

Abstract
Cells often migrate in vivo in an extracellular matrix that is intrinsically three-dimensional (3D) and the role of actin filament architecture in 3D cell migration is less well understood. Here we show that, while recently identified linkers of nucleoskeleton to cytoskeleton (LINC) complexes play a minimal role in conventional 2D migration, they play a critical role in regulating the organization of a subset of actin filament bundles – the perinuclear actin cap - connected to the nucleus through Nesprin2giant and Nesprin3 in cells in 3D collagen I matrix. Actin cap fibers prolong the nucleus and mediate the formation of pseudopodial protrusions, which drive matrix traction and 3D cell migration. Disruption of LINC complexes disorganizes the actin cap, which impairs 3D cell migration. A simple mechanical model explains why LINC complexes and the perinuclear actin cap are essential in 3D migration by providing mechanical support to the formation of pseudopodial protrusions.
The roles of actin filament dynamics and network organization in conventional cell migration and morphology on flat substrates have been studied extensively1,2. However, fibroblasts often migrate in vivo in an extracellular matrix that is intrinsically three-dimensional (3D) and the role of actin filament architecture in 3D cell migration is less well-understood3,4,5. In particular, whether recently discovered connections between nucleus and cytoskeleton mediated by Linkers of the Nucleoskeleton to the Cytoskeleton (LINC) complexes6 play any role in cell shape, cell migration, and associated protrusion activity in 3D extracellular matrices is unknown7. This question is important since protrusion activity plays a central role in 3D migration8,9, as pseudopodial protrusion processes allow cells to probe the pericellular matrix, locally attach to and pull on surrounding fibers, and detach from them dynamically10. Corresponding local remodelling of the 3D matrix, which does not occur in conventional 2D migration, is required for effective cell migration inside a 3D matrix11. This question is also important because cells on planar substrates display a flattened fan-like morphology, while cells completely embedded in a more physiological 3D matrix environment often adopt a spindle-like morphology well suited to negotiate tight matrices11,12,13,14,15.
LINC complexes are protein assemblies that span the nuclear envelope and mediate physical connections between the nuclear lamina and the cytoskeleton6. These connections are mediated by interactions between SUN (Sad1/UNC-84) domain–containing proteins and KASH (Klarsicht/ANC-1/Syne-1 homology) domain–containing proteins at the outer nuclear membrane16,17,18,19,20,21. The down-regulation of both Sun1 and Sun2 prevents the localization of Nesprin-2 giant at the nuclear envelope6,22. The expression of either the recombinant SUN domain of Sun1 and Sun2 within the ER lumen or the KASH domain of Nesprins (nuclear envelope spectrins; also Syne) 1, 2, and 3 results in the displacement of all Nesprins from the NE to the ER6,22,23. The KASH domain of Nesprins 1, 2, and 3 can interact promiscuously with either Sun1 or Sun223. Whether Nesprins and interactions between Nesprins and Sun proteins play a role in 3D cell migration are unknown.
Here we use quantitative functional live-cell assays to show that LINC complexes play a critical role in regulating 3D actin architecture in cells in 3D matrix, and mediate protrusion dynamics, which in turn drive 3D matrix remodeling and 3D cell migration. We first show how lamin A/C deficiency, which disrupts LINC complexes at the nuclear envelope24,25, induces a migration phenotype only in 3D collagen matrices, not on conventional 2D substrates. To establish the molecular mechanism underlying this 3D matrix-specific defect, we examined the role of LINC complexes and found that LINC complex molecules Nesprin 2 giant and Nesprin 3 differentially modulate 3D migration by directly regulating the nuclear connections and organization of a subset of actin filament bundles attached to the nuclear envelope, the perinuclear actin cap26,27,28. This three-dimensional version of the recently characterized perinuclear actin cap observed in a wide range of human and rodent cells on 2D substrates28 in turn directs the formation and dynamics of pseudopodial protrusions, the main protrusions that control the ability of a cell to pull on and remodel its surrounding matrix. A simple mechanical model suggests why the nucleus and nucleo-cytoskeleton connections play no significant role in 2D migration, but are central in 3D migration.
Results
Three-dimensional, not two-dimensional, cell migration depends on nuclear lamin A/C
Here, we investigate the potential role of lamin A/C in the spontaneous migration of fibroblasts, which in vivo involves individual cells moving within a 3D collagen I-rich matrix29. We compared the migration of single Lmna−/− and Lmna+/+ mouse embryonic fibroblasts (MEFs) on conventional collagen I-coated substrates to the migration of these cells fully embedded inside a 3D collagen I matrix. We avoided the use of the in vitro wound-healing assay where cells are first grown to confluence to measure cell migration as this assay is of limited physiological relevance since fibroblasts migrate as singles in vivo30. On conventional planar substrates, single Lmna−/− and Lmna+/+ cells showed no significant difference in speed (Fig. 1A) and persistence time of migration (Fig. 1B), the average time it took for a cell to deviate from a straight trajectory8,31. However, the speed of Lmna−/− cells was significantly lower and the persistence time of migration was significantly longer than those of Lmna+/+ cells when these cells were embedded inside a 3D collagen matrix (Fig. 1, E and F). We note that only matrix-embedded cells at least 100 µm away from the bottom coverslip were examined to avoid confounding edge effects32. These results were confirmed with lamin A/C-depleted C2C12 mouse myoblasts. Similar to MEFs, lamin A/C-depleted C2C12 cells exhibited the same velocity and persistence time of migration on planar substrates (Fig. 1 C and D) but significantly lower velocity and a slightly longer persistence time of migration in 3D when compared to their control counterparts (WT cells transfected with scrambled shRNA) (Fig. 1, G and H).
Figure 1. Lamin deficiency modulates cell motility and morphology when cells are embedded in a 3D matrix, not on planar substrates.

(A) and (B). Averaged speed (A) and persistence time of migration (B) of individual Mouse Embryonic Fibroblast (MEF) Lmna+/+ and Lmna−/− cells placed on collagen I-coated 2D substrates. Persistence time is the averaged time it takes for the direction of cell motility to become de-correlated. (C) and (D). Averaged speed (C) and persistence time of migration (D) of individual MEF Lmna+/+ and Lmna−/− cells inside a 3D collagen I matrix. (E) and (F). Averaged speed (E) and persistence time of migration (F) of individual mouse myoblasts (C2C12) transfected with scrambled shRNA or shRNA-depleted of lamin A/C inside a 3D collagen matrix. (G) and (H). Typical time-dependent overall morphology of Lmna+/+ (G) and Lmna−/− (H) MEFs embedded inside a collagen matrix. While arrows point to elongated morphology, black arrows point to rounded morphology. Scale bar, 20 μm. (I). Averaged number of actively growing membrane protrusions per cell per 90 min measured under high-magnification phase-contrast microscopy. Protrusions are deemed active when they are associated with significant traction of the matrix in their vicinity. Protrusion remnants at the rear end of motile cells are not counted. (J) and (K). Fraction of the time when cellular morphology of MEFs (A) or C2C12s (B) with or without lamin A/C is elongated as opposed to rounded up or collapsed. N = 10 cells per type of cell in triplicate for a total of 30 cells for each condition. Measurements in panels A–D, I and J are mean ± SEM. (***): P<0.001 (student t-test; N = 33 cells in triplicate for a total of 99 cells for each condition). Measurements in panels E, F, and K are mean ± SEM. (***): P<0.001 (one way ANOVA test; N = 33 cells in triplicate for a total of 99 cells).
