pooled from at least three separate trials (*p < 0.05; ANOVA). (E) Representative recordings of [Ca2+]ER in permeabilized cells during store filling initiated by addition of 200 nM Ca2+ and 1.5 mM MgATP and store depletion by the application of 10 μM InsP3. Each trace represents mean of 15–20 cells in a single image field. (F–G) Summary data (mean ± SEM) for 116–146 cells pooled from at least three independent trials. (F) The steady-state uptake capacity of [Ca2+]ER after store filling and (G) the amplitude of [Ca2+]ER release in the presence of InsP3 (*p < 0.05; ANOVA).
Because steady-state [Ca2+]ER is determined by the balance between ER Ca2+ leak and Ca2+ uptake by sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), [Ca2+]ER was further evaluated by monitoring [Ca2+]i in response to inhibition of ER Ca2+ uptake by the SERCA inhibitor cyclopiazonic acid (CPA) in the absence of external Ca2+. A smaller Ca2+ response was evoked by CPA in wild-type cells than in Bcl-x-KO cells (Figure 5C). The ER Ca2+ release was quantified by calculating the integral of the [Ca2+]i transient, since the total released [Ca2+] more closely reflects the steady-state store content (Figure 5D). Again, the phenotype of wild-type cells was observed only in Bcl-x-KO cells reexpressing Bcl-xL localized at the ER but not Bcl-xL localized at mitochondria. To further confirm the effects of organelle-targeted Bcl-xL on ER Ca2+ store, we also carried direct measurements of [Ca2+]ER using the low-affinity Ca2+ indicator mag-fura-2. Because mag-fura-2 compartmentalizes in the ER as well as in the cytoplasm, the plasma membrane was permeabilized to remove cytoplasmic indicator, and the cells were perfused with a Ca2+-free solution to allow passive depletion of ER Ca2+ store (Eckenrode et al., 2010). After equilibration, Ca2+ uptake into the store was induced by switching the perfusate to the solution containing 1.5 mM MgATP and 200 nM free [Ca2+]. Once a steady state had been reached, the solution was switched to one containing saturating [InsP3] (10 μM) without MgATP (Foskett et al., 2007). As depicted in Figure 5E, application of InsP3 induced depletion of the ER Ca2+ store. This was a unidirectional measure of Ca2+ flux, since the MgATP required for SERCA pump function was not present during InsP3 addition. Consistent with the previous experiments, Bcl-xL deficiency elevated steady-state Ca2+ in the ER that was rescued by Bcl-xL reexpression on the ER (Figure 5, E and F). In addition, the magnitudes of Ca2+ release evoked by saturating [InsP3] mirrored the filling state of the ER (Eckenrode et al., 2010; Figure 5G) and are in agreement with the data from intact cells (Figure 5A). Taken together, these data show that expression of ER-targeted Bcl-xL produces phenotypes that are entirely consistent with our present understanding of how this protein functions at the ER to regulated Ca2+ signaling.
Endogenous Bcl-xL is essential for antiapoptotic activities of Bcl-xL localized at the ER
Overexpressing ER-localized Bcl-xL in the breast cancer line MCF-7 and Rat-1 fibroblasts exhibits broader antiapoptotic activities than Bcl-xL targeted at mitochondria (Fiebig et al., 2006). To explore the discrepancy between this finding and our observations that ER-localized Bcl-xL failed to protect against apoptosis in Bcl-x-KO cells (Figure 4), we stably reexpressed Bcl-xL-cb5 in wild-type and Bcl-x-KO MEF cells at similar levels and obtained uncloned populations of cells (Figure 6A). Cells were then exposed to various death stimuli (etoposide, doxorubicin, actinomycin D, staurosporine, and H2O2), and cell viability was measured. Whereas Bcl-xL-cb5 did not possess any antiapoptotic activities in Bcl-x-KO cells, expressing the same protein in wild-type cells markedly protected against cell death in all apoptotic paradigms examined, indicating that antiapoptotic activities of Bcl-xL targeted at ER depends on endogenous Bcl-xL (Figure 6, B and C). Examination of caspase 3/7 activity of cells treated with different apoptosis triggers further confirmed the indispensable role of endogenous Bcl-xL in the antiapoptotic function of ER-targeted Bcl-xL (Figure 6, D and E).
