

Abstract
Dibutylphthalate (DBP), di(2-ethylhexyl)phthalate (DEHP), and di(2-ethylhexyl)adipate (DEHA) are used as plasticizers. Their metabolites activate peroxisome proliferator-activated receptor (PPAR) α, which may be related to their toxicities. However, species differences in the receptor functions between rodents and human make it difficult to precisely extrapolate their toxicity from animal studies to human. In this paper, we compared the species differences in the activation of mouse and human hepatic PPARα by these plasticizers using wild-type (mPPARα) and humanized PPARα (hPPARα) mice. At 12 weeks old, each genotyped male mouse was classified into three groups, and fed daily for 2 weeks per os with corn oil (vehicle control), 2.5 or 5.0 mmol/kg DBP (696, 1392 mg/kg), DEHP (977, 1953 mg/kg), and DEHA (926, 1853 mg/kg), respectively. Generally, hepatic PPARα of mPPARα mice was more strongly activated than that of hPPARα mice when several target genes involving β-oxidation of fatty acids were evaluated. Interestingly, all plasticizers also activated hepatic constitutive androstane receptor (CAR) more in hPPARα mice than in mPPARα mice. Taken together, these plasticizers activated mouse and human hepatic PPARα as well as CAR. The activation of PPARα was stronger in mPPARα mice than in hPPARα mice, while the opposite was true of CAR.
1. Introduction
Dibutylphthalate (DBP), di(2-ethylhexyl)phthalate (DEHP), and di(2-ethylhexyl)adipate (DEHA) are used as representative industrial plasticizers, though the use of the first two considerably decreased recently. These chemicals are involved in peroxisome proliferations, similar to endogenous fatty acids, exogenous fibrates, and thiazolidinediones [1–4]. Once most plasticizers are taken into the body, they are metabolized by lipase in several organs such as liver and small intestine, and their metabolites, especially mono-carboxylic acids, activate peroxisome proliferator-activated receptor alpha (PPARα), and influence the receptor-related lipid metabolism, anti-inflammation, glucose metabolism, and ketogenesis [5].
Peroxisome proliferators (PPs) cause hepatocarcinogenesis in rodents, and PPARα is involved in the mode of action [6]. However, the lower expression of PPARα in human liver [7] and ligand affinity for the agonists [2, 3] has been discussed within the context of how the risk of these chemicals is extrapolated to human from the animal data [8]. Indeed, the International Agency for Research on Cancer downgraded the DEHP carcinogenicity potential from 2B to 3, which produced some conflicting views over the past decade [9–13], but then restored the potential to the 2B grade in 2011 [14]. In addition, recent results showed that not only mouse but also human PPARα was eventually activated by several activators, such as trichloroacetic acid [15] or perfluorooctanoic acid [16], with species differences in PPARα-related gene activation [17]. These results further complicated the risk assessment of peroxisome proliferators.
PPARα-humanized (hPPARα) mice, so-called hPPARα Tet-OFF, that express human PPARα only in the liver of PPARα-null mice were recently established [18]. This mouse line expresses human PPARα considerably higher than mouse PPARα in wild-type mice and is a useful tool to elucidate the former function: 0.1, 0.3 mg/kg b.w. of ammonium perfluorooctanoate-activated mouse PPARα, but not human PPARα, suggesting that the activation of the latter may be weaker than the former [16]. In contrast, when 0.1% Wy-14,643 (which is estimated at about 100 ~ 130 mg/kg b.w.) was administered to wild-type and hPPARα mice, the functional activations of the target genes such as mitochondrial and peroxisomal β-oxidation enzymes were almost the same or slightly less in the latter than in the former [18–20]. Taken together, the activation of human PPARα may be weaker than that of mouse PPARα. However, it is doubtful whether the findings are always similar to the other peroxisome proliferators such as DEHP.
Constitutive androstane receptor (CAR) is a representative transcriptional regulator for drug-metabolizing enzymes such as cytochrome P450 (CYP), UDP-glucuronosyl transferase (UGT), or sulfotransferase and activated by xenobiotic ligand phenobarbital (PB) or 1,4-bis [2-(3,5-dichloropyridyloxyl)] benzene (TCPOBOP) [21–23]. Many peroxisome proliferators such as DEHP [24] or PFOA [25] are also xenobiotic ligands or activators. On the other hand, CAR plays an important role in lipid homeostasis because of the interactive action with PPARα and inhibition of PPARα-related oxidation of fatty acids [26]. Indeed, TCPOBOP treatment increased serum triglyceride (TG) [27] because of downregulation of β-oxidation and upregulation of fatty acid synthesis. However, there is no report whether other phthalates such as DBP and adipates activate CAR and influence lipid homeostasis. It is important to examine whether these phthalates act on CAR because CAR activation is related with liver toxicity, such as modulation of acetaminophen-induced hepatotoxicity [28] or PB-induced liver tumor development [29, 30].
In this study, we selected three plasticizers currently used worldwide, DBP, DEHP, and DEHA, to determine the differences among hepatic mouse and human PPARα and CAR activation in response to these plasticizers using two PPARα mouse lines, wild-type (mPPARα) and hPPARα mice. We also investigated how both receptor activations influence plasma and liver TG levels for detection of functional changes in hepatic PPARα and CAR by treatment of plasticizers.
2. Materials and Methods
2.1. Chemicals
Standard grades of DEHP (≥99.5%), DEHA (≥99.0%), and DBP (≥99.5%) were purchased from Wako Pure Chemical Industries (Osaka, Japan).
2.2. Experimental Animals
This study was conducted according to the Guidelines for Animal Experiments of The Nagoya University Animal Center. Two genotyped male mice with a Sv/129 genetic background, hPPARα [18] and wild-type mPPARα, were used to identify respective PPARα functions in the lipid metabolism. All mice were housed in a temperature- and light-controlled environment (25°C, 12 h light/dark cycle) and maintained on stock rodent chow and tap water ad libitum. At 12 weeks old, each genotyped mouse was classified into three groups: one group was treated with corn oil daily for two weeks by gavage (vehicle control group); the other two were treated with 2.5 or 5.0 mmol/kg DEHP (977, 1953 mg/kg), DEHA (926, 1853 mg/kg), or DBP (696, 1392 mg/kg), for two weeks. No significant differences were observed in the body weight at the start of the three plasticizer treatments (data not shown). On the next day after the last dose (18–20 hours later), all the mice were killed by decapitation, and the blood and livers were removed. The liver samples were stored at −80°C until use; as for the blood, after centrifuging at 3,500 g for 10 min, the plasma was stored at −80°C until use.
2.3. Nuclear Fraction
A nuclear fraction was extracted from a part of the frozen liver using a CelLytic NuCLEAR Extraction Kit (SIGMA, Tokyo, Japan).
