

Abstract
A unique heterocyclic carbamate prodrug of seco-CBI-indole2 that releases no residual byproduct is reported as a new member of a class of hydrolyzable prodrugs of the duocarmycin and CC-1065 family of natural products. The prodrug was designed to be activated by hydrolysis of a carbamate releasing the free drug without the cleavage release of a traceable extraneous group. Unlike prior carbamate prodrugs examined that are rapidly cleaved in vivo, the cyclic carbamate was found to be exceptionally stable to hydrolysis under both chemical and biological conditions providing a slow, sustained release of the exceptionally potent free drug. An in vivo evaluation of the prodrug found that its efficacy exceeded that of the parent drug, that its therapeutic window of efficacy versus toxicity is much larger than the parent drug, and that its slow free drug release permitted the safe and efficacious use of doses 150-fold higher than the parent compound.
Introduction
Duocarmycin SA (1)1 and CC-1065 (2)2 are two parent members of a class of highly potent naturally occurring antitumor agents that also include duocarmycin A3 and yatakemycin4 (Figure 1). This unique class of natural products derives its antitumor properties from their ability to alkylate DNA in a sequence selective manner.5,6 Comprehensive studies of the natural products, their synthetic unnatural enantiomers,7 and key analogues have defined many of the fundamental features that control the DNA alkylation selectivity, efficiency, and catalysis, resulting in a detailed understanding of the relationships between structure, reactivity, and biological activity.6,7,8
Figure 1.

Natural Products.
CBI (1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one) is one of the most extensively studied synthetic analogues of the family since we first introduced it in 1989.9 The CBI alkylation subunit is not only more synthetically accessible and participates in the now characteristic DNA alkylation reaction effectively,10 but it has also been found to be four times more stable and four times more potent than the naturally occurring alkylation subunit of 2, approaching the stability and potency of the duocarmycin SA (1) alkylation subunit. Since analogues incorporating the CBI alkylation subunit have also been established to exhibit efficacious in vivo antitumor activity in animal models, it is an excellent synthetic replacement on which to examine the structure–function features of the natural products, including new prodrug design and evaluation.11
During the course of the total syntheses of the natural products and related analogues including CBI-indole2 (5),11c it was established that the synthetic phenol precursors such as 4, which have yet to undergo the Winstein Ar-3' spirocyclization, are equipotent to and indistinguishable from their cyclized cyclopropane containing counterparts within in vitro cytotoxic assays, DNA alkylation studies, and in vivo antitumor models. Due to this indistinguishable behavior both in vitro and in vivo and because their extraordinary potency creates special precautions for their handling, protection of the phenol precursors not only permits safe handling during their preparation, but it also provides an effective site on which to create prodrugs that can be designed for controlled release in vivo.12 Such prodrugs incorporating phenol acylation have been developed to simultaneously improve solubility, pharmacokinetics, storage life, handling safety, and efficacy in vivo.12,13,14 Two such carbamate-based drugs, a derivative of duocarmycin A12c–d (t1/2 = 20 h, calf serum) and carzelesin (U-80,244, t1/2 < 1 h, human plasma),12a–b which are rapidly cleaved in vivo (1–20 h), entered clinical trials but have ultimately not progressed. In related studies, we disclosed the carbamate prodrugs 3a–f of (+)-CBI-indole2 as shown in Figure 2, many of which were found to be essentially equipotent to (+)-CBI-indole2 (5) in vitro.12e This work established that the free drug is rapidly released in a cellular assay and is able to spirocyclize, alkylate DNA, and express its biological activity efficiently in a manner essentially indistinguishable from the free drug itself. Herein, we describe a novel intramolecular heterocyclic carbamate (+)-CBI-indole2 prodrug (6, Figure 3) that is subject to an analogous hydrolysis mechanism of activation,15 but that is both substantially more stable and upon activation does not release any extraneous or traceable functionality into the surrounding cellular environment. Significantly, the resulting drug is accordingly less potent both in vitro and in vivo, but substantially safer to handle and more efficacious in vivo, effectively taming the extraordinary potency of this class of antitumor drugs.
Figure 2.

Carbamate prodrug design.
Figure 3.

Cyclic carbamate prodrug 6.
Chemistry
Synthesis
Prodrug (+)-6 was synthesized16 in 11 steps from known intermediate 717 as shown in Scheme 1. The phenol of 7 was protected as its benzyl ether and 8 was hydrolyzed using LiOH to provide the carboxylic acid 9 in good overall yield. Carboxylic acid 9 was subjected to a Curtius rearrangement using diphenylphosphoryl azide (DPPA) and Et3N in freshly distilled t-BuOH providing the Boc protected aniline 10 in 79% yield. The use of non-distilled t-BuOH resulted in low yields due to competing hydrolytic release of the free aniline. Regioselective C1 iodination of 10 and subsequent N-alkylation of 11 with 1,3-dichloropropene proceeded effectively, providing the cyclization precursor 12. Finally, a selective 5-exo-trig free radical cyclization18 of 12 using sub stoichiometric quantities of Bu3SnH (0.9 equiv) provided 13 in 83% yield with only trace amounts of further reduced (debrominated) material observed.
Scheme 1.

Compound 13, which has served as a key precursor in the divergent synthesis19 of a series of compounds,20 was further elaborated to aniline 15 using triphenylsilylamine21 as an ammonia surrogate for a Pd(0) catalyzed aryl amination22 with LiHDMS in THF and ligand 14 (Scheme 2). Fortunately, a solution of LiHMDS could be used in place of solid LiHMDS, which alleviated the need for use of a glove box as reported.22 Other amination reactions, including the use of benzophenone imine and copper-promoted couplings with acetamidine, yielded only trace amounts of the desired amination product. Bu4NF deprotection of the resulting amine and debenzylation of the phenol under hydrogenation conditions produced aniline 15. Aniline 15 was converted to the cyclic carbamate 16 by a double acylation with triphosgene, which proceeded cleanly and in quantitative yield. At this point, compound 16 was resolved into its two enantiomers using chiral phase HPLC with 20% i-PrOH/hexanes as the eluent. We chose to resolve 16 instead of 6 itself in order to permit access to additional resolved analogues and to avoid the lower solubility of the full prodrug 6 in the chromatography solvents. Each enantiomer of 16 was subjected to Boc deprotection with 4 N HCl in EtOAc and immediate N-acylation with 17, providing (+)-and ent-(−)-6 in 52% yield.
Scheme 2.

The parent compound of 6 was prepared as shown in Scheme 3 through a four step sequence. The aniline of intermediate 15 was differentially protected as a Fmoc carbamate. Subsequent Boc deprotection and coupling with carboxylic acid 17 gave 19, which was Fmoc deprotected and cyclized upon treatment with piperidine to provide the parent compound 20 as a racemic mixture.
Scheme 3.

Stability of the Cyclic Carbamate Prodrug
In order to determine the ability of the free drug to be released under physiological conditions, the chemical reactivity of N-Boc-prodrug 16 was assessed under a variety of acidic, basic, and nucleophilic conditions. The cyclic carbamate of 16 proved robust to hydrolysis under acidic conditions (1:1 TFA:CH2Cl2, 4 N HCl in EtOAc) and was stable over a period of 48 h at 23 °C, although the Boc protecting group was readily cleaved under such conditions. Similarly, compound 6 was also found to be stable to the above acidic conditions for up to 72 h at 23 °C. As shown in Figure 4, 16 was also stable to organic bases in aprotic solvents (entries 1–3), but the cyclic carbamate was slowly hydrolyzed in the presence of NaHCO3 in protic solvents in a reaction that proceeded at a greater rate as the polarity of the solution increased (entries 3–6). Compound 16 was found to be completely stable in the presence of the nucleophiles BnSH and BnOH (100 equiv) in MeOH and THF at 23 °C for 48 h, and was stable to BnNH2 in THF, but was rapidly cleaved with BnNH2 (100 equiv) in MeOH in 24 h.
Figure 4.

N-Boc prodrug 16 stability under basic conditions.
Finally, the stability of the full prodrug 6 was examined in pH 7.0 phosphate buffer (t1/2 > 4 weeks, no cleavage observed) and in human plasma (t1/2 > 48 h, 5% free drug release) indicating that the cyclic carbamate is remarkably stable under both conditions, but subject to slow release in human plasma. By contrast, the open chain carbamates explored in earlier studies, including those leading to carzelesin, were designed for much more rapid release (1–20 h). We also found that 6 is incapable of alkylating DNA in cell-free systems23 (see Supporting Information), indicating that any in vitro cytotoxic activity or in vivo antitumor activity of 16 or 6 is due to release of the free drug and not the prodrug itself.