Actin filament organization and protrusion activity of matrix-embedded cells depend on lamin A/C
To understand the difference in 3D migration induced by lamin A/C deficiency, we measured the protrusive activity of these cells in 3D matrix. Pseudopodial protrusions allow cells inside a 3D matrix to probe the pericellular matrix, and locally attach to and pull on the surrounding fibers10,33. High protrusion activity is indicative of high migration speed for cells embedded inside a matrix34,35. Since Lmna−/− and Lmna+/+ cells showed different speed when embedded inside the matrix, we compared the dynamics of pseudopodial protrusions of these cells in matrix (Fig. 1, I and J). Using time-lapsed light microscopy, we found that the number of actively growing protrusions (i.e. distinct from remnants at the cell's trailing edge) per unit time per cell was significantly lower in Lmna−/− cells than in Lmna+/+ cells (Fig. 1, K and L). Over long periods of time, Lmna−/− cells alternated equally between a relatively rounded shape and an elongated spindle-like morphology (Figs. 1J and 2D for Lmna−/− cells). In contrast, Lmna+/+ cells displayed an elongated shape 85% of the time (Figs. 1L and 2C). Accordingly, the global reduction of pseudopodial activity in Lmna−/− cells faithfully reflected the small fraction of time that these cells spent in the elongated morphology (compare results in Fig 2C to Fig. 2D). Similar differential protrusion activity was observed for the pair of control (scrambled shRNA) and shRNA lamin A/C-depleted C2C12 cells (Fig. 1M). Moreover, the speed of Lmna−/− cells in the elongated shape was not significantly different from that of Lmna+/+ cells in the elongated shape (data not shown), providing additional evidence that the difference in speed between Lmna−/− and Lmna+/+ cells stems from their difference in protrusion activity.
Figure 2. Lamin deficiency severely affects 3D actin filament architecture of cells in 3D matrix.
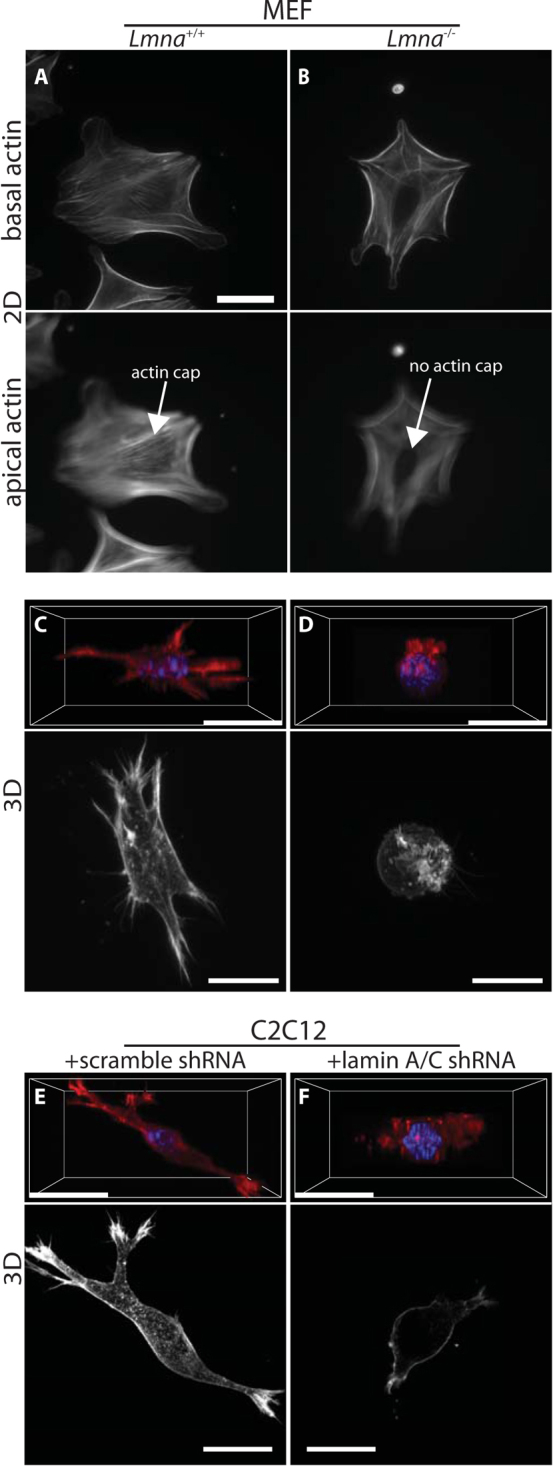
(A–B). Actin filament organization of Lmna+/+ (A) and Lmna−/− MEFs (B) on 2D collagen-coated substrates. Wild-type cells show an organized perinuclear actin cap that wraps around the interphase nucleus, while knockout cells lack an actin cap(bottom panels); however, basal actin organization is mostly unchanged (top panels). (C–D). 3D perspective (top) and maximum-intensity projections in XY-plane (bottom) of actin filament organization in Lmna+/+ (A) and Lmna−/− MEFs (B) in a 3D collagen matrix. (C–D). 3D perspective (top) and maximum-intensity projections in XY-plane (bottom) of actin filament organization in control scrambled (A) and lamin A/C-depleted (B) C2C12 cells inside a 3D collagen matrix. Scale bar, 20 μm.
Protrusion activity is driven by actin filament re-organization; therefore to understand the observed differences in protrusion activity, we investigated the actin filament organization of Lmna−/− and Lmna+/+ cells in 3D matrix. Actin filament organization of Lmna+/+ MEFs was not overtly different from that of Lmna−/− MEFs on 2D collagen substrates, with the exception of the perinuclear actin cap, a contractile actin filament structure that wraps around the nucleus and shapes it26,27,28, which was absent in Lmna−/− MEFs (Fig. 2, A and B). Moreover, our previous work has shown that the number, size, and shape of both focal adhesions were identical in these two cell lines, when they were placed on conventional flat substrates25. However, confocal light microscopy of Lmna+/+ and Lmna−/− MEFs embedded in 3D collagen I matrix revealed drastic differences in actin filament architecture induced by lamin A/C deficiency. When compared to wild-type cells (Fig. 2C), Lmna−/− MEFs appeared to lack any actin filament structure and the cells were collapsed around the nucleus (Fig. 2D). Three-dimensional confocal projections (Fig. 2, C and D, top panels) and maximum intensity projections from confocal images (Fig. 2, C and D, bottom panels) both revealed an extended actin filament organization in Lmna+/+ cells, while Lmna−/− cells were devoid of any elongated actin organization, in agreement with the above protrusion and elongation data (Fig. 1, G–J). Similar to MEFs, the 3D actin filament architecture of control and shRNA lamin A/C-depleted C2C12s were dramatically different (Fig. 2, E and F).
Together these results show that lamin A/C deficiency does not induce significant changes in single-cell migration on 2D substrates, but seems to mediate a major re-organization of the actin filament network of matrix-embedded cells, which would result in the observed significantly reduced protrusion activity and reduced cell migration speed in 3D matrix.
