

Abstract
Pharmacological targeting of individual ErbB receptors elicits antitumor activity, but is frequently compromised by resistance leading to therapeutic failure. Here, we describe an immunotherapeutic approach that exploits prevalent and fundamental mechanisms by which aberrant upregulation of the ErbB network drives tumorigenesis. A chimeric antigen receptor named T1E28z was engineered, in which the promiscuous ErbB ligand, T1E, is fused to a CD28 + CD3ζ endodomain. Using a panel of ErbB-engineered 32D hematopoietic cells, we found that human T1E28z+ T cells are selectively activated by all ErbB1-based homodimers and heterodimers and by the potently mitogenic ErbB2/3 heterodimer. Owing to this flexible targeting capability, recognition and destruction of several tumor cell lines was achieved by T1E28z+ T cells in vitro, comprising a wide diversity of ErbB receptor profiles and tumor origins. Furthermore, compelling antitumor activity was observed in mice bearing established xenografts, characterized either by ErbB1/2 or ErbB2/3 overexpression and representative of insidious or rapidly progressive tumor types. Together, these findings support the clinical development of a broadly applicable immunotherapeutic approach in which the propensity of solid tumors to dysregulate the extended ErbB network is targeted for therapeutic gain.
INTRODUCTION
The ErbB receptor family comprises four members, designated epidermal growth factor receptor (EGFR or ErbB-1), ErbB-2 (HER2 or neu), ErbB-3 and ErbB-4 (1–3). In the adult, all four receptors are widely expressed at low levels. However, overexpression of ErbB1 and/or ErbB2 was implicated in the pathogenesis of many carcinomas, including those derived from head and neck (4,5), breast (6,7), lung (8), gastrointestinal tract (4,9), prostate (10,11), gynecologic tract (12,13) and pancreas (14). Aberrant ErbB signaling promotes resistance to commonly used therapeutic modalities, including hormonal agents (15), chemotherapy (16) and radiotherapy (17). On the basis of the oncogene addiction concept, it is therefore logical that individual ErbB receptors have been targeted in patients with cancer. However, results have often failed to meet a priori expectations.
Rather than signaling in isolation, it is increasingly appreciated that ErbB receptors operate as a closely integrated spectrum of dimers (1–3). The synergistic and interdependent nature of the network is illustrated by the markedly greater transforming capacity of ErbB heterodimers than homodimers (18). Although all possible dimers have been detected, pairing is hierarchical (18). The ErbB2 orphan receptor lacks a high affinity ligand. Nonetheless, it is the preferred partner for other ErbB receptors and delivers potent tyrosine kinase-driven signaling, particularly when paired with ErbB1 or ErbB3 (7). Increasing evidence also supports the key importance of ErbB3 in transformation, a role that has been underestimated by the oncogene addiction paradigm (5,7,11,13,14). Although ErbB3 has a considerably diminished kinase activity, it is the primary hub by which the network recruits phosphatidylinositol 3-kinase signaling. In some models, ErbB3 is an obligate partner in tumorigenesis driven by ErbB1 and ErbB2 (19). In contrast to ErbB1-3, however, the role of ErbB4 in oncogenesis remains much less clear.
Pharmaceuticals directed individually against ErbB1 or ErbB2 have attracted considerable interest in the treatment of many cancer types. Inevitably, however, tumor cells acquire resistance to the selective pressure imposed by these agents. Often, this results from enhanced signaling by nontargeted ErbB receptor dimers (20–26). Recognition of this fact, coupled with the realization that the ErbB network is both integrated and dynamic, has resulted in the development of therapies directed against two or more family members (27–29). Even still, durable success may be difficult to attain owing to the profusion of ErbB dimers and panoply of alternative pathways that can provide necessary signals to tumor cells (30–32). To circumvent this, we developed a directly cytotoxic approach that couples T-cell activation with the detection of ErbB dimer overexpression on the tumor cell surface.
Chimeric antigen receptors (CARs) are fusion molecules in which a targeting moiety is coupled in series to hinge, transmembrane and activating endodomains (33,34). When expressed in T-cells, CARs engage a designated native “antigen” on the tumor cell, obviating the need for either HLA expression or antigen processing. Target binding is coupled with delivery of a tailored activating signal, leading to tumor cell destruction and incitement of secondary immune amplification mechanisms. To engineer a CAR with broad specificity for the ErbB network, we have exploited a promiscuous ErbB ligand named T1E as the targeting moiety (35). T1E is a chimeric polypeptide in which the N-terminal seven amino acids from human transforming growth factor (TGF)-α have been fused to the C-terminal 48 amino acids of epidermal growth factor (EGF) (Figure 1A). Like both parent cytokines, T1E binds with high affinity to the ErbB1-based homodimers and heterodimers. Uniquely, however, T1E also binds ErbB2/3 heterodimers with comparable affinity to the natural ligand (heregulins) but does not bind to ErbB2 or ErbB3 alone (35). We hypothesized that a T1E-based CAR would harness T-cell immunity against “driver” ErbB dimers implicated in the pathogenesis of several tumor types, leading to therapeutically beneficial responses.
Figure 1.
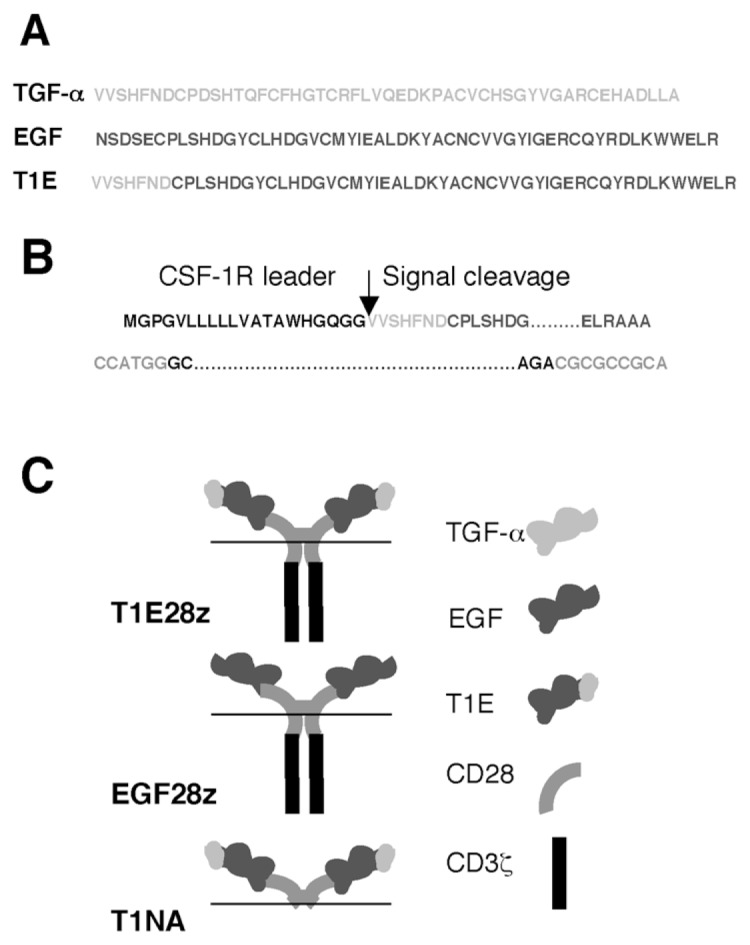
Structure of ErbB-specific CARs. (A) Amino acid sequence of mature forms of TGF-α, EGF and the derived chimeric T1E peptide. (B) Junction of T1E peptide with the leader derived from colony-stimulating factor-1 receptor (CSF-1R). (C) Cartoon structure of T1E28z and two control CARs (EGF28z, targeted with EGF; T1NA, T1E coupled with a truncated endodomain).