Biological Properties
In Vitro Cytotoxic Activity
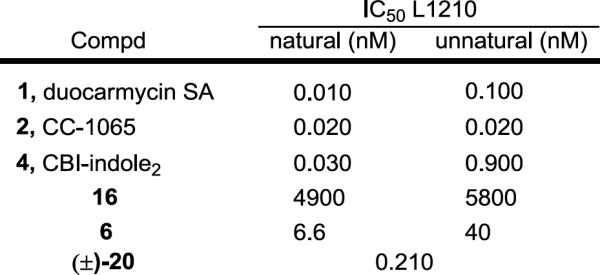
Both (+)- and ent-(−)-6 and their N-Boc precursors 16 were tested for cell growth inhibition in a cytotoxic assay with the L1210 murine leukemia cell line (Figure 5). The natural enantiomer of the prodrug (+)-6 was found to be approximately 200-fold less potent (IC50 of 6.6 nM) than the free drug seco-CBI-indole2 4 (IC50 of 30 pM) and 6-fold more potent that its unnatural enantiomer. The racemic parent drug (±)-20 was found to have an IC50 of 210 pM, suggesting that the active enantiomer is approximately 3–4 fold less active than 4, and indicating that the prodrug (+)-6 is 30–70 fold less potent than the parent drug 20. Consistent with expectations, the full prodrug 6 proved to be 100 to 1000 times more potent than its N-Boc precursor 16, which in turn is 50–100 fold less active than N-Boc-CBI (natural enantiomer IC50 = 80 nM).9 This data is consistent with the remarkable stability of the prodrug to chemical hydrolysis conditions, pH 7 phosphate buffer, and in human plasma, and its ineffective in vitro DNA alkylation reaction21 (not shown), indicating that the release of free drug is similarly slow under the conditions of an in vitro cellular assay as well. Despite the lower potency relative to the free drug 4 and the racemic parent compound 20, it is notable that the cyclic carbamate prodrug (+)-6 now displays an in vitro cellular potency (IC50 = 1–10 nM) on par with most clinically used antitumor drugs.
Figure 5.
In vitro cytotoxic activity.
In Vivo Antitumor Activity
Even though results of the in vitro cellular assay showed that (+)-6 is substantially less potent than its parent drug, the slow release of the compound could prove to be advantageous in vivo due to the inherent potency and toxicity of the parent compound. Therefore, the in vivo antitumor activity of (+)-6 was assessed alongside seco-CBI-indole2 (4) in an antitumor model consisting of L1210 murine leukemia cells implanted ip into DBA/2J mice (Figure 6), which has been used traditionally as an initial antitumor model for comparisons in this class.11,12,14,15 A dose range of 300 to 9000 μg/kg for prodrug (+)-6 (scaled to its in vitro cytotoxic activity IC50) and 60 to 500 μg/kg for seco-CBI-indole2 (4) and a dosing schedule (administered three times ip on days 1, 5, and 9) for both compounds was employed. A subtle, but additional important empirical observation made in the studies is that the prodrug administration is tolerated at the injection sites of the animals much better than the free drug.
Figure 6.

In vivo antitumor activity (L1210, ip).
The optimal does range for 4 was previously established (60–100 μg/kg) and was extended for the study herein to highlight its narrow therapeutic window versus the potential behavior of prodrug (+)-6. As anticipated, (+)-CBI-indole2 (4) proved toxic at doses of 100–500 μg/kg leading to premature death of the animals and productive antitumor activity was observed only at the dose of 60 μg/kg (T/C = 197), albeit producing only 1/10 long term (250 days) survivors in this extended study (Figure 6). By contrast, the prodrug (+)-6 exhibited productive antitumor activity over the entire and much larger dose range examined (30-fold range). The most efficacious activity was observed at the highest dose of 9000 μg/kg, producing 5/10 long term cures (>250 days, T/C > 980) and indicating that even higher doses may be not only tolerable, but potentially even more efficacious. This highest dose represents one that is 150 times greater than the optimal dose observed with (+)-4, in line with the 100–200 fold differences in their cytotoxic potencies. In addition, the dose range of over which (+)-6 exhibited productive activity was much larger, the in vivo antitumor activity was more efficacious (T/C > 980), and long term cures (5/10 > 250 day survivors) were observed even without an effort at dosing optimization.
Conclusions
A novel heterocyclic carbamate prodrug 6 of (+)-CBI-indole2, which can be released via hydrolysis, was synthesized and evaluated for its in vitro cytotoxic activity and in vivo antitumor activity. Compared to its open chain counterparts explored in earlier studies, the cyclic carbamate prodrug was found to be remarkably stable to chemical hydrolysis conditions as well as in pH 7.0 phosphate buffer and human plasma. Accordingly, 6 was less potent in vitro and in vivo compared to the parent drug 4, but was found to be substantially safer and more efficacious in vivo, being superior in extending life expectancy of tumor-bearing animals even at 150-fold higher doses. Notable elements of the cyclic carbamate prodrug behavior include not only its hydrolysis liberation of the free drug that releases no residual byproduct (CO2), but also its remarkable stability relative to its acyclic counterparts explored in early studies. This results in an apparent slow, sustained release of free drug that permits the safer and more efficacious use of larger doses of drug (as much as 150-fold), effectively taming the extraordinary potency of this class of antitumor drugs
Experimental Section
General
Reagents and solvents were purchased reagent-grade and used without further purification. Pooled human plasma, with sodium citrate as an anticoagulant, was purchased from Innovative Research and stored at −20 °C. THF was freshly distilled from sodium benzophenone ketyl. t-BuOH was freshly distilled from calcium hydride. All reactions were performed in oven-dried glassware under an Ar atmosphere. Evaporation and concentration in vacuo was performed at 20 °C. TLC was conducted using precoated SiO2 60 F254 glass plates from EMD with visualization by UV light (254 or 366 nm). Chiral phase HPLC was performed using a Shimadzu HPLC on a semi-preparative Diacel ChiralCel OD column (0.46 cm × 25 cm) with a flow rate of 7 mL/min and with UV detection at λ = 254 nm. Optical rotations were determined on a Rudolf Research Analytical Autopol III Automatic Polarimeter (λ = 589 nm, 25 °C). NMR (1H or 13C) were recorded on Bruker DRX-500 and DRX-600 NMP spectrophotometers at 298K. Residual solvent peaks were used as an internal reference. Coupling constants (J) (H,H) are given in Hz. Coupling patterns are designated as singlet (s), doublet (d), triplet (t), quadruplet (q), multiplet (m), or broad singlet (br). IR spectra were recorded on a Thermo Scientific Nicolet 380 FT-IR spectrophotometer and measured neat. High resolution mass spectral data were acquired on an Agilent Technologies high resolution LC/MSD-TOF, and the detected masses are given as m/z with m representing the molecular ion. The purity of each tested compound (>95%) was determined on an Agilent 1100 LC/MS instrument using a ZORBAX SB-C18 column (3.5 mm, 4.6 mm × 50 mm, with a flow rate of 0.75 mL/min and detection at 220 and 254 nm) with a 10–98% acetonitrile/water/0.1% formic acid gradient.
Ethyl 5-Bromo-4-hydroxy-2-naphthoate (7)
A solution of potassium tert-butoxide (20.0 g, 0.78 mol) at 55 °C in t-BuOH (249 mL) was treated with a premixed solution of diethyl succinate (40.4 mL, 0.243 mol) and 3-bromobenzaldehyde (18.9 mL, 0.162 mol) dropwise. Upon completion of the addition, the reaction mixture was warmed to 85 °C and stirred for 2 h. After 2 h, the reaction mixutre was cooled to 25 °C. The reaction mixture was acidified to pH < 4 with 2 N aqueous HCl and concentrated. The aqueous suspension was then extracted with ethyl acetate (3×). The organic layers were combined and washed with saturated aqueous NaHCO3 (5×). The basic aqueous washes were combined and reacidified with 2 N aqueous HCl to pH 1. Finally, the aqueous phase was extracted with ethyl acetate (3×). The organic layers were combined, dried over Na2SO4, and concentrated under reduced pressure, which afforded the desired half ester (39.1 g, 77%) as an orange oil. The half ester (39.1 g, 0.124 mol) was dissolved in acetic anhydride (178 mL) and NaOAc (18.7 g, 0.137 mol) was added. The reaction mixture was warmed to 140 °C and stirred for 6 h. Upon completion, the reaction mixture was cooled to 25 °C and poured into H2O. The aqueous layer was extracted with ethyl acetate (3×). The organic layers were combined, dried over Na2SO4, and concentrated under reduced pressure. The residue was dissolved in anhydrous ethanol (620 mL). K2CO3 (104 g, 0.624 mol) was added, and the reaction mixture was warmed at 80 °C for 1 h. The reaction mixture was cooled and acidified to pH 1 with 2 N aqueous HCl. The ethanol was removed under reduced pressure and the aqueous suspension was extracted with ethyl acetate (3×). The organic extracts were combined, dried over Na2SO4, and concentrated under reduced pressure. Flash chromatography (SiO2, 16 × 30 cm, 0–15% EtOAc/hexanes gradient elution) provided 7 (5.4 g, 15% over 3 steps) as a yellow solid and its 7-bromo isomer (12.4 g, 34% over 3 steps). 1H NMR (CDCl3, 500 MHz) δ 8.16 (s, 1H), 8.07 (s, 1H), 7.89 (d, J = 6.5 Hz, 1H), 7.73 (d, J = 6.5 Hz, 1H), 7.63 (s, 1H), 7.29 (t, J = 10 Hz, 1H), 4.43 (q, J = 6.0 Hz, 2H) 1.44 (t, J = 6.0 Hz, 3H). 13C NMR (CDCl3, 125 MHz) δ 165.9, 152.7, 136.3, 133.6, 130.6, 129.2, 126.7, 123.6, 122.7, 115.2, 112.3, 61.3, 14.3. IR (film) νmax 3367, 2979, 1690, 1227 cm−1. ESI-TOF HRMS m/z 294.9959 (M+H+, C13H11BrO3 requires 294.9964).
