

Abstract
Cancer development is complex and involves several layers of interactions and pleotropic signalling mechanisms leading to progression. Cancer cells associate with resident stromal fibroblasts, smooth muscle cells, macrophages, endothelium, neurons and migrating cells at metastatic sites and phenotypically and genotypically activate them. These become an integral part of the cancer cell community through activated cell signalling mechanisms. During this process, the cancer cells and cells in the cancer microenvironment “co-evolve” in part due to oxidative stress, and acquire the ability to mimic other cell types (which can be termed osteomimicry, vasculomimicry, neuromimicry and stem cell mimicry), and undergo transition from epithelium to mesenchyme with definitive behavioral modifications. In our laboratory, we demonstrated that prostate cancer cells co-evolve in their genotypic and phenotypic characters with stroma and acquire osteomimetic properties allowing them to proliferate and survive in the skeleton as bone metastasis. Several signalling interactions in the bone microenvironment, mediated by reactive oxygen species, soluble and membrane bound factors, such as superoxide, β2microglobulin and RANKL have been described. Targeting the signalling pathways in the cancer-associated stromal microenvironment in combination with known conventional therepeutic modalities could have a synergistic effect on cancer treatment. Since cancer cells are constantly interacting and acquiring adaptive and survival changes primarily directed by their microenvironment, it is important to delineate these interactions and co-target both cancer and stroma to improve the treatment and overall survival of cancer patients.
Keywords: prostate cancer, bone metastasis, stroma based plasticity, osteomimicry and cancer plasticity
1. Introduction
Prostate cancer is a complex disease. Cancer progression must involve both genetic and behavioral changes in cancer cells and these changes are in part driven by the cancer associated stromal cells and tumor microenvironment [1–2]. In this review, we focus our analysis on the following events in the primary and metastatic sites, using human prostate cancer as a model. These are: 1) oxidative stress and hypoxia 2) co-evolution of cancer and stromal cells; 3) exhibition of mimicry by cancer cells to gain increased functional and renewal diversities to survive in the “hostile” new microenvironment, defined by sites of dissemination; 4) activation of cancer cell growth, invasion and metastasis programs through the process of epithelial to mesenchymal transition (EMT); 5) bone metastasis defined by interactions between the bone microenvironment and prostate cancer cells, which is largely responsible for prostate cancer lethality; and 6) the biologic significance and therapeutic implications of understanding interactions between the tumor and its microenvironment. We summarize our recent laboratory approaches to tackle the problem of tumor-stroma microenvironment interaction with the hope of developing and advancing new therapeutic targeting of prostate cancer bone and soft tissue metastases.
2. Prostate carcinogenesis and oxidative stress within the tumor microenvironment
The normal prostate epithelium consists of prostatic ducts with four kinds of cells, the basal cell, stem cell, secretory luminal cells and neuroendocrine cells. The stromal component consists of smooth muscle, fibroblasts, vascular endothelial cells, nerve cells, inflammatory cells, insoluble matrix and soluble factors (Figure 1A). Studies by De Marzo et al. highlight the role of inflammation in prostate cancer, suggesting that atrophic lesions are an early event in prostate carcinogenesis. The simple atrophic lesions (prostate inflammatory atrophy or PIA) lack papillary infoldings, with decreased luminal cytoplasmic volume, with scattered mononuclear cells in the luminal and stromal compartments [3]. The macrophages in the tumor microenvironment produce ROS and reactive nitrogen species. The resulting increases in superoxide (O2.−), hydrogen peroxide (H2O2), hydroxyl radical and free iron, damage DNA causing genetic mutations and initiate cancer progression. Recent studies have identified some of the molecular changes associated with prostate atrophy and these include nonclonal p53 mutations [4] androgen receptor mutation [5], hypermethylation of the CpG island of gluthathione tranferase-P1 (GSTP1) promoter [6] (Figure 1B). These mutations initiate high grade prostate intraepithelial neoplasia and progressive prostate cancer [7].
Figure 1.
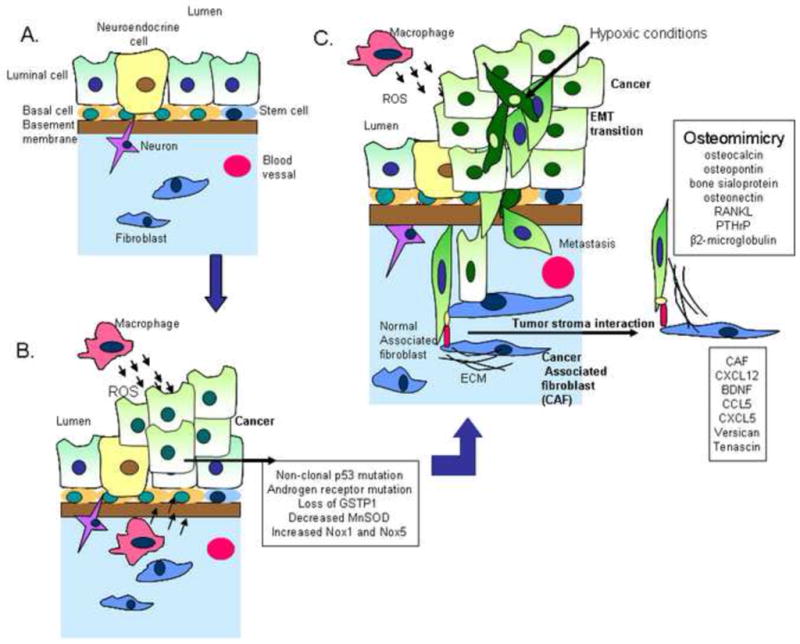
Tumor stroma interactions in the prostate microenvironment. A. Normal prostate microenvironment contains luminal cells, basal cells, neuroendocrine cells, stem cells and the stroma component contains fibroblast and neurons. B. Prostate cancer initiation occurs with increased reactive oxygen species generation and increased mutation in cancer cells. The mutations observed are non-clonal p53 and androgen receptor mutations and alterations in the redox environment such as decreased glutathione transferase (GSTP1) and manganese superoxide dismutase (MnSOD) function and increased NADPH oxidase 1. C. As the cancer advances it interacts with the microenvironment and these interactions enter a vicious cycle promoting cancer aggressiveness. As tumors grow large, they develop hypoxia and limited nutrients, conditions which can induce EMT and increase mobility. Additionally the cancer gains the characteristic of mimicry even before the cancer cells metastasize to the bone. Some of the genes involved in osteomimicry are β2-microglobulin, PTHrP, RANKL and other bone proteins secreted by the cancer cell, such as, osteocalcin, osteopontin, bone sialoprotein and osteonectin. Additionally cancer associated fibroblasts also undergo alterations, such as increases in, brain derived neurotrophic factor (BDNF), CCL5, CXCL5, CXCL12, versican and tenascin.