MATERIALS AND METHODS
Constructs
The leader sequence of the human colony-stimulating factor-1 receptor was placed upstream of T1E sequences to maximize probability of signal cleavage at the precise junction with the TGF-α N-terminus (SignalP 3.0 server; Figure 1B). The cDNA was synthesized as an Nco1-Not1 cassette by overlap extension polymerase chain reaction and was substituted for the scFv in SFG P28z (36) (Figure 1C).
The T1NA (T1E no activation domain) control CAR is identical, except that the endodomain has been truncated to contain only the membrane proximal three amino acids of CD28 (see Figure 1C). In the EGF28z construct (see Figure 1C), full-length mature EGF was placed downstream of a CD8α leader sequence (bases 1–60) to maximize the probability of signal cleavage at the junction with the EGF N-terminus (SignalP 3.0 server). The S28z CAR (37) contains an scFv derived from the SM3 hybridoma (MUC1 specific) and serves as an endodomain-matched control, since it does not bind MUC1 efficiently.
The 4αβ chimeric cytokine receptor was coexpressed with CARs using an intervening T2A peptide, placed downstream of a furin cleavage site (38). Firefly luciferase was expressed in the indicated tumor cell lines using the pBabe puro vector (37). Full-length MUC1 was expressed using the SFG retroviral vector as described (37).
ErbB cDNAs in pcDNA3 were gifts from Professor Y. Yarden (Weizmann Institute, Rehovot, Israel). Human ErbB4 cDNA was a gift from Dr. I. Hiles (Wellcome Foundation). Both ErbB2 and ErbB4 were cloned into the Nco1 site of the SFG retroviral vector.
Generation/Maintenance of ErbB-Expressing 32D Cells
The ErbB-negative murine myeloid cell line 32D was maintained in RPMI 1640 + 10% fetal calf serum (FCS) + 15% WEHI 3B conditioned media as a source of murine interleukin (IL)-3. Cells that express ErbB1, ErbB3, ErbB1 + 3 and ErbB2 + 3 were gifts of Prof. E. van Zoelen (Radboud University Nijmegen, The Netherlands). All other ErbB-engineered cell lines were derived from these and from parental 32D cells (a gift of B Guinn, King’s College London, UK) by retroviral transduction with SFG-ErbB2 and/or SFG-ErbB4 and/or by transfection with pcDNA3-ErbB1/3. Cells engineered to express ErbB2 were immunoselected using the Institute of Cancer Research (ICR) rat monoclonal antibody ICR12 followed by sheep anti-rat immunoglobulin (Ig)-coated paramagnetic beads (Invitrogen, Paisley, UK). Cells engineered to express ErbB4 were selected by IL-3–independent growth in heregulin (HRG)-1β (Sigma Aldrich, Poole, UK) (39).
Tumor Cell Lines
All tumor cell lines were maintained in Dulbecco’s modified Eagle’s medium + 10% FCS and were sourced as follows. HN3 and HN4 cell lines were obtained from the Ludwig Institute for Cancer Research, London. PJ34 cell lines were obtained from the European Collection of Cell Cultures. The 006/1 and 015B cell lines were established in-house (by C Box and SA Eccles). CAL27 cell lines were obtained from the American Type Culture Collection. Vβ6, H357, Detroit 562, HSC3, BiCR56 and BiCR6 were gifts from Professor G. Thomas (University of Southampton, UK). All breast/mammary cancer cell lines and MDA-MB-435 cells were from the Cancer Research U.K. organization (London, UK).
Culture and Transduction of Primary Human T Cells
Human peripheral blood mononuclear cells were obtained from the U.K. National Blood Service, healthy volunteers or patients with squamous cell carcinoma of the head and neck (SCCHN) under approval of the Guy’s and St Thomas’ Research Ethics Committee (references 09/H0804/92 and 09/H0707/86, respectively). Gene transfer was performed exactly as described (36). Thereafter, T cells were propagated in RPMI 1640 + 10% human AB serum (Sigma-Aldrich) + IL-2 (100 U/mL; Aldesleukin, Novartis, Camberley, UK) or, where indicated, IL-4 (30 ng/mL; GMP grade from Gentaur [Kampenhout, Belgium] or research grade from Peprotech [Rocky Hill, NJ, USA]).
Flow Cytometry Analysis
Flow cytometry was performed using a Coulter EPICS XL cytometer with Expo32 ADC software or a FACScalibur cytometer with Cellquest Pro software. Expression of T1E28z, T1NA and EGF28z was demonstrated using the following: (a) mouse antihuman EGF monoclonal antibody (clone 10825; R&D Systems, Abingdon, UK) followed by phycoethrin (PE)-conjugated goat anti-mouse Ig (Dako, Ely, UK) or (b) goat anti-EGF polyclonal antiserum (R&D Systems), followed by fluorescein isothiocyanate–conjugated rabbit–anti-goat IgG (Dako). Expression of human ErbB family members was demonstrated using rat anti-ErbB1 (ICR62 clone), rat anti-ErbB2 (ICR12 clone, both ICR antibodies were generated in-house at ICR, Sutton, Surrey, UK), mouse anti-ErbB3 (clone H3.105.5; Neomarkers, ThermoFisher Scientific, Loughborough, UK) and mouse anti-ErbB4 (clone H4.77.16; Neomarkers), followed by PE-conjugated secondary antibodies (Invitrogen, Paisley, UK). Blockade of Fc receptors was achieved using swine or rabbit serum (Dako). Conjugated antibodies were absorbed in the appropriate serum before addition to cells.
Protein Analysis
Electrophoresis was performed under reducing conditions using a 6% (ErbB3) or 8% acrylamide (CD3ζ) resolving gel. Western blots were probed with anti-CD3ζ (clone 8D3; BD Pharmingen, Oxford, UK [36]), rabbit anti-ErbB3 antiserum or goat anti-lamin (both from Santa Cruz Biotechnology, Heidelberg, Germany).
In Vitro Assessment of Antitumor Activity
Engineered T cells (1 × 106 cells) were cocultivated with confluent tumor monolayers in 24-well plates for the indicated period, after which monolayers were stained with crystal violet as described (38). Monolayers were viewed using a Zeiss Axiovert S100 microscope with an AxioCam HR camera. Supernatants harvested from T-cell tumor monolayer cocultures were analyzed for interferon (IFN)-γ or IL-2 using paired antibody sets (R&D Systems) or using cytokine bead arrays (BD human Th1/Th2/Th17 kit; BD Biosciences, Oxford, UK), as described by the manufacturers. Unless otherwise stated, T-cell cultures were maintained in IL-2 (100 U/mL, added from 24 h after initiation of cultures). Where indicated, T cells were restimulated periodically by culture with fresh tumor monolayers. Viable T-cell number was determined by trypan blue exclusion.
