

Abstract
Congenital myasthenic syndromes (CMSs) are neuromuscular disorders that can be caused by defects in acetylcholine receptor (AChR) function. Disease-associated point mutants can reveal the unsuspected functional significance of mutated residues. We identified two pathogenic mutations in the extracellular domain of the AChR α subunit (AChRα) in a patient with myasthenic symptoms since birth: a V188M mutation in the C-loop and a heteroallelic G74C mutation in the main immunogenic region. The G74C mutation markedly reduced surface AChR expression in cultured cells, whereas the V188M mutant was expressed robustly but had severely impaired kinetics. Single-channel patch-clamp analysis indicated that V188M markedly decreased the apparent AChR channel opening rate and gating efficiency. Mutant cycle analysis of energetic coupling among conserved residues within or dispersed around the AChRα C-loop revealed that V188 is functionally linked to Y190 in the C-loop and to D200 in β-strand 10, which connects to the M1 transmembrane domain. Furthermore, V188M weakens inter-residue coupling of K145 in β-strand 7 with Y190 and with D200. Cumulatively, these results indicate that V188 of AChRα is part of an interdependent tetrad that contributes to rearrangement of the C-loop during the initial coupling of agonist binding to channel gating.
Introduction
Congenital myasthenic syndromes (CMSs) are heterogeneous disorders in which the safety margin of neuromuscular transmission is impaired by one or more defects in motor endplate–associated proteins (1). Most defects arise from mutations that occur in subunits of the acetylcholine receptor (AChR) (reviewed in ref. 2); other deleterious mutations have been identified in choline acetyl-transferase (ChAT) (3); the collagenic tail subunit of the endplate species of acetylcholinesterase (AChE) (4, 5); the cytoplasmic protein rapsyn (6); agrin (7); the muscle-specific receptor tyrosine kinase (MuSK) (8); Dok-7, a muscle-intrinsic activator of MuSK (9); plectin (10); glutamine-fructose-6-phosphate transaminase 1 (GFPT1) (11); and the Nav1.4 sodium channel (12). The pathogenic mutations in AChR subunits either decrease the expression or alter the kinetics of activation of the receptor by nerve-released ACh. The mutations that alter activation kinetics result in physiologically opposite slow- and fast-channel syndromes that prolong or speed the decay of the synaptic current (13).
The AChR is a ligand-gated ion channel in which 5 homologous subunits align side by side to form a cylindrical structure lodged within the cell membrane. At the innervated adult endplate (EP) the stoichiometry of the subunits is (α1)2β1δε, whereas at denervated or developing EPs the fetal γ subunit is present in place of the ε subunit. Each subunit includes a large extracellular domain, 4 transmembrane domains, and a large intracellular domain (14). The extracellular domain consists of an inner core of 6 β-strands and an outer shell of 4 β-strands, whereas the transmembrane domains are α-helices (15). The 2 agonist-binding pockets of AChR are located at interfaces between the α subunits and the neighboring ε or δ subunits. The principal face of each binding site is formed by the α subunit, which contributes peptide loops A–C, while the complimentary face is formed by either the δ or the ε subunit contributing loops D–G (16). A structural transitional zone between the ligand-binding and channel domains transduces conformational changes resulting from agonist binding into opening of the ion channel.
Crystallographic studies of isolated binding domains of the mouse AChR (17) and of the ACh binding protein of the freshwater snail and the saltwater mollusk (18, 19) as well as cryoelectron microscopy analysis of Torpedo AChR at 4-Å resolution (15) rationalized previous observations on the functional properties of the receptor but did not reveal how different residues or domains of the receptor contribute to the transition from the closed to the open state. Subsequent studies, however, attained this goal by exploring energetic coupling among residues within or between subunits by combined mutagenesis studies, single-channel recordings, and thermodynamic mutant cycle analyses (20–24). These studies revealed that activation of the receptor begins by transition of the C-loop of each α subunit from an opened-up to a closed-in conformation (24–26); this leads to displacement of β-strand 10, which is contiguous with the pre-M1 domain, and results in conformational changes of the β1-β2 linker and the Cys-loop (Figure 1). Both the β1-β2 linker and the Cys-loop energetically couple with residues in the M2-M3 linker and in the course of channel opening displace the M2 helix away from the center of pore (21, 23, 27).
Figure 1. Structural model of the mouse AChR α subunit at 1.94-Å resolution (PDB 2QC1) showing positions of the identified mutations.

MIR, main immunogenic region.
In this study we identified a p.V188M mutation in the C-loop of the AChR α1 subunit that disrupts the initial coupling of agonist binding to channel gating. Unlike previously identified residues that affect coupling, αVal188 is conserved only in the muscle AChR α1 subunit but not in α subunits of other AChR subtypes. Single-channel recordings from HEK cells expressing the αV188M receptor reveal very brief channel openings flanked by prolonged closings. Estimates of activation rate constants for the αV188M AChR from single-channel recordings, obtained over a range of ACh concentrations, show that αV188M profoundly affects the apparent channel opening rate and gating efficiency of the receptor, effects that are independent of the size of the substituting residue. Finally, using mutant cycle analyses (MCA) we show that αVal188 is part of a network of residues that link agonist-mediated conformational changes in loop C to subsequent structural rearrangements that culminate in opening the AChR channel.
Results
Characteristics of the CMS patient.
The patient, a woman 42 years old at the time of the study, was born with floppy infant syndrome, with a feeble cry and poor sucking reflex. She had recurrent aspirations and droopy eyelids. She walked at 12 months but often fell. Later she had ocular muscle weakness, abnormal fatigability, and generalized weakness. Thymectomy at age 8 years was of no benefit. At age 14, repetitive stimulation of the femoral nerve at 2 Hz revealed a 66% decremental response of the first compared with the fourth evoked compound action potential from the rectus femoris muscle. She had no anti-AChR antibodies. Light and electron microscopy studies of intercostal muscle neuromuscular junctions at age 14 revealed no structural abnormality. The patient’s symptoms responded partially to pyridostigmine. There was no history of similarly affected family members.
Mutation analysis.