Ethyl 4-(Benzyloxy)-5-bromo-2-naphthoate (8)
Naphthol 7 (3.20 g, 11.0 mmol) was dissolved in anhydrous DMF (78 mL). K2CO3 (3.05 g, 22.0 mmol), benzyl bromide (1.59 mL, 13.2 mmol), and Bu4NI (163 mg, 0.440 mmol) were added. The solution was stirred at 25 °C for 16 h. The reaction mixture was poured into H2O and extracted with ethyl acetate (3×). The organic extracts were combined, dried over Na2SO4, and concentrated under reduced pressure. The solid was recrystallized with 5% EtOAc/hexanes and the mother liquor was further purified by flash chromatography (SiO2, 6 × 15 cm, 10–20% EtOAc/hexanes gradient elution) affording additional 8 (3.30 g combined, 77%) as a brown crystalline solid. 1H NMR (CDCl3 500 MHz) δ 8.17 (s, 1H), 7.87 (d, J = 7.5 Hz, 1H), 7.85 (d, J = 8.0 Hz, 1H), 7.61 (d, J = 7.5 Hz, 2H), 7.57 (s, 1H), 7.40 (t, J = 7.5 Hz, 2H), 7.35–7.32 (m, 1H), 7.29 (t, J = 7.5 Hz, 1H), 5.30 (s, 2H), 4.43 (q, J = 7.0 Hz, 2H), 1.44 (t, J = 7.0 Hz, 3H). 13C NMR (CDCl3, 125 MHz) δ 166.0, 154.5, 136.1, 136.0, 135.0, 129.3, 128.3, 128.0 (2C), 127.8, 126.8, 125.8, 124.1, 116.7, 106.9, 71.2, 61,1. IR (film) νmax 2980, 1712, 1413, 1236 cm−1. ESI-TOF HRMS m/z 385.0433 (M+H+, C20H17BrO3 requires 385.0434).
4-(Benzyloxy)-5-bromo-2-naphthoic Acid (9)
Ester 8 (2.29 g, 5.94 mmol) was dissolved in a 3:1:1 mixture of THF:CH3OH:H2O (0.1 M). LiOH•H2O was added and the reaction mixture was stirred at 25 °C for 24 h. Upon completion, the reaction mixture was acidified to pH 1 with the addition of 10% aqueous HCl. A precipitate formed during the acidification and it was collected by vacuum filtration. The remaining aqueous layer was then extracted with ethyl acetate (3×). The organic extracts were combined, dried over Na2SO4, and concentrated under reduced pressure. The filtered and extracted products were combined to give 9 (2.09 g, 100%) as a pale yellow solid. 1H NMR (DMSO-d6, 500 MHz) δ 8.23 (s, 1H), 8.10 (d, J = 6.0 Hz, 1H), 7.91 (d, J = 6.5 Hz, 1H), 7.61 (d, J = 7.0 Hz, 2H), 7.55 (s, 1H), 7.43–7.39 (m, 3H), 7.33 (t, J = 7.0 Hz, 1H), 5.35 (s, 2H). 13C NMR (DMSO-d6, 125 MHz) δ 166.7, 153.8, 136.2, 135.9, 135.0, 129.8, 128.8, 128.2, 127.8, 127.7, 127.5, 124.7, 123.8, 115.5, 107.0, 70.4. IR (film) νmax 3368, 2969, 1680 cm−1. ESI-TOF HRMS m/z 357.0125 (M+H+, C18H13BrO3 requires 357.0121).
tert-Butyl-(4-(benzyloxy)-5-bromonaphthalen-2-yl)carbamate (10)
Carboxylic acid 9 (950 mg, 2.66 mmol) was dissolved in freshly distilled t-BuOH (0.01 M) over 4Å molecular sieves. Et3N (467 μL, 3.35 mmol) and diphenylphosphoryl azide (602 μL, 2.79 mmol) were added. The reaction mixture was warmed to 85 °C under Ar and stirred for 14 h. Upon completion, the mixture was filtered through cotton to remove the molecular sieves and concentrated under reduced pressure. The residue was diluted with 10% aqueous HCl and extracted with EtOAc (3×). The organic extracts were combined and washed with H2O (2×) and saturated aqueous NaCl. The organic phase was dried over Na2SO4, and concentrated under reduced pressure. Flash chromatography (SiO2, 5 × 12 cm, 5% EtOAc/hexanes elution) provided 10 (1.02 g, 89%) as a tan solid. 1H NMR (CDCl3, 600 MHz) δ 7.62 (m, 2H), 7.58 (d, J = 7.8 Hz, 2H), 7.49 (s, 1H), 7.39 (t, J = 7.2 Hz, 2H), 7.33 (t, J = 6.0 Hz, 1H), 7.16 (t, J = 7.8 Hz, 1H), 7.03 (s, 1H), 6.58 (s, 1H), 5.22 (s, 2H), 1.54 (s, 9H). 13C NMR (CDCl3, 150 MHz) δ 155.2, 152.5, 137.6, 136.4, 136.3, 131.3, 130.0, 128.4, 127.9, 127.3, 126.9, 120.4, 120.2, 120.1, 116.6, 107.8, 101.8, 71.4, 28.3. IR (film) νmax 3325, 2977, 1702, 1156 cm−1. ESI-TOF HRMS m/z 428.0856 (M+H+, C22H22BrNO3 requires 428.0856).
tert-Butyl-(4-(benzyloxy)-5-bromo-1-iodonaphthalen-2-yl)carbamate (11)
Carbamate 10 (1.20 g, 2.80 mmol) was dissolved in freshly distilled THF (0.17 M) under Ar and in the absence of light, and TsOH•H2O (53 mg, 0.28 mmol) and N-iodosuccinamide (753 mg, 3.30 mmol) were added. The reaction mixture was allowed to stir at 25 °C for 2 h. After 2 h, the reaction was quenched with the addition saturated aqueous NaHCO3 and diluted with ethyl acetate. The organic layer was washed with saturated aqueous NaCl, dried over Na2SO4, and concentrated under reduced pressure. Flash chromatography (SiO2, 5 × 16 cm, 5% EtOAc/hexanes elution) provided 11 (1.47 g, 94%) as an orange solid. 1H NMR (CDCl3, 500 MHz) δ 8.16 (s, 1H), 8.09 (d, J = 8.0 Hz, 1H), 7.70 (d, J = 7.5 Hz, 1H), 7.62 (d, J = 7.0 Hz, 2H), 7.39 (t, J = 7.2 Hz, 2H), 7.34 (d, J = 7.0 Hz, 1H), 7.32 (s, 1H), 7.25–7.22 (m, 2H), 5.28 (s, 2H), 1.58 (s, 9H). 13C NMR (CDCl3, 125 MHz) δ 155.8, 152.4, 139.0, 136.7, 135.9, 132.2, 131.9, 130.0, 128.5, 128.3, 128.0, 127.9, 121.4, 120.2, 120.1, 117.0, 101.9, 81.3, 71.3, 28.3. IR (film) νmax 3378, 2978, 1730, 1225, 1145 cm−1. ESI-TOF HRMS m/z 553.9820 (M+H+, C22H21BrINO3 requires 553.9822).