FIGURE 6:

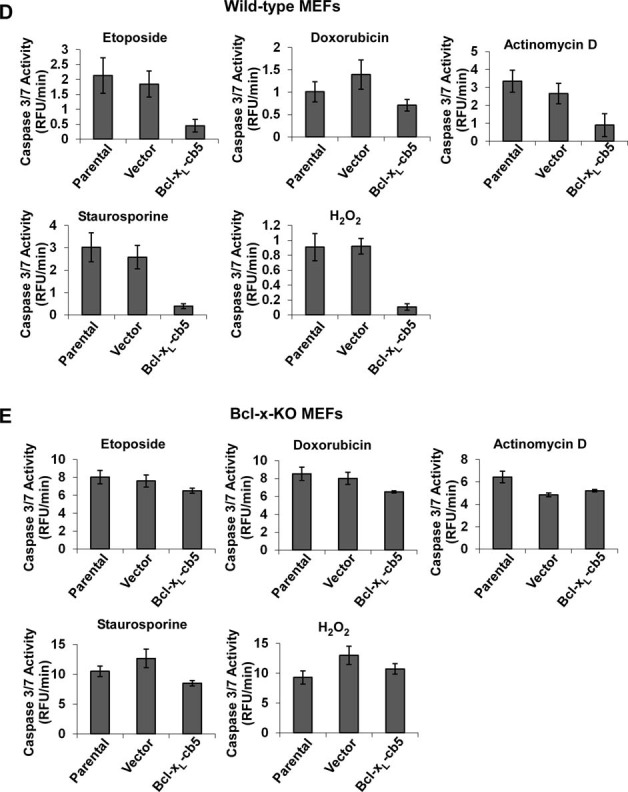
Antiapoptotic activities of ER-targeted Bcl-xL depend on endogenous Bcl-xL. (A) Bcl-xL was stably reexpressed in wild-type and Bcl-x-KO MEF cells, and expression of Bcl-xL in uncloned cell populations was determined by Western blot. Note that the molecular weights of Bcl-xL-cb5 and endogenous Bcl-xL are different. (B) Wild-type MEF cells with or without Bcl-xL-cb5 expressions were treated with the indicated death stimuli, and cell viability was determined. The data depict mean ± SD of three independent experiments. (C) The indicated Bcl-x-KO MEF cell lines were treated with the apoptotic stimuli as shown in B, and their effects on cell survival were examined. Mean ± SD of three independent experiments are shown. (D) On treatment with the indicated death stimuli for the indicated time points, caspase 3/7 activity of various wild-type MEF cells was determined using a fluorometric assay. Each experiment was performed in triplicate, and mean ± SD of three independent experiments is shown. (E) Different Bcl-x-KO MEF cells were treated with the indicated apoptotic stimuli for the indicated time points, and caspase 3/7 activity was measured. Data represent the mean ± SD of three independent experiments.
DISCUSSION
Antiapoptotic Bcl-xL is localized at both mitochondria and the ER (Hsu et al., 1997; Tagami et al., 2000; Fiebig et al., 2006). At mitochondria, Bcl-xL functions to prevent MOM permeabilization during apoptotic stress and also plays an important role in optimizing mitochondrial bioenergetic function through interactions with various membrane proteins (Vander Heiden et al., 2001; Alavian et al., 2011; Chen et al., 2011). In previous work, we demonstrated that Bcl-xL functioned at the ER to modulate basal Ca2+ signaling and steady-state ER Ca2+ store content by interacting with the InsP3 receptor (White et al., 2005; Li et al., 2007). Collectively, these studies reveal that Bcl-xL has diverse physiological roles that appear to depend on its intracellular localization. In the present study, we sought to better understand the relationship between intracellular localization of Bcl-xL and apoptotic protection. Our approach was to generate a Bcl-xL–deficient model in which Bcl-xL targeted at different organelles was reexpressed and its effects on apoptotic sensitivity were examined. MEF cells deficient in Bcl-xL expression were established from bcl-x heterozygous mice. Although alternative splicing of the bcl-x gene generates Bcl-xL and Bcl-xS, Bcl-xL is the major splicing product in MEF cells (Figure 1A). Unlike the effect of reducing Bcl-2 expression in LNCaP cells (Rubenstein et al., 2011), knocking out Bcl-xL expression in MEF cells failed to induce compensatory changes in other Bcl-2 family members (Figure 1, B and C). As expected, cells deficient in Bcl-xL expression were more sensitive to various death stimuli than were wild-type cells. Furthermore, reexpression of wild-type Bcl-xL in Bcl-x-KO cells restored cellular protection against apoptotic insults in a Bcl-xL dose-dependent manner (Figure 3). These data demonstrate the validity of this experimental model.