The role of nuclear lamin A/C in 3D matrix traction and remodeling
Matrix-embedded cells need to deform and remodel their surrounding microenvironment in order to move11. We hypothesized that the lower cell speed and reduced protrusion activity of Lmna−/− cells in collagen matrix was caused, in part, by a reduced ability of these cells to deform and pull on their surrounding matrix. To test this hypothesis, we used a recently developed 3D multiple-particle assay whereby large polystyrene beads (diameter, 3.6 μm) were embedded in the matrix and tracked with high spatial resolution in all three x-, y-, and z-directions with <10-nm lateral resolution and <100-nm vertical resolution9 (Fig. 3, A–D). Because these beads are carboxylated, they are strongly bound to the collagen fibers of the matrix and the movements of the beads faithfully report on the local movements of the matrix induced by the cells9. We verified that the spontaneous movements of the beads were negligible in the absence of cells (<10 nm). For both Lmna+/+ and Lmna−/− cells, beads mostly moved near actively growing protrusions at the leading edge of the cell, not in regions of the cellular periphery devoid of protrusions. Hence, the local movements of the matrix were not caused by their passive displacement by cells; rather the matrix was actively pulled, mediating cell movements. The total deformation and the magnitude of cell-mediated matrix traction, as measured by the maximum displacements of the beads, were drastically reduced for Lmna−/− cells compared to Lmna+/+ cells (Fig. 3, E–G). Moreover, lamin A/C deficiency affected the mechanical character of matrix deformation induced by the migrating cells, which was typically much more elastic (fully reversible deformation of the matrix) than irreversible (ratio of 0 corresponds to a purely elastic deformation of the matrix, a ratio of 1 corresponds to a completely irreversible deformation) in Lmna+/+ cells compared to Lmna−/− cells (Fig. 3H).
Figure 3. Lamin deficiency reduces the extent and modifies the rheological character of matrix remodeling during 3D migration.

(A) and (B). Typical deformation maps of the collagen matrix induced by an Lmna+/+ MEF (A) and an Lmna−/− MEF (B). These maps were obtained by simultaneously tracking the three-dimensional displacements of multiple 3.6 μm-diameter fiduciary carboxylated beads embedded in the collagen I matrix under phase contrast light microscopy. The lengths of the arrows represent three times the actual local magnitude of matrix deformation. The movie was recorded for 90 min. (C) and (D). Typical time-dependent three-dimensional x, y, and z coordinates of single fiduciary beads in the matrix denoted a and b in panels A and B, respectively, near a Lmna+/+ cell (C) and near a Lmna−/− cell (D). (E). Graphical definition of values used to assess matrix remodeling. (F) and (G). Total (Lt) and maximum movements (Lmax) of beads embedded in the matrix and surrounding Lmna+/+ and Lmna−/− cells. Beads were tracked for 90 min. (H). Percentage of permanent matrix deformation mediated by Lmna+/+ and Lmna−/− cells. Permanent deformation is measured by the ratio of the distance between first and final bead positions (Lf) divided by the total displacement (Lt). This number is 1 when cell-mediated deformation of the matrix is irreversible or permanent and 0 when the deformation is perfectly reversible or elastic. (I). Reflection confocal micrograph showing collagen fiber organization and “virtual beads” defined to help assess the magnitude of matrix remodeling induced by cell-induced traction and release of collagen fibers. (J). “Heat” map of matrix remodeling. Red regions denote areas of maximum displacement of the collagen matrix towards the cell body, while blue show regions of maximum release away from the cell body. Measurements in panels F–H, and K represent mean ± SEM. (***): P<0.001 (student t-test). Scale Bar: 20 μm.
As an alternative method to assess matrix remodeling, we used a new assay based on the time-dependent imaging of collagen fibers by reflection confocal microscopy (Fig. 3I). Using a custom software, we were able to analyze matrix deformations for the whole field of view during cell migration (Fig. 3, J and K). Analysis of matrix deformation by control (shRNA scramble) and lamin A/C-depleted C2C12 cells yielded similar results to those found for MEFs in the particle-tracking assay. Together, these results indicate that lamin A/C is required for matrix traction and remodeling, a necessary contributor of effective 3D cell migration.
To further test the hypothesis that nuclear lamin A/C specifically regulates actin filament organization, protrusion dynamics, and cell migration in a matrix (Figs. 1 and 2), we re-introduced EGFP-lamin A in Lmna−/− cells. Remarkably we found that the forced expression of lamin A largely rescued the migration phenotype and associated defect in protrusion activity presented by Lmna−/− cells (Fig. 4, A–C). Moreover, Lmna−/− transfected with EGFP-lamin A showed the extended organization shown by Lmna+/+ cells (Fig. 4D), as well as the restoration of a spindle-like morphology similar to Lmna+/+ cells. These results suggest that the effect of lamin A/C depletion on actin filament organization, protrusion activity, and cell speed in 3D matrix is specific.
Figure 4. Disruption of LINC complexes recapitulates the migratory phenotype caused by lamin deficiency.

(A–C). Cell speed (A), persistence time of migration (B), and fraction of time when the cellular morphology is elongated (C) for Lmna+/+ MEFs (first column), Lmna−/− MEFs transfected with EGFP-lamin A (second column), Lmna−/− MEFs (third column), and Lmna+/+ MEFs transfected with EGFP-KASH2, which displaces the LINC complexes from the nuclear envelope23 Measurements in panels A–C represent mean ± SEM. N = 13 cells per type of cell in triplicate for a total of 39 cells. *: P< 0.05 comparing with Lmna+/+ cells; ns: non-significant comparing with Lmna−/− MEFs. (D–F). Actin filament organization in Lmna−/− cells transfected with EGFP-lamin A (D), Lmna+/+ cells transfected with EGFP-KASH2 (E), and Lmna+/+ cells transfected with the control construct EGFP-KASHext (F) (G–I). Immuno-localization of lamin A/C (G), Nesprin 2 giant (H), and Nesprin 3 (I) in control C2C12 cells transfected with scrambled shRNA (top left), lamin A/C-depleted C2C12 cells (top right), Nesprin 2 giant-depleted C2C12 cells (bottom left), and Nesprin 3-depleted C2C12 cells (bottom right). Lamin A/C immunostaining is unaffected by shRNA-depletion of either Nesprin 2 giant or Nesprin 3. Both Nesprins, however, appear slightly mislocalized following Lamin A/C depletion (compare to cells still expressing lamin A/C; yellow arrows). Interestingly, each Nesprin appears more dense at the nuclear envelope when the other is knocked down (purple arrows). Insets in panels (H) and (I) show Nesprin 2 giant and Nesprin 3 fluorescence immunostaining, respectively. (J) Speed of matrix-embedded control scrambled, Nesprin 2 giant-depleted, and Nesprin 3-depleted C2C12s. (K–L). Actin filament organization in matrix-embedded Nesprin 2 giant-depleted C2C12s (K), and Nesprin 3-depleted C2C12s (L) embedded. (M). Fraction of time when cellular morphology of matrix-embedded control shRNA scramble cells, shRNA lamin A/C-depleted cells, shRNA Nesprin 2 giant-depleted cells, and shRNA Nesprin 3-depleted cells is elongated as opposed to rounded up or collapsed. Inset, relative distributions (quarter-binned) of time spent in the collapsed (0) and elongated (1) (N). 3D cell speed vs. fraction of time elongated for C2C12 cells embedded in a collagen matrix highlighting the anomalous behavior of Nesprin 2-depleted cells. Scale Bar: 20 μm.
