

Abstract
Incidence of nonalcoholic fatty liver disease is increasing with an estimated prevalence of 20–30% in developed nations. This is leading to increased incidence of chronic liver disease, cirrhosis, and hepatocellular cancer. It is critical to understand the etiology and pathogenesis of any disease to create therapeutic targets and develop new treatments. In this paper we discuss the etiology and pathogenesis of nonalcoholic steatohepatitis with special focus on obesity, role of insulin resistance, and molecular mechanisms of hepatotoxicity.
1. Introduction
The term non-alcoholic fatty liver disease (NAFLD) refers to the spectrum of diseases characterized by fatty infiltration of the liver ranging from steatosis, steatohepatitis, or cirrhosis. Hepatic steatosis with or without hepatitis, in the absence of alcohol use, was first described by Ludwig et al. and is referred to as non-alcoholic steatosis or non-alcoholic steatohepatitis (NASH) [1]. NAFLD is a common disease with an estimated prevalence in unselected population of developed nations around 20–30% [2]. The rapid rise in the incidence of the NAFLD might be explained by the recent epidemic of obesity and metabolic syndrome, which are manifested at hepatic level as NAFLD [3–5]. Most patients with NAFLD have simple hepatic steatosis without progression to steatohepatitis and fibrosis. However, in 2-3% of patients, NAFLD can progress to NASH that can eventually cause progressive fibrosis and lead to cirrhosis and related complications including hepatocellular carcinoma [3, 6, 7]. Once patients with simple steatosis develop NASH, up to 50% of them could develop advanced fibrosis [8].
A “two-hit hypothesis” was then proposed to explain the pathogenesis and progression of NAFLD, where the first hit causes accumulation of excess triglycerides in the liver leading to simple steatosis and the second hit causes the steatosis to progress to inflammation and fibrosis [9, 10]. The two-hit hypothesis was recently questioned as it was suggested that the hepatic accumulation of triglycerides in the liver might be instead protective towards further hepatic damage [11, 12].
Development of obesity or metabolic obesity, defined by isolated increase in visceral fat in people who are not obese, is often seen as the starting point for development of NAFLD [13], leading to the cascade of events ending in the formation of hepatic steatosis. How does increased visceral fat lead to increased fat accumulation in liver? What is the role of insulin resistance? What are the cellular and molecular mechanisms involved? What are the chemical mediators involved?
2. Factors Contributing to Development of Obesity
Development of obesity or metabolic obesity is seen as the initial step in the development of metabolic syndrome and non-alcoholic fatty liver disease. Obesity is likely due to contributions from multiple factors including but not limited to impaired central appetite regulation, genetic predisposition, and contribution from dietary factors and lack of physical activity [13].
Central nervous system plays a critical role in regulation of body weight via a negative feedback mechanism. Increase in body fat stores alert the appetite center in hypothalamus leading to appetite control and adipose tissue homeostasis [14]. Insulin and leptin are considered as prime mediators of this mechanism [15]. In overweight individuals, the amount of leptin in circulation is high but they develop resistance to leptin and so their appetite is not well controlled, leading to failure of the negative feedback mechanism [16, 17]. The molecular mechanisms leading to leptin resistance and its role in the development of obesity are discussed in detail elsewhere [18].
While increased caloric intake definitely has a critical role in the development of obesity, there has been considerable interest about various dietary components and their relative contribution to the development of obesity. Increased fructose consumption has been shown as a risk factor for development of NASH and that increased fructose consumption correlates with the severity of fibrosis in patients with NAFLD [19, 20]. The explanation is that fructose consumption leads to obesity or metabolic syndrome and that NAFLD is the hepatic manifestation but it is interesting to note that increased fructose consumption is an independent risk factor for development of fatty liver irrespective of metabolic syndrome [21, 22].
Patients with NALFD are shown to have increased percentage of dietary fat content and also get much lower percentage of their calories from fruits [23, 24]; not only total fat content in the diet but also the composition of fat has seen considerable interest in recent times. Current literature supports the fact that diet of patients with NAFLD might be high in saturated fatty acids and n-6 polyunsaturated fatty acids and low in n-3 polyunsaturated fatty acids and monounsaturated fatty acids [25–27]. Though much of the focus has been on diets with high percentage of fats, diet, rich in synthetic disaccharides have also been shown to induce hepatic fibrosis in rats [28].
Gut microbiota and its interaction with the consumed nutrients have also been the focus of research and microbiota could have a possible role in obesity by their influence on amount of nutrients absorbed. Several mechanisms were proposed including altered gut permeability and digesting the ingested polysaccharides thereby increasing the amount of energy absorbed [29].