2.4. Analysis of Protein Concentrations
Each tissue was homogenized with a three-fold volume of 10 mM phosphate buffer (pH 7.4) containing 0.25 M sucrose. Protein concentrations of the homogenate samples were measured using a Protein Assay Kit (Bio-Rad, Tokyo, Japan).
2.5. Lipid Concentrations in Plasma and Liver
Lipid from liver was extracted using the method of Folch et al. [31]. TG in the liver and plasma measured using a TG-IE kit (Wako, Osaka, Japan).
2.6. Histopathological Analysis
The organs fixed in 10% neutral buffered formalin were embedded in paraffin and sliced into 2 μm sections. Tissue sections of the livers were stained with hematoxylin and eosin and examined under a light microscope using the BZ-8000 (Keyence Corporation, Osaka, Japan). Histopathological findings were scored according to the degree of lipid accumulation and necrosis with inflammatory cell infiltration.
2.7. Real-Time Quantitative PCR
Total RNA was isolated using RNeasy Mini Kit (QIAGEN, Tokyo, Japan). Complementary DNA (cDNA) was synthesized from 1 μg of total RNA using Oligo(dT)20 primer. RNA quantity and quality were checked by a GeneQuant II RNA/DNA Calculator (Pharmacia Biotech, Framingham, MA). Primers were designed using Primer Express software (Applied Biosystems) based on the sequence of the respective GI number, as shown in the Supplemental Table available online at doi:10.1155/2012/201284. As for MTP and Cyp4a14, primers were used elsewhere [26, 32]. These mRNA levels were monitored by the ABI PRISM 7000 Sequence Detection system (Applied Biosystems, Foster City, CA), as described previously [16, 33, 34].
2.8. Western Blotting
Western blotting was conducted by the method described previously [35]. Briefly, the samples for electrophoresis adjusted to 10 μg protein in liver homogenates of nuclear fraction were subjected to 10% SDS-PAGE and transferred to the nitrocellulose membranes. After blocking with 3% skim milk, each membrane was incubated with the primary antibody, followed by incubation with alkaline phosphatase-conjugated goat anti-rabbit IgG (Jackson Immuno Research, West Grove, PA). The primary polyclonal antibody was prepared using purified medium-chain acyl-CoA dehydrogenase (MCAD) [36], keto-acyl-CoA thiolase (PT) [37], very long-chain acyl-CoA dehydrogenase (VLCAD) [38], and peroxisomal bifunctional protein (PH) [39]. These antibodies were already used elsewhere [15]. The primary polyclonal antibodies of PPARα were purchased from Santa Cruz Biotechnology, Inc. (CA). Each band was quantified using densitometry, the Lane & Spot Analyzer version 5.0 (ATTO Corporation, Tokyo, Japan) as described elsewhere [16, 33, 35]. Each band was normalized to the respective level of glyceraldehyde-3-phosphate dehydrogenase.
2.9. Electrophoretic Mobility Shift Assay (EMSA)
The following oligonucleotides, synthesized by Sigma Aldrich Japan (Tokyo, Japan), were used as probes based on the sequence of DR-4 nuclear-receptor-(NR-) binding sites reported by Kim et al. [40]: NR-1 probe, 5′-biotin-TCTGTACTTTCCTGACCTT-3′; NR-2 probe, 5′-biotin-TCAACTTGACTGACACC-3′.LightShift Chemiluminescent EMSA kit (Pierce Biotechnology, Rockford) was used with a slight modification. Sample mixture contained nuclear extract (4 μg), 0.2 mg/mL poly (dI-dC), 5% glycerol, 0.1% NP-40, 5 mM MgCl2, 0.2 mM EDTA, 2% Ficol (400), 47 mg/mL transfer RNA, and 2 μM biotin-labeled double-stranded oligonucleotide. The reaction samples were resolved on nondenaturing electrophoresis (4% acrylamide) and transferred to a positively charged nylon membrane (Roche Diagnostics, Mannheim, Germany). Constitutive androstane receptor (CAR)-NR-1 and CAR-NR-2 complexes were detected with a Chemiluminescent Nucleic Acid Detection Module (Pierce Biotechnology) and visualized using a Lumi Vision PRO HS II (Aisin Seiki Co., Ltd., Japan).
2.10. Statistical Analysis
Comparisons were made using the two-way analysis of variance (ANOVA) and the Tukey-Kramer HSD post hoc test. A logarithmic transformation was applied to MTP-mRNA before statistical analysis. Values of P < 0.05 were considered to indicate statistical significance.
3. Results
3.1. Body and Liver Weights
No significant differences were observed in body weight after the treatments (Table 1). Exposure to 2.5 (low-dose) and 5.0 mmnol/kg (high-dose) DEHP and DEHA increased both liver weight and liver/body weight ratio only in mPPARα mice, but high-dose DBP increased only the absolute liver weights (Table 1). In contrast, treatment with any plasticizer failed to influence either the liver weight or the liver/body ratio in hPPARα mice.
Table 1.
Body, liver weights and TG levels after treatment with plasticizers for 2 weeks.
| B.W. | Liver weight | Liver weight/ B.W. (%) | Plasma TG | Liver TG | ||
|---|---|---|---|---|---|---|
| mPParα | Control | 23.9 ± 0.91 | 0.88 ± 0.11 | 3.68 ± 0.38 | 79.4 ± 16.3 | 14.8 ± 1.53 |
| DBP 2.5 | 25.9 ± 2.05 | 1.08 ± 0.13 | 4.14 ± 0.17 | 89.9 ± 24.8 | 12.5 ± 2.76 | |
| DBP 5.0 | 26.7 ± 2.01 | 1.20 ± 0.10* | 4.49 ± 0.40 | 113.9 ± 40.4 | 11.4 ± 1.68 | |
| DEHP 2.5 | 22.1 ± 1.82 | 1.13 ± 0.11* | 5.09 ± 0.24* | 82.6 ± 13.8 | 11.6 ± 1.56 | |
| DEHP 5.0 | 22.9 ± 0.92 | 1.26 ± 0.06* | 5.54 ± 0.33* | 84.0 ± 24.5 | 6.8 ± 0.90* | |
| DEHA 2.5 | 25.9 ± 0.85 | 1.20 ± 0.07* | 4.63 ± 0.22* | 136.9 ± 15.9 | 11.4 ± 0.90 | |
| DEHA 5.0 | 24.2 ± 1.81 | 1.28 ± 0.18* | 5.27 ± 0.35* | 119.5 ± 36.3 | 7.5 ± 1.76* | |
|
| ||||||
| hPParα | Control | 22.7 ± 2.20 | 1.04 ± 0.06 | 4.59 ± 0.25 | 97.0 ± 23.6 | 24.4 ± 5.51# |
| DBP 2.5 | 25.0 ± 2.32 | 1.07 ± 0.08 | 4.29 ± 0.18 | 127.0 ± 35.0 | 22.6 ± 4.66 | |
| DBP 5.0 | 23.1 ± 4.51 | 1.05 ± 0.28 | 4.76 ± 0.29 | 95.1 ± 26.0 | 31.9 ± 19.31 | |
| DEHP 2.5 | 23.8 ± 2.58 | 1.12 ± 0.17 | 4.69 ± 0.25 | 111.5 ± 28.0 | 20.6 ± 4.66 | |
| DEHP 5.0 | 21.6 ± 2.58 | 1.03 ± 0.17 | 4.52 ± 0.37 | 67.8 ± 35.0 | 30.9 ± 4.24* | |
| DEHA 2.5 | 24.9 ± 1.03 | 1.12 ± 0.08 | 4.48 ± 0.14 | 142.3 ± 59.9 | 23.1 ± 1.98 | |
| DEHA 5.0 | 24.7 ± 2.94 | 1.23 ± 0.17 | 4.98 ± 0.25 | 176.0 ± 41.0* | 28.4 ± 2.73 | |
B.W: body weight.