Bcl-2 proteins have been reported to be localized at both mitochondria and the ER (Germain and Shore, 2003). To distinguish between the functions of Bcl-xL localized at different organelles, we targeted Bcl-xL to a particular organelle by replacing the carboxyl transmembrane region of Bcl-xL with known organelle-targeting sequences. Given that mitochondria and the ER have been proposed to dynamically interact with each other inside cells (Sano et al., 2009), it is challenging to exclusively target proteins to a given organelle. Unlike previous studies in which antiapoptotic Bcl-2 proteins targeted at a particular organelle were overexpressed (Zhu et al., 1996; Fiebig et al., 2006), in the present study we carefully selected stable cell lines so that mitochondria- or ER-localized Bcl-xL was expressed at levels similar to that observed in wild-type cells (Figure 4). It was expected that lower expression levels would also help ensure more exclusive targeting. The colocalization of the targeted Bcl-xL proteins with their respective organelles was assessed using confocal microscopy, and the data show correct targeting of our constructs, at least within the spatial limits of confocal microscopy (Supplemental Figure S1).
We observed that in the Bcl-x-KO background mitochondrially localized Bcl-xL functioned as effectively as wild-type protein in inhibiting apoptosis. On the other hand, ER-targeted Bcl-xL failed to exhibit any antiapoptotic activity in Bcl-x-KO cells (Figure 4). It is intriguing, however, that Bcl-xL expression at the ER in Bcl-x-KO cells completely rescued abnormal Ca2+-signaling phenotypes (Figure 5). It is well documented that steady-state ER Ca2+ correlates closely with apoptotic sensitivity—increased ER Ca2+, increased apoptosis—and vice versa. Here increased ER Ca2+ content sensitizes cells to apoptotic stimuli by providing a greater pool of releasable Ca2+ that enhances mitochondrial Ca2+ uptake and susceptibility to MOM permeabilization (Oakes et al., 2006; Pinton and Rizzuto, 2006; Rong and Distelhorst, 2008). In support of such a model, lowering ER Ca2+ content alone protects against apoptotic stimuli (Pinton et al., 2001), and restoring Ca2+ levels in ER-depleted stores resensitizes cells to apoptosis (Scorrano et al., 2003). In the present study, however, restoring ER Ca2+ content by targeting Bcl-xL to the ER in Bcl-x-KO cells had no effect on apoptosis. This was also true of staurosporine and H2O2, which are stimuli predicted to be proapoptotic because of their ability to mobilize ER Ca2+ through InsP3R activation (Zheng and Shen, 2005; Khan et al., 2007; Takada et al., 2011).
Both overexpression of Bcl-2 and Bcl-xL have been shown to lower ER store Ca2+ (Lam et al., 1994; Foyouzi-Youssefi et al., 2000; Pinton et al., 2001; White et al., 2005; Li et al., 2007). In previous studies, we provided evidence for a molecular mechanism that accounts for Bcl-xL–regulated ER store content. We found that Bcl-xL interacts directly with the InsP3R channel to increase the functional affinity for its agonist InsP3. Although the interaction has no effect on InsP3R-dependent Ca2+ release in response to saturating [InsP3], it dramatically increases channel activity at low [InsP3], effectively increasing the basal level of channel activity in resting cells and reducing steady-state ER Ca2+ content (White et al., 2005; Li et al., 2007). Only saturating [InsP3] was assessed in the present study, and so increased InsP3R sensitivity with Bcl-xL expression is not apparent. Nevertheless, the data show elevated steady-state ER Ca2+ in Bcl-x-KO cells, consistent with our previous models. Moreover, these data demonstrate that endogenous Bcl-xL is an absolute requirement for normal ER Ca2+ homeostasis.
Although our previous studies concluded that reduced ER store content was not necessarily a requirement for Bcl-xL to be antiapoptotic, they did confirm that Bcl-xL-InsP3R interactions at the ER were an essential component of maximal apoptotic protection (White et al., 2005; Li et al., 2007). Why then in the present study did restoring ER Ca2+ homeostasis by targeted expression of Bcl-xL at the ER have no effect on apoptosis sensitivity? Indeed, this is in direct contradiction to previous studies showing that overexpression of ER-localized Bcl-xL did confer antiapoptotic activity against several apoptotic insults (Fiebig et al., 2006). We reasoned that the discrepancy could be accounted for if Bcl-xL localized at other organelles was essential for it to be antiapoptotic at the ER. Consistent with this, expression of ER-targeted Bcl-xL was fully effective in protecting against apoptosis in wild-type cells (Figure 6). Thus the present study provides molecular evidence that the protective function of Bcl-xL at the ER requires Bcl-xL localization at other cellular compartments. We speculate that this compartment is mitochondria, but further studies are needed to assess exactly how mitochondrially targeted Bcl-xL is required to sense physiological changes conferred by Bcl-xL at the ER, and how these signals are translated into apoptotic protection. Our previous studies demonstrated that Bcl-xL interacted with InsP3Rs to enhance Ca2+ signals, which were sensed by neighboring mitochondria to promote Ca2+-dependent mitochondrial bioenergetics (White et al., 2005). In this scenario, it is possible that mitochondrial Bcl-xL is required to facilitate ER-to-mitochondrial Ca2+ transfer, and enhanced mitochondrial bioenergetics subsequently renders cells better able to withstand apoptotic challenges.