To determine the molecular basis of the patient’s illness, we sequenced each AChR subunit gene. This revealed two heterozygous mutations in CHRNA1: p.αG74C (c.280G>T) and p.αV188M (c.622G>A). (Nucleotide numbers start from the translational start site, with +1 corresponding to the A of the ATG translation initiation codon [NM_000079.3]; codon numbers start from the first codon of the mature peptide [NP_000070.1]). αG74C is in the main immunogenic region (MIR) and αV188M in the C-loop of the agonist-binding domain of the α subunit (Figure 1). The mutated Gly74 is conserved across the AChR α1 and β subunits of different species and in all human AChR α subunits except in α2 and α4. The mutated Val188 is conserved across α1 subunits of different species (Figure 2) but not in α subunits of other AChR subtypes. Neither mutation is present in 400 normal alleles of 200 unrelated subjects. Family analysis indicated that each mutation is recessive, the paternal and maternal alleles harboring αG74C and αV188M, respectively.
Figure 2. Sequence alignment of the α1 AChR subunits in different species.

Expression studies in HEK cells.
To assess pathogenicity of the observed mutations, we engineered each mutation into the human α subunit and co-transfected cDNAs encoding either mutant or wild-type α subunits with complementary wild-type β, δ, and ε subunits into HEK cells (Figure 3A). To assess expression of the AChR on the cell surface, we measured the binding of [125I]α-bungarotoxin ([125I]α-bgt) to intact cells (Figure 3A). Surface expression of αG74C AChR was reduced to 14% that of wild-type and that of αV188M AChR was close to normal. Therefore, αV188M determines the phenotype.
Figure 3. α-bgt binding studies and single-channel currents elicited by ACh.

(A) [125I]α-bgt binding to surface receptors on intact HEK cells transfected with wild-type and mutant AChRs. The results are normalized for α-bgt binding to wild-type AChR and represent mean ± SD of 3–6 experiments. (B) Single-channel currents elicited by ACh from HEK cells expressing wild-type and mutant AChRs. Left: Representative channel openings shown as upward deflections. Right: Logarithmically binned burst duration histograms fitted to the sum of exponentials. Arrows indicate mean durations of dominant burst components.
Activation kinetics of the αV188M mutant.
The safety margin of neuromuscular transmission depends critically on the ability of nerve-released ACh to rapidly and efficiently activate AChR channels. To identify elementary kinetic steps in AChR activation altered by αV188M, we first compared the activation of wild-type and mutant receptors expressed in HEK cells by analyzing the single-channel currents elicited by limiting low concentrations of ACh. A concentration of 50 nM ACh elicited a sufficient number of channel openings from wild-type AChR, but mutant AChR required 1 μM ACh (Figure 3B). The resulting channel openings appeared either as isolated openings or as several openings in quick succession, called bursts, the duration of which mirrors the rate of decay of the postsynaptic current. Construction of histograms of burst dwell times and fitting them with sums of exponentials revealed 2 components for αV188M and 3 components for wild-type AChR (Figure 3B and Table 1). For the wild-type AChR, the mean duration of the longest and predominant burst component was 3.31 ms, but for the mutant it was only 0.68 ms.
Table 1.
Burst durations of wild-type and mutant AChRs in HEK cells
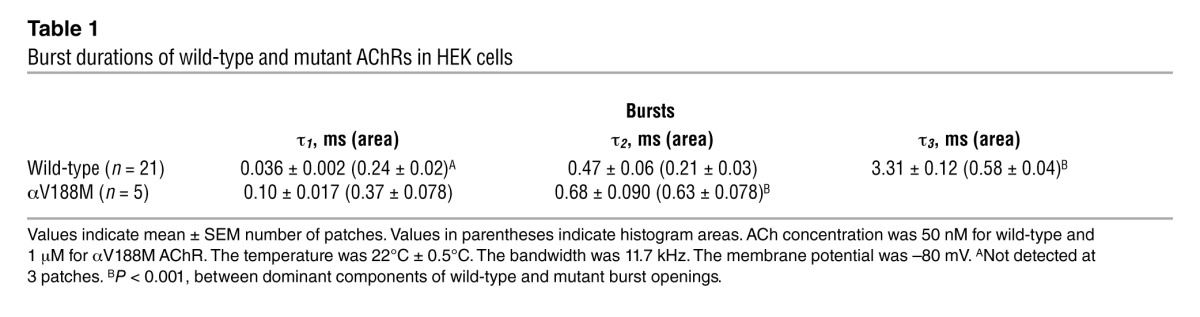
To identify the kinetic steps responsible for the short-lived bursts of the mutant receptor, we recorded single-channel currents over a range of ACh concentrations and constructed histograms of the resulting open and closed dwell times (see Methods). For wild-type AChR, ACh concentrations higher than 10 μM elicited well-defined clusters of events, all from the same AChR channel, but for the αV188M AChR the threshold for forming clusters was 30 μM ACh (Figure 4). For both wild-type and mutant receptors, the dominant component of closed dwell times shifted to shorter durations with increasing ACh concentrations, but for the mutant receptor the shift toward brief closed durations was much smaller, and at saturating concentrations the closed durations remained prolonged (Figure 4). Because the ACh-dependent shift from long to brief closed durations signals a process driven by binding of ACh, the prolonged closed durations for αV188M AChR indicate a change in one or more elementary kinetic steps that precede opening of the channel. The open duration histograms changed little across the range of ACh concentrations and remained consistently briefer for the mutant than the wild-type receptor (Figure 4). Thus, αV188M destabilizes the di-liganded open channel state.
Figure 4. Activation kinetics of wild-type and αV188M AChR.

Left: Representative single-channel currents at the indicated ACh concentrations recorded from HEK cells expressing (A) wild-type and (B) αV188M AChRs. Currents are shown as upward deflections; bandwidth, 10 kHz. Center and right: Histograms of closed and open durations corresponding to each ACh concentration are shown with the probability density functions (smooth curves) generated from a global fit of the scheme to dwell times obtained for the entire range of ACh concentrations. Fitted rate constants are shown in Table 2. On the y axes, each histogram entry is the probability of occurrence on a square root (Sqrt) scale.