Each value represents mean ± S.D. *Significantly different from respective controls (P < 0.05). #Significantly different from mPPARα controls (P < 0.05).
3.2. TG in the Plasma and Liver
The plasma TG level in mPPARα control mice was similar to that in hPPARα controls (Table 1). High-dose DEHA increased plasma TG levels in hPPARα mice, but not in mPPARα mice. In contrast, the other plasticizers did not influence the levels. In each of the control mice, hepatic TG levels were significantly greater in hPPARα mice than in the mPPARα mice (Table 1). High-dose DEHP and DEHA decreased the levels in the liver of mPPARα mice. High-dose DEHP increased the levels in hPPARα mice, whereas DEHA did not. DBP did not influence the TG levels in both genotyped mice. Thus, the TG decrease due to the accelerated lipid metabolism was seen in mPPARα mice treated with DEHP or DEHA. In contrast, hepatic TG accumulation was seen in DEHP-treated hPPARα mice.
3.3. Histopathological Changes
In the control animals, no obvious differences in the scores of lipid accumulation, inflammatory and necrotic cell infiltrations were observed in the liver between both genotyped mice (Figure 1, scores not shown). As mentioned above, hepatic TG levels were greater in hPPARα controls than mPPARα controls; however no obvious histopathological differences in lipid accumulation were found between the two genotyped mice. The hepatocellular enlargements were prominently observed in mPPARα mice of the high-dose DEHP group and slightly in those of high-dose DEHA and DBP groups. Cytoplasmic vacuoles due to lipid accumulation were seen in hPPARα mice exposed to the three plasticizers, though the changes were not dose dependent. A focal necrosis with inflammatory cells was seen in two of five hPPARα mice exposed to high-dose DEHP, all animals exposed to high-dose DEHA and three of five animals exposed to low-dose DEHA. Moderate eosinophilic cytoplasm which may result from the increase in peroxisome or mitochondria was observed in all mPPARα mice treated with high-dose DEHP; however, the finding was minimal in those on the low dose. In contrast, only two of five animals on high-dose DBP and DEHA exhibited minimal or mild eosinophilic cytoplasm, respectively. Taken together, popular histopathological changes caused by peroxisome proliferators such as liver enlargement and eosinophilic cytoplasm were prominent in mPPARα mice treated with high-dose DEHP. On the other hand, focal necrosis was seen mainly in hPPARα mice exposed to high-dose DEHA.
Figure 1.
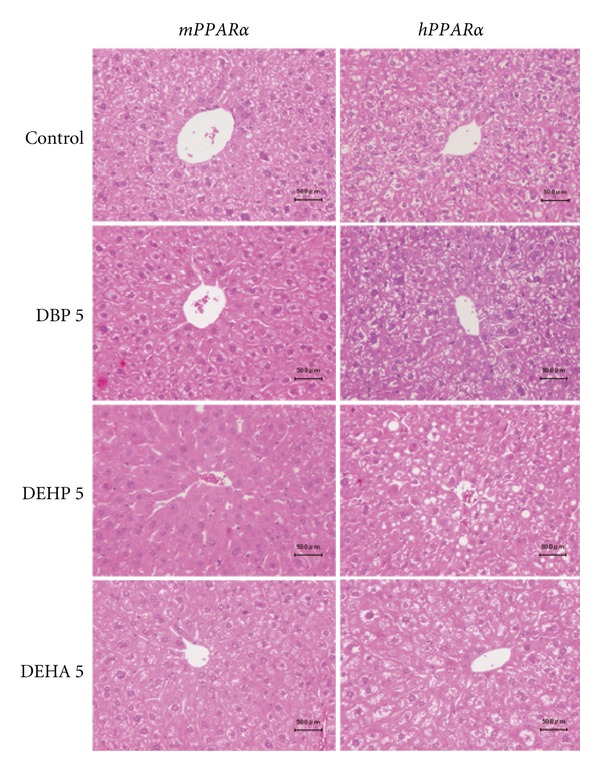
Histopathological changes in livers in mPPARα and hPPARα mice treated with control, high-dose DBP, DEHP, and DEHP for 2 weeks. Hepatocellular enlargements were prominently observed in mPPARα mice of DEHP group and slightly in those of DEHA and DBP. Moderate eosinophilic cytoplasm was observed in mPPARα mice treated with DEHP. Cytoplasmic vacuoles due to lipid accumulation were seen in hPPARα mice exposed to three plasticizers. Each scale bar indicates 50 μm.
3.4. PPARα and Target Genes
Low-dose DBP significantly increased PH- and PT-mRNA levels (2.7-fold and 2.0-fold, resp.) in mPPARα mice (Figure 2), whereas low-dose DEHP and DEHA did not. In high-dose groups, all plasticizers increased hepatic peroxisomal PH- and PT-mRNA in mPPARα mice, while DBP alone induced PT-mRNA in hPPARα mice. The increases were greatest in DEHP-treated mPPARα mice (7.1-fold and 4.1-fold, resp.), and those by DBP and DEHA treatments were almost the same (2.6-fold, 2.5-fold and 3.0-fold, 2.9-fold, resp.). All plasticizers at low dose did not influence hepatic mitochondrial MCAD- and VLCAD-mRNA levels. High-dose DEHP, however, increased both mRNA levels only in mPPARα mice, but only marginally (1.8-fold and 1.4-fold, resp.).
Figure 2.