tert-Butyl-(4-(benzyloxy)-5-bromo-1-iodonaphthalen-2-yl)-(3-chloroallyl)carbamate (12)
Compound 11 (1.65 g, 2.99 mmol) and Bu4NI (55 mg, 0.15 mmol) were dissolved in anhydrous DMF (0.16 M) and the solution was cooled to 0 °C. Once cooled, 60% NaH in mineral oil (239 mg, 5.98 mmol) was added and the reaction mixture was allowed to stir at 0 °C for 30 min. 1,3-Dichloropropene (0.84 mL, 8.97 mmol) was added dropwise and the solution was warmed to room temperature. After 1 h, the reaction mixture was quenched with the addition of saturated aqueous NH4Cl and diluted with ethyl acetate. The organic layer was washed with H2O, saturated aqueous NaCl, dried over Na2SO4, and concentrated under reduced pressure. Flash chromatography (SiO2, 5 × 8 cm, 10% EtOAc/hexanes elution) provided an E/Z mixture of alkene 12 (1.818 g, 96%) as a yellow foam. 1H NMR (acetone-d6, 600 MHz) δ 8.35 (m, 2H), 7.92 (d, J = 7.2 Hz, 2H), 7.61 (br, 4H) 7.45 (t, J = 8.4 Hz, 2H), 7.42–7.40 (m, 4H), 7.35–7.33 (m, 2H), 7.18 (d, J = 18.0 Hz, 2H), 6.21–6.08 (m, 3H), 5.39 (s, 4H), 4.60 (dd, J = 18.9, 5.4 Hz, 1H), 4.42 (dd, J = 15.0, 7.2 Hz, 1H), 4.27 (dd, J = 15.6, 6.6 Hz, 1H), 3.99 (dd, J = 14.1, 6.6 Hz, 1H), 1.55 (br, 4H), 1.28 (br, 14H). 13C NMR (acetone-d6, 150 MHz) δ 157.28, 157.27, 154.8, 154.6, 145.9, 145.8, 139.2, 139.1, 138.2, 138.1, 136.13, 136.12, 135.4 (2C), 130.9, 130.2, 130.1, 129.9, 129.86, 129.81, 129.7, 129.4, 126.2, 125.4, 123.2, 122.2, 118.2, 112.4, 112.3, 98.0, 97.2, 82.2, 81.8, 72.8, 72.6, 50.6, 47.3, 29.3. IR (film) νmax 2974, 2928, 1697, 1156, 749 cm−1. ESI-TOF HRMS m/z 627.9750 (M+H+, C25H24BrClINO3 requires 627.9746).
tert-Butyl 1,2-Dihydro-5-(benzyloxy)-6-bromo-1-(chloromethyl)-1H-benzo[e]indole-3(2H)-carboxylate (13)
Alkene 12 (1.81 g, 2.89 mmol) and AIBN (140 mg, 0.86 mmol) were dissolved in benzene (0.05 M). Freshly prepared Bu3SnH (701 μL, 2.60 mmol) was added and the system was purged of oxygen using Ar and the freeze/pump/thaw method. The reaction mixture was warmed to 80 °C for 12 h. Upon completion, the reaction mixture was concentrated under reduced pressure and purified by flash chromatography (10% w/w KF fused SiO2, 5 × 16 cm, 0–10% EtOAc/hexanes gradient elution) to provide 13 (1.32 g, 90%) as a white solid. 1H NMR (acetone-d6, 600 MHz) δ 7.98 (br, 1H), 7.81 (d, J = 8.4 Hz, 1H), 7.65–7.63 (m, 3H), 7.41 (t, J = 7.2 Hz, 2H), 7.35–7.30 (m, 2H), 5.31 (s, 2H), 4.21–4.16 (m, 2H), 4.12–4.09 (m, 1H), 3.96 (dd, J = 11.1, 3.0 Hz, 1H), 3.71 (dd, J = 8.4, 11.4 Hz, 1H), 1.58 (s, 9H). 13C NMR (acetone-d6, 150 MHz) δ 157.8, 153.8, 144.3, 138.4, 135.0, 132.6, 130.1 129.7, 129.4, 124.9, 124.3, 121.6, 119.4, 117.2, 100.7, 82.4, 72.8, 54.4, 48.6, 43.1, 29.5. IR (film) νmax 2926, 1692, 1330, 1135, 752 cm−1. ESI-TOF HRMS m/z 502.0772 (M+H+, C25H25BrClNO3 requires 502.0779).
tert-Butyl 1,2-Dihydro-6-amino-1-(chloromethyl)-5-hydroxy-1H-benzo[e]indole-3(2H)-carboxylate (15)
An oven-dried microwave vial was charged with Pd2(dba)3 (10.9 mg, 11 μmol), 2-dicyclohexylphosphinobiphenyl (14, 8.3 mg, 0.023 mmol), and (C6H5)3SiNH2 (72.1 mg, 0.261 mmol). The vial was evacuated and filled with Ar. Compound 13 (120 mg, 0.238 mmol) was added and the vial was evacuated again. Toluene (2.3 mL) was added and the vessel was purged with Ar. Finally, LiHMDS (0.29 mL, 1 M in THF) was added and the vessel was sealed. The reaction was submerged in a 100 °C oil bath for 24 h. After 24 h, the reaction mixture was cooled to room temperature, diluted with diethyl ether, filtered through a plug of Celite, and concentrated. The residue was dissolved in THF (15 mL) and cooled to 0 °C. Bu4NF (0.36 mL, 1 M in THF) was added dropwise. The reaction mixture was allowed to stir for 30 min before being quenched with the addition of saturated aqueous NH4Cl and diluted with ethyl acetate. The organic layer was washed with saturated aqueous NaCl, dried over NasSO4, and concentrated. The residue was purified by flash chromatography (SiO2, 4 × 8 cm, 5–10% EtOAc/hexanes gradient elution). The product was carried on to the next reaction mixture without characterization due to co-elution of triphenyl byproduct. The amine (104 mg theoretical, 0.238 mmol) was dissolved in anhydrous CH3OH (6 mL) under Ar. 10% Pd/C (29 mg, 0.024 mmol) was added and the atmosphere was exchanged with H2. The reaction mixture was allowed to stir at 25 °C for 5 h. The reaction mixture was diluted with diethyl ether, filtered through Celite, and concentrated under reduced pressure. Flash chromatography (SiO2, 3 × 8 cm, 50–70% Et2O/hexanes gradient elution) provided 15 (56 mg, 67% over 3 steps) as a tan solid. 1H NMR (acetone-d6, 600 MHz) δ 7.48 (br, 1H), 7.12 (t, J = 7.8 Hz, 1H), 6.84 (d, J = 7.8 Hz, 1H), 6.44 (d, J = 6.6 Hz, 1H), 4.13–4.05 (m, 2H), 3.92–3.87 (m, 2H), 3.55 (t, J = 10.8 Hz, 1H), 1.54 (s, 9H). 13C NMR (acetone-d6, 150 MHz) δ 158.6, 153.7, 148.8, 134.8, 130.0, 126.8, 115.3, 112.5, 111.4, 108.7, 99.6, 81.7, 54.1, 48.3, 43.4, 29.3. IR (film) νmax 3391, 2974, 1706, 1583, 1406, 1142 cm−1. ESI-TOF HRMS m/z 349.1323 (M+H+, C18H21ClN2O3 requires 349.1313).
tert-Butyl 10-(Chloromethyl)-5-oxo-9,10-dihydro-4H-pyrrolo[3',2':5,6]naphtho[1,8-de][1,3]oxazine-8(5H)-carboxylate (16)
Naphthol 15 (56 mg, 0.160 mmol) and triphosgene (47 mg, 0.160 mmol) were dissolved in toluene (3.2 mL) at 25 °C. The reaction mixture was stirred for 1 h before being diluted with H2O and ethyl acetate. The organic layer was washed with saturated aqueous NaCl, dried over Na2SO4, and concentrated under reduced pressure. Flash chromatography (SiO2, 2 × 6 cm, 20–50% EtOAc/hexanes gradient elution) provided 16 (60 mg, 100%) as a yellow solid. 1H NMR (acetone-d6, 600 MHz) δ 9.86 (s, 1H), 7.66 (br, 1H), 7.37 (t, J = 8.4 Hz, 1H), 7.32 (d, J = 8.4 Hz, 1H), 6.66 (d, J = 7.8 Hz, 1H), 4.19–4.18 (m, 2H), 4.07–4.05 (m, 1H), 3.98 (dd, J = 11.1, 3.6 Hz, 1H), 3.77 (dd, J = 8.2, 11.4 Hz, 1H), 1.58 (s, 9H). 13C NMR (acetone-d6, 150 MHz) δ 178.5, 153.7, 152.8, 148.37, 148.31, 146.1, 137.0, 136.9, 131.8, 131.2, 118.8, 116.8, 110.0, 105.5, 100.3, 82.7, 54.5, 48.5, 42.5, 29.4. IR (film) νmax 2924, 1701, 1606, 1405, 1332, 1140 cm–1. ESI-TOF HRMS m/z 375.1105 (M+H+, C19H19ClN2O4 requires 375.1106).
The enantiomers were resolved on a semi-preparative Diacel chiralcel OD column (0.46 cm × 25 cm) with 20% i-PrOH/hexanes elution; α = 1.38.
(1S)-16: [α]23D −31 (c 0.75, THF), natural enantiomer. (1R)-16: [α]23D +32 (c 0.80, THF), unnatural enantiomer.