To quantify changes in elementary rate constants underlying activation of the αV188M AChR, we next analyzed the open and closed dwell times according to a linear scheme of receptor activation that recognizes 3 closed and 1 open state of the receptor (Figure 5). In this scheme, 2 agonists (A) bind to the receptor (R) with association rate constants k+1 and k+2, and dissociate with rate constants k–1 and k–2. The receptor occupied by 2 agonists opens with rate constant β and closes with rate constant α. At high concentrations, ACh blocks the open channel with rate k+b, and the channel unblocks with rate k–b. Owing to bandwidth limitations, this scheme omits the recently described intermediate closed state between A2R and A2R* (asterisk indicates the open state of the receptor) (28), so the fitted channel gating rate and equilibrium constants presented here are apparent constants.
Figure 5. Scheme of AChR activation.

For both wild-type and mutant αV188M AChRs, probability density functions computed from the fitted rate constants describe the dwell time distributions for the entire range of ACh concentrations. The analysis provides estimates of rate constants for ACh association and dissociation and of apparent rate constants for opening and closing of the channel (Table 2). The most significant effect of αV188M was an approximately 70-fold decrease in the channel gating efficiency θ, defined by the ratio of rate constants β/α, which was largely due to an approximately 80-fold decrease in β. The subtle decrease in the closing rate constant α in the mutant receptor, shown in Table 2, suggests that the briefer mutant receptor open times in Figure 4 derive from fewer missed closings in the mutant than wild-type receptor.
Table 2.
Kinetic analysis of AChRs with mutations in the α subunit
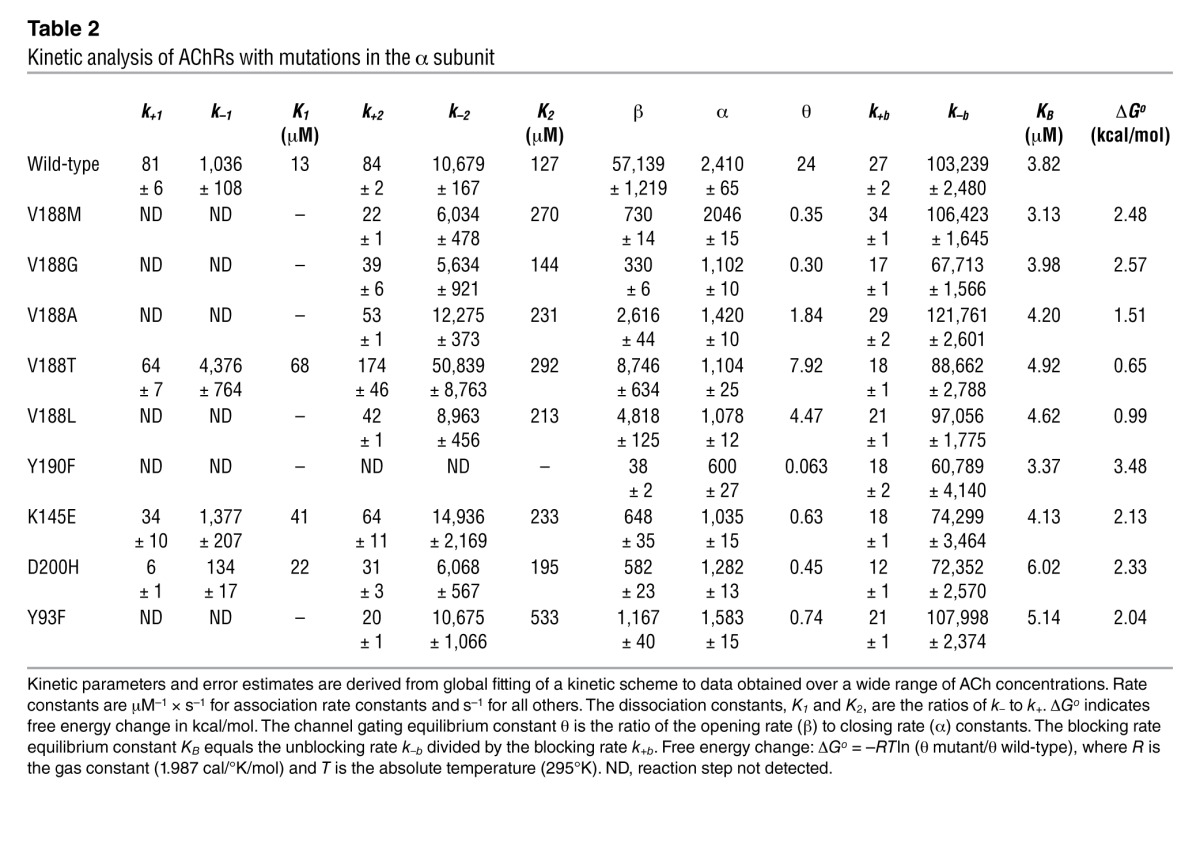
The fitted rate constants in Table 2 allow calculation of the channel open probability (Popen) as a function of ACh concentration. A plot of Popen over 3 orders of magnitude of ACh concentration revealed a marked decrease and rightward shift of Popen of the mutant relative to the wild-type AChR (Figure 6). The plotted points are well described by the Popen curve computed from the fitted rate constants and support the validity of the estimated rate constants shown in Table 2. The fitted rate constants in Table 2 also allow prediction of the mean burst duration of the αV188M receptor, approximated by (1 + β/k–2)/α. The predicted value of 0.55 ms agrees well with the independently determined burst duration of 0.68 ms in the presence of 1 μM ACh (Table 1). The shortened burst duration predicts an abnormally fast decay of synaptic currents typical of a fast-channel myasthenic syndrome (13). The probability the channel opens once it has bound two molecules of ACh, obtained from β/(β + k–2), was 0.84 for wild-type AChR and 0.11 for αV188M AChR, predicting a reduced peak of the synaptic response to nerve impulse.
Figure 6. Popen as a function of ACh concentration.

Symbols and vertical lines indicate mean ± SD and represent Popen computed from 29 to 300 clusters in a single patch. Smooth curves indicate the Popen predicted by the fitted rate constants shown in Table 2.