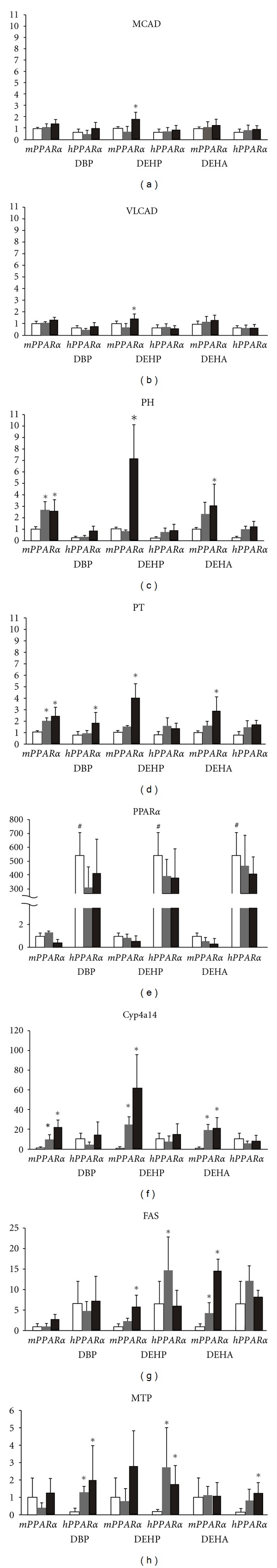
mRNA expressions of hepatic PPARα and its related genes in duplicate analyses. Expressions of mRNA were analyzed by quantitative real-time PCR. Each mRNA was normalized to the level of GAPDH-mRNA expression in the same preparation, and mean of control in mPPARα mice was assigned a value of 1.0. White, gray, and black columns represent control values, 2.5 mM- and 5.0 mM-treated group, respectively. Each column and bar represents mean ± S.D., respectively. A logarithmic transformation was applied to MTP-mRNA before statistical analysis. *Significantly different from respective controls (P < 0.05). #Significantly different among genotypes (P < 0.05).
All plasticizers at low dose increased PH and PT protein in the liver of both genotyped mice except PH in DEHA-treated hPPARα mice and PT in DBP-treated mPPARα mice (Figures 3(a) and 3(b)). All plasticizers at high dose also increased PH and PT protein in the livers of both mPPARα and hPPARα mice. The inductions of PH were slightly stronger in mPPARα exposed to DBP and DEHP (DBP, 5.9-fold; DEHP, 6.0-fold; DEHA, 5.3-fold) than in hPPARα mice (3.9-fold, 1.9-fold, 5.1-fold, resp.). The increases of PT by DEHP or DEHA treatments were also stronger in mPPARα (2.8-fold and 1.8-fold, resp.) than in hPPARα mice (1.3-fold and 1.4-fold, resp.), although those by DBP were almost the same in both mPPARα and hPPARα mice.
Figure 3.
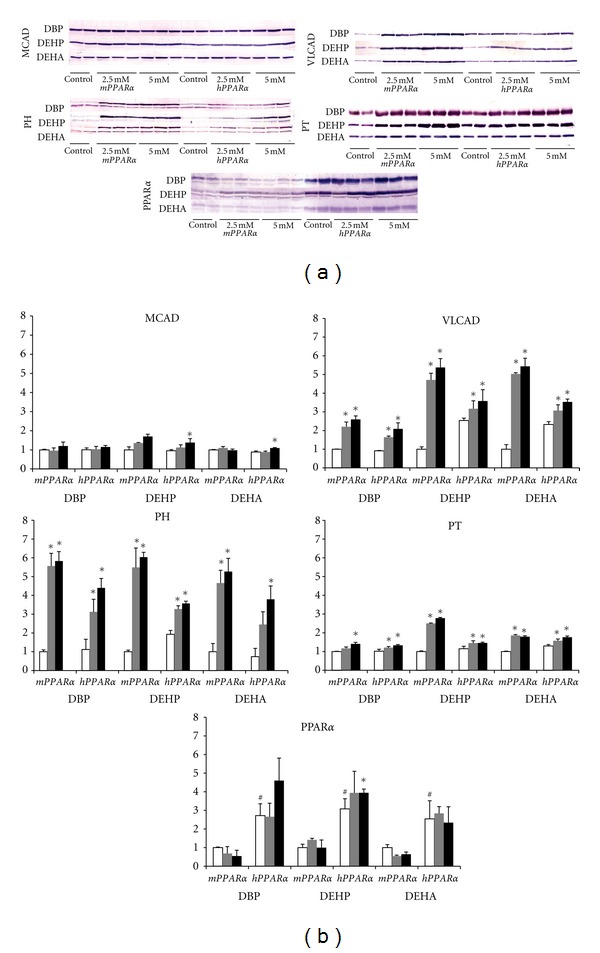
(a) Western blotting analysis of hepatic PPARα and related genes. All mice from each treatment and genotype were examined across two gels, one of which is shown here. (b) Western blotting analysis of hepatic PPARα and related genes. Each band was quantified by densitometric analysis as described in Materials and Methods, and mean strength of control in mPPARα mice was assigned a value of 1.0. White, gray, and black columns represent control values, 2.5 mM- and 5.0 mM-treated group, respectively. Each column and bar represents mean ± S.D., respectively. *Significantly different from respective controls (P < 0.05). #Significantly different among genotypes (P < 0.05).
In mitochondrial enzymes, three plasticizers at any doses increased hepatic VLCAD protein expressions in both mPPARα and hPPARα mice. The inductions appeared to be stronger in mPPARα mice exposed to DEHP and DEHA (DBP: 2.6-fold, DEHP: 5.4-fold, DEHA: 5.4-fold) than in corresponding hPPARα mice (2.3-fold, 1.4-fold, 1.5-fold, resp.), similar to peroxisomal enzyme PH. High-dose DEHP and DEHA increased hepatic MCAD levels in mPPARα and hPPARα mice, and in hPPARα mice, respectively, whereas DBP did not affect the levels in either mPPARα mice or hPPARα mice.
Low- and high-dose DEHA, DEHP, and DBP also increased hepatic Cyp4a14, a microsomal enzyme involved in ω-oxidation of many plasticizers, expressions only in mPPARα mice but not in hPPARα mice (Figure 2). Inductions in the former mice were 23-fold, 62-fold, and 21-fold at high-dose DBP, DEHP, and DEHA, respectively.
In the control group, the expression of PPARα was significantly greater in hPPARα mice than in mPPARα mice either in the mRNA (540-fold) or protein (about 3-fold) levels (Figures 2, 3(a), and 3(b)). No treatments elevated mouse and human PPARα-mRNAs. High-dose DEHP increased only PPARα protein expression in hPPARα mice, but other treatments did not.
Low- and high-dose DEHA and high-dose DEHP significantly increased FAS-mRNA to 4.4-fold and 14.7-fold, and 5.8-fold in mPPARα mice, respectively (Figure 2). Low-dose DEHP also increased it to 14.9-fold in hPPARα mice. However, DBP treatment did not influence FAS-mRNA in both genotype mice. We also measured MTP-mRNA levels in the liver: low- and high-dose DBP and DEHP increased the mRNA to 8.8-fold and 13.5-fold, and 18.8-fold and 11.8-fold, respectively, in hPPARα mice but not in mPPARα mice. Similarly, high-dose DEHA increased MTP-mRNAs (8.5-fold) only in hPPARα mice.