2.1 Oxidative stress in prostate cancer
Cancer cells have a pro-oxidant environment due to increased ROS such as O2.− and H2O2 [8]. This is a consequence of increasing superoxide generating enzymes and down regulation on superoxide scavenging enzyme. NADPH oxidase (Nox) is a superoxide generating enzyme. Several Nox members exist of which Nox1 and Nox5 are upregulated in prostate cancer. Ectopic expression of Nox1 in prostate cancer cells has been shown to enhance growth, angiogenesis and tumorigenicity [9]. Nelson et al observed loss of glutathione tranferase-P1 (GSTP1) function in almost all prostate cancer cases examined [10]. GSTP1 is a detoxification enzyme and conjugates glutathione to toxic electrophilic compounds such as xenobiotics and chemotherapeutic agents. Decrease in GSTP1 increases toxic compounds and induces oxidative stress [11]. Another superoxide scavenging enzyme which is down regulated in cancer cells is the mitochondrial manganese superoxide dismutase (MnSOD) [12, 13]. Overexpression of MnSOD has been shown to inhibit the growth of androgen independent prostate cancer cells [14]. Alterations in these enzymes play a crucial role in prostate cancer development.
2.2 Hypoxic microenvironment
The tumor microenvironment is constantly changing and cancer cells adapt, evolve and survive during this process. When cancer cells divide uncontrollably, they form larger tumors. As a consequence there is limited availability of nutrients and oxygen in the microenvironment. Cancer cells are exposed to intermittent hypoxic (lack of oxygen) conditions. The ROS signaling mechanisms in the cancer cells determine the fate of cancer cells in response to hypoxia. Recent studies highlight the importance of superoxide signaling in hypoxic conditions [15]. Cancer cells with downregulated MnSOD have increased superoxide, these cells upregulate hypoxia inducible factor (HIF-1a) transcription factor and induce angiogenesis and tumorigenicity [15, 16]. Whereas MnSOD overexpressing cancer cells, do hot have high superoxide levels and do not upregulate HIF-1a in response to hypoxia [15, 17]. As a result these cells were shown to have significantly decreased angiogenesis and tumorigenicity [16]. These results demonstrate that MnSOD and superoxide play a crucial role in tumor progression in response to changes such as hypoxic conditions within the tumor microenvironment. HIF-1a also plays an essential role in the angiogenic-osteogenic coupling in the bone microenvironment [18].
3 Prostate microenvironment and cancer associated fibroblasts
Tissue and cell recombination studies demonstrate the important regulatory role of fibromuscular stroma and stromal fibroblasts in prostate development and prostate carcinogenesis. In these studies, urogenital sinus mesenchyme (UGM) or embryonic/adult stromal fibroblasts were shown to drive the growth of UG epithelium [19–22] and prostate cancer [23]. Using a tissue recombination technique, it was demonstrated that in AR-defective testicular feminized mice (Tfm) the UGM tissue was not able to generate a normal prostate gland, whereas the wild type AR expressing UGM did. These studies suggest that AR signaling from the stroma regulates the development and differentiation of the normal prostate epithelium [20]. Using cell recombination studies, the progression of prostate cancer from androgen-dependent to androgen-independent states and the subsequent progression to bone metastatic phenotypes can be achieved by cellular interactions between prostate cancer and prostate or bone stromal cells in mice in vivo or when co-cultured under three dimensional (3D) conditions [1, 2]. These findings, taken together, emphasized the important role of the stromal and tumor microenvironment in prostate cancer progression.
3.1 Cancer-stroma interactions
Fibroblasts associated with cancer tissue or cancer associated fibroblasts (CAF) are structurally and functionally different from fibroblasts adjacent to normal epithelium [24]. These cells exhibit marked differences in gene expression profiles and have been shown to predict the progression of prostate cancer [25]. We demonstrated the reciprocal cellular interaction between prostate cancer and CAF or stromal fibroblasts from different zonal origin [2, 26, 27]. Using LNCaP, an androgen responsive marginally tumorigenic human prostate cancer cell line, co-cultured with microcarrier beads previously seeded with prostate or bone stromal cells of the human prostate gland or human bone, under 3D culture system, we demonstrated permanent nonrandom genetic and phenotypic changes. LNCaP cells underwent androgen independent and metastatic progression [2]. Cancer cells and stromal cells interact through physical contact, or through soluble factors or insoluble ECM factors. CAFs from clinical patient samples and from prostate and bone origin from 3D studies were assayed for gene expression changes. These stromal fibroblast which interacted with cancer cells, has increased levels of brain derived neurotropic factor (BDNF), chemokines, CCL5 and CXCL5, versican, tenascin, connective tissue growth factor, stromal cell derived factor-1 (SDF-1/CXCL12), and HIF-1a [26] (Figure 1C). These were validated using clinical tissue or serum samples obtained from bone metastatic prostate cancer patients [26]. Other studies demonstrate the role of stromal soluble factors interacting with receptors on prostate cancer cells. The stromal factors include VEGF, bFGF, HGF/SF, TGF-β, IGF-1, IL-6 and KGF [28]. These studies highlight the bidirectional interactions and co-evolution of tumor stroma in cancer progression.