Disruption of LINC complexes and depletion of Nesprins recapitulate 3D migratory and protrusive defects induced by lamin A/C deficiency
To establish the molecular mechanism underlying the lamin A/C-induced difference in actin filament organization, protrusion activity, and cell migration in 3D matrix, we examined the role of LINC complexes in these processes. We have previously shown that lamin A/C deficiency in MEFs disrupts LINC complexes at the nuclear envelope25. LINC complexes mechanically connect the nuclear lamina to the actin cytoskeleton network23 via Nesprin 2 giant which directly binds F-actin36 and Nesprin 3 which binds actin filaments through the multi-domain protein plectin37. Furthermore, we have shown previously that for cells on 2D substrates, the targeted disruption of LINC complexes has the same effect as lamin A/C deficiency in preventing the formation of the perinuclear actin cap, altering nuclear shape, and preventing regulation of nuclear shape by cell shape26. Therefore, we hypothesized that the above 3D-matrix-specific functional defects – fewer pseudopodial protrusions and lower 3D cell speed – were caused by the disruption of the LINC complexes in cells lacking lamin A/C. We assayed cells transfected with either the EGFP-KASH2 domain of Nesprins, which displaces Nesprins from the nuclear envelope to the ER/cytoplasm, or the control construct EGFP-KASH2ext, which localizes to the nuclear envelope but has no effect on LINC complexes23. We found that the speed of cells transfected with the KASH2 domain and placed inside a 3D matrix was reduced to the same level as the speed of Lmna−/− cells in matrix (Fig. 4A). The persistence time of migration of these cells was increased (Fig. 4B) and their protrusion activity was decreased (Fig. 4C) to the same values as those of Lmna−/− cells.
Moreover, the organization of actin filament bundles in Lmna−/− cells transfected by EGFP-hLMNA was similar to Lmna+/+ cells (Fig. 4D). Importantly, the actin filament network organization of KASH2-transfected cells was similar to the actin filament organization in Lmna−/− cells, consisting of mostly cortical actin and few actin filament bundles that could mediate protrusion activity (Fig. 4E). Finally, the actin filament organization and overall morphology of control KASH2ext-transfected Lmna+/+ cells in matrix were similar to those of Lmna+/+ cells (Fig. 4F). These results indicate that the disruption of LINC complexes recapitulates the migration/protrusion phenotype displayed by Lmna−/− cells in 3D matrix.
To delineate the separate roles of Nesprin 2 giant and Nesprin 3 of the LINC complex, these molecules were stably downregulated in C2C12 cells using shRNA. Actin filament organization, cell speed, and protrusion activity of the resulting cells in a 3D collagen matrix were then characterized (Fig. 4, G–N). First, as expected, immunofluorescence microscopy showed that shRNA-mediated depletion of either Nesprin 2 giant or Nesprin 3 had no effect on lamin A/C localization (Fig. 4G). Using new custom antibodies against Nesprin 2 giant and Nesprin 3, we found that shRNA-mediated depletion of lamin A/C caused partial mislocalization of Nesprin 2 giant and Nesprin 3, respectively (Fig. 4 H and I; top right panels). Interestingly, the depletion of Nesprin 2 giant seemed to induce a higher density of Nesprin 3 at the nuclear envelope (Fig. 4H; bottom right panel). Vice versa, the depletion of Nesprin 3 seemed to induce a higher density of Nesprin 2 giant at the nuclear envelope (Fig. 4I; bottom left panel), indicative of a possible competition for SUN-domain binding sites between Nesprin 3 and Nesprin 2 giant. Indeed, previous results detailing the “fluid” nature of LINC complexes38 would support the notion that either Nesprin 2 giant or Nesprin 3 would be able to flow in and occupy unbound SUN-domains.
Time-lapsed microscopy of cells in 3D matrix showed that, to a greater extent than lamin A/C-depleted cells, Nesprin 2 giant- and Nesprin 3-depleted cells both exhibited reduced 3D cell migration, with Nesprin 3-depleted cells showing significantly slower speed than Nesprin 2 giant-depleted cells (Fig. 4J). Maximum-intensity projections of confocal z-stacks of the actin filament network in Nesprin 2 giant- and Nesprin 3-depleted cells embedded in 3D matrix provided a possible explanation for the observed difference in cell speed. Nesprin 2 giant-depleted cells tended to be elongated (Fig. 4K), while Nesprin 3-depleted cells were often rounded and showed disorganized actin filament architecture (Fig. 4L). Quantification of the movies showed that Nesprin 2 giant-depleted cells spent ~80% of the time elongated, while Nesprin 3-depleted cells only spent ~15% in the same state (Fig. 4M). Interestingly, Nesprin 2 giant-depleted cells spent more time elongated than control cells did, while exhibiting a significantly slower 3D cell speed—seemingly contradictory results (Fig. 4, J and M). However, further analysis showed that while Nesprin 2 giant-depleted cells did indeed spend more time elongated, they remained elongated showing no dynamic shape changes (Fig 4M, histograms in bar insets showing fractions of time spent elongated with .25 bin size). Plotting 3D cell speed against fraction of time elongated shows the differential ability of Nesprin 2 giant and Nesprin 3 to regulate 3D cell migration (Fig. 4N). These results suggest that the presence and dynamic nature of protrusions are both necessary for effective cell migration in 3D matrix. These results also highlight the importance of LINC complexes components, specifically Nesprin 2 giant and Nesprin 3, in mediating 3D cell migration speed and persistence. Interestingly, we also show somewhat divergent roles for Nesprin 2 giant and Nesprin 3.
Discussion
Fibroblasts deficient in lamin A/C show no intrinsic single-cell motility defect when placed on collagen-coated glass substrates, but display a severe cell motility defect once fully embedded inside a more physiological 3D collagen I matrix. Our results suggest that this defect results from the diminished ability of these cells to pull on and remodel their surrounding matrix because these cells do not dynamically protrude into the surrounding matrix as readily as control cells (Fig. 5, A and B). A previous study indicated that fibroblasts deficient in lamin A/C showed a reduced ability to close a wound in a (less physiologically relevant) planar wound-healing assay29. Since our results show that lamin A/C deficiency does not cause a defect in single-cell motility, this previous result could suggest that lamin A/C may affect cell-cell adhesion, a contributing factor to rates of wound-healing. This 3D cell migration phenotype is similar in fibroblasts in which the LINC complexes are specifically disrupted and in lamin A/C-depleted myoblasts. Taken together, our results suggest that this 3D matrix-specific combined motility/protrusion defect is caused by a major re-organization of the actin filament network seemingly due to the disrupted connections between the nuclear envelope and the actin filament network.
Figure 5. Nucleus-cytoskeleton connections are essential to 3D migration.

(A) and (B). Cartoons of 3D embedded cells (A) with and (B) without Lamin A/C. Cells expressing lamin A/C are able to able to support actin-based protrusions and pulling because of intact nucleo-cytoskeletal connections. In cells devoid of lamin A/C, actin filaments are no longer held in place at the nuclear envelope by LINC complexes, removing nuclear support of actin-based movement. (C). Modeling the actin fibers in a cell on a 2D substrate. Lamellipodial protrusions are supported by connections to the stiff collagen-coated glass substrate. (D). Modeling the actin fibers in a cell embedded in a soft 3D matrix. Protrusions are supported by nucleo-cytoskeletal (N–S) connections in cells with intact LINC complexes. (E). For cells embedded in a 3D matrix without intact LINC complexes, the cell is unable to sustain protrusions as actin fibers are pushing through a soft matrix.