In summary, we developed Bcl-xL–deficient MEF cells suitable for the study of Bcl-xL–regulated apoptotic pathways. We demonstrated that localization at mitochondria is necessary for endogenous levels of Bcl-xL to provide apoptotic protection. Furthermore, we showed that localization of Bcl-xL at the ER is required for normal Ca2+ homeostasis, but that targeting Bcl-xL at the ER provides increases apoptotic protection only if Bcl-xL is also present at other organelles.
MATERIALS AND METHODS
Reagents
Etoposide, doxorubicin, H2O2, and actinomycin D were purchased from Sigma-Aldrich (St. Louis, MO), and staurosporine was purchased from Enzo (Farmingdale, NY). All reagents were dissolved in dimethyl sulfoxide. Lipofectamine 2000 was purchased from Invitrogen (Carlsbad, CA). Unless otherwise stated, all other reagents were purchased from Sigma-Aldrich. Antibodies (Abs) used for Western blot analysis were anti–Bcl-xS/L S-18 polyclonal Ab (pAb; Santa Cruz Biotechnology, Santa Cruz, CA), anti–β-actin monoclonal Ab (mAb; Sigma-Aldrich), anti–Bak pAb (Upstate, Lake Placid, NY), anti–Bax N20 pAb (Santa Cruz Biotechnology), anti–Mcl-1 pAb (Epitomics, Burlingame, CA), antixBcl-2 mAb (Santa Cruz Biotechnology), anti–Bcl-w pAb (Cell Signaling Technology, Danvers, MA), anti–type 1 InsP3R and type 2 InsP3R pAbs (gifts from Kevin Foskett), anti–type 3 InsP3R mAb (BD Transduction Laboratory, San Jose, CA), anti-calreticulin pAb (Enzo), anti-Tom20 pAb (a gift from Brain Wattenberg), peroxidase-conjugated goat anti-rabbit immunoglobulin G (IgG; Thermo Scientific, Waltham, MA), peroxidase-conjugated goat anti–mouse IgG (Thermo Scientific), and Alexa Fluor 594 goat anti–rabbit IgG (Molecular Probes, Eugene, OR).
DNA plasmids
Human Bcl-xL cDNA was cloned into the mammalian expression vector pEF6/myc-His A (Invitrogen) to generate the plasmid pEF6-Bcl-xL. A stop codon was introduced after amino acid 210 by PCR to generate the plasmid pEF6-Bcl-xL-ΔC. The mitochondrial targeting sequence ActA and the ER targeting sequence cb5 were also generated by PCR as described previously (Zong et al., 2003) and subsequently subcloned after amino acid 210 of Bcl-xL to make pEF6-Bcl-xL-ActA and pEF6-Bcl-xL-cb5, respectively. The constructs used for intracellular localization experiments were generated by subcloning appropriate DNA fragments from pEF6 into pEGFP-C1 (Clontech, Madison, WI).
Generation of cell lines
Embryos from an 11-d-pregnant Bcl-x heterozygous mouse were isolated. Primary embryonic cells were dissociated and plated on gelatin-coated dishes. Seven days later, adherent embryonic cells were plated and immortalized by transfecting the pCDNA3 plasmid encoding SV40 large T antigen. The limiting dilution method was used to isolate single clones from each of the 10 original embryos. After stable cell lines had been established, the expression of Bcl-xL was examined by Western blot. To reexpress Bcl-xL in Bcl-x-KO cells, we transfected pEF6 plasmids encoding wild-type or mutant Bcl-xL cDNAs into wild-type or Bcl-x-KO MEF cells using Lipofectamine 2000 reagent (Invitrogen). Cells stably expressing Bcl-xL proteins were obtained by growing transfected cells in the medium containing the antibiotic blasticidin (1.5 μg/ml; Invitrogen) and subcloned by a limited dilution approach. Western blot analysis was used to determine the expression level of different Bcl-xL proteins in Bcl-x-KO MEFs. All cells were cultured in DMEM medium (Mediatech, Manassas, VA) supplemented with 10% fetal bovine serum (FBS; Gemini, West Sacramento, CA), 100 U/ml penicillin (Mediatech), and 100 μg/ml streptomycin (Mediatech) in an incubator with 95% humidity and 5% CO2 at 37°C.