3.2 Osteomimicry
Prostate cancer cells express and secrete several proteins which are highly restricted to bone, such as osteocalcin (OC), osteopontin (OPN), bone sialoprotein (BSP) and osteonectin (ON). This is termed osteomimicry (Figure 1C). It has been observed that prostate cancer cells can form mineralizing bone under certain cell culture conditions [29–31]. Prostate cancer cells have been shown to increase receptor activator NF-3B ligand (RANKL) and parathyroid hormone related peptide (PTHrP), which play a role in bone turnover [32, 33]. Further studies demonstrated that β2-Microglobulin (β2M) is a key regulator of osteomimicry [34]. β2M is overexpressed in prostate cancer bone metastatic tissues and induces osteomimetic properties in cancer cells by activating the cAMP-PKA pathway, leading to the phosphorylation of CREB. pCREB was shown to bind to CRE sites on OC and BSP promoters [34].
3.3 Epithelial to mesenchymal transition (EMT) in prostate cancer
EMT is an highly conserved embryonic program where polarized immotile epithelial cells transition to motile mesenchymal cells [35]. EMT is commonly associated with cancer migration, invasion and metastasis. This process was also shown to be reversible in morphology, termed mesenchymal to epithelial transition (MET) [36], but not necessarily in phenotype and behavior. In other words, cancer cells undergoing MET may acquire a more rather than a less aggressive phenotype. The canonical mediators of EMT in embryogenesis are the TGF-β superfamily and the WNT family [37]. The common feature of EMT is the loss of E-cadherin and an increase in vimentin. In cancer, EMT occurrence allows benign tumors to infiltrate surrounding tissues and metastasize (Figure 1C). In prostate cancer, EMT has been described in the androgen refractory prostate cancer (ARCaP) cell model [38, 39]. Several subclones of ARCaP cells were isolated with variable phenotypic features. Of these ARCaPE (ARCaP cells with epithelial phenotype) has a cobblestone morphology with expression of epithelial markers such as E-cadherin, cytokeratin 18 and cytokeratin 19 [39]. ARCaPM (ARCaP cells with mesenchymal phenotype) was derived form ARCaPE cells after treatment with soluble growth factors such as TGFβ1, EGF, IGF and β2M [39–42]. ARCaPE cells have low propensity for bone metastasis. ARCaPE cells after injecting intracardially underwent permanent mesenchymal transition. We tested and confirmed the roles of some soluble factors such a β2M, which is not only responsible for driving EMT and bone metastasis of human prostate cancer cells but also exerted the same effects by promoting EMT and bone metastasis in human breast, renal and lung cancer cells (Josson, S., Nomura, T., et al. unpublished data). The resulting ARCaPM cells had high levels of the mesenchymal markers such as vimentin, N-cadherin and Snail and exhibit 100% incidence of bone metastasis when injected intra-cardially [39, 40]. Thus the ARCaP model is a unique isogenic model for prostate cancer EMT and bone metastasis. Other factors which were differentially expressed in the ARCaP model are RANK-RANKL-OPG signaling which plays an important role in bone metabolism and metastasis [40, 41].
4. Bone microenvironment and prostate cancer interactions
Bone metastases are considered lethal and have been an active area of research for therapeutic development. This section highlights approaches that determine prostate cancer-bone cell interaction and potential therapeutic developments targeting cancer bone metastases.
4.1 Interactions of cancer cells with osteoblasts and osteoclasts
The organic bone matrix consists largely of type 1 collagen (95%) and minor amounts of proteoglycans and non-collagenous proteins (5%) [43]. The collagen matrix inter-disperses with non-collagenous matrix proteins, OPN, BSP, and ON [44, 45]. Osteoblasts (OB) and osteoclasts (OCl) cells are critical for bone formation and remodeling [46]. OBs are derived from mesenchymal stem cells (MSC). Bone morphogenetic proteins (BMPs) and growth factors stimulate MSC to form pre-osteoblasts and osteoblasts [43]. They also secrete a variety of growth factors such as IGFs, TGF-β, FGFs, and BMPs that are incorporated into the bone matrices [47] (Figure 2). Osteoclasts are derived from monocytes [43]. The osteoblasts express RANKL, which binds receptor actrivator NF-3B (RANK) on monocytes, which in the presence of macrophage-colony stimulating factor and RANKL promotes the fusion of several monocytes to form a multinucleated osteoclast. Osteoclasts are responsible for bone resorption. They secrete acid and proteases to degrade the bone matrix [43]. The OCl plough through the bone and release the growth factors, nutrients, calcium ions, deposited by the OB and also chemokines, cytokines and cell adhesion molecules. Once the cancer cells have established themselves in the bone microenvironment, the cancer cells, osteoblasts and osteoclasts enter into a “vicious cycle” resulting in increased bone turnover and enhancing the metastatic ability of cancer cells. Bone metastasis can be osteoblastic or osteolytic. Prostate cancer bone metastasis is often characterized as osteoblastic, with increased mineral density at the site of the cancer [48]. At the molecular level, prostate cancer bone metastasis has both osteoblastic as well as, osteolytic features with, osteoblastic lesions predominate.
Figure 2.