Our results also suggest that the highly organized actin filament structure that is specifically connected to the apical surface of the nuclear envelope of cells on 2D substrates through LINC complexes, i.e. the perinuclear actin cap26, has a 3D analogue. On 2D substrates, a wide range of cells (mouse and human, embryonic fibroblasts, lung fibroblasts, myoblasts, and endothelial cells) feature highly ordered array of thick contractile acto-myosin fibers that are dynamically anchored to the nuclear envelope through LINC complexes28. In a 3D extracellular matrix, cells form nucleus-bound actin filament bundles that allow for productive traction forces on the surrounding matrix. Disrupting these connections through the forced expression of KASH2 construct, lamin A/C deficiency depletion or shRNA depletion of Nesprins all lead to defective shape and migration of cells in matrix. While the perinuclear actin cap appears to play no significant role in 2D migration, its 3D version - nuclear-bound stress fibers (Fig. 5A) - seems to be involved in protrusion activity, matrix remodeling, and cell migration in 3D matrix.
Differences between 2D migration on (stiff) collagen gel-coated substrates and 3D migration in (soft) collagen gels could be caused by a change in stiffness39,40,41. Of course, differences in 3D cell migration induced by disruption of LINC complexes or lamin deficiency could be explained by such a change in stiffness. Nevertheless, in two previous studies, we have carefully compared the cell migration phenotypes of cells in 3D collagen matrix vs. on 2D collagen substrates of high compliance using engineered substrates. We found, for instance, that the regulation of cell speed and persistence by focal adhesion proteins could not be recapitulated using compliant flat surfaces35. We have also shown that the remarkable oscillatory migration phenotype observed in cells depleted of zyxin observed in 3D matrices could not be recapitulated on soft flat substrates, but could be recapitulated on 1D (stiff) patterns34. We have to conclude that the qualitative differences between cell migration in 3D and 2D observed here are not merely due to the fact that matrices are more compliant than collagen-coated glass substrates, but are in large part induced by a change in dimensionality of the environment.
A possible explanation for the significant difference in 3D cell speed between Lmna+/+ and Lmna−/− cells could be that these two cells have a different size. Indeed, Lmna−/− cells appear smaller than Lmna+/+ cells (Figs. 1 and 2, 5 A and B); however, careful examination shows that their apparent difference in size only results from the presence/absence of protrusions. If cell migration was not mediated by protrusion activity, an apparent smaller size/surface area of Lmna−/− cells would a priori reduce their friction with the surrounding matrix and allow them move faster, which is the opposite of what is observed. The reduced persistence time of migration of Lmna−/− cells may also be caused by reduced protrusion activity. The shorter lifetime of protrusions in Lmna−/− cells does not provide these cells with sufficient time to sustain persistent movements. Together, these results suggest that significantly reduced pseudopodial activity is likely to be responsible for diminished 3D cell speed, which, in turn, is strongly correlated with the loss of nuclear lamin A/C and associated disruption of LINC-anchored perinuclear actin cap.
A priori lamin A/C deficiency could affect 3D cell migration indirectly, through differential gene regulation, as lamin A/C interacts indirectly with the genome42. Here, we present evidence that mechanical effects might also contribute to the migration defects of lamin-deficient cells. Furthermore, the lamin A/C mutant phenotype is recapitulated by displacing the LINC complexes from the nuclear envelope using the EGFP-KASH2 construct and differential gene regulation does not readily explain why lamin deficiency does not affect 2D cell migration, while it affects 3D migration. Therefore, dismantling nucleus-cytoskeleton connections alone has the same quantitative and qualitative effect on cell migration and protrusion activity as a total lamin A/C knockout. Hence, our results support a mechanical explanation for the observed defect in 3D migration.
More specific disruptions of the LINC complexes, i.e. shRNA-mediated depletion of either Nesprin 2 giant or Nesprin 3, showed a similar migration defect as either the shRNA-induced depletion of Lamin A/C or the disruption of LINC complexes using KASH domains. In Nesprin 2 giant-depleted cells, however, this migratory defect was not correlated with a rounded morphology, as seen in every other case. This could be a clue about possibly divergent functions for Nesprin 2 giant and Nesprin 3. It has been shown that Nesprins bind promiscuously to SUN domains43,44. Thus, depletion of Nesprin, and its subsequent loss from the nuclear envelope, should free SUN binding sites, leading to the enrichment of other nesprins at the nuclear envelope, a model supported by our results (Fig. 3, H and I). An enrichment at the nuclear envelope of either Nesprin 2 giant, which has an actin binding domain, or Nesprin 3, which through plectin binds all three cytoskeletal proteins, would show drastically different concentrations and ratios of these cytoskeletal elements near the nucleus45. Nevertheless, both Nesprin 2 giant and Nesprin 3 are critical for effective 3D cell migration. Since plectin binds intermediate filaments and microtubules and cell polarization depends on these proteins30,46,47, the effect of Nesprins depletion on 3D migration could be also partially attributed to the disruption of these cytoskeletal proteins.
Recent work by Emerson et al. showed that matrix-embedded LmnaR401C mutant cells generated enhanced global contraction of collagen I matrices compared to WT cells48, while we observed decreased local contraction of such matrices in the presence of Lmna−/− cells. A possible reason for this apparent discrepancy may be due to the difference in mutants analyzed in both studies. It is well-known that different lamin mutants can lead to vastly different human diseases7. It is therefore rather expected that migration of Lmna−/− and LmnaR401C cells in 3D matrix can be different, potentially due to the different status of LINC complexes at the nuclear envelope in these two cells. Additionally, Scharner et al. recently characterized several novel mutations in the Lmna gene responsible for Emery-Dreyfuss Muscular Dystrophy (EDMD)49. These mutations caused major nuclear deformation, similar to those observed with other laminopathic mutations, but did not appear to change the perinuclear actin cap in 2D26, which illustrates, once again, that the status of LINC complexes at the nuclear envelope correlates with the status of nuclear envelope-bound actin fibers (i.e. the status of the perinuclear actin cap), those that mediate 3D migration.
We propose a simple mechanical model that could explain the dimensionality-dependent effect of the disruption of LINC complexes at the nuclear envelope on cell migration. In the 2D case, actin filaments form both the perinuclear actin cap, which is connected to the nucleus through LINC complexes, and basal stress fibers, which are not connected to the nucleus. In the 3D case, our results could suggest that the actin filament fibers that generate the major pseudopodial protrusions span the cell length, are those encompassing and connecting to the nucleus through LINC complexes (Fig. 3 and 5). In 2D, protrusions are supported by actin fibers terminating at focal adhesions on collagen-coated glass, which is much stiffer than the nucleus. Elimination of LINC complexes that connect actin-cap fibers to the nucleus minimally impact protrusion-based migration because only the mechanically weaker link (i.e. the nucleus) in the assembly is eliminated (Fig. 5C). In 3D, the substrate becomes the weaker link since the collagen matrix is much softer than the nucleus (Fig. 5D)50,51. For cell migration to occur, protrusions extend into the matrix and effective protrusion extension with minimal dissipation require anchorage of actin fibers to the nucleus so that these fibers can push within the cell against a stiff base (i.e. the nucleus). In cells deficient in lamin A/C, actin filaments are no longer held in place at the nuclear envelope by LINC complexes, removing nuclear support of actin-based movement. Therefore, the interphase nucleus and its connections to fibers of the perinuclear actin cap that direct pseudopodia are critical to 3D cell migration.