While low physical activity might not directly contribute to NAFLD in otherwise healthy patients, increase in physical activity coupled with weight loss has been shown to improve liver profile in overweight patients with chronic liver disease [30].
3. Obesity Is a Proinflammatory State: Results in Insulin Resistance
Two types of adipose tissue are recognized in humans: brown adipose tissue and white adipose tissue. brown adipose tissue, mainly found in neonates, helps with heat production and has a protective effect against hypothermia. White adipose tissue, present in adults, consists of adipocytes, endothelial cells, fibroblasts, leukocytes, and bone marrow derived macrophages. The only function of white adipose tissue was initially thought to be energy store. Instead, new research is pointing towards adipose tissue having a more complex endocrine function mediated by the production of numerous proinflammatory cytokines called adipocytokines [31, 32]. It should also be noted that not all white adipose tissue might be the same; increasing volume of visceral adipose tissue and their production of pro-inflammatory cytokines seems to play an important role in development of insulin resistance compared to subcutaneous adipose tissue [33].
Adipocytokines produced by the adipose tissue include adiponectin, leptin, resistin, visfatin, tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), monocyte chemoattractant protein-1 (MCP-1; also known as CCL2 or CC-chemokine ligand 2), plasminogen activator inhibitor-1, angiotensinogen, retinol-binding protein-4, and serum amyloid A [34–38]. Adipocytokines are not exclusively produced by adipocytes but some, like TNF-α, are mainly produced by the macrophages in the adipose tissue. MCP-1 produced by adipocytes is a major factor contributing to macrophage recruitment to the adipose tissue [39]. Adipose tissue in obese individuals is associated with increased macrophage activity, which is responsible for almost all of the TNF-α and major part of the IL-6 expressed by the adipose tissue [40]. Thus, obesity is state of chronic inflammation characterized by abnormal cytokine (adipocytokines) production and activation of pro-inflammatory signaling pathways [41]. The following sequence of events has been proposed: development of obesity leads to increased volume of adipose tissue, followed by increased production of MCP-1 by the adipocytes, which attracts more macrophages to the adipose tissue itself. Once the macrophages in the adipose tissue are activated, a self-perpetuating inflammatory cascade is triggered by secretion of pro-inflammatory cytokines like TNF-α and IL-6 [31].
As noted above, the distribution of fat is also important in the pathogenesis of metabolic syndrome and visceral adipose tissue is considered a better indicator of insulin resistance and cardio vascular disease [42]. This could be due to either release of greater amounts of adipocytokines by visceral fat tissue compared to subcutaneous tissue in obese individuals [43] or could be due to the fact that visceral fat has direct access to portal circulation and thereby having stronger impact on liver [34].
Sodium salicylate an antiinflammatory medication has been used to decrease glycosuria associated with diabetes many years before the discovery of association between type-2 diabetes mellitus and increased inflammatory markers [44]. Since then more studies have shown increased levels of inflammatory mediators like C-reactive protein, interleukin-6, and plasminogen activator inhibitor-1 in patients with type-2 diabetes [45–48].
Obesity is a proinflammatory state with high levels of circulating pro-inflammatory cytokines and diabetes is also a state of chronic inflammation; how are these two conditions related? The answer to this question was provided by a study that has shown that TNF-α can induce insulin resistance in obese rodents and also that neutralization of TNF-α can decrease the insulin resistance with resulting increased peripheral uptake of glucose [49]. Since then similar findings of elevated TNF-α were also found in humans with increased insulin resistance and impaired glucose tolerance [50–52].
High TNF-α levels can induce insulin resistance in animal models through the activation of I-kappa-B-kinase-β (IKKβ)/nuclear-factor-kappa-B (NF-κB) and Jun N-terminal kinase (JNK) pathways [53]. JNK can cause insulin resistance through the phosphorylation of serine residues in insulin receptor substrate-1 (IRS-1) [54, 55]. IKKβ activation leads to activation of NF-κB via transcription and sub-sequent increased expression of markers and mediators of inflammation causing insulin resistance. Increasing obesity will lead to increased production of adipocytokines like TNF-α, IL-6 that lead to perpetuating cycle of JNK, and NF-κB activation leading to worsening insulin resistance. Detailed review of signaling pathways associated with insulin resistance due to inflammation was discussed elsewhere [56].