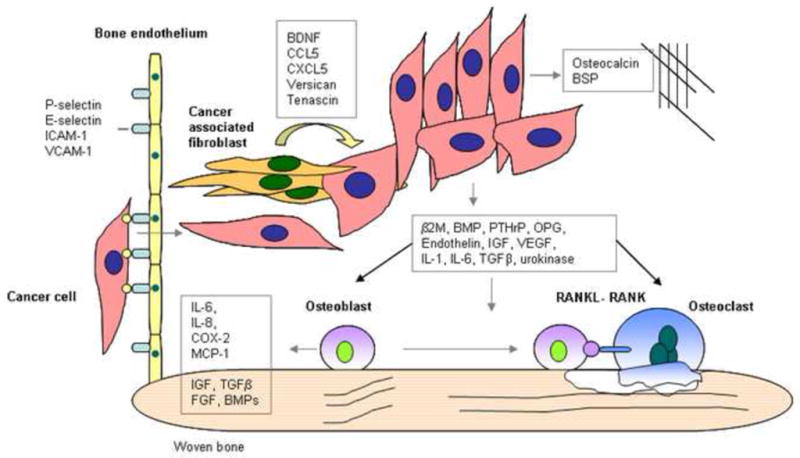
Cancer-stromal interaction in the bone microenvironment. Cancer cells gain access to bone through the bone endothelium which expresses specific adhesion molecules such as P-selectin, E-selectin, ICAM-1 and VCAM-1, which interact with cancer cells. Cancer cells modulate cancer-associated fibroblasts in the bone microenvironment. Cancer cells release bone specific proteins and other proteins which induce bone turnover, these include β2microgloblulin, BMPs, endothelin, IGF, IL-1, IL-6, OPG, PTHrP, TGFβ, urokinase and VEGF.
Prostate cancer cells produce several factors that have osteomimetic and osteoblastic growth stimulatory properties, such as β2M [34], BMPs [49] and endothelin [50] (Figure 2). β2M has been shown to activate the transcription of OC and BSP in prostate cancer cells. Whereas BMPs and endothelin can directly affect the maturation of osteoblasts, β2M, ET-1 and TGF-β signaling activation are all increased in bone metastatic prostate cancer patients. Other factors that work indirectly are PTHrP, which inhibits osteoblast apoptosis [51] and stimulates osteoclast maturation. Proteins such as prostate specific antigen and urinary plasminogen activator, activate latent forms of TGFβ, which is osteogenic [52]. The bone marrow endothelial cells express several adhesion molecules constitutively and simultaneously. These are P-selectin, E-selectin, ICAM-1 and VCAM-1, which are only expressed in other tissues during inflammation [43] (Figure 2).
4.2 RANKL-RANK-Osteoprotergerin (OPG) axis and communication between cancer and bone cells
The RANKL/RANK/OPG triad represents one of the best studied regulatory mechanisms in immunity and bone remodeling. As a member of the TNF superfamily of cytokines, RANKL functions by binding to its cognate receptor RANK, leading to trimerization and activation of the receptor [53]. Activated RANK interacts with TNF receptor associated proteins, a scaffold platform linking RANK receptor to NF3B and MAPK signaling pathways [54]. The function of RANKL/RANK signaling mainly affects differentiation and survival, especially differentiation of hematopoietic precursors to osteoclastic lineage and survival of matured osteoclasts during bone remodeling [55]. OPG contains an extracellular domain for binding to RANKL, but lacks intracellular domains for eliciting signaling transduction. OPG is thus a decoy receptor, which interferes with the osteoclastic RANKL/RANK signaling to prevent bone loss [56]. Approaches have been pursued for the development of a RANKL monoclonal antibody, denosumab, to prevent bone loss in prostate and breast cancer bone metastases by slowing down bone turnover, hence prostate and breast cancer growth on the skeleton. The RANKL/RANK/OPG system is highly relevant to prostate cancer progression since osteoblastic activity is initiated by osteoclastic resorption of the bone. The RANKL/RANK/OPG triad can be “hijacked” by tumor cells to promote prostate cancer bone metastasis. Some bone metastatic prostate cancer cells express RANKL [57] that can interact directly with RANK on hematopoietic stem cells in the bone microenvironment for osteoclast differentiation and maturation. As the bone is rich with growth factors and cytokines, bone resorption by matured osteoclasts causes the release of additional tropic factors in support of prostate cancer cell growth and survival. RANKL was found to be expressed in clinical prostate cancer specimens, and human prostate cancer cell lines of LNCaP lineage, found to express RANKL, induced osteoclastogenesis which can be blocked with OPG [33]. Some prostate cancer cells, however, may not express RANKL. It has been shown that these cells express growth factors such as PTHrP, TGFβ, and IL-1, which induce RANKL expression and facilitate osteoclastogenesis and bone metastases [58]. We showed that expression of RANKL in cancer cells may be induced by cancer-stromal interaction in the tumor microenvironment [40, 41]. In the ARCaP EMT model, RANKL expression is increased in the ARCaPM cells compared to the ARCaPE cells, suggesting a role in the EMT process [40, 41]. In summary, RANKL/RANK/OPG autocrine/paracrine communication represents a complex intercellular regulatory mechanism involving both cancer cells and constituents within the tumor microenvironment, and is an attractive therapeutic target for the control of bone turnover for the treatment of osteoporosis and cancer bone metastases (Figure 2).