Four-hour cytotoxicity assays were performed by measurement of lactate dehydrogenase release (Roche Applied Science, Burgess Hill, UK), as described (36). Tumor cell killing was also quantified using a Cell Proliferation Kit II (Roche Applied Science). In that case, 1 × 106 T cells were cocultivated with a confluent monolayer (24-well plate, 1.9 cm2) of tumor cells for the indicated period. After the removal of nonadherent T cells, viable tumor cells were then incubated with 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5- carboxanilide inner salt (XTT) as described by the manufacturer. Comparison was made with a confluent monolayer that had not been cultivated with T cells. The percentage of viable cells remaining was determined by measurement of absorbance measured at 492 nm, using 690 nm as the reference wavelength.
In Vivo Assessment of Efficacy
All in vivo experimentation strictly adhered to current U.K. Home Office guidelines, as stated in the project license (license number 70/6677) and personal license (license number 70/21581) that governed this work.
Model 1
Firefly luciferase–expressing HN3 tumor cells (5 × 106 cells) were inoculated into 25 SCID Beige mice using the intraperitoneal (IP) route. After 8 d, tumor engraftment was confirmed by bioluminescence imaging (BLI), and mice were sorted into groups with similar average signal intensity. On d 13, engineered T-cells (2 × 107 cells each or phosphate-buffered saline [PBS]) were administered intraperitoneally. No cytokines were administered to animals treated in this manner. To assess ErbB status of HN3 xenografts, excised tumors were mechanically disaggregated before immunostaining as above.
Model 2
The MDA-MB-435 cell line was retrovirally engineered to coexpress firefly luciferase, MUC1 and ErbB2. Tumor cells (5 × 106 cells) were inoculated intraperitoneally into 20 SCID Beige mice. Human T cells were engineered to express T4 and then expanded for 2 wks by using IL-4 (38). After 3 d, mice were treated with PBS, IL-4 (125 ng × three doses per week), 2 × 107 T4+ T cells or the combination of T4+ T cells + IL-4.
In each case, BLI was performed at the indicated time points using Xenogen IVIS Imaging System (Xenogen) with Living Image software (Xenogen). Mice were injected intraperitoneally with D-luciferin (150 mg/kg; Xenogen) and imaged under isoflurane anesthesia after 10 min. Image acquisition was conducted on a 15- or 25-cm field of view with small binning for exposure times of 1–300 s. Animals were inspected daily and sacrificed when experimental endpoints had been achieved or when symptomatic as a result of tumor progression (whichever sooner).
Imaging of HN3 Tumor Xenografts by Using Positron Emission Tomography
Mice with IP HN3 xenografts were deprived of food for 1 h, anesthetized using isoflurane and maintained on a heating pad for 20 min before intravenous (IV) injection of 4–7.6 mBq 18F-fluorodeoxy glucose. Imaging was performed using a Siemens Inveon MicroPET/CT (positron emission tomography/computerized tomography). A CT attenuation scan was performed followed by a 200-min PET scan. Images were coregistered, and data were reconstructed to generate a three-dimensional image using an OSEM2D reconstruction algorithm (Inveon acquisition workshop software package).
Statistical Analysis
Datasets were analyzed with Graphpad Prism software (version 5), using two-way analysis of variance (ANOVA) (for grouped analyses) or two-tailed Student t test (for comparison of two groups).
All supplementary materials are available online at www.molmed.org.
RESULTS
Engineering of a Chimeric Antigen Receptor with Reactivity against Multiple ErbB Dimer Species
To engineer a broad specificity CAR targeted against relevant ErbB dimers, T1E sequences were placed upstream of CD28 (hinge/transmembrane/endodomain) and CD3ζ endodomain motifs (36). The resultant fusion receptor (T1E28z) was compared with two control CARs, in which an EGF targeting moiety (EGF28z) or a truncated endodomain were used (T1E no activation domain [T1NA]; see Figure 1C).
Expression of T1E28z and control CARs was detected in T cells by using an anti-EGF antibody (Figure 2A) at proportionate levels in CD4+ and CD8+ subsets (Figure 2B). Integrity and molecular mass of the CAR was determined by Western blotting (Figure 2C).
Figure 2.
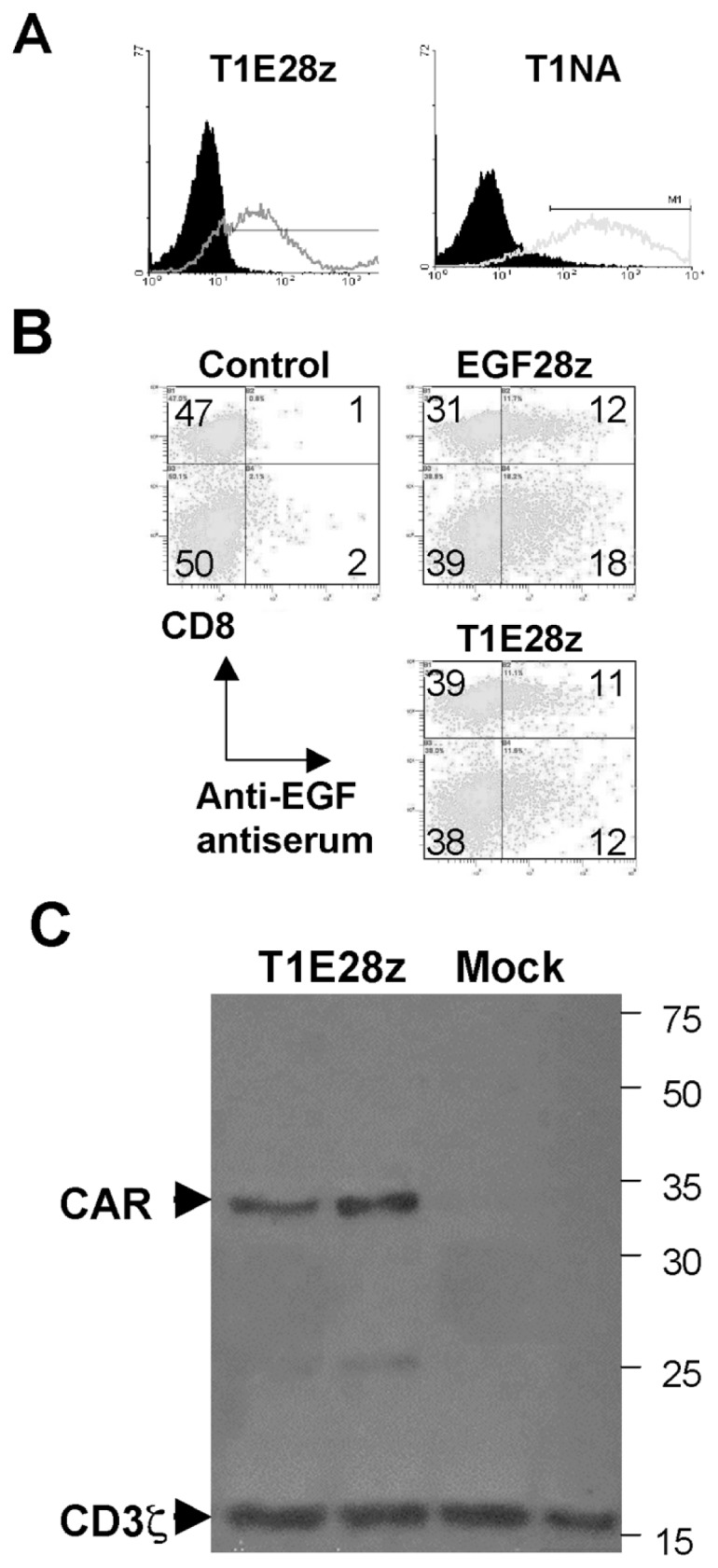
Expression of ErbB-specific CARs in human T cells. Expression of ErbB targeted CARs in transduced T cells was detected by the fluorescence-activated cell sorter (FACS) method by using monoclonal (A; control shown as filled histogram) or polyclonal (B) anti-EGF antibody or by Western blotting (C; probed under reducing conditions with anti-CD3ζ). In each case, controls represent untransduced T-cells.