In summary, patch-clamp analysis of the kinetic properties of αV188M AChR revealed reduced gating efficiency, abnormally brief channel opening events, a decreased probability of channel opening by ACh, and a decreased amplitude of the synaptic response to ACh. The overall physiological consequence is a markedly reduced safety margin for neuromuscular transmission.
Other substitutions of α1 Val at codon 188.
Substituting Met for Val at codon 188 increases side chain size. To determine whether this can account for the adverse effects of the mutation, we replaced αVal188 by Gly, Ala, and Thr, all smaller than Val, and by Leu, which is larger than Val. Each substitution reduced rather than enhanced the apparent channel opening rate constant and gating efficiency, with Gly having the greatest and Thr the smallest effect (Table 2).
Mutant cycle analysis.
The available structural data show that the C-loop changes its conformation in the course of agonist binding. Without agonist, the C-loop adopts a range of opened-up conformations that allow agonist access to the binding pocket, but when the agonist binds the C-loop closes inward and traps the agonist (24, 26, 29, 30). Concomitantly, electrostatic interactions among local conserved residues change: without agonist, αAsp200 on β-strand 10 adjacent to the C-loop and αLys145 on β-strand 7 form a salt bridge, but upon binding the agonist, αTyr190 on the C-loop approaches αLys145, forming a hydrogen bond (20, 24) (Figure 7A). Mutation analysis combined with single-channel kinetic analysis revealed that these 3 residues, conserved among muscle and most neuronal AChRs, are energetically coupled in contributing to channel gating, and thus initiate the chain of events that ultimately open the receptor ion channel (20). Because high-resolution structural data showed that αVal188 lodges between αAsp200 and αTyr190 (Figure 7A), we used mutant cycle analysis (MCA) to determine whether αVal188 is part of the network of residues involved in the initial coupling of agonist binding to channel gating.
Figure 7. The α1 AChR subunit binding site and MCA of potentially interacting residues.

(A) Stereo view of the binding site at 1.9-Å resolution (PDB 2QC1) shows spatial disposition of the potentially interacting residues. (B) Cubic mutant cycle of energetic interactions between αV188, αY190, and αK145. (C) Cubic MCA of energetic interactions among αV188, αD200, and αK145. (D) Two-dimensional mutant cycle of energetic interaction among αV188 and αY93. In B–D, numbers on arrows show difference in free energy change (ΔΔG) between 2 different AChRs in kcal/mol. The indicated diagonal lines in planes show first-order coupling free energy (ΔΔGint) for a given pair of mutants. For the front planes in B and C, ΔΔGint = –0.78 kcal/mol; for the back planes in B and C, ΔΔGint = 0.13 and 0.78 kcal/mol, respectively.
A founding principle in MCA is that the change in free energy caused by a mutation depends on other residues in the protein (31). If mutation of a second residue affects the free energy change caused by mutation of the first, the two residues are energetically coupled and potentially interact directly. Evidence for direct interaction depends on independent structural data showing physical proximity, which for the α1 extracellular domain show that αVal188 is adjacent to several conserved residues essential for gating (17). For the present application of MCA, the free energy change caused by each mutation is computed from the channel gating equilibrium constant θ derived from kinetic fitting of single-channel dwell times, which equals –RTlnθ, where R is the gas constant and T absolute temperature. To compute energetic coupling between 2 residues, free energy changes were computed for wild-type, each single mutant, and the double mutant AChR, and the differences in free energy between pairs of residues are shown along 4 lines that form a square. If the free energy changes along parallel lines of the square are the same, the 2 residues are not coupled, whereas if they differ, they are coupled. For compact illustration, the present MCA data are cast as cubes, with each face of the cube representing the free energy of coupling between pairs of residues.
We first tested for energetic coupling between αVal188 and either αTyr190 or αAsp200. To this end, we generated mutations of each residue individually and of pairs of residues in the same subunit, recorded single-channel currents for each mutant AChR, computed the apparent channel gating equilibrium constant that yielded gating free energy, and constructed 2-dimensional mutant cycles shown as faces of a 3-dimensional cubic cycle (Figures 7, B and C). The analyses indicate that αVal188 couples strongly to both αTyr190 and αAsp200, with first-order coupling free energies (ΔΔGint) of –2.47 kcal/mol (Figure 7C, top face) and –2.53 cal/mol (Figure 7C, top face), respectively (Table 3). Thus, the contribution of Val188 to channel gating depends strongly on both αTyr190 and αAsp200. Because Val188 directly contacts αTyr190 and αAsp200 (17), the interdependence of the residues likely originates from direct inter-residue contact.
Table 3.