N-(2-(10-(Chloromethyl)-5-oxo-5,8,9,10-tetrahydro-4H-pyrrolo[3',2':5,6]naphtho[1,8-de][1,3]oxazine-8-carbonyl)-1H-indol-5-yl)-1H-indole-2-carboxamide (6)
Compound 16 (7.5 mg, 0.020 mmol) was dissolved in 4 N HCl in EtOAc (0.5 mL) and the mixture was allowed to stir at room temperature for 25 min. The solvent was removed under a stream of nitrogen and the residue was redissolved in anhydrous DMF (0.4 mL). EDCI (11.4 mg, 0.06 mmol) and 17 (7.0 mg, 0.022 mmol) were added and the reaction mixture was allowed to stir at 25 °C for 24 h. The reaction mixture was quenched with the addition of H2O and diluted with ethyl acetate. The organic phase was washed with 2 N aqueous HCl (3×), saturated aqueous NaHCO3 (5×), and saturated aqueous NaCl. The organic extract was dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by PTLC (SiO2, 40% THF/toluene) to provide 6 (6.08 mg, 52%, typically 52–60%) as a tan solid. 1H NMR (DMSO-d6, 600 MHz) δ 11.85 (s, 1H), 11.75 (s, 1H), 11.14 (br, 1H), 10.20 (s, 1H), 8.25 (s, 1H), 7.91 (s, 1H), 7.67 (d, J = 8.4 Hz, 1H), 7.59 (dd, J = 9.0, 1.8 Hz, 1H), 7.48 (t, J = 9.0 Hz, 2H), 7.43 (m, 4H), 7.27 (s, 1H), 7.21 (t, J = 7.8 Hz, 1H), 7.07 (t, J = 7.8 Hz, 1H), 6.66 (dd, J = 5.7, 3.0 Hz, 1H), 4.87 (t, J = 10.2 Hz, 1H), 4.61 (dd, J = 10.8, 2.4 Hz, 1H), 4.03–4.02 (m, 1H), 4.00–3.98 (m, 2H). 13C NMR (DMSO-d6, 150 MHz) δ 160.2, 159.4, 149.8, 146.5, 143.4, 136.6, 134.8, 133.3, 131.8, 131.7, 130.7, 129.5, 129.1, 127.9, 127.03, 127.00, 126.9, 123.4, 121.5, 119.5, 119.4, 118.7, 115.3, 112.8, 112.29, 112.21, 108.7, 106.1, 104.4, 103.3, 99.8, 54.7, 47.2, 40.8. IR (film) νmax 3255, 1731, 1603, 1514, 1400, 1232, 794, 733 cm−1. ESI-TOF HRMS m/z 576.1431 (M+H+, C32H22ClN5O4 requires 576.1433).
(1S)-6: [α]23D +18.4 (c 0.21, THF), natural enantiomer. (1R)-6: [α]23D −18.5 (c 0.24, THF), unnatural enantiomer.
N-(2-(5-Amino-4-oxo-1,2,9,9a-tetrahydrocyclopropa[c]benzo[e]indole-2-carbonyl)-1H-indol-5-yl)-1H-indole-2-carboxamide (20)
Intermediate 15 (10 mg, 0.028 mmol) was suspended in H2O (0.4 mL) and cooled to 0 °C. Fmoc-Cl (9.6 mg, 0.037 mmol) in dioxane (0.2 mL) was added and the reaction mixture was allowed to slowly warm to room temperature over 17 h. The reaction mixture was diluted with H2O and extracted with EtOAc (2×). The organic layers were combined, dried over Na2SO4, and concentrated under reduced pressure. The residue was dissolved in 4 N HCl in EtOAc (0.8 mL) and the mixture was allowed to stir at room temperature for 25 min. The solvent was removed under a stream of nitrogen and the residue was redissolved in anhydrous DMF (0.8 mL). EDCI (10.7 mg, 0.056 mmol) and 17 (10.7 mg, 0.34 mmol) were added and the reaction mixture was allowed to stir at 25 °C for 24 h. The reaction mixture was quenched with the addition of H2O and diluted with EtOAc. The organic phase was washed with 2 N aqueous HCl (3×), saturated aqueous NaHCO3 (5×), and saturated aqueous NaCl. The organic extract was dried over Na2SO4 and concentrated under reduced pressure. The crude residue was dissolved in DMF (0.8 mL) and piperidine (160 μL) was added. The reaction mixture was allowed to stir at room temperature for 1 h after which the solvent was removed under reduced pressure. The residue was purified by PTLC (SiO2, 60% THF/toluene) to provide 20 (4.1 mg, 29% over 4 steps) as a yellow solid. 1H NMR (DMSO-d6, 600 MHz) δ 11.81 (s, 1H), 11.72 (s, 1H), 10.17 (s, 1H), 8.21 (s, 1H), 7.67 (d, J = 7.8 Hz, 1H), 7.60 (d, J = 9.0 Hz, 1H), 7.47 (d, J = 9.0 Hz, 2H), 7.42 (s, 1H), 7.25 (s, 1H), 7.21 (t, J = 7.8 Hz, 1H), 7.17 (t, J = 8.4 Hz, 1H) 7.07 (t, J = 7.8 Hz, 1H), 6.81 (s, 1H), 6.58 (d, J = 8.4 Hz, 1H), 6.20 (d, J = 7.2 Hz, 1H), 4.60–4.57 (m, 1H), 4.45 (d, J = 10.2 Hz, 1H), 3.07 (m, 1H), 1.61 (t, J = 4.8 Hz, 1H), 1.51–1.49 (m, 1H). 13C NMR (DMSO-d6, 150 MHz) δ 188.8, 161.1, 159.2, 158.4, 150.5, 142.1, 136.4, 133.4, 132.4, 131.5, 131.4, 129.7, 126.7, 126.6, 123.2, 121.2, 119.6, 119.5, 113.6, 112.9, 112.6, 112.0, 110.5, 107.7, 106.8, 103.1, 63.1, 53.6, 32.4, 29.8, 24.1. ESI-TOF HRMS m/z 514.1872 (M+H+, C31H23N5O3 requires 514.1874).
In Vivo Antitumor Activity
B6D2F1 mice were injected intraperitoneal (i.p.) with syngeneic L1210 cells (1 × 106) on day 0. Ten mice were randomly assigned to control vehicle or treatment groups for compounds (+)-4 and (+)-6 at doses of 60, 100, 250, and 500 g/kg/inj for (+)-4 or 300, 1000, 3000, and 9000 μg/kg/inj for (+)-6. Compounds (+)-4 and (+)-6 were formulated in 100% DMSO, and injected i.p. on study days 1, 5, and 9. Following injection of tumor cells, animals were monitored daily and weighed two times per week. Percent survival (T/C) for treated and control groups were determined by dividing the total survival days for each treatment group by the total survival days for the control group and multiplying × 100. All animal studies were carried out in the animal facilities of The University of Kansas Medical Center with strict adherence to the guidelines of the IACUC Animal Welfare Committee of KUMC (IACUC approval # 2009–1837).
Supplementary Material
Acknowledgment
We gratefully acknowledge the financial support of the National Institutes of Health (CA041986), the Skaggs Institute for Chemical Biology, and a grant from the Kansas Biotech Authority. A.L.W. and K.K.D are Skaggs fellows. A.L.W. is the recipient of a predoctoral NSF Fellowship (2009–2012).
Abbreviations
- CBI
1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one
- DPPA
diphenylphosphoryl azide
- LiHMDS
lithium bis(trimethylsilyl)amide
- THF
tetrahydrofuran
- BnSH
benzyl thiol
- BnOH
benzyl alcohol
- BnNH2
benzylamine
- EtOAc
ethyl acetate
- DMF
dimethylformamide
- MeOH
methanol
- Fmoc-Cl
fluorenylmethyloxycarbonyl chloride
Footnotes
Supporting Information Available: A gel figure examining the DNA alkylation properties of 6, the synthesis and characterization of N-Boc-ACBI (21) and N-CO2Me-ACBI (24), and reactivity (solvolysis) data for 21 and 24.