Our results highlight the fundamentally different requirements for functional nucleo-cytoskeletal connections in cell migration on flat substrates and in 3D extracellular matrix. Moreover, while the nucleus does not seem to play a direct role in migration on flat substrates, it takes central stage for migration within more physiologically relevant 3D matrices. At a collagen concentration of 2 mg/ml, the mean matrix pore size (<0.4 μm; see Materials and Methods) is significantly smaller than the cell body and the interphase nucleus. Therefore, a priori, lamin A/C-deficient cells and cells with disrupted LINC complexes should display facilitated migration though the matrix compared to their control counterparts since their nucleus is significantly more compliant, i.e. more deformable52,53. Here our results suggest that the active deformation of the nucleus by the contractile fibers of the perinuclear actin cap to move the cell through the tortuous matrix plays a more important role than the intrinsic lamin A/C-dependent stiffness of the nucleus. These observations add to the increasing list of critical and specific role played by the LINC-anchored perinuclear actin cap, including nuclear shaping26 and stem cell differentiation28: 3D protrusion activity, cell shape, matrix remodeling and traction forces, and cell migration.
Lamin A/C-deficient mice display a severe muscular dystrophy phenotype54. Our results could point to new ways of understanding this disease, as well as the numerous other laminopathies, as well as point to new clinically relevant routes for the development of new therapeutic strategies for these diseases7.
Methods
Cell culture, generation of cell lines and antibodies
Lmna−/− and Lmna−/− MEFs (generous gift from Colin Stewart, Astar, Singapore) were cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% (v/v) fetal bovine calf serum (ATCC), 100 U penicillin and 100 µg streptomycin (Sigma, St. Louis, MO). Culture medium for C2C12 mouse myoblasts was supplemented with 10 μg/ml puromycin (Sigma). Cells were maintained at 37°C and 5% CO2 in a humidified environment during culture and imaging. The cells were passaged every 2–3 days for a maximum of six passages for wild type and two weeks for transfected cells. We found no enhanced apoptosis induced in Lmna−/− cells and transfected cells both on and in collagen matrices, which show the same low cell death rate as control cells.
Four to five individual shRNA from Mission shRNA Bacterial glycerol stocks (Sigma, St Louis, MO) directed either against mouse Nesprin 2 giant (NM_001005510) or Nesprin 3 (NM_172500) were individually transfected – along with a non-targeting control shRNA (SCH002, Sigma) - into C2C12 myoblasts with Lipofectamine PLUS and selected at 10 μg/mL of Puromycin in DMEM supplemented with 10% FBS and Penicillin/Streptomycin. Downregulation was assessed over a four-week period by comparing immunofluorescence and immunoblotting patterns of downregulated cells with control shRNA cells. Individual clones were isolated using cloning cylinders and the level of stable downregulation was assessed in Western blots and Immunofluorescence. Nesprin 2 giant and Nesprin 3 were stably downregulated using shRNA NM_001005510.2-21409s21c1 and shRNA NM_172500.2-2830s21c1, respectively.
To generate antibodies against Nesprin 2 giant or Nesprin 3, total RNA of C2C12 cells was reverse transcribed and PCR amplified using primers to amplify either the C-terminal region of Nesprin 2 just upstream from the transmembrane domain or the entire coding sequence of the cytoplasmic region of Nesprin 3. PCR products were cloned into the pcr8 gw topo vector and sequenced. LR clonase reactions were carried out according to the manufacturer's protocol (Invitrogen) to obtain N-terminal GST fusion proteins. Bacterial synthesis of recombinant proteins was induced using IPTG in BL21 E. coli and proteins were purified on an AKTA FPLC with GSTrapp FF columns (GE Healthcare). Eluted fusion proteins were pooled, dialyzed against urea, separated on a superdex200 column and concentrated using an Amicon Ultracel 10 k. Polyclonal antibodies were produced by immunizing rabbits with the purified GST fusion proteins and affinity-purified on a CNBr sepharose column (PRIMM Biotech).
Collagen I-coated substrates and 3D collagen I gels
Cell culture plates were prepared by adding rat tail type I collagen in acetic acid (BD Biosciences) to reach a coverage density of 33 μg/cm2 and incubated at room temperature for 2 h. Plates were then washed three times with PBS and cells were plated at low density. Cell-impregnated 3D collagen matrices were prepared by mixing cells in culture medium and 10X reconstitution buffer, 1:1 (v/v) with type I collagen, for a final concentration of 2 mg/ml. This concentration was chosen so that the mean matrix pore size (<0.4 μm) was significantly smaller than the cell body and nucleus (see Materials and Methods). 1 M NaOH was added to normalize pH (pH 7.0, 15–30 μl 1 M NaOH) and the mixture was placed in coverslip-bottom culture plates (LabTek, Campbell, CA). All ingredients were kept chilled to avoid premature collagen polymerization. Special care was taken during mixing to avoid the introduction of bubbles into the mixtures. Gels were allowed to solidify overnight in an incubator at 37°C and 5% CO2. 0.5 ml culture medium was added on top of the gels prior to the experiments. The cell density was maintained low to ensure that single cell migration measurements were accurate. The same procedure was used to coat 2D substrates with a thin layer of collagen I. C2C12 cell culture medium was supplemented with 100 ng/ml PDGF to stimulate movement in matrices.
Effective pore size of the collagen gel
The pore size of the 3D matrix was evaluated by high-resolution multiple-particle tracking55,56 of matrix-embedded 2-μm diameter fluorescent beads, which were coated with polyethylene glycol (PEG) to avoid direct interaction with collagen. From the measurements of both the diffusion coefficients of the beads in collagen and their diffusion coefficient in culture medium, the pore size was evaluated using the formula Dg/D0 = exp[(−π/4)(a+rcol)/(rpore+rcol)]257. Here Dg/D0 are the diffusion coefficients of the beads in the gel and in medium, a = 1 μm in the bead radius, rcol is the diameter of a collagen I fiber (~20 nm), and rpore is the effective pore size of the collagen gel. We found Dg = 0.001 μm2/s while Do was estimated using Stokes Einstein relationship. The pore size of the matrix ranged between 0.2 μm and 3.5 μm with an average value of 0.4 μm.
Light microscopy
To visualize actin in cells plated on the substrates and inside the gels, cells were fixed, permeabilized, and incubated with Alexa Fluor 568 phalloidin (Invitrogen) for 1 h. Images of cells were collected using a Zeiss Confocor II Confocal Microscope with Zeiss LSM software (Integrated Imaging Center, JHU), a Nikon A1 Confocal Microscope with Nikon Elements Software, or a Cascade 1 K CCD campera (Roper Scientific) mounted on a Nikon TE2000E epiflorescence microscope equipped with a 60x oil-immersion objective (Nikon) and controlled by Metamorph imaging software (Universal Imaging).
Cell speed and persistence
Cells embedded in 3D collagen I matrices were monitored by low magnification (10X) phase contrast microscopy for 16 h. Speed was determined by tracking single cells using an image recognition software (Metamorph/Metavue), with distance/time measurements were collected every 2 min and averaged for each cell. A persistent move is defined as the length (≥10 μm) traveled by a cell before it makes a significant change in direction (angle between previous direction and new direction <70°). These definitions were deduced so as to exclude noise incorporated by any microscope stage drift, etc. The speed, persistence time, and persistence distance of at least 35 cells were computed on three different days for each condition.