Inter-residue energetic coupling determined by MCA
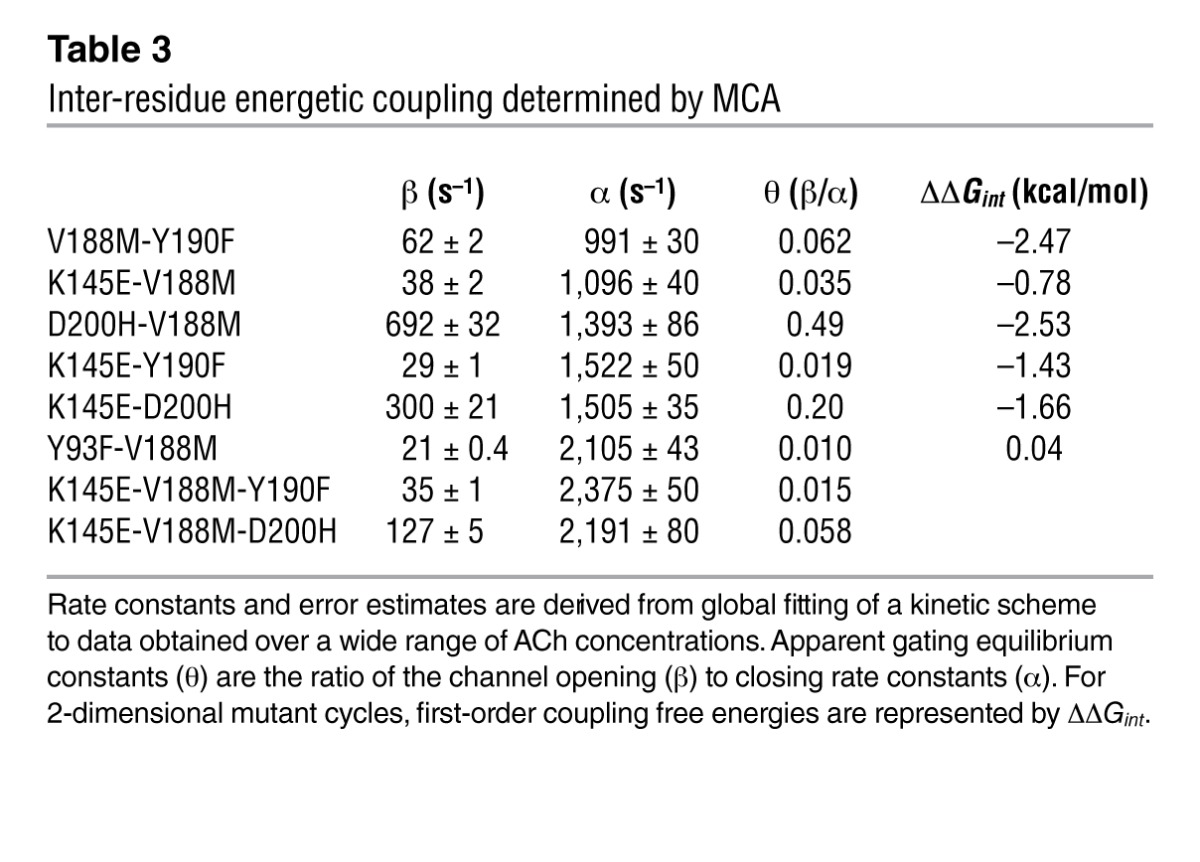
To determine whether a third residue affects coupling of 2 residues, we compared the coupling free energies for the 2 residues with and without a third mutated residue. Given that both αTyr190 and αAsp200 couple to αLys145 (20), we asked whether mutation of Lys145 affects the strong coupling of Val188 with either of these 2 residues. The analyses indicated that the αK145E mutation weakened coupling between αVal188 and αTyr190, with ΔΔGint decreasing from –2.47 (Figure 7C, top face) to –1.56 kcal/mol (Figure 7C, bottom face). Furthermore, the αK145E mutation also weakened coupling between αVal188 and αAsp200, with ΔΔGint decreasing from –2.53 (Figure 7C, top face) to –0.97 kcal/mol (Figure 7C, bottom face). Thus, the magnitude of energetic coupling between the pairs αV188/αY190 and αV188/αD200 depends on αLys145.
Conversely, we asked whether energetic coupling of αLys145 with αTyr190 and αAsp200 depends on αVal188. The analyses indicate that coupling between αLys145 and αTyr190 was weakened by αV188M, with ΔΔGint decreasing from –1.43 without αV188M (Figure 7B, left face, and Table 3) to –0.52 kcal/mol in the presence of αV188M (Figure 7B, right face). Furthermore, the αV188M mutation also weakened coupling between αLys145 and αAsp200, with ΔΔGint decreasing from –1.66 without αV188M (Figure 7C, left face, and Table 3) to –0.10 kcal/mol in the presence of αV188M (Figure 7C, right face). Thus, energetic coupling between the pairs αK145/αY190 and αK145/αD200 depends on αVal188.
Within the crystal structure of the α1 extracellular domain, αTyr190, αVal188, and αAsp200 form a contiguous chain, but αVal188 does not directly contact αLys145 (Figure 7A). Thus, we assessed energetic coupling between αVal188 and αLys145. Energetic coupling in this residue pair, shown by the front faces of the cubes in Figure 7, B and C, was low, with a ΔΔGint of only –0.78 kcal/mol (Table 3). Although αVal188 and αLys145 were weakly coupled, we found that the coupling free energy was affected by mutations of either αTyr190 or αAsp200. Comparison of the front and back faces (Figure 7B) shows that mutation of αTyr190 reduces coupling from –0.78 to 0.13 kcal/mol, while comparison of the front and back faces (Figure 7C) shows that mutation of αAsp200 altered coupling from –0.78 to 0.78 kcal/mol. Our collective findings show that residue pairs that exhibit strong energetic coupling make direct contact, whereas those that couple weakly do not. Moreover, αVal188 joins αTyr190, αAsp200, and αLys145 to form an interdependent tetrad that links agonist-mediated changes in the C-loop to channel gating.
A recently reported crystal structure of the agonist-occupied chimeric α7 ligand-binding domain provided evidence for a π-cation interaction between residues equivalent to αTyr93 and αLys145 in the α1 subunit (30). We therefore tested whether αVal188 is energetically coupled with αTyr93 in human α1 AChR. The 2-dimensional mutant cycle in Figure 7D shows that αVal188 and αTyr93 were not energetically coupled (Table 3). Thus, although mutation of either residue alone markedly alters channel gating, the combined mutations produce additive changes and thus αVal188 and αTyr93 are independent of each other. The lack of inter-residue coupling again correlates with the structural data showing that αTyr93 and αVal188 are spatially separate (Figure 7A).
Discussion
We identify 2 heteroallelic mutations in the AChR α1 subunit in a patient with severe myasthenic symptoms since birth. The first mutation, αG73C, markedly reduces surface expression of AChR in HEK cells. The second mutation, αV188M, allows robust AChR expression in HEK cells but has a profound effect on receptor activation. Thus, αV188M determines the phenotype. The functional role of αVal188 has not been investigated in previous mutagenesis studies. To our knowledge, αV188M is the first naturally occurring mutation in the C-loop in the Cys-loop receptor superfamily.
αV188M decreases gating efficiency approximately 70-fold and decreases the receptor’s affinity for ACh approximately 2-fold.
The fitted rate constants underlying activation of αV188M AChR are validated by close correspondence of the predicted burst duration with that recorded with 1 μM ACh for the mutant receptor and also by agreement between the predicted Popen and that derived from direct recordings (Figure 6). The findings indicate a compromised safety margin of neuromuscular transmission owing to a reduced Popen, an abnormally fast decay of the synaptic current, and a decreased amplitude of synaptic response.