Gene expression analysis
Total RNA from two wild-type and two Bcl-x-KO MEFs was isolated using a RNeasy kit (Qiagen, Germantown, MD). The quality of total RNA was confirmed using a 2100 Bioanalyzer (Agilent, Santa Clara, CA). cRNA was generated using a WT Expression Kit (Ambion, Austin, TX) and labeled using a GeneChip WT Terminal Labeling Kit (Affymetrix, Santa Clara, CA). Hybridization to GeneChip Mouse Gene 1.0 ST Array (Affymetrix) was carried out according to the manufacturer's protocol. After array processing and scanning, the resulting raw data were summarized and normalized using RMA in Partek Genomics Suite 6.5 (Partek, St. Louis, MO) by the University of Louisville Microarray Facility. Exon-level signals were summarized to gene expression levels using mean values. The significance of the changes was determined using analysis of variance and applying false discovery rate as multiple-test correction.
Cell viability assay
MEF cells were plated in 48-well tissue culture plates with ∼1 × 104 cells per well 24 h before treatment with apoptotic stimuli. At the indicated time points, cells were trypsinized and resuspended in growth medium containing 1.0 μg/ml propidium iodide (PI; Invitrogen). Cell viability was measured by PI exclusion using flow cytometry (FACSCalibur; Beckon Dickinson, San Jose, CA) as described previously (Olberding et al., 2010). The experiments were performed in triplicate. Viability of the treated cells was presented as percentage of those of untreated control cells.
Caspase 3/7 assay
The Sensolyte Homogeneous R110 Caspase 3/7 Assay Kit (AnaSpec, San Jose, CA) was used to directly measure caspase 3/7 activity in cells. Twenty-four hours before the treatment, cells were plated in white-walled 96-well plates. At the indicated time points, measurement was performed according to the manufacturer's protocols. The fluorescence signal (excitation/emission, 496/520 nm) was measured kinetically over 2 h at an interval of 1 min by a Gemini EM microplate spectrofluorometer (Molecular Devices, Sunnyvale, CA). Data were presented as relative fluorescence units (RFUs) versus time (RFU/min). The slope was calculated and normalized to those of untreated cells as described previously (Olberding et al., 2010).
Western blot analysis
Cell pellets were collected, and whole-cell lysates were generated by resuspending cells in RIPA lysis buffer (150 mM sodium chloride, 1.0% [vol/vol] Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, and 50 mM Tris, pH 8.0) containing protease inhibitors (Complete; Roche, Indianapolis, IN). To remove cell debris and nuclear materials, we first sonicated cells for 30 s at 10% amplitude (Sonic Dismembrator, Model 500; Fischer Scientific, Hampton, NH) before centrifugation at 13,200 rpm at 4°C for 10 min and collected the supernatant . The protein concentration was determined using the BCA Assay (Pierce, Rockford, IL). Twenty micrograms of protein sample was loaded on a 4–12% Bis-Tris gel (Bio-Rad, Hercules, CA) and transferred to polyvinylidene fluoride (Millipore, Billerica, MA). The membrane filters were incubated in buffer containing 1× phosphate-buffered saline (PBS), 0.2% (vol/vol) Tween 20, and 10% (wt/vol) nonfat dry milk (Bio-Rad) with appropriate primary antibodies at 4°C overnight or at 25°C for about 3 h. After three 10-min washes with 1× PBS/0.2% Tween solution, the membrane filters were then incubated with the appropriate peroxidase-coupled secondary antibodies at 4°C overnight or for at 25°C for about 3 h. Proteins were detected using the enhanced chemiluminescence detection system (Pierce) as described previously (Wang et al., 2011a).
Immunofluorescence
Bcl-x-KO MEF cells were plated on 18-mm coverslips in a six-well tissue culture plate and grown for 24 h. Cells were then transiently transfected with the pEGFP-C1 plasmids carrying the cDNAs of various Bcl-xL mutants using the jetPRIME transfection reagent (Polyplus-transfection SA, Illkirch, France) following the manufacturer's instructions. Forty-eight hours posttransfection, coverslips were transferred into a 12-well tissue culture plate and washed twice with 1.0 ml of 1× Hanks balanced salt solution (HBSS). After 15 min of fixation with 0.4 ml of 0.4% paraformaldehyde, cells were washed three times with 1.0 ml of 1× HBSS and then permeabilized with 0.4 ml of 0.2% Triton X-100 for 15 min. After three washes with 1.0 ml of incubation solution (0.02% Triton X-100 and 1.5% FBS in 1× HSSB), cells were incubated with primary antibodies in incubation solution for 1 h at room temperature. After three 10-min washes with incubation solution, cells were incubated with secondary antibodies for 1 h at room temperature and again washed three times with incubation solution. The coverslips were then immersed in mounting medium (Dako, Carpinteria, CA), put on slides, and sealed with nail polish. The fluorescence images were visualized and acquired using a PlanApo 60×, 1.40 numerical aperture oil immersion objective on a Nikon Eclipse Ti confocal microscope (Nikon, Melville, NY).