4.4 Molecular approaches to co-target prostate cancer and cancer associated bone cells
Cancer host interactions play a fundamental role in directing cancer plasticity and progression and response to treatments such as hormone therapy, chemotherapy and radiation. The future development of novel therapies should co-target the cancer host interactions for better treatment efficacy. Since cancer and its associated stroma co-evolve, targeting only the cancer cells may not be sufficient. Several examples of co-targeting have been shown to be effective. Thalidomide, an anti-angiogenic drug, was used to target the endothelium along with chemotherapy to treat prostate cancer cells. Endothelin-1 is secreted by the prostate cancer cells and acts on osteoblasts via the ET-1A receptor. This interaction is co-targeted by the ET-1 receptor antagonist atrasentan in prostate cancer [59]. Antibodies have been used to co-target interactions between paracrine/ autocrine growth factor/ receptor interactions in the tumor and stroma. Additionally, cancer cell-ECM interactions have also been targeted using antibodies. These include antibodies against IGF-1R, PDGFR, integrins and stem cell hedgehog signaling [28, 60, 61]. Prostate cancer and bone cell interaction were co-targeted by using bisphosphonates to target bone turnover and a decoy RANK receptor, OPG, or an anti-RANKL monoclonal antibody, denosumab, to target the osteoblast/ prostate cancer interphase [32]. The use of radioactive nuclides, strontium-89 and samarium-153, has met with some limited success in prostate cancer patients with hormone-refractory bony metastases. Radium-223 has been shown to have a significant effect on bone metastasis [62, 63]. Adenoviral gene therapy was used to co-target prostate cancer and bone stroma using therapeutic cytotoxic herpes simplex-thymidine kinase and viral replication controlled by tissue specific and tumor restrictive human non-collgenous bone matrix protein promoters, hOC or hBSP [64].
5. Conclusions and future prospective
The field of tumor-stroma biology has expanded our understanding of cancer as more than a single cell disease. Rather, cancer involves reciprocal interaction and co-evolution between cancer cells and host stroma with ROS, soluble growth factors, chemokines and extracellular matrices serving as the key mediators. Our work and the work of others have shown that cancer cells and their associated stroma are remarkably plastic and capable of expressing genes mimicking the tumor microenvironment. These new understandings of cancer-stroma interaction raise the possibility of co-targeting not only the cancer cell component but also cancer-associated stroma, and blocking not only autocrine but also paracrine cell signaling. Further expansion of our understanding of tumor-stroma biology could lead to the successful development of more effective animal models to study the mechanisms of prostate cancer metastases. This will be a step toward the discovery of more effective therapeutic interventions for prostate cancer metastases through the interruption of cancer-stromal fibroblasts, cancer-bone, cancer-endothelium, cancer-nervous system and cancer-immune system communications.
Acknowledgments
We would like to thank Gary Mawyer and Murali Gururajan for helping with the editing of the article.
Abbreviations
- AR
androgen receptor
- ARCaP
androgen refractory prostate cancer
- β2M
β2-microglobulin
- BDNF
brain derived neurotropic factor
- BMP
bone morphogenetic protein
- BSP
bone sialoprotein
- CAF
cancer associated fibroblast
- CCL5
chemokine (C-C) ligand 5
- CXCL5
chemokine (C-X-C) ligand 5
- CXCL12
chemokine (C-X-C) ligand 12
- ECM
extracellular matrix
- EMT
epithelial mesenchymal transition
- FGF
fibroblast growth factor
- GSH
glutathione
- GSTP1
glutathione transferase-P1
- H2O2
hydrogen peroxide
- HIF
1α-hypoxia inducible factor-1alpha
- HGF/SF
hepatocyte growth factor/scatter factor
- IL-1
interleukin 1
- IL-6
interleukin 6
- KGF
keratinocyte growth factor
- MAPK
mitogen activated protein kinase
- MnSOD
manganese superoxide dismutase
- MSC
mesenchymal stem cells
- NF-κB
nuclear factor kappaB
- Nox
NADPH oxidase
- O2.−
superoxide
- OB
osteoblast
- OCl
osteoclast
- OC
osteocalcin
- ON
osteonectin
- OPG
osteoprotegerin
- OPN
osteopontin
- PDGFR
platelet derived growth factor receptor
- PKC
protein kinase C
- RANK
receptor activated NF-kappaB
- RANKL
receptor activated NF-kappaB ligand
- ROS
reactive oxygen species
- SDF-1
stromal derived factor-1
- TGFβ
transforming growth factor β
- TNF
tumor necrosis factor
- UGM
urogenital sinus mesenchyme
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thalmann GN, Anezinis PE, Chang SM, Zhau HE, Kim EE, Hopwood VL, et al. Androgen-independent cancer progression bone metastasis in the LNCaP model of human prostate cancer. Cancer Res. 1994;54:2577–2581. [PubMed] [Google Scholar]
- 2.Rhee HW, Zhau HE, Pathak S, Multani AS, Pennanen S, Visakorpi T, et al. Permanent phenotypic and genotypic changes of prostate cancer cells cultured in a three-dimensional rotating-wall vessel. In Vitro Cell Dev Biol Anim. 2001;37:127–140. doi: 10.1290/1071-2690(2001)037<0127:PPAGCO>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 3.De Marzo AM. The pathology of human prostatic atrophy and inflammation. In: Chung LWK, Simons JW, editors. Prostate Cancer: Biology, Genetics, and the New Therapeutics. 2. New Jersey: Humana Press; 2007. p. 34. [Google Scholar]