The consequences of αV188M cannot be attributed to the larger bulk of Met than Val because substitutions with smaller (Gly, Ala, and Thr) or larger (Leu) residues all reduced gating efficiency. However, that Leu and Thr, which like Val have branched side-chains, reduce gating efficiency to a lesser extent than Gly and Ala, suggests that a branched side-chain residue at position α188 is important for the initial transduction of agonist binding to channel gating.
MCA showed that the functional consequences of the mutation αV188M are altered if either αTyr190 or αAsp200 is mutated simultaneously. Pairing αV188M with mutants of either αTyr190 or αAsp200 revealed large coupling free energies, around –2.5 kcal/mol, which is similar to the originally reported inter-residue coupling free energy for the pairs αD200/αK145 and αY190/αK145 (20). Analysis of the crystal structure of the α1 extracellular domain revealed that αTyr190, αVal188, and αAsp200 form a contiguous chain, so their mutual interdependence likely originates from direct interactions.
The crystal structure of the α1 extracellular domain is a reasonable model for the conformation of the resting state of AChR in the region encompassing αV188. This is likely because the α1 structure was obtained with bound α-bgt, and the local region is similar to that of acetylcholine-binding protein without bound agonist. In particular, in the α1 structure, αTyr190 on the C-loop is far from αLys145 on β-strand 7. In the available structures with agonist bound, the C-loop adopts a closed conformation that brings αTyr190 into close register with Lys145, forming a strong hydrogen bond (18, 19, 29, 30). Furthermore, in the crystal structure of an α7 chimeric ligand-binding domain with bound agonist, an Arg residue at the position equivalent to αVal188 establishes close register with the residue equivalent to αTyr190 through a π-cation interaction (30). Combined with these structural data, our findings suggest that αVal188, through an ideal steric fit, enables the proper interaction of αTyr190 with αLys145. Furthermore, in both the resting and agonist-bound conformations, the steric properties of αVal188 enable formation of a stable salt bridge between αAsp200 and αLys145. Thus, in the context of the available crystallography and mutagenesis data, our functional studies indicate that αVal188 is a key residue within the network of residues that initiates coupling of agonist binding to channel gating.
αVal188 is not conserved among subtypes of nicotinic AChR α subunits, with neuronal AChRs containing Ile, Leu, Arg, or Lys at equivalent positions.
Our findings show that αVal188 is an essential contributor to the triad of conserved residues that initiate coupling of agonist binding to channel gating, suggesting that among different types of nicotinic AChRs, sequence variations at positions equivalent to αVal188 enable tuning of the synaptic response according to physiological needs.
Methods
Mutation analysis.
We directly sequenced AChR α1, β1, δ, and ε subunit genes using genomic DNA. The mutations were traced with allele-specific PCR in family members and in 400 normal alleles of 200 unrelated controls.
Construction and expression of wild-type and mutant AChRs.
Sources of human α, β, ε, and δ subunit cDNAs were as previously described (32). All 4 cDNAs were subcloned into the CMV-based expression vector pRBG4 for expression in HEK293 cells (32). The artificial mutations were engineered into wild-type AChR subunit cDNAs in pRBG4 using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). The presence of each mutation and absence of unwanted mutations were confirmed by sequencing the entire inserts. HEK cells were transfected with plasmids comprising pRBG4-α, -β, -ε, and -δ and pEGFP-N1 at a ratio of 2:1:1:1:1, using FuGENE 6 transfection reagent (Roche).
α-bgt binding measurements.
The total number of [125I]α-bgt sites on the surface of transfected HEK cells was determined as previously described (32).
Patch-clamp recordings and single-channel kinetic analysis.
Recordings were obtained in the cell-attached configuration at a membrane potential of –80 mV at 22°C and with bath and pipette solutions containing (in mM): KCl, 142; NaCl, 5.4; CaCl2, 1.8; MgCl2, 1.7; HEPES, 10, pH 7.4. Single-channel currents were recorded using an Axopatch 200A amplifier (Axon Instruments) at a bandwidth of 50 kHz, digitized at 5-μs intervals using a Digidata 1322A (Axon Instruments) and recorded to hard disk using the program Clampex 8.2 (Axon Instruments). Recordings obtained with ACh at 1 μM or less were analyzed at a uniform bandwidth of 11.7 kHz with dead time of 15.3 μs imposed. Recordings obtained with ACh at 10 μM or more were analyzed with dead time at 25 μs at 10 kHz with TACx4.0.9 software (Bruxton). Dwell-time histograms were plotted on a logarithmic abscissa and fitted by the sum of exponentials by maximum likelihood (33).
To estimate rate constants underlying AChR activation, we employed desensitizing concentrations of ACh that cause events from a single channel to cluster into identifiable activation episodes (34). Clusters were identified as a series of closely spaced openings preceded and followed by closed intervals greater than a defined critical time. The critical time was determined by a method that misclassifies an equal number of events between 2 adjacent closed-time components (35). For each receptor, the critical time that provided the best fit for the closed-time histogram was chosen for the final analysis. Clusters with fewer than 5 openings were excluded from analysis. Individual clusters were examined for homogeneity by determination of the mean Popen and open duration for each cluster, and clusters within 2 SDs of the mean were accepted for further analysis (36, 37). The resulting global set of open and closed dwell times of wild-type and mutant AChRs were analyzed using the program MIL (QuB suite; http://www.qub.buffalo.edu/wiki/index.php/Main_Page), which uses an interval-based maximum likelihood method that also corrects for missed events (38). The kinetic analysis of wild-type and mutant receptors yielded a set of rate constants that were fitted to a scheme of receptor activation shown in Figure 5.
For each type of AChR, single-channel dwell times obtained over a range of ACh concentrations were fitted simultaneously. Data were obtained over a range of ACh concentrations from 10 to 1,000 μM for wild-type, αV188A, and αK145E AChRs; from 30 to 1,000 μM for αV188M, αV188G, αV188T, αV188L, and αD200H AChRs; from 100 to 1,000 μM for αY93F and αK145E/αD200H AChRs; and 1,000 μM for others. The final set of rate constants was checked by superimposing probability density functions calculated from the rate constants on the experimental dwell time histograms and by the ability of the rate constants to predict burst length at low ACh concentrations and the Popen (39, 40).