References
- 1.Ichimura M, Ogawa T, Takahashi K, Kobayashi E, Kawamoto I, Yasuzawa T, Takahashi I, Nakano H. Duocarmycin SA, A New Antitumor Antibiotic From Streptomyces sp. J. Antibiot. 1990;43:1037–1038. doi: 10.7164/antibiotics.43.1037. [DOI] [PubMed] [Google Scholar]
- 2.Martin DG, Biles C, Gerpheide SA, Hanka LJ, Krueger WC, McGovren JP, Mizsak SA, Neil GL, Stewart JC, Visser J. CC-1065 (NSC 298223), A Potent New Antitumor Agent. Improved Production and Isolation, Characterization and Antitumor Activity. J. Antibiot. 1981;34:1119–1125. doi: 10.7164/antibiotics.34.1119. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi I, Takahashi K, Ichimura M, Morimoto M, Asano K, Kawamoto I, Tomita F, Nakano H. Duocarmycin, A New Antitumor Antibiotic From Streptomyces. J. Antibiot. 1988;41:1915–1917. doi: 10.7164/antibiotics.41.1915. [DOI] [PubMed] [Google Scholar]
- 4.Igarashi Y, Futamata K, Fujita T, Sekine A, Senda H, Naoki H, Furumai T. Yatakemycin, A Novel Antifungal Antibiotic Produced by Streptomyces sp. TP-A0356. J. Antibiot. 2003;56:107–113. doi: 10.7164/antibiotics.56.107. [DOI] [PubMed] [Google Scholar]
- 5.For duocarmycin SA, see: Boger DL, Johnson DS, Yun W. (+)- and ent-(−)-Duocarmycin SA and (+)- and ent-(−)-N-BOC-DSA DNA Alkylation Properties. Alkylation Site Models That Accommodate the Offset AT-Rich Adenine N3 Alkylation Selectivity of the Enantiomeric Agents. J. Am. Chem. Soc. 1994;116:1635–1656. doi: 10.1016/s0968-0896(00)82007-6.. For yatakemycin, see: Parrish JP, Kastrinsky DB, Wolkenberg SE, Igarashi Y, Boger DL. DNA Alkylation Properties of Yatakemycin. J. Am. Chem. Soc. 2003;125:10971–10976. doi: 10.1021/ja035984h.. Trzupek JD, Gottesfeld JM, Boger DL. Alkylation of Duplex DNA in Nucleosome Core Particles by Duocarmycin SA and Yatakemycin. Nat. Chem. Biol. 2006;2:79–82. doi: 10.1038/nchembio761.. Tichenor MS, MacMillan KS, Trzupek JD, Rayl TJ, Hwang I, Boger DL. Systematic Exploration of the Structural Features of Yatakemycin Impacting DNA Alkylation and Biological Activity. J. Am. Chem. Soc. 2007;129:10858–10869. doi: 10.1021/ja072777z.. For CC-1065, see: Hurley LH, Lee C-S, McGovren JP, Warpehoski MA, Mitchell MA, Kelly RC, Aristoff PA. Molecular Basis for Sequence-Specific DNA Alkylation by CC-1065. Biochemistry. 1988;27:3886–3892. doi: 10.1021/bi00410a054.. Boger DL, Johnson DS, Yun W, Tarby CM. Molecular Basis for Sequence Selective DNA Alkylation by (+)- and ent-(−)-CC-1065 and Related Agents: Alkylation Site Models that Accommodate the Offset AT-Rich Adenine N3 Alkylation Selectivity. Bioorg. Med. Chem. 1994;2:115–135. doi: 10.1016/s0968-0896(00)82007-6.. Boger DL, Munk SA, Zarrinmayeh H. (+)-CC-1065-DNA Alkylation: Key Studies Demonstrating a Noncovalent Binding Selectivity Contribution to the Alkylation Selectivity. J. Am. Chem. Soc. 1991;113:3980–3983.. Boger DL, Zarrinmayeh H, Munk SA, Kitos PA, Suntornwat O. Demonstration of a Pronounced Effect of Noncovalent Binding Selectivity on the (+)-CC-1065 DNA Alkylation and Identification of the Pharmacophore of the Alkylation Subunit. Proc. Nat. Acad. Sci. U.S.A. 1991;88:1431–1435. doi: 10.1073/pnas.88.4.1431.. Boger DL, Coleman RS, Invergo BJ, Sakya SM, Ishizaki T, Munk SA, Zarrinmayeh H, Kitos PA, Thompson SC. Synthesis and Evaluation of Aborted and Extended CC-1065 Functional Analogs: (+)- and (−)-CPI-PDE-I1, (+)- and (−)-CPI-CDPI1, and (+)- and (−)-CPI-CDPI3. Preparation of Key Partial Structures and Definition of an Additional Functional Role of the CC-1065 Central and Right-Hand Subunits. J. Am. Chem. Soc. 1990;112:4623–4632.. Boger DL, Munk SA, Ishizaki T. (+)-CC-1065 DNA Alkylation: Observation of An Expected Relationship Between Cyclopropane Electrophile Reactivity and the Efficiency of DNA Alkylation. J. Am Chem. Soc. 1991;113:2779–2780.. Boger DL, Coleman RS, Invergo BJ, Zarrinmayeh H, Kitos PA, Thompson SC, Leong T, McLaughlin LW. A Demonstration of the Intrinsic Importance of Stabilizing Hydrophobic Binding and Noncovalent van der Waals Contacts Dominant in the Noncovalent CC-1065:DNA Binding. Chem.-Biol. Interactions. 1990;73:29–52. doi: 10.1016/0009-2797(90)90107-x.. For duocarmycin A, see: Boger DL, Ishizaki T, Zarrinmayeh H, Munk SA, Kitos PA, Suntornwat O. Duocarmycin-Pyrindamycin DNA Alkylation Properties and Identification, Synthesis, and Evaluation of Agents Incorporating the Pharmacophore of the Duocarmycin-Pyrindamycin Alkylation Subunit. Identification of the CC-1065 Duocarmycin Common Pharmacophore. J. Am. Chem. Soc. 1990;112:8961–8971.. Boger DL, Ishizaki T, Zarrinmayeh H. Isolation and Characterization of the Duocarmycin-Adenine DNA Adduct. J. Am. Chem. Soc. 1991;113:6645–6649.. Boger DL, Yun W, Terashima S, Fukuda Y, Nakatani K, Kitos PA, Jin Q. DNA Alkylation Properties of the Duocarmycins: (+)-Duocarmycin A, epi-(+)-Duocarmycin A, ent-(−)-Duocarmycin A and epi,ent-(−)-Duocarmycin A. Bioorg. Med. Chem. Lett. 1992;2:759–765.. Boger DL, Yun W. Reversibility of the Duocarmycin A and SA DNA Alkylation Reaction. J. Am. Chem. Soc. 1993;115:9872–9873.
- 6.Reviews: Boger DL, Johnson DS. CC-1065 and the Duocarmycins: Understanding Their Biological Function Through Mechanistic Studies. Angew. Chem., Int. Ed. Engl. 1996;35:1438–1474.. Boger DL. The Duocarmycins: Synthetic and Mechanistic Studies. Acc. Chem. Res. 1995;28:20–29.. Boger DL, Johnson DS. CC-1065 and the Duocarmycins: Unraveling the Keys to a New Class of Naturally Derived DNA Alkylating Agents. Proc. Natl. Acad. Sci. U.S.A. 1995;92:3642–3649. doi: 10.1073/pnas.92.9.3642.. Boger DL, Garbaccio RM. Shape-Dependent Catalysis: Insights into the Source of Catalysis for the CC-1065 and Duocarmycin DNA Alkylation Reaction. Acc. Chem. Res. 1999;32:1043–1052.. Tichenor MS, Boger DL. Yatakemycin: Total Synthesis, DNA Alkylation, and Biological Properties. Natural Prod. Rep. 2008;25:220–226. doi: 10.1039/b705665f.. MacMillan KS, Boger DL. Fundamental Relationships Between Structure, Reactivity, and Biological Activity for the Duocarmycins and CC-1065. J. Med. Chem. 2009;52:5771–5780. doi: 10.1021/jm9006214.. Searcey M. Duocarmycins: Nature's Prodrugs? Curr. Pharm. Des. 2002;8:1375–1389. doi: 10.2174/1381612023394539.. Tse WC, Boger DL. Sequence-Selective DNA Recognition: Natural Products and Nature's Lessons. Chem. Biol. 2004;11:1607–1617. doi: 10.1016/j.chembiol.2003.08.012.