Collectively, inductions of peroxisomal, mitochondrial, and microsomal enzymes involved in β-oxidation were stronger in mPPARα mice than in hPPARα mice treated with plasticizers in terms of mRNA levels, whereas transporter enzyme was induced only in hPPARα mice exposed to plasticizers.
3.5. CAR and Target Gene
Low- and high-dose DEHA and high-dose DEHP and DBP decreased CAR-mRNA levels in mPPARα mice, but the levels in hPPARα mice were not affected at any dose (Figure 4(a)). In contrast, high-dose DEHP strongly induced typical CAR target gene, Cyp2b10-mRNA, in hPPARα mice (48.3-fold). Low- and high-dose DEHA induced Cyp2b10-mRNA levels in hPPARα mice (31.2-fold and 24.5-fold, resp.). The high-dose DEHA also elevated the mRNA levels in mPPARα mice (9.2-fold), but only marginally compared with those in hPPARα mice. In contrast, DBP did not influence the levels in both genotyped mice.
Figure 4.
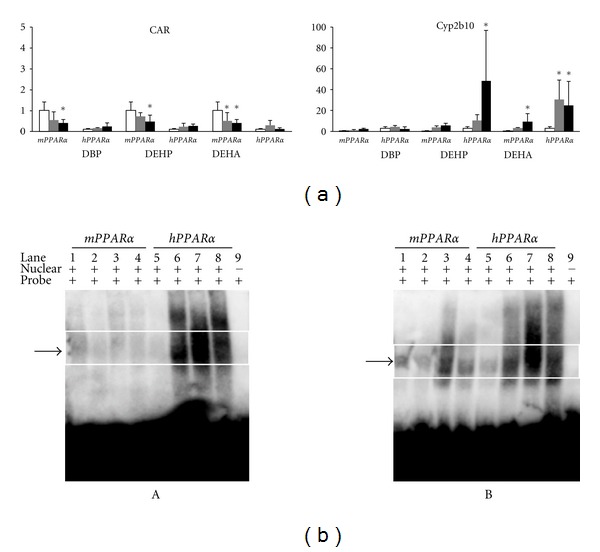
(a) Effects on hepatic expressions of CAR and Cyp2b10-mRNA levels. Each mRNA level was normalized to the level of GAPDH mRNA in the same preparation, and the mean of the control group in wild-type (mPPARα) mice was assigned a value of 1.0. White, gray and black columns represent control values, 2.5 mM- and 5.0 mM-treated group, respectively. Values are expressed as mean ± S.D. *Significantly different from respective control group (P < 0.05). (b) Electrophoresis mobility shift assays of CAR-NR-1 (A) and CAR-NR-2 (B) complexes in liver nuclear fraction from control or treated-mPPARα (wild-type) and hPPARα mice. Lanes 1 and 5, control of wild-type, respectively; lanes 2 and 6, wild-type and hPPARα mice treated with 5.0 mM DBP, respectively; lanes 3 and lane 7, wild-type and hPPARα mice treated with 5.0 mM DEHP, respectively; lanes 4 and lane 8, wild-type and hPPARα mice treated with 5.0 mM DEHA, respectively; lane 9, oligonucleotide for NR-1 or NR-2 only. Arrows indicate the shifted CAR-NR complex.
The treatments with all plasticizers dramatically induced NR-1 (Figure 4(b) A) and NR-2 (Figure 4(b) B) DNA-binding activity of hepatic CAR in hPPARα mice at high dose. The high-dose DEHP also induced NR-2-binding activity in mPPARα mice, but DBP or DEHA did not. The activities in hPPARα mice were strongest in the DEHP-treated group, followed by the DEHA- and DBP-treated group.
In summary, plasticizers, especially in DEHP or DEHA, bind to hepatic CAR and markedly induce CAR-target gene mainly in hPPARα mice.
4. Discussion
The present study clearly shows that three plasticizers (DEHP, DEHA, and DBP) significantly activated mouse hepatic PPARα in mPPARα mice, but the activation of human hepatic PPARα in hPPARα mice was weaker than that of the former mouse line even at the high-dose exposure, especially in peroxisomal β- or ω-oxidation. Among the three plasticizers, DEHP is the strongest from the standpoint of PPARα-mediated gene responses. These results are consistent with in vitro studies [3, 4] which demonstrated that mono (2-ethylhexyl) phthalate (MEHP) activated mouse PPARα at lower concentrations and exhibited a stronger response than those of human PPARα [4], and MEHP activated mouse and human PPARα at a lower concentration than the respective monoesters of DBP and DEHA [3, 4]. Interestingly, these species differences in PPARα activation were most prominent in microsomal PPARα-target gene, Cyp4a14, followed by mitochondrial (MCAD, VLCAD) or peroxisomal enzymes (PH, PT). Notably, all the plasticizers also activated CAR preferentially in hPPARα mice. The activation was also stronger in DEHP than DEHA judging from the target gene (Cyp2b10) as well as the DNA-binding (NR-1 and 2) activity analysis.
As mentioned above, DEHP and DEHA activated PPARα and CAR preferentially in mPPARα and hPPARα mice, respectively. Our finding is very similar to the fact that DEHP induced Cyp2b10 more strongly in the livers of PPARα-null mice than mPPARαones [24, 41]. Although the reason why CAR induction was stronger in hPPARα mice than in mPPARα mice remains unclear, it is likely that CAR is more easily activated when the function of PPARα is weak, as with human PPARα in hPPARa mice [15] or lack of PPARα in Pparα-null mice [41]. CAR was reported to crosstalk with PPARα and suppress its related gene expressions such as Cyp4a14 and carnitine palmitoyltransferase 1α in the liver of mice [26, 27]. It is of interest that DEHP activated both receptors more than DEHA. However, the chemical form of the activator for each receptor may be different; since MEHP did not induce Cyp2b10 in JWZ-CAR cell line [42], the parent substance itself may be an activator of CAR. No report on DEHA indicated that either the parent substance itself or the metabolite(s) is a preferential activator for CAR. In the present study, DBP also induced binding activity of CAR in hPPARα mice but did not increase Cyp2b10-mRNA in that strain, though DBP has been reported to activate CAR in the liver of rats [43]. Interestingly, the CAR2 splice variant of human CAR is activated by DEHP [44], which suggests that human CAR may also play an important role in DEHP toxicity. Taken together, CAR-mediated effects by plasticizers should be noted as a novel aspect of their toxicities to provide a new rationale to evaluate toxicity correctly.