Cell protrusion dynamics
Pseudopodial protrusions of at least 5 μm in length and diameter were monitored using a Roper Scientific Cascade 1 K CCD camera mounted on a Nikon TE2000E microscope. High- magnification images were collected 5 min apart for 16 h. The protrusions of at least 35 cells were characterized on three different days for each condition.
Cell-mediated 3D local traction of the matrix
To quantify matrix remodeling by the embedded cells, mechanical traction of the collagen matrix induced by cell migration was measured by tracking matrix-impregnated 3.6-μm carboxylated polystyrene beads (Duke Scientific, Palo Alto, CA) in the x, y, and z, directions as previously described9. Beads were sonicated and added to the collagen solutions prior to gelation. The following quantities were computed: the maximum displacement of the beads from their original location, the total distance traveled by the beads, and the final displacement divided by the total distance to measure the percentage of permanent deformation of the collagen gel. The local matrix traction in the vicinity of five cells (representing about 30 beads per cell) was measured on three different days for each condition.
PIV tracking of cell-mediated matrix deformation
Particle imaging velocimetry has been extensively used to characterize flows at a micro- or macro-scales58,59. MATPIV©, an open-access platform for particle imaging velocimetry was used to track the spatio-temporal deformation of collagen gels induced by cells. Correlation between successive images was formulated as
where I is the intensity recorded at pixels denoted with super-indices and time is denoted with sub-indices. The peak of the correlation, obtained h and s pixels removed from the reference position gives the displacement of the interrogation area.
To utilize PIV in studying the deformation of collagen induced by cells, a 2 mg/ml collagen gel embedded with live cells was imaged at 2 min intervals, using the reflection mode on a Nikon A1 confocal microscope. It was ensured that the images captured the deformation of gel over a z-thickness of up to 10 μm. Subsequently, taking a maximum-intensity projection in z-direction, we ensured that the deformation in z-direction is included in the analysis. Using a signal-to-noise ratio of 3 (to avoid the occurrence of false-vectors) and linearly interpolating the deformations into regions with sparse features, a full-field deformation map for the image was obtained at every time point. Random locations on the gel were manually specified in order to obtain the trajectory of these regions over time. The location of each region was updated at each time point by linearly interpolating the displacement of the region from the nearest 10 neighbors of any single point on the region. Finally, in order to quantify the nature of gel-deformation, the ratio between the net-displacement and the total path of the trajectory of individual regions was obtained for each of the regions and averaged.
Statistics
Mean values, standard error of measurement (SEM), and statistical analysis were calculated using Microsoft Excel (Microsoft Corporation, Redmond, WA) and plotted using Graphpad Prism (Graphpad Software, San Diego, CA). Two-tailed unpaired t tests and ANOVA tests were conducted to determine significance, which was indicated using standard Michelin Guide scale (*** for P < 0.001, ** for P < 0.01, and * for P < 0.05).
Author Contributions
SBK, RJB, DH, and DW designed the experiments. SBK, RJB, SB, AG, PHW, JM, SXS, and DW conducted the experiments and analyzed the data. SBK, DR, SZ, CMH, and DH provided new reagents. SB, AC, JM, and PHW provided new analytical tools. SBK and DW wrote the paper. SBK, RJB, DH, and DW edited the paper.
Acknowledgments
This work was supported by NIH grants GM084204 (DH, DW) and CA143868 (DW), and a Research Grant from the Muscular Dystrophy Association (DH). SBK is supported by an IGERT-NSF graduate fellowship.
References
- Pollard T. D. & Borisy G. G. Cellular motility driven by assembly and disassembly of actin filaments. Cell 112, 453–465 (2003). [DOI] [PubMed] [Google Scholar]
- Ridley A. J. et al. Cell migration: integrating signals from front to back. Science 302, 1704–1709 (2003). [DOI] [PubMed] [Google Scholar]
- Sahai E. & Marshall C. J. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat Cell Biol 5, 711–719 (2003). [DOI] [PubMed] [Google Scholar]
- Symons M. & Segall J. E. Rac and Rho driving tumor invasion: who's at the wheel? Genome Biol 10, 213 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedrin D., van Rheenen J., Hernandez L., Condeelis J. & Segall J. E. Cell motility and cytoskeletal regulation in invasion and metastasis. J Mammary Gland Biol Neoplasia 12, 143–152 (2007). [DOI] [PubMed] [Google Scholar]
- Crisp M. et al. Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol 172, 41–53 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart C. L., Roux K. J. & Burke B. Blurring the boundary: the nuclear envelope extends its reach. Science 318, 1408–1412 (2007). [DOI] [PubMed] [Google Scholar]
- Zaman M. H. et al. Migration of tumor cells in 3D matrices is governed by matrix stiffness along with cell-matrix adhesion and proteolysis. Proc Natl Acad Sci U S A 103, 10889–10894 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom R. J., George J. P., Celedon A., Sun S. X. & Wirtz D. Mapping local matrix remodeling induced by a migrating tumor cell using three-dimensional multiple-particle tracking. Biophys J 95, 4077–4088 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshel A. S., Wei Q., Adelstein R. S. & Sheetz M. P. Basic mechanism of three-dimensional collagen fibre transport by fibroblasts. Nat Cell Biol 7, 157–164 (2005). [DOI] [PubMed] [Google Scholar]
- Sabeh F., Shimizu-Hirota R. & Weiss S. J. Protease-dependent versus -independent cancer cell invasion programs: three-dimensional amoeboid movement revisited. J Cell Biol 185, 11–19 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walpita D. & Hay E. Studying actin-dependent processes in tissue culture. Nat Rev Mol Cell Biol 3, 137–141 (2002). [DOI] [PubMed] [Google Scholar]
- Zhou X. et al. Fibronectin fibrillogenesis regulates three-dimensional neovessel formation. Genes Dev 22, 1231–1243 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotary K. B. et al. Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell 114, 33–45 (2003). [DOI] [PubMed] [Google Scholar]
- Wolf K. et al. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat Cell Biol 9, 893–904 (2007). [DOI] [PubMed] [Google Scholar]
- Starr D. A. & Han M. ANChors away: an actin based mechanism of nuclear positioning. J Cell Sci 116, 211–216 (2003). [DOI] [PubMed] [Google Scholar]
- Starr D. A. et al. unc-83 encodes a novel component of the nuclear envelope and is essential for proper nuclear migration. Development 128, 5039–5050 (2001). [DOI] [PubMed] [Google Scholar]
- McGee M. D., Rillo R., Anderson A. S. & Starr D. A. UNC-83 IS a KASH protein required for nuclear migration and is recruited to the outer nuclear membrane by a physical interaction with the SUN protein UNC-84. Mol Biol Cell 17, 1790–1801 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone C. J. et al. The C. elegans hook protein, ZYG-12, mediates the essential attachment between the centrosome and nucleus. Cell 115, 825–836 (2003). [DOI] [PubMed] [Google Scholar]
- Kracklauer M. P., Banks S. M., Xie X., Wu Y. & Fischer J. A. Drosophila klaroid encodes a SUN domain protein required for Klarsicht localization to the nuclear envelope and nuclear migration in the eye. Fly (Austin) 1, 75–85 (2007). [DOI] [PubMed] [Google Scholar]