- 7.(a) Boger DL, Coleman RS. Total Synthesis of (+)-CC-1065 and ent-(−)-CC-1065. J. Am. Chem. Soc. 1988;110:1321–1323. [Google Scholar]; (b) Boger DL, Coleman RS. Total Synthesis of CC-1065, and the Precise, Functional Agent CPI-CDPI2. J. Am. Chem. Soc. 1988;110:4796–4807. [Google Scholar]; (c) Boger DL, Machiya K. Total Synthesis of (+)-Duocarmycin SA. J. Am. Chem. Soc. 1992;114:10056–10058. [Google Scholar]; (d) Boger DL, Machiya K, Hertzog DL, Kitos PA, Holmes D. Total Synthesis and Preliminary Evaluation of (+)- and ent-(−)-Duocarmycin SA. J. Am. Chem. Soc. 1993;115:9025–9036. [Google Scholar]; (e) Boger DL, McKie JA, Nishi T, Ogiku T. Enantioselective Total Synthesis of (+)-Duocarmycin A, epi-(+)-Duocarmycin A, and Their Unnatural Enantiomers. J. Am. Chem. Soc. 1996;118:2301–2302. [Google Scholar]; (f) Boger DL, McKie JA, Nishi T, Ogiku T. Total Synthesis of (+)-Duocarmycin A and epi-(+)-Duocarmycin A and Their Unnatural Enantiomers: Assessment of Chemical and Biological Properties. J. Am. Chem. Soc. 1997;119:311–325. [Google Scholar]; (g) Tichenor MS, Kastrinsky DB, Boger DL. Total Synthesis, Structure Revision, and Absolute Configuration of (+)-Yatakemycin. J. Am. Chem. Soc. 2004;126:8396–8398. doi: 10.1021/ja0472735. [DOI] [PubMed] [Google Scholar]; (h) Tichenor MS, Trzupek JD, Kastrinsky DB, Shiga F, Hwang I, Boger DL. Asymmetric Total Synthesis of (+)- and ent-(−)-Yatakemycin and Duocarmycin SA. Evaluation of Yatakemycin Key Partial Structures and Its Unnatural Enantiomer. J. Am. Chem. Soc. 2006;128:15683–15696. doi: 10.1021/ja064228j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) MacMillan KS, Nguyen T, Hwang I, Boger DL. Total Synthesis and Evaluation of iso-Duocarmycin SA and iso-Yatakemycin. J. Am. Chem. Soc. 2009;131:1187–1194. doi: 10.1021/ja808108q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Boger DL, Hertzog DL, Bollinger B, Johnson DS, Cai H, Goldberg J, Turnbull P. Duocarmycin SA Shortened, Simplified, and Extended Agents: A Systematic Examination of the Role of the DNA Binding Subunit. J. Am. Chem. Soc. 1997;119:4977–4986. [Google Scholar]; (b) Boger DL, Bollinger B, Hertzog DL, Johnson DS, Cai H, Mesini P, Garbaccio RM, Jin Q, Kitos PA. Reversed and Sandwiched Analogs of Duocarmycin SA: Establishment of the Origin of the Sequence-Selective Alkylation of DNA and New Insights into the Source of Catalysis. J. Am. Chem. Soc. 1997;119:4987–4998. [Google Scholar]; (c) Boger DL, Garbaccio RM. Catalysis of the CC-1065 and Duocarmycin DNA Alkylation Reaction: DNA Binding Induced Conformational Change in the Agent Results in Activation. Bioorg. Med. Chem. 1997;5:263–276. doi: 10.1016/s0968-0896(96)00238-6. [DOI] [PubMed] [Google Scholar]
- 9.(a) Boger DL, Ishizaki T, Kitos PA, Suntornwat O. Synthesis of N-(tert-Butyloxycarbonyl)-CBI, CBI, CBI-CDPI1, and CBI-CDPI2: Enhanced Functional Analogs of CC-1065 Incorporating the 1,2,9,9a-Tetrahydrocyclopropa[c]benz[e]indol-4-one (CBI) Left-Hand Subunit. J. Org. Chem. 1990;55:5823–5832. [Google Scholar]; (b) Boger DL, Ishizaki T, Wysocki RJ, Jr., Munk SA, Kitos PA, Suntornwat O. Total Synthesis and Evaluation of (±)-N-(tert-Butoxycarbonyl)-CBI, (±)-CBI-CDPI1, and (±)-CBI-CDPI2: CC-1065 Functional Agents Incorporating the Equivalent 1,2,9,9a-Tetrahydrocyclopropa[1,2-c]benz[1,2-e]indol-4-one (CBI) Left-Hand Subunit. J. Am. Chem. Soc. 1989;111:6461–6463. [Google Scholar]; (c) Boger DL, Ishizaki T. Resolution of a CBI Precursor and Incorporation into the Synthesis of (+)-CBI, (+)-CBI-CDPI1, (+)-CBI-CDPI2: Enhanced Functional Analogs of (+)-CC-1065. A Critical Appraisal of a Proposed Relationship Between Electrophile Reactivity, DNA Binding Properties, and Cytotoxic Potency. Tetrahedron Lett. 1990;31:793–796. [Google Scholar]; (d) Boger DL, Ishizaki T, Zarrinmayeh H, Kitos PA, Suntornwat O. A Potent, Simple Derivative of an Analog of the CC-1065 Alkylation Subunit. Bioorg. Med. Chem. Lett. 1991;1:55–58. [Google Scholar]; (e) Boger DL, Munk SA. DNA Alkylation Properties of Enhanced Functional Analogs of CC-1065 Incorporating the 1,2,9,9a-Tetrahydrocyclopropa[1,2-c]benz[1,2-e]indol-4-one (CBI) Alkylation Subunit. J. Am. Chem. Soc. 1992;114:5487–5496. [Google Scholar]; (f) Boger DL, Yun W. Role of the CC-1065 and Duocarmycin N2 Substituent: Validation of a Direct Relationship Between Solvolysis Chemical Stability and in vitro Biological Potency. J. Am. Chem. Soc. 1994;116:5523–5524. [Google Scholar]; (g) Boger DL, Yun W, Cai H, Han N. CBI-CDPBO1 and CBI-CDPBI1: CC-1065 Analogs Containing Deep-Seated Modifications in the DNA Binding Subunit. Bioorg. Med. Chem. 1995;3:761–775. doi: 10.1016/0968-0896(95)00066-p. [DOI] [PubMed] [Google Scholar]; (h) Boger DL, Yun W. CBI-TMI: Synthesis and Evaluation of a Key Analog of the Duocarmycins. Validation of a Direct Relationship between Chemical Solvolytic Stability and Cytotoxic Potency and Confirmation of the Structural Features Responsible for the Distinguishing Behavior of Enantiomeric Pairs of Agents. J. Am. Chem. Soc. 1994;116:7996–8006. [Google Scholar]; (i) Parrish JP, Katrinsky DB, Stauffer F, Hedrick MP, Hwang I, Boger DL. Establishment of Substitution Effects in the DNA Binding Subunit of CBI Analogues of the Duocarmycins and CC-1065. Bioorg. Med. Chem. 2003;11:3815–3838. doi: 10.1016/s0968-0896(03)00194-9. [DOI] [PubMed] [Google Scholar]; (j) Parrish JP, Hughes TV, Hwang I, Boger DL. Establishing the Parabolic Relationship Between Reactivity and Activity for Derivatives and Analogues of the Duocarmycin and CC-1065 alkylation Subunit. J. Am. Chem. Soc. 2004;126:80–81. doi: 10.1021/ja038162t. [DOI] [PubMed] [Google Scholar]
- 10.(a) Boger DL, Yun W, Teegarden BR. An Improved Synthesis of 1,2,9,9a-Tetrahydrocyclopropa[c]benz[e]indol-4-one (CBI): A Simplified Analog of the CC-1065 Alkylation Subunit. J. Org. Chem. 1992;57:2873–2876. [Google Scholar]; (b) Boger DL, McKie JA. An Efficient Synthesis of 1,2,9,9a-Tetrahydrocyclopropa[c]benz[e]indol-4-one (CBI): An Enhanced and Simplified Analog of the CC-1065 and Duocarmycin Alkylation Subunits. J. Org. Chem. 1995;60:1271–1275. [Google Scholar]; (c) Boger DL, McKie JA, Boyce CW. Asymmetric Synthesis of the 1,2,9,9a-Tetrahydrocyclopropa[c]benz[e]indol-4-one (CBI) Alkylation Subunit of CC-1065 and Duocarmycin Analogues. Synlett. 1997:515–517. [Google Scholar]; (d) Kastrinsky DB, Boger DL. Effective Asymmetric Synthesis of 1,2,9,9a-Tetrahydrocyclopropa[e]benz[e]indol-4-one (CBI) J. Org. Chem. 2004;69:2284–2289. doi: 10.1021/jo035465x. [DOI] [PubMed] [Google Scholar]; (e) Lajiness JP, Boger DL. Asymmetric Synthesis of 1,2,9,9a-Tetrahydrocyclopropa[c]benz[e]indol-4-one (CBI) J. Org. Chem. 2010;76:583–587. doi: 10.1021/jo102136w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Boger DL, Yun W, Han N. 1,2,9,9a-Tetrahydrocyclopropa[c]benz[e]indol-4-one (CBI) Analogs of CC-1065 and the Duocarmycins: Synthesis and Evaluation. Bioorg. Med. Chem. 1995;3:1429–1453. doi: 10.1016/0968-0896(95)00130-9. [DOI] [PubMed] [Google Scholar]; (b) Boger DL, Ishizaki T, Sakya S, Munk SA, Kitos PA, Jin Q, Besterman JM. Synthesis and Preliminary Evaluation of (+)-CBI-indole2: An Enhanced Functional Analog of CC-1065. Bioorg. Med. Chem. Lett. 1991;1:115–120. [Google Scholar]
- 12.For Carzelesin, see: Li L, DeKoning TF, Kelly RC, Krueger WC, McGovren JP, Padbury GE, Petzold GL, Wallace TL, Ouding RJ, Prairie MD, Gebhard I. Cytotoxicity and Antitumor Activity of Carzelesin, a Prodrug Cyclopropylpyrroloindole Analogue. Cancer Res. 1992;52:4904–4913.. van Tellingen O, Nooijen WJ, Schaaf LJ, van der Valk M, van Asperen J, Henrar REC, Beijnen JH. Comparative Pharmacology of the Novel Cyclopropylpyrroloindole-Prodrug Carzelesin in Mice, Rats, and Humans. Cancer Res. 1998;58:2410–2416.. For KW-2189, see: Kobayashi E, Okamoto A, Asada M, Okabe M, Nagamura S, Asai A, Saito H, Gomi K, Hirata T. Characteristics of Antitumor Activity of KW-2189, A Novel Water-Soluble Derivative of Duocarmycin, Against Murine and Human Tumors. Cancer Res. 1994;54:2404–2410.. Nagamura S, Kanda Y, Kobayashi E, Gomi K, Saito H. Synthesis and Antitumor Activity of Duocarmycin Derivatives. Chem. Pharm. Bull. 1995;43:1530–1535. doi: 10.1248/cpb.43.1530.. For other CBI carbamate prodrugs: Boger DL, Boyce CW, Garbaccio RM, Searcey M, Jin Q. CBI Prodrug Analogs of CC-1065 and the Duocarmycins. Synthesis. 1999:1505–1509.. Li LS, Sinha SC. Studies Toward the Duocarmycin Prodrugs for the Antibody Prodrug Therapy Approach. Tetrahedron Lett. 2009;50:2932–2935. doi: 10.1016/j.tetlet.2009.03.205.