To evaluate targeting specificity, ErbB-negative 32D hematopoietic cells were engineered to express ErbB1–4 (39), either individually or in all possible binary combinations (Figure 3A). Owing to weak cross-reactivity of the anti-ErbB3 antibody with ErbB2 on 32D cells (for example, row 3, column 9), expression of this family member was confirmed by Western blotting (Figure 3B). Cocultivation experiments demonstrated that T1E28z mediates T-cell recognition of 32D cells that express all possible ErbB1-based homodimers and heterodimers, leading to production of IL-2 and IFN-γ (Figure 3C). Targeting of ErbB1/2 heterodimers was consistently achieved most efficiently (P < 0.0001). Importantly, we observed that T1E28z also directed T-cell specificity against the ErbB2/3 heterodimer, but not against ErbB2 or ErbB3 alone. Weaker targeting of ErbB4 and other ErbB4-containing dimers was also observed. By contrast, EGF28z only targets T cells against ErbB1+ 32D cells and was less efficient in this respect than T1E28z (P < 0.0001). As anticipated, control (untransduced or T1NA+) T-cell populations were not activated under these conditions. Targeting specificity was confirmed further by the ability of EGF and HRG-1β to inhibit competitively the activation of T1E28z+ T cells by ErbB1 and ErbB2/3 heterodimers, respectively (Figure 3D). Together, these data demonstrate that T1E28z effectively targets human T cells against several ErbB dimer species that are commonly over-represented in diverse solid tumors.
Figure 3.

ErbB specificity of the T1E28z CAR. (A) ErbB null 32D myeloid cells were engineered to express all 10 ErbB receptor/dimer combinations. Expression of the indicated ErbB receptor was detected by FACS (filled histogram represents staining of parental 32D cells with the same antibody combination) and, in the case of ErbB3, by Western blotting (B). (C) To define targeting specificity, 1 × 106 of the indicated engineered T-cell populations was cocultivated with an equal number of 32D cells that express the specified ErbB receptor(s). Supernatants were harvested at 24 h (IL-2) and 72 h (IFN-γ) for ELISA analysis. Data shown are mean ± standard deviation (SD) of three (IL-2) or six (IFN-γ) replicates after correction for transduction efficiency between groups. ***P < 0.001, **P < 0.01, comparing T1E28z (black) or EGF28z (gray) with other control groups. (D) 32D cells that express ErbB1 (32D:1) or ErbB2 + 3 (32D:23) were cocultivated with T1E28z+ T cells in the presence of exogenous EGF (left) or HRG-1β (right). Supernatants harvested at 72 h were analyzed for IFN-γ content.
Human T Cells Transduced with T1E28z Exhibit Broad Reactivity against Head and Neck Cancer Cell Lines
To evaluate antitumor activity of T1E28z, we focused initially on SCCHN. A panel of 12 SCCHN tumor lines was characterized for expression of ErbB1–4 (Figure 4A). As anticipated from studies of SCCHN in humans (5), all cell lines expressed ErbB1 and were commonly positive for ErbB2 and/or ErbB3 but not ErbB4.
Figure 4.

T1E28z engineered T cells destroy human head and neck cancer cells. (A) Twelve human SCCHN cell lines (and MDA-MB-435 control cells) were analyzed for cell surface expression of ErbB1–4 by FACS. Staining with the indicated ErbB-specific (open histogram) or isotype control (filled histogram) antibody is indicated. (B) Confluent monolayers of the indicated tumor cell lines (24-well plate, 1.9 cm2) were cocultivated with 1 × 106 T1E28z+ T cells. Control T cells expressed T1NA (a matched endodomain-truncated CAR) or were untransduced (Untrans.). After 72 h, residual monolayers were fixed and stained with crystal violet. Punctate staining in T1E28z wells represents adherent lymphocytes. (C) One million of the indicated T-cell populations were cocultivated overnight with a confluent 1.9-cm2 monolayer of luciferase (LUC)-expressing HN3 cells or Vβ6 cells. After the specified interval, T cells were removed and residual viable tumor cells were quantified using XTT (mean ± SD, n = 3). Percentage lysis was calculated as (100 – cell viability).
T-cells that express T1E28z or appropriate controls were cocultivated with a confluent monolayer of all SCCHN tumor cells. In each case, selective destruction of ErbB+ tumor monolayers was achieved by T1E28z+ but not by control T cells (Figures 4B, C). Activation of T1E28z+ T cells was accompanied by production of IFN-γ (Figure 5A), proliferation (Figure 5B and Supplementary Figure 1) and enrichment (Figure 5C), in a manner that required an ErbB-permissive tumor monolayer and a signaling-competent CAR endodomain (Figure 5D). By contrast, tumor cells that express ErbB3 together with low levels of ErbB1 and ErbB2 (for example, MDA-MB-435; Figure 4A) were recognized poorly (Figure 4B), leading to very-low-level cytokine production (Supplementary Figure 2; note that y axis is calibrated in pg/mL).
Figure 5.

T1E28z engineered T cells are activated by human head and neck cancer cells. (A) Confluent head and neck tumor monolayers (24-well plate, 1.9 cm2) were cocultivated with 1 × 106 of the indicated T-cell populations. Supernatants were harvested after 72 h and analyzed for IFN-γ (mean ± SD). The number of independent replicate experiments is indicated: *** P < 0.001, **P < 0.01, *P < 0.05. (B) Engineered T cells were cocultivated with HN3 tumor cell monolayers where indicated by overhead arrows. T cells were enumerated at specified intervals. (C) T cells in (B) were analyzed by FACS for cell surface expression of T1E28z at the indicated time points. Untransduced T cells served as the negative control. (D) Engineered T cells were stimulated on a HN3 monolayer and cultured in the presence of IL-2. Control cultures were maintained in IL-2 alone without monolayer stimulation. Cultures were analyzed for CAR expression using monoclonal anti-EGF antibody at baseline and after 7 d. Filled histograms show staining of untransduced T cells. Proliferation/enrichment data are representative of at least three experiments.
Use of an IL-4–Regulated System to Expand and Enrich T1E28z+ T Cells Ex Vivo
We have recently described a system whereby IL-4 can be harnessed to achieve selective expansion of gene-modified cells, acting via an ectopically expressed IL-4Rα/IL-2Rβ chimeric cytokine receptor (4αβ) (38). When T1E28z is coexpressed with 4αβ, the resultant T4+ T cells are activated by a similar spectrum of ErbB combinations (Figure 6A) and SCCHN tumor cells (Figure 6B and Supplementary Figure 3). Marked enrichment of ex vivo propagated 4αβ+ T cells is achieved by IL-4 (38). Consequently, complete SCCHN tumor monolayer destruction is achieved using as few as 0.25 million T4+ T cells, representing an effector:target ratio of <1 (Figure 6C). Once again, MDA-MB-435 tumor cells elicited very weak or no activation of T4+ T cells (Figures 6C, E). Although expanded in IL-4, T4+ T cells do not exhibit type 2 polarization. Upon activation by SCCHN tumor monolayers, they produce much IFN-γ and IL-2, but small amounts of IL-4 (Figure 6D). Moreover, production of IFN-γ by activated T4+ T cells is further enhanced by exogenous IL-4 (Figure 6E).