- Technau M. & Roth S. The Drosophila KASH domain proteins Msp-300 and Klarsicht and the SUN domain protein klaroid have no essential function during oogenesis. Fly (Austin) 2, (2008). [DOI] [PubMed] [Google Scholar]
- Padmakumar V. C. et al. The inner nuclear membrane protein Sun1 mediates the anchorage of Nesprin-2 to the nuclear envelope. J Cell Sci 118, 3419–3430 (2005). [DOI] [PubMed] [Google Scholar]
- Stewart-Hutchinson P. J., Hale C. M., Wirtz D. & Hodzic D. Structural requirements for the assembly of LINC complexes and their function in cellular mechanical stiffness. Exp Cell Res 314, 1892–1905 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejat A. et al. Lamin A/C-mediated neuromuscular junction defects in Emery-Dreifuss muscular dystrophy. J Cell Biol 184, 31–44 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale C. M. et al. Dysfunctional connections between the nucleus and the actin and microtubule networks in laminopathic models. Biophys J 95, 5462–5475 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatau S. B. et al. A perinuclear actin cap regulates nuclear shape. Proc Natl Acad Sci U S A 106, 19017–19022 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatau S. B., Kim D. H., Hale C. M., Bloom R. J. & Wirtz D. The perinuclear actin cap in health and disease. Nucleus 1, 337–342 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatau S. B., Khatau S. B., K S., Hanjaya-Putra D., Mali P., Cheng L., Lee J. S. H., Gerecht S. & Wirtz D. The differential formation of the LINC-mediated perinuclear actin cap in pluripotent and somatic cells PLoS ONE (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. S. et al. Nuclear lamin A/C deficiency induces defects in cell mechanics, polarization, and migration. Biophys J 93, 2542–2552 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale C. M. et al. SMRT analysis of MTOC and nuclear positioning reveals the role of EB1 and LIC1 in single-cell polarization. J Cell Sci 124, 4267–4285 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. D. et al. Epidermal growth factor-induced enhancement of glioblastoma cell migration in 3D arises from an intrinsic increase in speed but an extrinsic matrix- and proteolysis-dependent increase in persistence. Mol Biol Cell 19, 4249–4259 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraley S. I., Feng Y., Wirtz D. & Longmore G. D. Reply: reducing background fluorescence reveals adhesions in 3D matrices. Nat Cell Biol 13, 5–7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom R. J., George J. P., Celedon A., Sun S. X. & Wirtz D. Mapping local matrix remodeling induced by a migrating tumor cell using three-dimensional multiple-particle tracking. Biophysical Journal 95, 4077–4088 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraley S. I., Feng Y., Giri A., Longmore G. D. & Wirtz D. Dimensional and temporal controls of three-dimensional cell migration by zyxin and binding partners. Nat Commun 3, 719 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraley S. I. et al. A distinctive role for focal adhesion proteins in three-dimensional cell motility. Nat Cell Biol 12, 598–604 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libotte T. et al. Lamin A/C-dependent localization of Nesprin-2, a giant scaffolder at the nuclear envelope. Mol Biol Cell 16, 3411–3424 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketema M. et al. Requirements for the localization of nesprin-3 at the nuclear envelope and its interaction with plectin. J Cell Sci 120, 3384–3394 (2007). [DOI] [PubMed] [Google Scholar]
- Ostlund C. et al. Dynamics and molecular interactions of linker of nucleoskeleton and cytoskeleton (LINC) complex proteins. J Cell Sci 122, 4099–4108 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyton S. R. & Putnam A. J. Extracellular matrix rigidity governs smooth muscle cell motility in a biphasic fashion. J Cell Physiol 204, 198–209 (2005). [DOI] [PubMed] [Google Scholar]
- Ulrich T. A., de Juan Pardo E. M. & Kumar S. The mechanical rigidity of the extracellular matrix regulates the structure, motility, and proliferation of glioma cells. Cancer Res 69, 4167–4174 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehfeldt F. et al. Hyaluronic acid matrices show matrix stiffness in 2D and 3D dictates cytoskeletal order and myosin-II phosphorylation within stem cells. Integr Biol (Camb) 4, 422–430 (2012). [DOI] [PubMed] [Google Scholar]
- Jaalouk D. E. & Lammerding J. Mechanotransduction gone awry. Nat Rev Mol Cell Biol 10, 63–73 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisp M. & Burke B. The nuclear envelope as an integrator of nuclear and cytoplasmic architecture. FEBS Lett 582, 2023–2032 (2008). [DOI] [PubMed] [Google Scholar]
- Razafsky D. & Hodzic D. Bringing KASH under the SUN: the many faces of nucleo-cytoskeletal connections. J Cell Biol 186, 461–472 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postel R., Ketema M., Kuikman I., de Pereda J. M. & Sonnenberg A. Nesprin-3 augments peripheral nuclear localization of intermediate filaments in zebrafish. J Cell Sci 124, 755–764. [DOI] [PubMed] [Google Scholar]
- Dupin I., Sakamoto Y. & Etienne-Manneville S. Cytoplasmic intermediate filaments mediate actin-driven positioning of the nucleus. J Cell Sci 124, 865–872 (2011). [DOI] [PubMed] [Google Scholar]
- Lee J. S., Chang M. I., Tseng Y. & Wirtz D. Cdc42 mediates nucleus movement and MTOC polarization in Swiss 3T3 fibroblasts under mechanical shear stress. Mol Biol Cell 16, 871–880 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson L. J. et al. Defects in cell spreading and ERK1/2 activation in fibroblasts with lamin A/C mutations. Biochim Biophys Acta 1792, 810–821 (2009). [DOI] [PubMed] [Google Scholar]
- Scharner J. et al. Novel LMNA mutations in patients with Emery-Dreifuss muscular dystrophy and functional characterization of four LMNA mutations. Hum Mutat 32, 152–167 (2011). [DOI] [PubMed] [Google Scholar]
- Tseng Y., Lee J. S., Kole T. P., Jiang I. & Wirtz D. Micro-organization and visco-elasticity of the interphase nucleus revealed by particle nanotracking. J Cell Sci 117, 2159–2167 (2004). [DOI] [PubMed] [Google Scholar]
- Tseng Y. et al. How actin crosslinking and bundling proteins cooperate to generate an enhanced cell mechanical response. Biochem Biophys Res Commun 334, 183–192 (2005). [DOI] [PubMed] [Google Scholar]
- Celedon A., Hale C. M. & Wirtz D. Magnetic manipulation of nanorods in the nucleus of living cells. Biophys J 101, 1880–1886 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl K. N., Kahn S. M., Wilson K. L. & Discher D. E. The nuclear envelope lamina network has elasticity and a compressibility limit suggestive of a molecular shock absorber. J Cell Sci Pt (2004). [DOI] [PubMed] [Google Scholar]
- Sullivan T. et al. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol 147, 913–920 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panorchan P., Tseng Y. & Wirtz D. Structure-function relationship of biological gels revealed by multiple particle tracking and differential interference contrast microscopy: The case of human lamin networks. Phys Rev E 70, 041906 (2004). [DOI] [PubMed] [Google Scholar]
- Apgar J. et al. Multiple-particle tracking measurements of heterogeneities in solutions of actin filaments and actin bundles. Biophys. J. 79, 1095–1106 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amsden B. An obstruction-scaling model for diffusion in homogeneous hydrogels. Macromolecules 32, (1999). [Google Scholar]
- Westerweel J., Vol. PhD (Delft University of Technology, Delft, 1993).
- Adrian R. J. Particle-imaging techniques for experimental fluid mechanics. Annual Review of Fluid Mechanics 23, 261–304 (1991). [Google Scholar]