Fluorescent image of live cells
Bcl-x-KO MEF cells were transiently transfected with the pEGFP-C1 plasmids carrying the various Bcl-xL mutants using an Amaxa Nucleofector device (Lonza, Allendale, NJ) following the manufacturer's instructions and seeded into 35-mm glass coverslips. To label endoplasmic reticulum, we cotransfected cells with pDsRed-ER plasmid (Clontech). To visualize mitochondria, we labeled cells with CellLight Mitochondria-RFP (Invitrogen) 16–24 h postnucleofection according to the manufacturer's instructions and imaged them after a further 24 h in culture. Coverslips were placed in a chamber (Warner Instruments, Hamden, CT) and mounted onto an Olympus IX71 inverted microscope (Olympus, Center Valley, PA). Individual cells were visualized using a PlanApo 60×, 1.42 numerical aperture oil immersion objective and confocal images acquired using a VT-Infinity 3 (VisiTech International, Sunderland, United Kingdom).
Cytoplasmic and ER [Ca2+] measurements
For cytoplasmic [Ca2+], cells cultured on glass coverslips were loaded with 2 μM fura-2AM (Invitrogen) by incubation at room temperature for 45 min and mounted in a recording chamber positioned on the stage of an inverted microscope (IX71; Olympus). Fura-2 was alternately excited at 340 and 380 nm, and the emitted fluorescence was filtered at 510 nm and collected and recorded using a charge-coupled device–based imaging system running SimplePCI software (Hamamatsu, Sewickley, PA). The chamber was continuously perfused with HBSS (pH 7.4) at room temperature. A rapid solution changer was used to switch the composition of the solution bathing the cells under study. To measure ER [Ca2+], we loaded cells with mag-fura-2AM (5 μM) for 60 min at room temperature and perfused them with intracellular-like medium (ICM) containing 125 mM KCl, 19 mM NaCl, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, and 1 mM ethylene glycol tetraacetic acid (pH 7.3 with KOH) and permeabilized them by a 2- to 3-min exposure to ICM containing 20 μM digitonin. After 20–30 min of incubation in ICM, stores were Ca2+ loaded by switching to ICM solution with a free [Ca2+] adjusted to 200 nM and 1.5 mM MgATP. To induce Ca2+ release, we applied InsP3 in the same Ca2+-containing ICM solution but without MgATP to prevent reuptake. Data were acquired as described for fura-2, and ratiometric data (R) were normalized to basal store-depleted levels (R0). For experiments using flash photolysis of caged-InsP3, cells were loaded with the membrane-permeable caged InsP3 compound ci-InsP3/PM (d-2,3-O-isopropylidene-6-O-(2-nitro-4,5-dimethoxy)benzyl-myo-inositol 1,4,5-trisphosphate-hexakis(propionoxymethyl) ester; SiChem, Bremen, Germany) by incubation with 1 μM for 60 min at room temperature. InsP3 was photoreleased by brief pulses (250 ms) of UV light (350–400 nm) delivered uniformly throughout the image field. The application of UV pulses precluded the use of fura-2 as a [Ca2+] indicator, and therefore cells were coloaded with the longer-wavelength dye fluo-2AM (5 μM; TEFLabs, Austin, TX) and fluorescence data (F) normalized to basal levels (F0).
Statistical analysis
Statistical analysis was performed using Student's t test and the two-way analysis of variance (ANOVA). p < 0.05 was considered significant.
Supplementary Material
Acknowledgments
We are grateful to John Eaton for advice and Kevin Foskett and Brain Wattenberg for providing antibodies. We also thank the Calcium Imaging Research Support Laboratory, Department of Physiology and Biophysics, Rosalind Franklin University; Sabine Waigel, Yinlu Chen, and Vennila Arumugam at the University of Louisville Microarray facility for microarray experiments; and Christopher Worth at the University of Louisville Brown Cancer Center Flow Cytometry Lab for cell sorting. This work was supported by the Schweppes Foundation and Rosalind Franklin University (C.W.), as well as National Institutes of Health Grants CA106599 and RR018733 and funding from the J. G. Brown Cancer Center (C.L.).