Figure 6.

In vitro activation of T4+ human T cells. Human T cells were engineered to coexpress the IL-4–responsive 4αβ chimeric cytokine receptor together with the T1E28z CAR (combination called “T4”). After expansion in IL-4, comparison was made with control T cells that were untransduced (Untrans) or that express T1NA (truncated endodomain). (A) To define targeting specificity for ErbB receptors/dimers, 1 × 106 of the indicated T-cell populations were cocultivated with an equal number of 32D cells that express the specified ErbB receptor(s). Supernatants were harvested at 72 h and analyzed for interferon (IFN)-γ (mean ± SD; ***P < 0.001; **P < 0.01; *P < 0.05). (B) 1 × 106 T cells were cocultivated with confluent monolayers of the indicated head and neck tumor cells cultured in 24-well (1.9-cm2) plates. Supernatants were harvested at 72 h and analyzed for IFN-γ (mean ± SD of three independent experiments; ***P < 0.001; LUC, luciferase). (C) T cells (number indicated above each well) were cocultivated for 72 h with the indicated head and neck tumor cell monolayers, cultured in 24-well plates. Nonadherent cells were then removed and, after fixation, residual tumor monolayers were stained using crystal violet (representative of three experiments). (D) T cells were established in triplicate cultures as described in (C). Cytokine analysis was performed on supernatants harvested after 24 h (IL-2, IL-4) and 72 h (IFN-γ). (E) T-cells (1 × 106 cells) were established in triplicate cultures with the indicated monolayers, in the presence or absence of IL-4 (30 ng/mL). Supernatants were analyzed after 72 h for IFN-γ.
Therapeutic Efficacy of T1E28z-Transduced T Cells against a Slowly Progressive SCCHN Xenograft
To test efficacy in vivo, human SCCHN was modeled in SCID Beige mice using firefly luciferase–expressing HN3 tumor cells (HN3-LUC) (37). After IP inoculation, discrete tumors (typically one to three) grew insidiously. By using PET scanning, we observed that these tumors were poorly avid for 18F-fluorodeoxyglucose, and a thin rim of peripheral uptake was observed, reflecting their tendency for indolent growth and central necrosis (Figure 7A). By contrast, HN3-LUC tumors could be readily imaged using bioluminescence (Figures 7A, B). Importantly, established tumors retained expression of ErbB1 and ErbB2 (Figure 7C). Although HN3-LUC xenografts also grew after subcutaneous inoculation, this model proved unsuitable for testing therapeutic efficacy. The reason was that tumors progressed with unpredictable kinetics and were commonly fractured after intratumoral injection.
Figure 7.

In vivo antitumor activity of T1E28z-transduced T cells against a slowly progressive head and neck tumor xenograft. (A) HN3 tumor cells were engineered to express firefly luciferase (HN3-LUC) and were propagated as IP xenografts. In this representative example, a large established tumor was imaged by PET using 18F-fluorodeoxyglucose (tumor center indicated by crosshairs) or BLI, following administration of luciferin. (B) Five mice were inoculated intraperitoneally with 5 × 106 HN3-LUC tumor cells and analyzed by BLI at the indicated intervals. (C) HN3-LUC tumors were recovered from mice described in (B) and analyzed by FACS for expression of ErbB1 and ErbB2. Comparison was made with HN3-LUC cells cultured in vitro. Filled histograms show staining with isotype control alone. (D) Twenty-five SCID Beige mice were inoculated with 5 × 106 HN3-LUC tumor cells, administered by the IP route. Tumor take was confirmed on d 8 by BLI. On d 13, mice were treated with T cells transduced with T1E28z (n = 7), T1NA (n = 6), T4 (combination of 4αβ and T1E28z; n = 6) or PBS (n = 6). Analysis of T cells before infusion is shown in Supplementary Figure 4. Mean bioluminescence for each group is shown over the course of the study. ***P < 0.001 (T1E28z versus PBS and T4 versus PBS); *P < 0.05 (T1E28z versus PBS and T4 versus PBS); ‡P < 0.001 (T4 versus PBS) and P < 0.01 (T1E28z versus PBS); ‡‡P < 0.001 (T4 versus T1NA) and P < 0.01 (T1E28z versus T1NA). (E) Serial imaging of the same mouse (median representative of each group) is shown at selected time points over the course of the study. (F) Bioluminescence data at all time points for each mouse are shown.
To test therapeutic efficacy, mice with 13 d established HN3-LUC tumors were treated intraperitoneally with PBS or 20 million T1E28z+ or T1NA+ T cells. A fourth group received T4+ T cells, administered at a dose of 20 or 7.5 million cells. Characterization of T cells before infusion confirmed that they were transduced and functionally active in vitro (Supplementary Figure 4). After adoptive transfer of immunotherapy, each group was examined using bioluminescence imaging over 4 months (mean values shown in Figure 7D). Data pertaining to the T4+ T cell–treated group were pooled, since animals treated with either dose responded similarly (Supplementary Figure 5). Figure 7E shows serial imaging of the same mouse in each group, selected as that which gave the median bioluminescence emission for the longest period throughout the study. Observations from each mouse at all time points are shown in Figure 7F. One mouse in the T1E28z group developed respiratory distress on d 78 and was culled. Necropsy revealed a centrally placed intrathoracic tumor (presumed thymoma) in the absence of macroscopically evident intra peritoneal disease or bioluminescence signal (Figure 7F).
Significant differences in bioluminescence emission between mice treated with PBS and either T1E28z+ or T4+ T cells became manifest on d 100 and increased thereafter. Control T1NA+ T-cells exerted intermediate antitumor activity, which may reflect the capacity of this truncated CAR to promote T-cell colocalization with tumor cells. Significant differences between mice treated with T1NA+ T cells versus either T1E28z+ or T4+ T cells were observed at d 128 (Figure 7D). Upon completion of this endpoint, the study was terminated.
Antitumor Activity of T1E28z-Transduced T Cells in Breast Cancer
To investigate potential utility in other malignancies, in vitro studies were performed with human breast and murine mammary carcinoma cell lines that express diverse ErbB profiles (Supplementary Figure 6). In addition to previously described controls, comparison was also made with an endodomain-matched control CAR named S28z (37). Activation of T1E28z+ but not control T cells was observed upon culture with several of the breast cancer cell lines, resulting in monolayer destruction (Figure 8A), cytotoxicity (Figure 8B), T-cell proliferation (Figure 8C and Supplementary Figure 7) and cytokine production (Figure 8D). By contrast, ErbBl° murine target cells (410.4, Supplementary Figure 6) only weakly activated T1E28z+ T cells (Supplementary Figure 2) and were not killed efficiently (Figure 8B). Owing to IL-4–mediated preenrichment (for example, Supplementary Figure 4), destruction of breast tumor cell monolayers was consistently achieved more efficiently by T4+ than T1E28z+ T cells (Figure 8A). Activated T4+ T cells once again produced a predominance of type 1 cytokines (Figure 8D and Supplementary Figure 8).
Figure 8.