4. Mechanisms of Hepatic Fat Accumulation: Role of Insulin Resistance
The liver plays a key role in lipid metabolism; its role includes uptake and de novo synthesis of free fatty acids (FFAs) followed by conversion of FFAs into triglycerides by esterification. These triglycerides are then released into the circulation as very low-density lipoproteins (VLDL) or stored as triglyceride vacuoles in hepatocytes [57]. FFAs that are not esterified into triglycerides will be metabolized in the liver by β-oxidation [58] (Figure 1).
Figure 1.
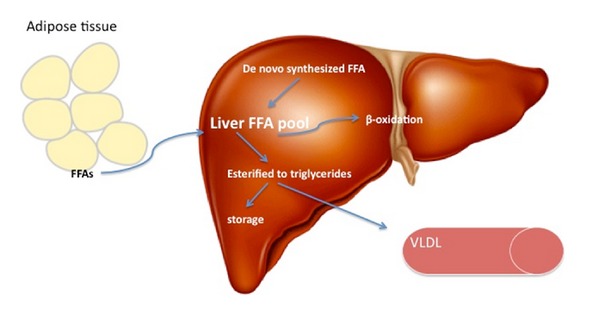
Lipid metabolism in liver. Liver FFA pool is derived from uptake of circulating FFAs and de novo synthesis. The FFAs are then either oxidized or esterified into triglycerides. Triglycerides are then released into circulation as VLDL or stored as vacuoles leading to hepatic steatosis.
In NAFLD, there is disruption of this cascade of events since the amount of FFAs delivered/synthesized in the liver exceeds its oxidative capacity. This leads to increased triglyceride synthesis and as the triglyceride synthesis continues to rise and exceed the amount that can be released as VLDLs, triglycerides accumulate in hepatocytes causing hepatic steatosis [58, 59]. This step of development of hepatic steatosis is considered as “first hit” in the pathogenesis of NAFLD [60, 61].
This raises the questions: what causes increased availability of FFAs to liver, is it increased delivery or is it due to increased de novo synthesis of FFAs in liver? What is the role of insulin resistance? Other than increased FFA availability, does disruption of other mechanisms like β-oxidation or VLDL synthesis contribute to hepatic lipid accumulation?
As much as 59% of hepatic triglyceride content is derived from free fatty acids and only 26.1% of the hepatic triglyceride was due to de novo synthesis as shown in this study, where isotope tracers were used to track hepatic fat content [62]. This increased delivery of FFAs to liver is due to insulin resistance because insulin resistance increases the total serum FFAs levels due to increased lipolysis in peripheral adipose [63, 64]. Cluster differentiation 36 pathway activation leads to increased FFA uptake by liver [65]. Increased expression of this pathway is seen in patients with insulin resistance and is implicated in pathogenesis of NAFLD [66]. Defective oxidation of the FFAs and dysfunctional VLDL synthesis were also thought to be a key factor in pathogenesis of NAFLD [67]. Though delivery of increased amounts of FFAs beyond the capacity of liver metabolism seems to be the primary cause of hepatic fat accumulation, it should be noted that disruption of other pathways could have a role and more importantly that insulin resistance is implicated in most of these mechanisms [13, 68, 69].
As discussed earlier, increased visceral adipose tissue is a risk factor for development of metabolic syndrome and visceral adipose tissue is more prone to insulin resistance when compared to peripheral fat. Insulin resistance in visceral fat leads to increased lipolysis and subsequent delivery of FFAs to the liver increases in an exponential manner due to its direct drainage into portal circulation [70].
5. Molecular Mechanisms and Mediators of Hepatotoxicity from Excess Lipids
Hepatic steatosis [61] was considered as first hit in the pathogenesis of NAFLD but it later became clear that accumulation of triglycerides is actually protective and that free fatty acids are the toxic substances that lead to steato-hepatitis and fibrosis [71, 72]. Diacylglycerol acyltransferase 2 (DGAT2) is an enzyme responsible for esterification of FFAs into triglycerides; inhibition of triglyceride synthesis by genetically deleting this enzyme has reduced hepatic steatosis in mouse model but made fibrosis worse due to FFA toxicity [11]. Interruption of triglyceride synthesis could be the initiating event for FFAs-mediated lipotoxicity (cellular toxicity due to accumulated fat) in liver cells [73]. As such, hepatic triglycerides are called the “good fat” and FFAs are called the “bad fat” [74].