Abbreviations used:
- BH
Bcl-2 homology
- ER
endoplasmic reticulum
- InsP3R
inositol 1,4,5-trisphosphate receptor
- MEF
mouse embryonic fibroblast
- MOM
mitochondrial outer membrane
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-02-0090) on May 9, 2012.
REFERENCES
- Alavian KN, et al. Bcl-x(L) regulates metabolic efficiency of neurons through interaction with the mitochondrial F(1)F(O) ATP synthase. Nat Cell Biol. 2011;13:1224–1233. doi: 10.1038/ncb2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nunez G, Thompson CB. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- Boyce M, Degterev A, Yuan J. Caspases: an ancient cellular sword of Damocles. Cell Death Differ. 2004;11:29–37. doi: 10.1038/sj.cdd.4401339. [DOI] [PubMed] [Google Scholar]
- Chen YB, et al. Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J Cell Biol. 2011;195:263–276. doi: 10.1083/jcb.201108059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization. Trends Cell Biol. 2008;18:157–164. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- Eckenrode EF, Yang J, Velmurugan GV, Foskett JK, White C. Apoptosis protection by Mcl-1 and Bcl-2 modulation of inositol 1,4,5-trisphosphate receptor-dependent Ca2+ signaling. J Biol Chem. 2010;285:13678–13684. doi: 10.1074/jbc.M109.096040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiebig AA, Zhu W, Hollerbach C, Leber B, Andrews DW. Bcl-XL is qualitatively different from and ten times more effective than Bcl-2 when expressed in a breast cancer cell line. BMC Cancer. 2006;6:213. doi: 10.1186/1471-2407-6-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foskett JK, White C, Cheung KH, Mak DO. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev. 2007;87:593–658. doi: 10.1152/physrev.00035.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foyouzi-Youssefi R, Arnaudeau S, Borner C, Kelley WL, Tschopp J, Lew DP, Demaurex N, Krause KH. Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc Natl Acad Sci USA. 2000;97:5723–5728. doi: 10.1073/pnas.97.11.5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain M, Shore GC. Cellular distribution of Bcl-2 family proteins. Sci STKE. 2003;2003:e10. doi: 10.1126/stke.2003.173.pe10. [DOI] [PubMed] [Google Scholar]
- Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- Hardwick JM, Youle RJ. SnapShot: BCL-2 proteins. Cell. 2009;138:404, 404. doi: 10.1016/j.cell.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YT, Wolter KG, Youle RJ. Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc Natl Acad Sci USA. 1997;94:3668–3672. doi: 10.1073/pnas.94.8.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annu Rev Biochem. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- Khan MT, Bhanumathy CD, Schug ZT, Joseph SK. Role of inositol 1,4,5-trisphosphate receptors in apoptosis in DT40 lymphocytes. J Biol Chem. 2007;282:32983–32990. doi: 10.1074/jbc.M705183200. [DOI] [PubMed] [Google Scholar]
- Kuwana T, Newmeyer DD. Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr Opin Cell Biol. 2003;15:691–699. doi: 10.1016/j.ceb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Lam M, Dubyak G, Chen L, Nunez G, Miesfeld RL, Distelhorst CW. Evidence that BCL-2 represses apoptosis by regulating endoplasmic reticulum-associated Ca2+ fluxes. Proc Natl Acad Sci USA. 1994;91:6569–6573. doi: 10.1073/pnas.91.14.6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Wang X, Vais H, Thompson CB, Foskett JK, White C. Apoptosis regulation by Bcl-x(L) modulation of mammalian inositol 1,4,5-trisphosphate receptor channel isoform gating. Proc Natl Acad Sci USA. 2007;104:12565–12570. doi: 10.1073/pnas.0702489104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoyama N, et al. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science. 1995;267:1506–1510. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- Muchmore SW, et al. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature. 1996;381:335–341. doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]
- Newmeyer DD, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 2003;112:481–490. doi: 10.1016/s0092-8674(03)00116-8. [DOI] [PubMed] [Google Scholar]
- Oakes SA, Lin SS, Bassik MC. The control of endoplasmic reticulum-initiated apoptosis by the BCL-2 family of proteins. Curr Mol Med. 2006;6:99–109. doi: 10.2174/156652406775574587. [DOI] [PubMed] [Google Scholar]