- 13.For glycosidic prodrugs: Tietze LF, Lieb M, Herzig T, Haunert F, Schuberth I. A Strategy for Tumor-Selective Chemotherapy by Enzymatic Liberation of seco-duocarmycin SA-derivatives from Nontoxic Prodrugs. Bioorg. Med. Chem. 2001;9:1929–1939. doi: 10.1016/s0968-0896(01)00098-0.. Tietze LF, Major F, Schuberth I. Antitumor Agents: Development of Highly Potent Glycosidic Duocarmycin Analogues for Selective Cancer Therapy. Angew. Chem. Int. Ed. 2006;45:6574–6577. doi: 10.1002/anie.200600936.. Tietze LF, Schuster HJ, Schmuck K, Schuberth I, Alves F. Duocarmycin-based Prodrugs for Cancer Prodrug Monotherapy. Bioorg. Med. Chem. 2008;16:6312–6318. doi: 10.1016/j.bmc.2008.05.009.. For reductively activated prodrugs: Hay MP, Anderson RF, Ferry DM, Wilson WR, Denny WA. Synthesis and Evaluation of Nitroheterocyclic Carbamate Prodrugs for Use with Nitroreductase-Mediated Gene-Directed Enzyme Prodrug Therapy. J. Med. Chem. 2003;46:5533–5545. doi: 10.1021/jm030308b.. Hay MP, Sykes BM, Denny WA, Wilson WR. A 2-Nitroimidazole Carbamate Prodrug of 5-Amino-1-(chloromethyl)-3-[(5,6,7-trimethoxyindol-2-yl)carbonyl]-1,2-dihydro-3H-benz[e]indole (Amino-seco-CBI-TMI) for Use With ADEPT and GDEPT. Bioorg. Med. Chem. Lett. 1999;9:2237–2242. doi: 10.1016/s0960-894x(99)00381-9.. Tercel M, Atwell GJ, Yang S, Ashoorzadeh A, Stevenson RJ, Botting KJ, Gu Y, Mehta SY, Denny WA, Wilson WR, Pruijn FB. Selective Treatment of Hypoxic Tumor Cells In Vivo: Phosphate Pre-Prodrugs of Nitro Analogues of the Duocarmycins. Angew. Chem. Int. Ed. 2011;50:2606–2609. doi: 10.1002/anie.201004456.. Townes H, Summerville K, Purnell B, Hooker M, Madsen E, Hudson S, Lee M. Investigation of a Novel Reductively-Activatable Anticancer Prodrug of seco-CBI-TMI, an Analog of Duocarmycin SA. Med. Chem. Res. 2002;11:248–253.. Boger DL, Garbaccio RM. A Novel Class of CC-1065 and Duocarmycin Analogues Subject to Mitomycin-Related Reductive Activation. J. Org. Chem. 1999;64:8350–8362. doi: 10.1021/jo991301y.. For other prodrugs: Wang Y, Li L, Tian Z, Jiang W, Larrick J. Synthesis and Antitumor Activity of CBI-Bearing Ester and Carbamate Prodrugs of CC-1065. Bioorg. Med. Chem. 2006;14:7854–7861. doi: 10.1016/j.bmc.2006.07.062.. Zhao RH, Erickson HK, Leece BA, Reid EE, Goldmacher VS, Lambert JM, Chari RVJ. Synthesis and Biological Evaluation of Antibody Conjugates of Phosphate Prodrugs of Cytotoxic DNA Alkylators for the Targeted Treatment of Cancer. J. Med. Chem. 2012;55:766–782. doi: 10.1021/jm201284m.
- 14.(a) Jin W, Trzupek JD, Rayl TJ, Broward MA, Vielhauer GA, Weir SJ, Hwang I, Boger DL. A Unique Class of Duocarmycin and CC-1065 Analogues Subject to Reductive Activation. J. Am. Chem. Soc. 2007;129:15391–15397. doi: 10.1021/ja075398e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lajiness JP, Robertson WM, Dunwiddie I, Broward MA, Vielhauer GA, Weir SJ, Boger DL. Design, Synthesis, and Evaluation of Duocarmycin O-Amino Phenol Prodrugs Subject to Tunable Reductive Activation. J. Med. Chem. 2010;53:7731–7738. doi: 10.1021/jm1010397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wolkenberg SE, Boger DL. Mechanisms of in situ Activation for DNA Targeting Antitumor Agents. Chem. Rev. 2002;102:2477–2495. doi: 10.1021/cr010046q. [DOI] [PubMed] [Google Scholar]
- 16.Boger DL, Boyce CW, Garbaccio RM, Goldberg JA. CC-1065 and the Duocarmycins: Synthetic Studies. Chem. Rev. 1997;97:787–828. doi: 10.1021/cr960095g. [DOI] [PubMed] [Google Scholar]
- 17.Boger DL, Han N, Tarby CM, Boyce CW, Cai H, Jin Q, Kitos PA. Synthesis, Chemical Properties, and Preliminary Evaluation of Substituted CBI Analogs of CC-1065 and the Duocarmycins Incorporating the 7-Cyano-1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one Alkylation Subunit: Hammett Quantitation of the Magnitude of Electronic Effects on Functional Reactivity. J. Org. Chem. 1996;61:4894–4912. doi: 10.1021/jo952033g. [DOI] [PubMed] [Google Scholar]
- 18.Boger DL, Boyce CW, Garbaccio RM, Searcey M. Synthesis of CC-1065 and Duocarmycin Analogs via Intramolecular Aryl Radical Cyclization of a Tethered Vinyl Chloride. Tetrahedron Lett. 1998;39:2227–2230. [Google Scholar]
- 19.Boger DL, Brotherton CE. Total Synthesis of Azafluoranthene Alkaloids: Refescine and Imelutine. J. Org. Chem. 1984;49:4050–4055. [Google Scholar]
- 20.Wolfe AL. unpublished studies.
- 21.Huang X, Buchwald SL. New Ammonia Equivalents for the Pd-Catalyzed Amination of Aryl Halides. Org. Lett. 2001;3:3417–3419. doi: 10.1021/ol0166808. [DOI] [PubMed] [Google Scholar]
- 22.(a) Boger DL, Panek JS. Palladium(0)-Mediated β-Carboline Synthesis: Preparation of the CDE Ring System of Lavendamycin. Tetrahedron Lett. 1984;25:3175–3178. [Google Scholar]; (b) Boger DL, Duff SR, Panek JS, Yasuda M. Inverse Electron Demand Diels-Alder Reactions of Heterocyclic Azadienes. Studies on the Total Synthesis of Lavendamycin: Investigative Studies on the Preparation of the CDE β-Carboline Ring System and AB Quinoline-5,8-quinone Ring System. J. Org. Chem. 1985;50:5782–5789. [Google Scholar]; (c) Boger DL, Panek JS, Duff SR, Yasuda M. Total Synthesis of Lavendamycin Methyl Ester. J. Org. Chem. 1985;50:5790–5795. [Google Scholar]
- 23.Boger DL, Munk SA, Zarrimayeh H, Ishizaki T, Haught J, Bina M. An Alternative and Convenient Strategy for Generation of Substantial Quantities of Singly 5'-P32-End-Labeled Double-Stranded DNA for Binding Studies. Development of a Protocol for Examination of Functional Features of (+)-CC-1065 and the Duocarmycins That Contribute to Their Sequence-Selective DNA Alkylation Properties. Tetrahedron. 1991;47:2661–2682. [PubMed] [Google Scholar]; See also: Boger DL, Wysocki RJ, Ishizaki T. Synthesis of N-Phenylsulfonyl-CI, N-Boc-CI, CI-CDPI1 and CI-CDPI2: CC-1065 Functional Analogs Incorporating the Parent 1,2,7,7a-Tetrahydrocyclopropa[1,2-c]indol-4-one (CI) Left-Hand Subunit. J. Am. Chem. Soc. 1990;112:5230–5240.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.