- 4.Tsujimoto Y, Takayama H, Nonomura N, Okuyama A, Aozasa K. Postatrophic hyperplasia of the prostate in Japan: histologic and immunohistochemical features and p53 gene mutation analysis. Prostate. 2002;52:279–287. doi: 10.1002/pros.10116. [DOI] [PubMed] [Google Scholar]
- 5.Tsujimoto Y, Takakuwa T, Takayama H, Nishimura K, Okuyama A, Aozasa K, et al. In situ shortening of CAG repeat length within the androgen receptor gene in prostatic cancer and its possible precursors. Prostate. 2004;58:283–290. doi: 10.1002/pros.10333. [DOI] [PubMed] [Google Scholar]
- 6.Nakayama M, Bennett CJ, Hicks JL, Epstein JI, Platz EA, Nelson WG, et al. Hypermethylation of the human glutathione S-transferase-pi gene (GSTP1) CpG island is present in a subset of proliferative inflammatory atrophy lesions but not in normal or hyperplastic epithelium of the prostate: a detailed study using laser-capture microdissection. Am J Pathol. 2003;163:923–933. doi: 10.1016/s0002-9440(10)63452-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Marzo AM. The pathology of human prostatic atrophy and inflammation. In: Chung LWK, Simons JW, editors. Prostate Cancer: Biology, Genetics, and the New Therapeutics. 2. New Jersey: Humana Press; 2007. p. 42. [Google Scholar]
- 8.Oberley LW, Buettner GR. Role of superoxide dismutase in cancer: a review. Cancer Res. 1979;39:1141–1149. [PubMed] [Google Scholar]
- 9.Lim SD, Sun C, Lambeth JD, Marshall F, Amin M, Chung L, et al. Increased Nox1 and hydrogen peroxide in prostate cancer. Prostate. 2005;62:200–207. doi: 10.1002/pros.20137. [DOI] [PubMed] [Google Scholar]
- 10.Nelson WG, De Marzo AM, Deweese TL, Lin X, Brooks JD, Putzi MJ, et al. Preneoplastic prostate lesions: an opportunity for prostate cancer prevention. Ann N Y Acad Sci. 2001;952:135–144. doi: 10.1111/j.1749-6632.2001.tb02734.x. [DOI] [PubMed] [Google Scholar]
- 11.DeWeese TL, Hruszkewycz AM, Marnett LJ. Oxidative stress in chemoprevention trials. Urology. 2001;57:137–140. doi: 10.1016/s0090-4295(00)00959-6. [DOI] [PubMed] [Google Scholar]
- 12.Liu B, Chen Y, St Clair DK. ROS and p53: a versatile partnership. Free Radic Biol Med. 2008;44:1529–1535. doi: 10.1016/j.freeradbiomed.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123–127. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Venkataraman S, Jiang X, Weydert C, Zhang Y, Zhang HJ, Goswami PC, et al. Manganese superoxide dismutase overexpression inhibits the growth of androgen-independent prostate cancer cells. Oncogene. 2005;24:77–89. doi: 10.1038/sj.onc.1208145. [DOI] [PubMed] [Google Scholar]
- 15.Kaewpila S, Venkataraman S, Buettner GR, Oberley LW. Manganese superoxide dismutase modulates hypoxia-inducible factor-1 alpha induction via superoxide. Cancer Res. 2008;68:2781–2788. doi: 10.1158/0008-5472.CAN-07-2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang HJ, Yan T, Oberley TD, Oberley LW. Comparison of effects of two polymorphic variants of manganese superoxide dismutase on human breast MCF-7 cancer cell phenotype. Cancer Res. 1999;59:6276–6283. [PubMed] [Google Scholar]
- 17.Wang M, Kirk JS, Venkataraman S, Domann FE, Zhang HJ, Schafer FQ, et al. Manganese superoxide dismutase suppresses hypoxic induction of hypoxia-inducible factor-1alpha and vascular endothelial growth factor. Oncogene. 2005;24:8154–8166. doi: 10.1038/sj.onc.1208986. [DOI] [PubMed] [Google Scholar]
- 18.Riddle RC, Khatri R, Schipani E, Clemens TL. Role of hypoxia-inducible factor-1alpha in angiogenic-osteogenic coupling. J Mol Med. 2009;87:583–590. doi: 10.1007/s00109-009-0477-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cunha GR, Lung B, Reese B. Glandular epithelial induction by embryonic mesenchyme in adult bladder epithelium of BALB/c mice. Invest Urol. 1980;17:302–304. [PubMed] [Google Scholar]
- 20.Cunha GR, Chung LW. Stromal-epithelial interactions--I. Induction of prostatic phenotype in urothelium of testicular feminized (Tfm/y) mice. J Steroid Biochem. 1981;14:1317–1324. doi: 10.1016/0022-4731(81)90338-1. [DOI] [PubMed] [Google Scholar]
- 21.Cunha GR, Chung LW, Shannon JM, Taguchi O, Fujii H. Hormone-induced morphogenesis and growth: role of mesenchymal-epithelial interactions. Recent Prog Horm Res. 1983;39:559–598. doi: 10.1016/b978-0-12-571139-5.50018-5. [DOI] [PubMed] [Google Scholar]
- 22.Chung LW, Cunha GR. Stromal-epithelial interactions: II. Regulation of prostatic growth by embryonic urogenital sinus mesenchyme. Prostate. 1983;4:503–511. doi: 10.1002/pros.2990040509. [DOI] [PubMed] [Google Scholar]