In vitro antitumor activity of T1E28z+ T cells against breast cancer. (A) 1 × 106 human T cells engineered to express the indicated CARs were cocultivated with confluent breast cancer monolayers, cultured in 24-well (1.9-cm2) plates. After 24 h, nonadherent cells were removed and monolayers were fixed and stained with crystal violet. (B) Four-hour lactate dehydrogenase release cytotoxicity assay in which T1E28z+ or control S28z+ T cells were cultured with BT20 breast cancer cells or ERBBl° 410.4 mammary carcinoma cells. (C) A representative experiment in which T1E28z+ and S28z+ T cells were cultured with BT20 breast cancer cells. After 24 h, IL-2 was added. Restimulation on a confluent BT20 monolayer was performed where indicated by the overhead arrows. T cells were enumerated at the indicated intervals. (D) Transduced T cells were cocultivated in 24-well plates with confluent monolayers or suspension cells as follows: 1) Jurkat (control; 1 × 106 cells); 2) MDA-MB-435; 3) BT20; 4) BT474; 5) MCF7; 6) SKBR3; 7) T47D; 8) medium alone. Supernatants were analyzed for the indicated cytokines after 24 h.
Therapeutic Activity against an Aggressive ErbB2/3-Expressing Tumor Xenograft
Next, we evaluated therapeutic efficacy in vivo in a tumor model characterized by overexpression of the potently mitogenic ErbB2/3 heterodimer. We previously showed that luciferase/MUC1 coexpressing MDA-MB-435 (MUC-LUC) cells establish a robust and aggressive IP cancer model (37). However, because these tumor cells only express ErbB3 together with low levels of ErbB2 (Figure 4A), they are targeted inefficiently by T1E28z+ and T4+ T cells (Figure 8A; Figure 8D, tumor cell number 2). On the basis of our in vitro studies, we hypothesized that efficient targeting would be achieved if these tumor cells coexpressed high levels of ErbB2. To test this, ErbB2-overexpressing MUC-LUC tumor cells were engineered (Figure 9A) and then established as IP xenografts. Animals with established tumors were treated with T4+ T cells after ex vivo expansion in clinically compliant conditions using IL-4 (38). Comparison was made between groups treated with T4+ T cells alone or together with exogenous IL-4. Control animals received PBS or IL-4 alone. We observed that T4+ T cells achieved striking control of this highly aggressive tumor (Figure 9B). Further benefit was gained when a combination of T4+ T cells and IL-4 were used, with mice harboring a significantly lower tumor burden than animals treated with T4+ T cells alone (Figure 9C). The reason for this result is currently unclear. One possibility is that the provision of IL-4 favors in vivo persistence of the T4+ T cells, thus permitting further growth retardation of the tumor. However, because the use of IL-4 alone induced some (although not significant) therapeutic activity (Figure 9B), improved tumor control may alternatively reflect an additive effect of the two therapies.
Figure 9.

In vivo activity of T1E28z+ T cells against an ErbB2/3-expressing aggressive tumor xenograft. (A) MDA-MB-435 cells were engineered to coexpress MUC1, ErbB2 and firefly luciferase (MUC-LUC ERBB2). FACS plots show staining with ErbB-specific (open histograms) and isotype control antibodies (filled histograms). (B) MUC-LUC ErbB2 tumor cells were inoculated intraperitoneally in 20 SCID Beige mice. After 3 d, mice (n = 5 per group) were treated with PBS, IL-4 (125 ng × 3 doses per wk), 2 × 107 T4+ T cells or the combination of T4+ T cells + IL-4. Tumor status was monitored by bioluminescence imaging. Data shown are mean ± standard error of the mean of each group. (***P < 0.0001 comparing either T4 or T4 + IL-4 versus either IL-4 or PBS; **P < 0.0001 comparing either T4 or T4 + IL-4 against PBS). (C) Serial bioluminescence emission by mice in the T4 and T4 + IL-4 groups (mean ± SEM), represented on a semi-log plot (*P = 0.015 on d 35; two-way ANOVA).
DISCUSSION
Coordinated activation of the ErbB family plays a fundamental role in development and is commonly subverted during tumorigenesis. Most often, such aberrant ErbB activity results from the upregulated expression and/or activation of one or more family members, notably ErbB1 and/or ErbB2. Increased signaling by mutually cooperative ErbB heterodimers (particularly ErbB2/ErbB3) is also a frequent property of solid tumors, notably HER2-amplified breast cancer (7). Tumors that display ErbB up-regulation are generally associated with worsened prognosis (1–3). Consequently, targeted therapies directed against this family occupy an increasing niche in cancer therapy and drug development.
In this study, we have pursued an immunotherapeutic approach to exploit the prevalence of ErbB overexpression in cancer. We hypothesized that potent and yet safe antitumor immunity could be achieved by directing T-cell specificity simultaneously against key ErbB dimer units that commonly drive tumorigenesis. This strategy contrasts with previously reported immunotherapeutic approaches directed against individual ErbB family members (40,41) and was chosen for two main reasons. First, tumors commonly acquire therapeutic resistance to ErbB-directed therapies through upregulated activation of non-targeted family members (20–25,42). In keeping with this, several precedents illustrate how combined targeting of two or more ErbB receptors can achieve improved antitumor activity (24,25,27). Second, we reasoned that harnessing of the multiple effector mechanisms deployed by T cells would elicit more potent and prompt antitumor efficacy, when compared with the cytostatic or indirectly lytic capabilities of other ErbB targeted therapies. As a result, less time is granted to tumor cells to evolve ErbB- independent resistance mechanisms.
To test this concept, we developed a CAR named T1E28z that incorporates a promiscuous ErbB ligand as a targeting moiety. We demonstrated that the T1E28z fusion receptor can engage ErbB1 homodimers and heterodimers in addition to the ErbB2/3 mitogenic unit. The greatest targeting activity was demonstrated against cells that coexpress ErbB1 and ErbB2. Unexpectedly, when tested using cells that express ErbB1 alone, T1E28z mediated enhanced T-cell activation compared with a matched fusion in which specificity was directed by EGF. Targeting of tumor cells by the T1E28z CAR was repeatedly demonstrated both in vitro and in vivo, using several cell lines representative of two distinct ErbB-associated solid tumors, namely head and neck cancer and breast cancer. In unpublished studies, we have shown that T-cells engrafted with this fusion can also destroy cell lines derived from other common solid tumor types (data not shown). Furthermore, antitumor activity was apparent against target cells that express a spectrum of ErbB expression profiles. The recognition and destruction of the T47D cell line, which expresses ErbB2 and ErbB3, but minimal ErbB1 (43), strengthens the evidence that T1E28z can engage ErbB2/3 hetero dimers on tumor cells. In vivo therapeutic efficacy was demonstrated in models of both slow-growing and rapidly progressive tumor types, driven by ErbB1 or ErbB2/3 signaling, respectively. Together, these findings demonstrate the flexible therapeutic value of this approach.
A key concern with systemic therapies targeted against ErbB receptors is the risk of unacceptable toxicity. Four precedents provide reassurance in this regard. First, ErbB-specific T cells are present at increased frequency in patients with diverse solid tumors (44). Second, immunization against ErbB2 has been optimized to a point where clinical and immunologic responses occur in the absence of significant toxicity (45). Third, extensive clinical experience has confirmed the safety of monoclonal antibodies and small molecules directed against the ErbB family, particularly ErbB1 and ErbB2 (46). Fourth, IV infusion of ErbB2-specific CD8+ T-cell clones has been conducted in patients with metastatic HER2-positive breast cancer, without significant toxicity (47).