Species differences of mouse and human PPARα activation by Wy-14,643 have been investigated using mPPARα and hPPARα mice fed 0.1% Wy-14,643-containing feed for 2 weeks ad libitum [18], at a dose roughly estimated to be 0.3 ~ 0.4 mmol/kg/day. This dose significantly induced peroxisomal and mitochondrial fatty acid-metabolizing enzymes such as acyl-CoA oxidase, VLCAD, and MCAD, followed by a similar decrease in serum triglycerides in both mouse lines. Even a lower dose of Wy-14,643 than the plasticizers used in this study was presumed to activate mouse and human PPARα to a similar extent along with decreased plasma TG levels. This result suggests that there may not be a species difference in the activation by Wy-14,643. Since all plasticizers induced PPARα-related enzymes involved in β- or ω-oxidation in mPPARα mice but none of them influenced the plasma TG level, the PPARα activation by Wy-14,643 is not coincident with the present study from the standpoint of PPARα-target gene induction as well as plasma TG levels.
DEHP was the strongest inducer of PPARα-related β-oxidation enzymes in mPPARα mice among the three chemicals. It was also the strongest activator for CAR in both mPPARαand hPPARα mice in our study. However, Wy-14,643 did not activate CAR [41]. In this regard, the effect of Wy-14,643 on the nuclear receptors is different from that of DEHP. TCPOBOP, a CAR potent agonist, was suggested to cause an accumulation of serum TG [26, 27], whereas the PPARα agonist Wy-14,643 decreased it. These opposite actions by CAR and PPARα in TG homeostasis [45] may reflect the plasma TG unchanged by DEHP, because DEHP induced both PPARα and CAR. In contrast, the hPPARα mice exposed to high-dose DEHA had elevated plasma TG. In these mice, MTP-mRNA, which was involved in the transport of TG from liver to blood, was induced and may partly be the reason for the increased plasma TG, even though CAR was also induced by DEHA treatment.
As for TG levels in livers, the high dose of DEHP or DEHA decreased the levels in mPPARα mice, whereas DEHP increased the levels in hPPARα mice. The increase in hPPARα mice, as different from that in mPPARα mice, may be ascribed to the weaker inductions of enzymes involved in β- and ω-oxidation in hPPARα mice than in mPPARα mice. MEHP increased TG in hepatocyte culture of guinea pig because of the weak induction of β-oxidation and lauric acid hydroxylation, whereas it decreased TG in rat hepatocytes due to the significant induction of these enzymes [46]. The degree of β-oxidation-related enzyme inductions by DEHP was comparable between mice and rats [34]. Taken together, the difference in mouse and human PPPARα functions presumably produced the different effects of DEHP or DEHA on hepatic TG accumulation between mPPARαand hPPARα mice.
In the present study, we only investigated the effects of three kinds of plasticizers on the lipid metabolism and did not investigate DEHP- or DEHA-caused tumors in relation to PPARα. CAR is thought to mediate the hepatocarcinogenic effects of xenobiotics [29], suggesting that it may contribute to the PPARα-independent hepatocarcinogenesis observed in PPARα-null mice following chronic DEHP exposure [35]. DEHP at a 1150 mg/kg dose for 4 days induced CAR and Cyp2b10-mRNAs only in PPARα-null mice, and 200 mg/kg DEHP induced them in both wild-type and PPARα-null mice [41]. The induced rate was greater in the latter than the former mice, suggesting that PPARα-null mice are more susceptible to DEHP-induced CAR signaling compared to that of mPPARα mice. DEHP activated not only PPARα but also CAR, though Wy-14,643 did not activate CAR [41]. This different signaling suggests that the molecular mechanism of carcinogenicity in phthalates may not always be the same as that of Wy-14,643.
Finally, hepatic mRNAs of cell cycle-related genes such as cyclin D1, protooncogene such as c-jun, and apoptosis-related gene Bax, were measured using mPPARα and hPPARα mice exposed to the plasticizers, but these mRNA levels did not increase in both genotyped mice; instead, decreases of cell cycle-related genes were observed in both genotyped mice (unpublished data), which is not consistent with the case of Wy-14,643 [19]. These results again suggest that DEHP-induced molecular signalings are not always the same as those by Wy-14,643. The reason for this is unclear, but the weaker affinity of DBP, DEHP, and DEHA for human and mouse PPARα than Wy-14,643 may be a possible explanation [4].
In conclusion, these plasticizers activated not only mouse and human hepatic PPARα but also CAR, and the activation of PPARα was stronger in mPPARα mice than in hPPARα mice, while that of CAR was the opposite. Thus, DEHP is not only a PPARα agonist but also a CAR activator, which may trigger each function.
Supplementary Material
Primer lists.
Acknowledgment
This study was supported in part by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (B. 14370121, 17390169), Food Safety Commission, Japan (1002), and Health and Labour Sciences Research Grants from Research on Food Safety of the Ministry of Health, Labour and Welfare in Japan.
Conflict of Interests
The authors declare that they have no conflict of interests.
Abbreviations
- ANOVA:
Analysis of variance
- CAR:
Constitutive androstane receptor
- CV:
Central vein
- DBP:
Dibutylphthalate
- DEHP:
Di(2-ethylhexyl)phthalate
- DEHA:
Di(2-ethylhexyl)adipate
- DGAT:
Diacylglycerol acyltransferase
- EMSA:
Electrophoretic mobility shift assay
- hPPARα:
Humanized PPARα mouse
- MCAD:
Medium-chain acyl-CoA dehydrogenase
- MEHP:
Mono(2-ethylhexyl)phthalate
- mPPARα:
Wild-type mouse
- MTP:
Microsomal triacylglycerol transfer protein
- NR:
DR-4 nuclear receptor binding site
- PB:
Phenobarbital
- PH:
Peroxisomal bifunctional protein
- PPARα:
Peroxisome proliferator-activated receptor α
- PT:
Keto-acyl-CoA thiolase
- TG:
Triglyceride
- VLCAD:
Very long-chain acyl-CoA dehydrogenase.