- Olberding KE, Wang X, Zhu Y, Pan J, Rai SN, Li C. Actinomycin D synergistically enhances the efficacy of the BH3 mimetic ABT-737 by downregulating Mcl-1 expression. Cancer Biol Ther. 2010;10 doi: 10.4161/cbt.10.9.13274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinton P, Ferrari D, Rapizzi E, Di VF, Pozzan T, Rizzuto R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J. 2001;20:2690–2701. doi: 10.1093/emboj/20.11.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinton P, Rizzuto R. Bcl-2 and Ca2+ homeostasis in the endoplasmic reticulum. Cell Death Differ. 2006;13:1409–1418. doi: 10.1038/sj.cdd.4401960. [DOI] [PubMed] [Google Scholar]
- Reed JC, Jurgensmeier JM, Matsuyama S. Bcl-2 family proteins and mitochondria. Biochim Biophys Acta. 1998;1366:127–137. doi: 10.1016/s0005-2728(98)00108-x. [DOI] [PubMed] [Google Scholar]
- Rong Y, Distelhorst CW. Bcl-2 protein family members: versatile regulators of calcium signaling in cell survival and apoptosis. Annu Rev Physiol. 2008;70:73–91. doi: 10.1146/annurev.physiol.70.021507.105852. [DOI] [PubMed] [Google Scholar]
- Rong YP, et al. Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2's inhibition of apoptotic calcium signals. Mol Cell. 2008;31:255–265. doi: 10.1016/j.molcel.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong YP, Bultynck G, Aromolaran AS, Zhong F, Parys JB, De SH, Mignery GA, Roderick HL, Bootman MD, Distelhorst CW. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc Natl Acad Sci USA. 2009;106:14397–14402. doi: 10.1073/pnas.0907555106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein M, Hollowell CM, Guinan P. Effects of BCL-2 suppression by antisense oligonucleotides on additional regulators of apoptosis compensatory change in non-targeted protein expression. In Vivo. 2011;25:725–732. [PubMed] [Google Scholar]
- Sano R, Annunziata I, Patterson A, Moshiach S, Gomero E, Opferman J, Forte M, d'Azzo A. GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca(2+)-dependent mitochondrial apoptosis. Mol Cell. 2009;36:500–511. doi: 10.1016/j.molcel.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz PS, Hockenbery DM. Bcl-2-related survival proteins. Cell Death Differ. 2006;13:1250–1255. doi: 10.1038/sj.cdd.4401982. [DOI] [PubMed] [Google Scholar]
- Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300:135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- Tagami S, Eguchi Y, Kinoshita M, Takeda M, Tsujimoto Y. A novel protein, RTN-XS, interacts with both Bcl-XL and Bcl-2 on endoplasmic reticulum and reduces their antiapoptotic activity. Oncogene. 2000;19:5736–5746. doi: 10.1038/sj.onc.1203948. [DOI] [PubMed] [Google Scholar]
- Takada M, Noguchi A, Sayama Y, Kurohane KY, Ishikawa T. Inositol 1,4,5-trisphosphate receptor-mediated initial Ca(2+) mobilization constitutes a triggering signal for hydrogen peroxide-induced apoptosis in INS-1 beta-cells. Biol Pharm Bull. 2011;34:954–958. doi: 10.1248/bpb.34.954. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Li XX, Gottleib E, Hill RB, Thompson CB, Colombini M. Bcl-xL promotes the open configuration of the voltage-dependent anion channel and metabolite passage through the outer mitochondrial membrane. J Biol Chem. 2001;276:19414–19419. doi: 10.1074/jbc.M101590200. [DOI] [PubMed] [Google Scholar]
- Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- Wang X, Eno CO, Altman BJ, Zhu Y, Zhao G, Olberding KE, Rathmell JC, Li C. ER stress modulates cellular metabolism. Biochem J. 2011a;435:285–296. doi: 10.1042/BJ20101864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Olberding KE, White C, Li C. Bcl-2 proteins regulate ER membrane permeability to luminal proteins during ER stress-induced apoptosis. Cell Death Differ. 2011b;18:38–47. doi: 10.1038/cdd.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, Foskett JK. The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat Cell Biol. 2005;7:1021–1028. doi: 10.1038/ncb1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Shen X. H2O2 directly activates inositol 1,4,5-trisphosphate receptors in endothelial cells. Redox Rep. 2005;10:29–36. doi: 10.1179/135100005X21660. [DOI] [PubMed] [Google Scholar]
- Zhu W, Cowie A, Wasfy GW, Penn LZ, Leber B, Andrews DW. Bcl-2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types. EMBO J. 1996;15:4130–4141. [PMC free article] [PubMed] [Google Scholar]
- Zong WX, Li C, Hatzivassiliou G, Lindsten T, Yu QC, Yuan J, Thompson CB. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J Cell Biol. 2003;162:59–69. doi: 10.1083/jcb.200302084. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.