Against this favorable background, Morgan et al. (48) recently treated a patient with metastatic colorectal cancer using autologous T cells, retargeted with a high affinity ErbB2-specific CAR that contained a fused CD28/4-1BB/CD3ζ endodomain. Treatment involved lympho-depletion following by IV administration of 1010 cells (79% CAR+). Unfortunately, however, the patient rapidly succumbed to adult respiratory distress syndrome followed by multiorgan failure—an event that was clearly attributable to the infused T-cells. This incident mandates a highly careful approach to the systemic administration of ErbB2- targeted T cells in humans.
Mindful of these issues, we set out here to develop a distinct immunotherapeutic approach to deliver selective toxicity against cancers that are driven by aberrant ErbB signaling. Importantly, whereas the T1E28z CAR can engage several tumor-associated ErbB dimers, it does not bind to ErbB2 or ErbB3 alone. Furthermore, it is not activated efficiently by cells that express low levels of one or more ErbB receptors (for example, MDA-MB-435 cells). In keeping with this, we observed no overt clinical evidence of toxicity in any of over 40 mice treated with human T1E28z-engineered T-cells. This observation is important for two reasons. First, similar to all known human ErbB ligands, human T1E and the derived human T1E28z CAR can engage murine ErbB receptors (data not shown). Second, despite many species-specific differences, activated human T cells can trigger lethal immunopathology in mice, for example, during xenogeneic graft versus host disease. To evaluate this further, we are currently undertaking rigorous toxicity studies to investigate possible “on-target, off-organ” effects in vivo, using both immune competent and immunocompromised hosts. The former approach is of particular importance, since several potentially immunosuppressive mechanisms cannot be modeled in SCID Beige mice. These include the influence of myeloid suppressor cells and regulatory T cells.
In vivo studies were performed using IP tumor models, whereby T-cells were administered to the peritoneal cavity. We selected this option because we previously found that IV-administered human T-cells migrate with delayed kinetics via the pulmonary circulation and thereafter redistribute to liver, spleen and lymph nodes. However, trafficking to either subcutaneous or intraperitoneal xenografts was poor. This challenge to clinical translation is discussed extensively by Parente-Pereira et al. (49). In light of this, we are planning clinical studies in which these T cells are delivered using a regional approach.
CONCLUSION
In summary, the conceptual and therapeutic advance reported here is a flexible and robust immunotherapy directed against ErbB dimers that drive several solid tumor types and commonly mediate therapeutic resistance. Therapeutic efficacy is consistently achieved both in vitro and in vivo against tumors of various lineages, highlighting the potential applicability of this approach to a wide range of malignancies. A clinical trial will follow shortly in patients with SCCHN, treated by intratumoral delivery after locoregional recurrence. Such studies will pave the way for systemic targeting, thereby harnessing for therapeutic gain the primacy of ErbB overexpression in several human tumors.
Supplemental Data
ACKNOWLEDGMENTS
This work was supported by the Association for International Cancer Research (Project Grant 08-0419), Breast Cancer Campaign (Project Grant 2006NovPR18), Guy’s and St Thomas’ Charity, Experimental Cancer Medicine Centre (King’s College London). Phase 1 testing of this immunotherapy was supported by the Moulton Charitable Foundation and by Guy’s and the Department of Health via the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy’s and St Thomas’ National Health Service (NHS) Foundation Trust and King’s College London. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. C Box was funded by the Oracle Cancer Trust at the Royal Marsden Hospital. The research of SA Eccles was funded by ICR and Cancer Research UK grant CA309/A8274 and funding provided via the NIHR specialist BRC award to the Royal Marsden NHS Foundation Trust in partnership with the Institute of Cancer Research. We thank colleagues who have generously provided materials that facilitated this work, as indicated in the text.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Arteaga CL. ErbB-targeted therapeutic approaches in human cancer. Exp Cell Res. 2003;284:122–30. doi: 10.1016/s0014-4827(02)00104-0. [DOI] [PubMed] [Google Scholar]
- 2.Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–16. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 3.Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009;21:177–84. doi: 10.1016/j.ceb.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 4.Morgan S, Grandis JR. ErbB receptors in the biology and pathology of the aerodigestive tract. Exp Cell Res. 2009;315:572–82. doi: 10.1016/j.yexcr.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rogers SJ, Harrington KJ, Rhys-Evans P, Charoenrat O, Eccles SA. Biological significance of c-erbB family oncogenes in head and neck cancer. Cancer Metastasis Rev. 2005;24:47–69. doi: 10.1007/s10555-005-5047-1. [DOI] [PubMed] [Google Scholar]
- 6.Foley J, et al. EGFR signaling in breast cancer: bad to the bone. Semin Cell Dev Biol. 2010;21:951–60. doi: 10.1016/j.semcdb.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holbro T, et al. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci U S A. 2003;100:8933–8. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirsch FR, Varella-Garcia M, Cappuzzo F. Predictive value of EGFR and HER2 overexpression in advanced non-small-cell lung cancer. Oncogene. 2009;28(Suppl 1):S32–S37. doi: 10.1038/onc.2009.199. [DOI] [PubMed] [Google Scholar]
- 9.Lockhart AC, Berlin JD. The epidermal growth factor receptor as a target for colorectal cancer therapy. Semin Oncol. 2005;32:52–60. doi: 10.1053/j.seminoncol.2004.09.036. [DOI] [PubMed] [Google Scholar]
- 10.Hammarsten P, et al. Low levels of phosphorylated epidermal growth factor receptor in nonmalignant and malignant prostate tissue predict favorable outcome in prostate cancer patients. Clin Cancer Res. 2010;16:1245–55. doi: 10.1158/1078-0432.CCR-09-0103. [DOI] [PubMed] [Google Scholar]
- 11.Jathal MK, Chen L, Mudryj M, Ghosh PM. Targeting ErbB3: the new RTK(id) on the prostate cancer block. Immunol Endocr Metab Agents Med Chem. 2011;11:131–49. doi: 10.2174/187152211795495643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bellone S, et al. Overexpression of epidermal growth factor type-1 receptor (EGF-R1) in cervical cancer: implications for Cetuximab- mediated therapy in recurrent/metastatic disease. Gynecol Oncol. 2007;106:513–20. doi: 10.1016/j.ygyno.2007.04.028. [DOI] [PubMed] [Google Scholar]
- 13.Sheng Q, et al. An activated ErbB3/NRG1 autocrine loop supports in vivo proliferation in ovarian cancer cells. Cancer Cell. 2010;17:298–310. doi: 10.1016/j.ccr.2009.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liles JS, et al. ErbB3 expression promotes tumorigenesis in pancreatic adenocarcinoma. Cancer Biol Ther. 2010;10:555–63. doi: 10.4161/cbt.10.6.12532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jhabvala-Romero F, Evans A, Guo S, Denton M, Clinton GM. Herstatin inhibits heregulin-mediated breast cancer cell growth and overcomes tamoxifen resistance in breast cancer cells that overexpress HER-2. Oncogene. 2003;22:8178–86. doi: 10.1038/sj.onc.1206912. [DOI] [PubMed] [Google Scholar]