References
- 1.Gonzalez FJ, Peters JM, Cattley RC. Mechanism of action of the nongenotoxic peroxisome proliferators: role of the peroxisome proliferator-activated receptor. Journal of the National Cancer Institute. 1998;90(22):1702–1709. doi: 10.1093/jnci/90.22.1702. [DOI] [PubMed] [Google Scholar]
- 2.Maloney EK, Waxman DJ. trans-Activation of PPARalpha and PPARgamma by structurally diverse environmental chemicals. Toxicology and Appllied Pharmacology. 1999;161(2):209–218. doi: 10.1006/taap.1999.8809. [DOI] [PubMed] [Google Scholar]
- 3.Hurst CH, Waxman DJ. Activation of PPARα and PPARγ by environmental phthalate monoesters. Toxicological Sciences. 2003;74(2):297–308. doi: 10.1093/toxsci/kfg145. [DOI] [PubMed] [Google Scholar]
- 4.Bility MT, Thompson JT, McKee RH, et al. Activation of mouse and human peroxisome proliferator-activated receptors (PPARs) by phthalate monoesters. Toxicological Sciences. 2004;82(1):170–182. doi: 10.1093/toxsci/kfh253. [DOI] [PubMed] [Google Scholar]
- 5.Mandard S, Muller M, Kersten S. Peroxisome proliferator-activated receptor α target genes. Cellular and Molecular Life Sciences. 2004;61(4):393–416. doi: 10.1007/s00018-003-3216-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klaunig JE, Babich MA, Baetcke KP, et al. PPARα agonist-induced rodent tumors: modes of action and human relevance. Critical Reviews in Toxicology. 2003;33(6):655–780. doi: 10.1080/713608372. [DOI] [PubMed] [Google Scholar]
- 7.Palmer CNA, Hsu MH, Griffin KJ, Raucy JL, Johnson EF. Peroxisome proliferator activated receptor-α expression in human liver. Molecular Pharmacology. 1998;53(1):14–22. [PubMed] [Google Scholar]
- 8.Rusyn I, Corton JC. Mechanistic considerations for human relevance of cancer hazard of di(2-ethylhexyl) phthalate. Mutation Research. 2012;750(2):141–158. doi: 10.1016/j.mrrev.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Melnick RL. Is peroxisome proliferation an obligatory precursor step in the carcinogenicity of di(2-ethylhexyl)phthalate (DEHP)? Environmental Health Perspectives. 2001;109(5):437–442. doi: 10.1289/ehp.01109437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guyton KZ, Chiu WA, Bateson TF, et al. A reexamination of the PPAR-α activation mode of action as a basis for assessing human cancer risks of environmental contaminants. Environmental Health Perspectives. 2009;117(11):1664–1672. doi: 10.1289/ehp.0900758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Melnick RL, Brody C, DiGangi J, Huff J. The IARC evaluation of DEHP excludes key papers demonstrating carcinogenic effects. International Journal of Occupational and Environmental Health. 2003;9(4):400–402. [PubMed] [Google Scholar]
- 12.Melnick RL. The IARC evaluation of di(2-ethylhexyl)phthalate (DEHP): a flawed decision based on an untested hypothesis. International Journal of Occupational and Environmental Health. 2002;8(3):284–286. doi: 10.1179/107735202800338803. [DOI] [PubMed] [Google Scholar]
- 13.Huff J. IARC and the DEHP quagmire. International Journal of Occupational and Environmental Health. 2003;9(4):402–404. doi: 10.1179/oeh.2003.9.4.402. [DOI] [PubMed] [Google Scholar]
- 14.Grosse Y, Baan R, Secretan-Lauby B, et al. Carcinogenicity of chemicals in industrial and consumer products, food contaminants and flavourings, and water chlorination byproducts. The lancet oncology. 2011;12(4):328–329. doi: 10.1016/s1470-2045(11)70088-2. [DOI] [PubMed] [Google Scholar]
- 15.Ramdhan DH, Kamijima M, Wang D, et al. Differential response to trichloroethylene-induced hepatosteatosis in wild-type and PPARα-humanized mice. Environmental Health Perspectives. 2010;118(11):1557–1563. doi: 10.1289/ehp.1001928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakamura T, Ito Y, Yanagiba Y, et al. Microgram-order ammonium perfluorooctanoate may activate mouse peroxisome proliferator-activated receptor α, but not human PPARα . Toxicology. 2009;265(1-2):27–33. doi: 10.1016/j.tox.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shah YM, Morimura K, Yang Q, Tanabe T, Takagi M, Gonzalez FJ. Peroxisome proliferator-activated receptor α regulates a microRNA-mediated signaling cascade responsible for hepatocellular proliferation. Molecular and Cellular Biology. 2007;27(12):4238–4247. doi: 10.1128/MCB.00317-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheung C, Akiyama TE, Ward JM, et al. Diminished hepatocellular proliferation in mice humanized for the nuclear receptor peroxisome proliferator-activated receptor α . Cancer Research. 2004;64(11):3849–3854. doi: 10.1158/0008-5472.CAN-04-0322. [DOI] [PubMed] [Google Scholar]
- 19.Morimura K, Cheung C, Ward JM, Reddy JK, Gonzalez FJ. Differential susceptibility of mice humanized for peroxisome proliferator-activated receptor α to Wy-14,643-induced liver tumorigenesis. Carcinogenesis. 2006;27(5):1074–1080. doi: 10.1093/carcin/bgi329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perrone CE, Shao L, Williams GM. Effect of rodent hepatocarcinogenic peroxisome proliferators on fatty acyl-CoA oxidase, DNA synthesis, and apoptosis in cultured human and rat hepatocytes. Toxicology and Applied Pharmacology. 1998;150(2):277–286. doi: 10.1006/taap.1998.8413. [DOI] [PubMed] [Google Scholar]
- 21.Tzameli I, Pissios P, Schuetz EG, Moore DD. The xenobiotic compound 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene is an agonist ligand for the nuclear receptor CAR. Molecular and Cellular Biology. 2000;20(9):2951–2958. doi: 10.1128/mcb.20.9.2951-2958.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zelko I, Negishi M. Phenobarbital-elicited activation of nuclear receptor CAR in induction of cytochrome P450 genes. Biochemical and Biophysical Research Communications. 2000;277(1):1–6. doi: 10.1006/bbrc.2000.3557. [DOI] [PubMed] [Google Scholar]
- 23.Honkakoski P, Zelko I, Sueyoshi T, Negishi M. The nuclear orphan receptor CAR-retinoid X receptor heterodimer activates the phenobarbital-responsive enhancer module of the CYP2B gene. Molecular and Cellular Biology. 1998;18(10):5652–5658. doi: 10.1128/mcb.18.10.5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eveillard A, Mselli-Lakhal L, Mogha A, et al. Di-(2-ethylhexyl)-phthalate (DEHP) activates the constitutive androstane receptor (CAR): a novel signalling pathway sensitive to phthalates. Biochemical Pharmacology. 2009;77(11):1735–1746. doi: 10.1016/j.bcp.2009.02.023. [DOI] [PubMed] [Google Scholar]