- 23.Camps JL, Chang SM, Hsu TC, Freeman MR, Hong SJ, Zhau HE, et al. Fibroblast-mediated acceleration of human epithelial tumor growth in vivo. Proc Natl Acad Sci U S A. 1990;87:75–79. doi: 10.1073/pnas.87.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dean JP, Nelson PS. Profiling influences of senescent and aged fibroblasts on prostate carcinogenesis. Br J Cancer. 2008;98:245–249. doi: 10.1038/sj.bjc.6604087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dakhova O, Ozen M, Creighton CJ, Li R, Ayala G, Rowley D, et al. Global gene expression analysis of reactive stroma in prostate cancer. Clin Cancer Res. 2009;15:3979–3989. doi: 10.1158/1078-0432.CCR-08-1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sung SY, Hsieh CL, Law A, Zhau HE, Pathak S, Multani AS, et al. Coevolution of prostate cancer and bone stroma in three-dimensional coculture: implications for cancer growth and metastasis. Cancer Res. 2008;68:9996–10003. doi: 10.1158/0008-5472.CAN-08-2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thalmann GN, Rhee H, Sikes RA, Pathak S, Multani A, Zhau HE, et al. Human Prostate Fibroblasts Induce Growth and Confer Castration Resistance and Metastatic Potential in LNCaP Cells. Eur Urol. 2009 doi: 10.1016/j.eururo.2009.08.026. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chung LW, Baseman A, Assikis V, Zhau HE. Molecular insights into prostate cancer progression: the missing link of tumor microenvironment. J Urol. 2005;173:10–20. doi: 10.1097/01.ju.0000141582.15218.10. [DOI] [PubMed] [Google Scholar]
- 29.Koeneman KS, Yeung F, Chung LW. Osteomimetic properties of prostate cancer cells: a hypothesis supporting the predilection of prostate cancer metastasis and growth in the bone environment. Prostate. 1999;39:246–261. doi: 10.1002/(sici)1097-0045(19990601)39:4<246::aid-pros5>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 30.Lecrone V, Li W, Devoll RE, Logothetis C, Farach-Carson MC. Calcium signals in prostate cancer cells: specific activation by bone-matrix proteins. Cell Calcium. 2000;27:35–42. doi: 10.1054/ceca.1999.0083. [DOI] [PubMed] [Google Scholar]
- 31.Thomas R, True LD, Bassuk JA, Lange PH, Vessella RL. Differential expression of osteonectin/SPARC during human prostate cancer progression. Clin Cancer Res. 2000;6:1140–1149. [PubMed] [Google Scholar]
- 32.Keller ET. The role of osteoclastic activity in prostate cancer skeletal metastases. Drugs Today (Barc) 2002;38:91–102. doi: 10.1358/dot.2002.38.2.820105. [DOI] [PubMed] [Google Scholar]
- 33.Zhang J, Dai J, Qi Y, Lin DL, Smith P, Strayhorn C, et al. Osteoprotegerin inhibits prostate cancer-induced osteoclastogenesis and prevents prostate tumor growth in the bone. J Clin Invest. 2001;107:1235–1244. doi: 10.1172/JCI11685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang WC, Wu D, Xie Z, Zhau HE, Nomura T, Zayzafoon M, et al. beta2-microglobulin is a signaling and growth-promoting factor for human prostate cancer bone metastasis. Cancer Res. 2006;66:9108–9116. doi: 10.1158/0008-5472.CAN-06-1996. [DOI] [PubMed] [Google Scholar]
- 35.Greenburg G, Hay ED. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J Cell Biol. 1982;95:333–339. doi: 10.1083/jcb.95.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yates C, Shepard CR, Papworth G, Dash A, Beer Stolz D, Tannenbaum S, et al. Novel three-dimensional organotypic liver bioreactor to directly visualize early events in metastatic progression. Adv Cancer Res. 2007;97:225–246. doi: 10.1016/S0065-230X(06)97010-9. [DOI] [PubMed] [Google Scholar]
- 37.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 38.Zhau HY, Chang SM, Chen BQ, Wang Y, Zhang H, Kao C, et al. Androgen-repressed phenotype in human prostate cancer. Proc Natl Acad Sci U S A. 1996;93:15152–15157. doi: 10.1073/pnas.93.26.15152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu J, Wang R, Xie ZH, Odero-Marah V, Pathak S, Multani A, et al. Prostate cancer metastasis: role of the host microenvironment in promoting epithelial to mesenchymal transition and increased bone and adrenal gland metastasis. Prostate. 2006;66:1664–1673. doi: 10.1002/pros.20488. [DOI] [PubMed] [Google Scholar]
- 40.Odero-Marah VA, Wang R, Chu G, Zayzafoon M, Xu J, Shi C, et al. Receptor activator of NF-kappaB Ligand (RANKL) expression is associated with epithelial to mesenchymal transition in human prostate cancer cells. Cell Res. 2008;18:858–870. doi: 10.1038/cr.2008.84. [DOI] [PubMed] [Google Scholar]
- 41.Zhau HE, Odero-Marah V, Lue HW, Nomura T, Wang R, Chu G, et al. Epithelial to mesenchymal transition (EMT) in human prostate cancer: lessons learned from ARCaP model. Clin Exp Metastasis. 2008;25:601–610. doi: 10.1007/s10585-008-9183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Graham TR, Zhau HE, Odero-Marah VA, Osunkoya AO, Kimbro KS, Tighiouart M, et al. Insulin-like growth factor-I-dependent up-regulation of ZEB1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res. 2008;68:2479–2488. doi: 10.1158/0008-5472.CAN-07-2559. [DOI] [PubMed] [Google Scholar]
- 43.Bussard KM, Gay CV, Mastro AM. The bone microenvironment in metastasis; what is special about bone? Cancer Metastasis Rev. 2008;27:41–55. doi: 10.1007/s10555-007-9109-4. [DOI] [PubMed] [Google Scholar]
- 44.Pinero GJ, Farach-Carson MC, Devoll RE, Aubin JE, Brunn JC, Butler WT. Bone matrix proteins in osteogenesis and remodelling in the neonatal rat mandible as studied by immunolocalization of osteopontin, bone sialoprotein, alpha 2HS-glycoprotein and alkaline phosphatase. Arch Oral Biol. 1995;40:145–155. doi: 10.1016/0003-9969(94)00144-z. [DOI] [PubMed] [Google Scholar]
- 45.Romanowski R, Jundt G, Termine JD, von der Mark K, Schulz A. Immunoelectron microscopy of osteonectin and type I collagen in osteoblasts and bone matrix. Calcif Tissue Int. 1990;46:353–360. doi: 10.1007/BF02554964. [DOI] [PubMed] [Google Scholar]