- 16.Wang S, Huang X, Lee CK, Liu B. Elevated expression of erbB3 confers paclitaxel resistance in erbB2-overexpressing breast cancer cells via upregulation of Survivin. Oncogene. 2010;29:4225–36. doi: 10.1038/onc.2010.180. [DOI] [PubMed] [Google Scholar]
- 17.Liang K, Ang KK, Milas L, Hunter N, Fan Z. The epidermal growth factor receptor mediates radioresistance. Int J Radiat Oncol Biol Phys. 2003;57:246–54. doi: 10.1016/s0360-3016(03)00511-x. [DOI] [PubMed] [Google Scholar]
- 18.Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000;19:3159–67. doi: 10.1093/emboj/19.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baselga J, Swain SM. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer. 2009;9:463–75. doi: 10.1038/nrc2656. [DOI] [PubMed] [Google Scholar]
- 20.Erjala K, et al. Signaling via ErbB2 and ErbB3 associates with resistance and epidermal growth factor receptor (EGFR) amplification with sensitivity to EGFR inhibitor gefitinib in head and neck squamous cell carcinoma cells. Clin Cancer Res. 2006;12:4103–11. doi: 10.1158/1078-0432.CCR-05-2404. [DOI] [PubMed] [Google Scholar]
- 21.Garrett JT, et al. Transcriptional and post-translational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc Natl Acad Sci U S A. 2011;108:5021–6. doi: 10.1073/pnas.1016140108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jain A, et al. HER kinase axis receptor dimer partner switching occurs in response to EGFR tyrosine kinase inhibition despite failure to block cellular proliferation. Cancer Res. 2010;70:1989–99. doi: 10.1158/0008-5472.CAN-09-3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kruser TJ, Wheeler DL. Mechanisms of resistance to HER family targeting antibodies. Exp Cell Res. 2010;316:1083–100. doi: 10.1016/j.yexcr.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 24.Ritter CA, et al. Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clin Cancer Res. 2007;13:4909–19. doi: 10.1158/1078-0432.CCR-07-0701. [DOI] [PubMed] [Google Scholar]
- 25.Sergina NV, et al. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007;445:437–41. doi: 10.1038/nature05474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou BB, et al. Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell. 2006;10:39–50. doi: 10.1016/j.ccr.2006.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schaefer G, et al. A two-in-one antibody against HER3 and EGFR has superior inhibitory activity compared with monospecific antibodies. Cancer Cell. 2011;20:472–86. doi: 10.1016/j.ccr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 28.Hickinson DM, et al. AZD8931, an equipotent, reversible inhibitor of signaling by epidermal growth factor receptor, ERBB2 (HER2), and ERBB3: a unique agent for simultaneous ERBB receptor blockade in cancer. Clin Cancer Res. 2010;16:1159–69. doi: 10.1158/1078-0432.CCR-09-2353. [DOI] [PubMed] [Google Scholar]
- 29.Burstein HJ, et al. Neratinib, an irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2-positive breast cancer. J Clin Oncol. 2010;28:1301–7. doi: 10.1200/JCO.2009.25.8707. [DOI] [PubMed] [Google Scholar]
- 30.Bianco R, et al. Vascular endothelial growth factor receptor-1 contributes to resistance to anti-epidermal growth factor receptor drugs in human cancer cells. Clin Cancer Res. 2008;14:5069–80. doi: 10.1158/1078-0432.CCR-07-4905. [DOI] [PubMed] [Google Scholar]
- 31.Guix M, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J Clin Invest. 2008;118:2609–19. doi: 10.1172/JCI34588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lai AZ, Abella JV, Park M. Crosstalk in Met receptor oncogenesis. Trends Cell Biol. 2009;19:542–51. doi: 10.1016/j.tcb.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 33.Davies DM, Maher J. Adoptive T-cell immunotherapy of cancer using chimeric antigen receptor-grafted T cells. Arch Immunol Ther Exp (Warsz) 2010;58:165–78. doi: 10.1007/s00005-010-0074-1. [DOI] [PubMed] [Google Scholar]
- 34.Sadelain M. T-cell engineering for cancer immunotherapy. Cancer J. 2009;15:451–5. doi: 10.1097/PPO.0b013e3181c51f37. [DOI] [PubMed] [Google Scholar]
- 35.Wingens M, et al. Structural analysis of an epidermal growth factor/transforming growth factor-alpha chimera with unique ErbB binding specificity. J Biol Chem. 2003;278:39114–23. doi: 10.1074/jbc.M305603200. [DOI] [PubMed] [Google Scholar]
- 36.Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor. Nat Biotechnol. 2002;20:70–5. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 37.Wilkie S, et al. Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol. 2008;180:4901–9. doi: 10.4049/jimmunol.180.7.4901. [DOI] [PubMed] [Google Scholar]
- 38.Wilkie S, et al. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J Biol Chem. 2010;285:25538–44. doi: 10.1074/jbc.M110.127951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang LM, et al. ErbB2 expression increases the spectrum and potency of ligand- mediated signal transduction through ErbB4. Proc Natl Acad Sci U S A. 1998;95:6809–14. doi: 10.1073/pnas.95.12.6809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Altenschmidt U, et al. Cytolysis of tumor cells expressing the Neu/erbB-2, erbB-3, and erbB-4 receptors by genetically targeted naive T lymphocytes. Clin Cancer Res. 1996;2:1001–8. [PubMed] [Google Scholar]
- 41.Stancovski I, et al. Targeting of T lymphocytes to Neu/HER2-expressing cells using chimeric single chain Fv receptors. J Immunol. 1993;151:6577–82. [PubMed] [Google Scholar]
- 42.Grovdal LM, et al. EGF receptor inhibitors increase ErbB3 mRNA and protein levels in breast cancer cells. Cell Signal. 2012;24:296–301. doi: 10.1016/j.cellsig.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 43.Contessa JN, Abell A, Valerie K, Lin PS, Schmidt-Ullrich RK. ErbB receptor tyrosine kinase network inhibition radiosensitizes carcinoma cells. Int J Radiat Oncol Biol Phys. 2006;65:851–8. doi: 10.1016/j.ijrobp.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 44.Andrade Filho PA, Lopez-Albaitero A, Gooding W, Ferris RL. Novel immunogenic HLA-A*0201-restricted epidermal growth factor receptor-specific T-cell epitope in head and neck cancer patients. J Immunother. 2010;33:83–91. doi: 10.1097/CJI.0b013e3181b8f421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mittendorf EA, Holmes JP, Ponniah S, Peoples GE. The E75 HER2/neu peptide vaccine. Cancer Immunol. Immunother. 2008;57:1511–21. doi: 10.1007/s00262-008-0540-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dalle S, Thieblemont C, Thomas L, Dumontet C. Monoclonal antibodies in clinical oncology. Anticancer Agents Med Chem. 2008;8:523–32. doi: 10.2174/187152008784533071. [DOI] [PubMed] [Google Scholar]
- 47.Bernhard H, et al. Adoptive transfer of autologous, HER2-specific, cytotoxic T lymphocytes for the treatment of HER2-overexpressing breast cancer. Cancer Immunol Immunother. 2008;57:271–80. doi: 10.1007/s00262-007-0355-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morgan RA, et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–51. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parente-Pereira AC, et al. Trafficking of CAR-engineered human T cells following regional or systemic adoptive transfer in SCID Beige mice. J Clin Immunol. 2011;31:710–8. doi: 10.1007/s10875-011-9532-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.