MCA.
We used MCA to determine whether the functional contributions of 2 residues in the AChR, X and Y, are interdependent. The analysis depends on examining the functional consequences of mutating residue X without and with mutation of residue Y and quantifying the free energy change associated with a given state change for the mutants AChRX, AChRY, and AChRXY. In the case of the AChR, the relevant state change was defined by the channel gating equilibrium constant of the di-liganded receptor, θ, which has an associated free energy change ΔG = –RTlnθ. Although recent studies show that θ is a composite of closed-state priming and channel gating steps (28) and thus is an apparent equilibrium constant, it still remains a highly sensitive measure of inter-residue coupling. The changes in gating free energy due to the mutations X, Y, and XY relative to the wild-type are designated ΔΔGX, ΔΔGY, and ΔΔGXY. These terms are related to the free energy of inter-residue interaction, ΔΔGint, as follows: ΔΔGXY= ΔΔGX + ΔΔGY + ΔΔGint (31, 41, 42). Given this relationship and noting that ΔΔGs for the individual mutant AChR is ΔGX – ΔGW, ΔGY – ΔGW, ΔGXY – ΔGW, ΔΔGint can be calculated from –RTln[(θW × θXY)/(θX × θY)], where θ is the measured channel gating equilibrium constant for wild-type (W) and mutant (X, Y, XY) AChRs. To determine interactions between 3 sets of residues, we used cubic MCA, where the faces of the cube are derived from 2-dimensional mutant cycles.
Statistics.
Wild-type and mutant values in Table 1 were compared by the 2-tailed Student’s t test. Standard error estimates of activation rate constant of wild-type and mutant receptors shown in Tables 2 and 3 were computed by the MIL program from the curvature of the likelihood function at its maximum (36). A P value less than 0.05 was considered significant.
Study approval.
All human studies were in accordance with the guidelines of, and approved by, the Institutional Review Board of the Mayo Clinic. The patient reported in this study provided informed consent to participate in the study.
Acknowledgments
This work was supported by NIH grants to A.G. Engel (NS-6277) and to S.M. Sine (NS-31744) and by a Muscular Dystrophy Association Grant to A.G. Engel.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(7):2613–2621. doi:10.1172/JCI63415.
See the related Commentary beginning on page 2356.
References
- 1.Engel AG. Current status of the congenital myasthenic syndromes. Neuromuscul Disord. 2012;22(2):99–111. doi: 10.1016/j.nmd.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Engel AG, Ohno K, Sine SM. Sleuthing molecular targets for neurological diseases at the neuromuscular junction. Nat Rev Neurosci. 2003;4(5):339–352. doi: 10.1038/nrn1101. [DOI] [PubMed] [Google Scholar]
- 3.Ohno K, et al. Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proc Natl Acad Sci U S A. 2001;98(4):2017–2022. doi: 10.1073/pnas.98.4.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ohno K, Brengman JM, Tsujino A, Engel AG. Human endplate acetylcholinesterase deficiency caused by mutations in the collagen-like tail subunit (ColQ) of the asymmetric enzyme. Proc Natl Acad Sci U S A. 1998;95(16):9654–9659. doi: 10.1073/pnas.95.16.9654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donger C, et al. Mutation in the human acetylcholinesterase-associated gene, COLQ, is responsible for congenital myasthenic syndrome with end-plate acetylcholinesterase deficiency. . Am J Hum Genet. 1998;63(4):967–975. doi: 10.1086/302059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohno K, et al. Rapsyn mutations in humans cause endplate acetylcholine receptor deficiency and myasthenic syndrome. Am J Hum Genet. 2002;70(4):875–885. doi: 10.1086/339465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huze C, et al. Identification of an agrin mutation that causes congenital myasthenia and affects synapse function. Am J Hum Genet. 2009;85(2):155–167. doi: 10.1016/j.ajhg.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chevessier F, et al. MUSK, a new target for mutations causing congenital myasthenic syndrome. Hum Mol Genet. 2004;13(24):3229–3240. doi: 10.1093/hmg/ddh333. [DOI] [PubMed] [Google Scholar]
- 9.Beeson D, et al. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science. 2006;313(5795):1975–1978. doi: 10.1126/science.1130837. [DOI] [PubMed] [Google Scholar]
- 10.Selcen D, et al. Myasthenic syndrome caused by plectinopathy. Neurology. 2011;76(4):327–336. doi: 10.1212/WNL.0b013e31820882bd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Senderek J, et al. Hexosamine biosynthetic pathway mutations cause neuromuscular transmission defect. Am J Hum Genet. 2011;88(2):162–172. doi: 10.1016/j.ajhg.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsujino A, et al. Myasthenic syndrome caused by mutation of the SCN4A sodium channel. . Proc Natl Acad Sci U S A. 2003;100(12):7377–7382. doi: 10.1073/pnas.1230273100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sine SM, Engel AG. Recent advances in Cys-loop receptor structure and function. Nature. 2006;440(7083):448–455. doi: 10.1038/nature04708. [DOI] [PubMed] [Google Scholar]
- 14.Galzi JL, Revah F, Bessis A, Changeux JP. Functional architecture of the nicotinic acetylcholine receptor: from electric organ to brain. Annu Rev Pharmacol Toxicol. 1991;31:37–72. doi: 10.1146/annurev.pa.31.040191.000345. [DOI] [PubMed] [Google Scholar]
- 15.Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4 Å resolution. J Mol Biol. 2005;346(4):967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 16.Sine SM. The nicotinic receptor ligand binding domain. J Neurobiol. 2002;53(4):431–446. doi: 10.1002/neu.10139. [DOI] [PubMed] [Google Scholar]
- 17.Dellisanti CD, Yao Y, Stroud JC, Wang ZZ, Chen L. Crystal structure of the extracellular domain of the nAChR α1 bound to α-bungarotoxin at 1.94 Å resolution. Nat Neurosci. 2007;10(8):953–962. doi: 10.1038/nn1942. [DOI] [PubMed] [Google Scholar]