- 46.Marks SC, Odgren PR. Structure and development of the skeleton. In: Bilezikian JP, Raisz LG, Rodan GA, editors. Principles of bone biology. Vol. 1. New York: Academic; 2002. pp. 3–15. [Google Scholar]
- 47.Hauschka PV, Mavrakos AE, Iafrati MD, Doleman SE, Klagsbrun M. Growth factors in bone matrix. Isolation of multiple types by affinity chromatography on heparin-Sepharose. J Biol Chem. 1986;261:12665–12674. [PubMed] [Google Scholar]
- 48.Urwin GH, Percival RC, Harris S, Beneton MN, Williams JL, Kanis JA. Generalised increase in bone resorption in carcinoma of the prostate. Br J Urol. 1985;57:721–723. doi: 10.1111/j.1464-410x.1985.tb07040.x. [DOI] [PubMed] [Google Scholar]
- 49.Autzen P, Robson CN, Bjartell A, Malcolm AJ, Johnson MI, Neal DE, et al. Bone morphogenetic protein 6 in skeletal metastases from prostate cancer and other common human malignancies. Br J Cancer. 1998;78:1219–1223. doi: 10.1038/bjc.1998.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nelson JB, Hedican SP, George DJ, Reddi AH, Piantadosi S, Eisenberger MA, et al. Identification of endothelin-1 in the pathophysiology of metastatic adenocarcinoma of the prostate. Nat Med. 1995;1:944–949. doi: 10.1038/nm0995-944. [DOI] [PubMed] [Google Scholar]
- 51.Karaplis AC, Vautour L. Parathyroid hormone-related peptide and the parathyroid hormone/parathyroid hormone-related peptide receptor in skeletal development. Curr Opin Nephrol Hypertens. 1997;6:308–313. doi: 10.1097/00041552-199707000-00002. [DOI] [PubMed] [Google Scholar]
- 52.Rabbani SA, Gladu J, Mazar AP, Henkin J, Goltzman D. Induction in human osteoblastic cells (SaOS2) of the early response genes fos, jun, and myc by the amino terminal fragment (ATF) of urokinase. J Cell Physiol. 1997;172:137–145. doi: 10.1002/(SICI)1097-4652(199708)172:2<137::AID-JCP1>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 53.Hofbauer LC, Khosla S, Dunstan CR, Lacey DL, Boyle WJ, Riggs BL. The roles of osteoprotegerin and osteoprotegerin ligand in the paracrine regulation of bone resorption. J Bone Miner Res. 2000;15:2–12. doi: 10.1359/jbmr.2000.15.1.2. [DOI] [PubMed] [Google Scholar]
- 54.Darnay BG, Besse A, Poblenz AT, Lamothe B, Jacoby JJ. TRAFs in RANK signaling. Adv Exp Med Biol. 2007;597:152–159. doi: 10.1007/978-0-387-70630-6_12. [DOI] [PubMed] [Google Scholar]
- 55.Jimi E, Akiyama S, Tsurukai T, Okahashi N, Kobayashi K, Udagawa N, et al. Osteoclast differentiation factor acts as a multifunctional regulator in murine osteoclast differentiation and function. J Immunol. 1999;163:434–442. [PubMed] [Google Scholar]
- 56.Theoleyre S, Wittrant Y, Tat SK, Fortun Y, Redini F, Heymann D. The molecular triad OPG/RANK/RANKL: involvement in the orchestration of pathophysiological bone remodeling. Cytokine Growth Factor Rev. 2004;15:457–475. doi: 10.1016/j.cytogfr.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 57.Chen G, Sircar K, Aprikian A, Potti A, Goltzman D, Rabbani SA. Expression of RANKL/RANK/OPG in primary and metastatic human prostate cancer as markers of disease stage and functional regulation. Cancer. 2006;107:289–298. doi: 10.1002/cncr.21978. [DOI] [PubMed] [Google Scholar]
- 58.Angelucci A, Garofalo S, Speca S, Bovadilla A, Gravina GL, Muzi P, et al. Arachidonic acid modulates the crosstalk between prostate carcinoma and bone stromal cells. Endocr Relat Cancer. 2008;15:91–100. doi: 10.1677/ERC-07-0100. [DOI] [PubMed] [Google Scholar]
- 59.Kopetz ES, Nelson JB, Carducci MA. Endothelin-1 as a target for therapeutic intervention in prostate cancer. Invest New Drugs. 2002;20:173–182. doi: 10.1023/a:1015630513908. [DOI] [PubMed] [Google Scholar]
- 60.Uehara H, Kim SJ, Karashima T, Shepherd DL, Fan D, Tsan R, et al. Effects of blocking platelet-derived growth factor-receptor signaling in a mouse model of experimental prostate cancer bone metastases. J Natl Cancer Inst. 2003;95:458–470. doi: 10.1093/jnci/95.6.458. [DOI] [PubMed] [Google Scholar]
- 61.Wu JD, Odman A, Higgins LM, Haugk K, Vessella R, Ludwig DL, et al. In vivo effects of the human type I insulin-like growth factor receptor antibody A12 on androgen-dependent and androgen-independent xenograft human prostate tumors. Clin Cancer Res. 2005;11:3065–3074. doi: 10.1158/1078-0432.CCR-04-1586. [DOI] [PubMed] [Google Scholar]
- 62.Tu SM, Kim J, Pagliaro LC, Vakar-Lopez F, Wong FC, Wen S, et al. Therapy tolerance in selected patients with androgen-independent prostate cancer following strontium-89 combined with chemotherapy. J Clin Oncol. 2005;23:7904–7910. doi: 10.1200/JCO.2005.01.2310. [DOI] [PubMed] [Google Scholar]
- 63.Nilsson S, Franzen L, Parker C, Tyrrell C, Blom R, Tennvall J, et al. Bone-targeted radium-223 in symptomatic, hormone-refractory prostate cancer: a randomised, multicentre, placebo-controlled phase II study. Lancet Oncol. 2007;8:587–594. doi: 10.1016/S1470-2045(07)70147-X. [DOI] [PubMed] [Google Scholar]
- 64.Hsieh CL, Gardner TA, Miao L, Balian G, Chung LW. Cotargeting tumor and stroma in a novel chimeric tumor model involving the growth of both human prostate cancer and bone stromal cells. Cancer Gene Ther. 2004;11:148–155. doi: 10.1038/sj.cgt.7700665. [DOI] [PubMed] [Google Scholar]