- 25.Cheng X, Klaassen CD. Perfluorocarboxylic acids induce cytochrome P450 enzymes in mouse liver through activation of PPAR-α and CAR transcription factors. Toxicological Sciences. 2008;106(1):29–36. doi: 10.1093/toxsci/kfn147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maglich JM, Lobe DC, Moore JT. The nuclear receptor CAR (NR1I3) regulates serum triglyceride levels under conditions of metabolic stress. Journal of Lipid Research. 2009;50(3):439–445. doi: 10.1194/jlr.M800226-JLR200. [DOI] [PubMed] [Google Scholar]
- 27.Rezen T, Tamasi V, Lovgren-Sandblom A, Bjorkhem I, Meyer UA, Rozman D. Effect of CAR activation on selected metabolic pathways in normal and hyperlipidemic mouse livers. BMC Genomics. 2009;10, article 384 doi: 10.1186/1471-2164-10-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang J, Huang W, Chua SS, Wei P, Moore DD. Modulation of acetaminophen-induced hepatotoxicity by the xenobiotic receptor CAR. Science. 2002;298(5592):422–424. doi: 10.1126/science.1073502. [DOI] [PubMed] [Google Scholar]
- 29.Huang W, Zhang J, Washington M, et al. Xenobiotic stress induces hepatomegaly and liver tumors via the nuclear receptor constitutive androstane receptor. Molecular Endocrinology. 2005;19(6):1646–1653. doi: 10.1210/me.2004-0520. [DOI] [PubMed] [Google Scholar]
- 30.Yamamoto Y, Moore R, Goldsworthy TL, Negishi M, Maronpot RR. The orphan nuclear receptor constitutive active/androstane receptor is essential for liver tumor promotion by phenobarbital in mice. Cancer Research. 2004;64(20):7197–7200. doi: 10.1158/0008-5472.CAN-04-1459. [DOI] [PubMed] [Google Scholar]
- 31.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. The Journal of Biological Chemistry. 1957;226(1):497–509. [PubMed] [Google Scholar]
- 32.Ameen C, Edvardsson U, Ljungberg A, et al. Activation of peroxisome proliferator-activated receptor alpha increases the expression and activity of microsomal triglyceride transfer protein in the liver. The Journal of Biological Chemistry. 2005;280(2):1224–1229. doi: 10.1074/jbc.M412107200. [DOI] [PubMed] [Google Scholar]
- 33.Yanagiba Y, Ito Y, Kamijima M, Gonzalez FJ, Nakajima T. Octachlorostyrene induces cytochrome P450, UDP-glucuronosyltransferase, and sulfotransferase via the aryl hydrocarbon receptor and constitutive androstane receptor. Toxicological Sciences. 2009;111(1):19–26. doi: 10.1093/toxsci/kfp130. [DOI] [PubMed] [Google Scholar]
- 34.Ito Y, Yamanoshita O, Kurata Y, Kamijima M, Aoyama T, Nakajima T. Induction of peroxisome proliferator-activated receptor alpha (PPARα)-related enzymes by di(2-ethylhexyl) phthalate (DEHP) treatment in mice and rats, but not marmosets. Archives of Toxicology. 2007;81(3):219–226. doi: 10.1007/s00204-006-0141-x. [DOI] [PubMed] [Google Scholar]
- 35.Ito Y, Yamanoshita O, Asaeda N, et al. Di(2-ethylhexyl)phthalate induces hepatic tumorigenesis through a peroxisome proliferator-activated receptor α-independent pathway. Journal of Occupational Health. 2007;49(3):172–182. doi: 10.1539/joh.49.172. [DOI] [PubMed] [Google Scholar]
- 36.Furuta S, Mayazawa S, Hashimoto T. Purification and properties of rat liver Acyl-CoA dehydrogenases and electron transfer flavoprotein. Journal of Biochemistry. 1981;90(6):1739–1750. doi: 10.1093/oxfordjournals.jbchem.a133651. [DOI] [PubMed] [Google Scholar]
- 37.Miyazawa S, Osumi T, Hashimoto T. The presence of a new 3-oxoacyl-CoA thiolase in rat liver peroxisomes. European Journal of Biochemistry. 1980;103(3):589–596. doi: 10.1111/j.1432-1033.1980.tb05984.x. [DOI] [PubMed] [Google Scholar]
- 38.Izai K, Uchida Y, Orii T, Yamamoto S, Hashimoto T. Novel fatty acid β-oxidation enzymes in rat liver mitochondria: I. Purification and properties of very-long-chain acyl-coenzyme A dehydrogenase. The Journal of Biological Chemistry. 1992;267(2):1027–1033. [PubMed] [Google Scholar]
- 39.Osumi T, Hashimoto T. Purification and properties of mitochondrial and peroxisomal 3-hydroxyacyl-CoA dehydrogenase from rat liver. Archives of Biochemistry and Biophysics. 1980;203(1):372–383. doi: 10.1016/0003-9861(80)90189-7. [DOI] [PubMed] [Google Scholar]
- 40.Kim J, Min G, Kemper B. Chromatin assembly enhances binding to the CYP2B1 phenobarbital-responsive unit (PBRU) of nuclear factor-1, which binds simultaneously with constitutive androstane receptor (CAR)/retinoid X receptor (RXR) and enhances CAR/RXR-mediated activation of the PBRU. The Journal of Biological Chemistry. 2001;276(10):7559–7567. doi: 10.1074/jbc.M008090200. [DOI] [PubMed] [Google Scholar]
- 41.Ren H, Aleksunes LM, Wood C, et al. Characterization of peroxisome proliferator-activated receptor α—independent effects of PPARα activators in the rodent liver: di-(2-ethylhexyl) phthalate also activates the constitutive-activated receptor. Toxicological Sciences. 2010;113(1):45–59. doi: 10.1093/toxsci/kfp251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eveillard A, Lasserre F, de Tayrac M, et al. Identification of potential mechanisms of toxicity after di-(2-ethylhexyl)-phthalate (DEHP) adult exposure in the liver using a systems biology approach. Toxicology and Applied Pharmacology. 2009;236(3):282–292. doi: 10.1016/j.taap.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 43.Wyde ME, Kirwan SE, Zhang F, et al. Di-n-butyl phthalate activates constitutive androstane receptor and pregnane X receptor and enhances the expression of steroid-metabolizing enzymes in the liver of rat fetuses. Toxicological Sciences. 2005;86(2):281–290. doi: 10.1093/toxsci/kfi204. [DOI] [PubMed] [Google Scholar]
- 44.DeKeyser JG, Stagliano MC, Auerbach SS, Prabhu KS, Jones AD, Omiecinski CJ. Di(2-ethylhexyl) phthalate is a highly potent agonist for the human constitutive androstane receptor splice variant CAR2. Molecular Pharmacology. 2009;75(5):1005–1013. doi: 10.1124/mol.108.053702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu C, Gilroy R, Taylor R, et al. Alteration of hepatic nuclear receptor-mediated signaling pathways in HCV patients with and without a history of alcohol drinking. Hepatology. 2011;54(6):1966–1974. doi: 10.1002/hep.24645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dirven HAAM, van den Broek PHH, Peeters MCE, et al. Effects of the peroxisome proliferator mono(2-ethylhexyl)phthalate in primary hepatocyte cultures derived from rat, guinea pig, rabbit and monkey. Relationship between interspecies differences in biotransformation and peroxisome proliferating potencies. Biochemical Pharmacology. 1993;45(12):2425–2434. doi: 10.1016/0006-2952(93)90223-j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primer lists.