- 18.Brejc K, van Dijk WV, Schuurmans M, van der Oost J, Smit AB, Sixma TK. Crystal structure of ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 2001;411(6835):269–276. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- 19.Hansen SB, Sulzenbacher G, Huxford T, Marchot P, Taylor P, Bourne Y. Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. . EMBO J. 2005;24(20):3635–3646. doi: 10.1038/sj.emboj.7600828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mukhtasimova N, Free C, Sine SM. Initial coupling of binding to gating mediated by conserved residues in muscle nicotinic receptor. J Gen Physiol. 2005;126(1):23–39. doi: 10.1085/jgp.200509283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee WY, Sine SM. Principal pathway coupling agonist binding to channel gating in nicotinic receptors. Nature. 2005;438(7065):243–247. doi: 10.1038/nature04156. [DOI] [PubMed] [Google Scholar]
- 22.Shen XM, Fukuda T, Ohno K, Sine SM, Engel AG. Congenital myasthenia-related AChR δ subunit mutation interferes with intersubunit communication essential for channel gating. J Clin Invest. 2008;118(5):1867–1876. doi: 10.1172/JCI34527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee WY, Free CR, Sine SM. Nicotinic receptor interloop proline anchors β1-β2 and Cys loops in coupling agonist binding to channel gating. J Gen Physiol. 2008;132(2):265–278. doi: 10.1085/jgp.200810014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang HL, et al. Single-channel current through nicotinic receptor produced by closure of binding C-loop. Biophys J. 2009;96(9):3582–3590. doi: 10.1016/j.bpj.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao F, et al. Agonist mediated conformational changes in acetylcholine-binding protein revealed by simulation and intrinsic tryptophan fluorescence. J Biol Chem. 2005;280(9):8443–8451. doi: 10.1074/jbc.M412389200. [DOI] [PubMed] [Google Scholar]
- 26.Cheng X, Wang H, Grant B, Sine SM, McCammon A. Targeted molecular dynamics study of C-loop closure and channel gating in nicotinic receptors. PLoS Comput Biol. 2006;2(9):e134. doi: 10.1371/journal.pcbi.0020134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee WY, Free CR, Sine SM. Binding to gating transduction in nicotinic receptors: Cys-loop energetically couples to pre-M1 and M2-M3 regions. J Neurosci. 2009;29(10):3189–3199. doi: 10.1523/JNEUROSCI.6185-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mukhtasimova N, Lee WY, Wang HL, Sine SM. Detection and trapping of intermediate states priming nicotinic receptor channel opening. Nature. 2009;459(7245):451–454. doi: 10.1038/nature07923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Celie PH, van Rossum-Fikkert SE, van Dijk WJ, Brejc K, Smit AB, Sixma TK. Nicotine and carbamycholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron. 2004;41(6):907–914. doi: 10.1016/S0896-6273(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 30.Li SX, et al. Ligand-binding domain of an α7-nicotinic receptor chimera and its complex with agonist. Nat Neurosci. 2011;14(10):1253–1259. doi: 10.1038/nn.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Horovitz A, Fersht A. Strategy for analyzing the co-operativity of intramolecular interactions in peptides and proteins. J Mol Biol. 1990;214(3):613–617. doi: 10.1016/0022-2836(90)90275-Q. [DOI] [PubMed] [Google Scholar]
- 32.Ohno K, et al. Congenital myasthenic syndrome caused by decreased agonist binding affinity due to a mutation in the acetylcholine receptor δ subunit. Neuron. 1996;17(1):157–170. doi: 10.1016/S0896-6273(00)80289-5. [DOI] [PubMed] [Google Scholar]
- 33.Sigworth FJ, Sine SM. Data transformation for improved display and fitting of single-channel dwell time histograms. Biophys J. 1987;52(6):1047–1054. doi: 10.1016/S0006-3495(87)83298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakmann B, Patlak J, Neher E. Single acetylcholine-activated channels show burst kinetics in the presence of desensitizing concentrations of agonist. Nature. 1980;286(5768):71–73. doi: 10.1038/286071a0. [DOI] [PubMed] [Google Scholar]
- 35.Colquhoun D, Sakmann B. Fast events in single channel currents activated by acetylcholine and its analogues at the frog muscle end-plate. J Physiol. 1985;369:501–557. doi: 10.1113/jphysiol.1985.sp015912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qin F, Auerbach A, Sachs F. Maximum likelihood estimation of aggregated Markov process. Pro Biol Sci. 1997;264(1380):375–383. doi: 10.1098/rspb.1997.0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen XM, et al. Mutation causing severe myasthenia reveals functional asymmetry of AChR signature Cys-loops in agonist binding and gating. J Clin Invest. 2003;111(4):497–505. doi: 10.1172/JCI16997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qin F, Auerbach A, Sachs F. Estimating single-channel kinetic parameters from idealized patch-clamp data containing missed events. Biophys J. 1996;70(1):264–280. doi: 10.1016/S0006-3495(96)79568-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Colquhoun D, Hawkes AG. Single–channel Recording. 1995. The principles of the stochastic interpretation of ion channel mechanisms. In: Sakmann B, Neher E, eds. pp. 397–482. New York, New York, USA: Plenum Press; [Google Scholar]
- 40.Colquhoun D, Hawkes AG. Single–channel Recording. 1995. A Q-matrix cookbook: how to write only one program to calculate the single-channel and macroscopic predictions for any kinetic mechanism. In: Sakmann B, Neher E, eds. pp. 589–633. New York, New York, USA: Plenum Press; [Google Scholar]
- 41.Wells JA. Additivity of mutational effects in proteins. Biochemistry. 1990;29(37):8509–8517. doi: 10.1021/bi00489a001. [DOI] [PubMed] [Google Scholar]
- 42.Horovitz A. Double-mutant cycles: a powerful tool for analyzing protein structure and function. Fold Des. 1996;1(6):R121–R126. doi: 10.1016/S1359-0278(96)00056-9. [DOI] [PubMed] [Google Scholar]