This raises the question: are all free fatty acids the same? Studies that looked at the composition of hepatic and circulating free fatty acids have revealed that patients with NAFLD have elevated levels of oleic acid (a monounsaturated fatty acid, MUFA) and palmitic acid (a saturated fatty acid, SFA) [75, 76]. On the other hand polyunsaturated fatty acids are not shown to be toxic to hepatocytes and could be protective in patients with NAFLD [13, 77]. Further information on this topic was provided by experimental studies that looked at the role of stearoyl-CoA desaturase-1 (SCD1), the enzyme that converts SFA to MUFA. Increased expression of SCD1 leads to more MUFA production, which was then incorporated into triglycerides and thus leading to well tolerated simple hepatic steatosis. But inhibition of SCD1 leads to accumulation of SFA and subsequent development of hepatocytes apoptosis and steatohepatitis [74, 78]. So for disease progression in NAFLD, the type of FFA accumulated is as important or may be more important than the quantity of FFAs accumulated in the hepatocytes [79].
Apoptosis is a process of programmed cell death and is considered an important mechanism in the progression of NAFLD [80–82]. Apoptosis is the key pathogenic mechanism noted in the biopsy specimens of the patients with NASH and in the spectrum of NAFLD presence of apoptosis distinguishes patients with simple steatosis from patients with NASH [83]. The extent and severity of the apoptosis correlates with the degree of inflammation and fibrosis, so patients with higher apoptosis rates will have advanced stage fibrosis [80]. Cytokeratin-18 fragments are markers for apoptotic hepatic cells and their circulating levels correlate with the severity of the fibrosis providing further evidence that apoptosis is an important feature of NASH [84]. Apoptosis mediated by FFAs is called lipoapoptosis [85] and the mediators of lipoapoptosis are further discussed here. Apoptotic pathways can be activated via extrinsic pathway mediated by receptors on cell surface or via intrinsic pathway mediated by intracellular organelles [86].
5.1. Toll-Like Receptors
Toll-like receptors (TLRs) are pattern recognition receptors that can identify pathogen-associated molecular patterns and in response, they activate the immune system via pro-inflammatory signaling pathways [87]. Saturated fatty acids like palmitic acid can activate TLR4-mediated upregulation of NF-κB with subsequent increased production of adipocytokines like TNF-α and IL-6 [88]. Decreased expression of TLR4 in mutant mouse model is shown to be protective against development of NASH [89].
In an experimental dextran sulfate sodium (DSS) colitis mouse model, mouse fed with high fat diet and DSS had increased levels of bacterial lipopolysaccharides in portal circulation, increased expression of TLR4, and severe hepatic inflammation when compared to controls [90]. TLR4 might be the crucial link in the gut microbiota-liver axis related to progression of NASH.
5.2. Death Receptors
Death receptors are cell surface receptors from the tumor necrosis factor family of receptors and play critical role in extrinsic apoptotic pathways [91]. The death receptors and their ligands expressed in liver include Fas, tumor necrosis factor receptor 1 (TNF-R1) and TNF-related apoptosis-inducing ligand receptor 1 and 2, TRAIL-R1 and TRAIL-R2, Fas ligand (FasL), TNF-α, and TRAIL [92]. In extrinsic pathway, death ligands activate their receptors forming adeath complex that in turn activates caspase-8 leading to apoptosis (caspases are death-inducing proteolytic enzymes). Overexpression of these death receptors and subsequent apoptosis is an important feature of NASH [80].
5.3. Mitochondrial Dysfunction and Reactive Oxygen Species
Reactive oxygen species (ROS) are a group of free radicals derived from molecular oxygen, and oxidative stress refers to the cellular damage done by these free radicals [93]. ROS are formed via oxidative reactions in intracellular organelles and mitochondria are a principal source of ROS, but in a normal healthy cell, the levels of ROS are very low due to various anti-oxidant defense mechanisms [94, 95].
In normal healthy subjects, mitochondrial β-oxidation is the preferential way to dispose of the FFAs by liver [96]. But in NAFLD, there is an excess of FFAs, and increased β-oxidation by mitochondria leads to increased delivery of electrons to the electron transport chain causing overreduction of electron transport chain and formation of ROS [95]. Mitochondrial DNA is vulnerable to damage by ROS; increased generation of ROS leads to damage of mitochondrial DNA leading to mitochondrial dysfunction, which further potentiates ROS formation [97].
Intracellular stress caused by accumulation of ROS leads to mitochondrial dysfunction resulting in release of proapoptotic proteins like cytochrome c into the cytosol. Cytochrome c then combines with apoptotic-protein activation factor-1 (Apaf-1) and caspase 9 to form an activation complex called the apoptosome. Apoptosome activates the downstream caspases 3, 6, and 7 to complete the final steps of apoptosis [98].
5.4. Lysosomal Permeabilization
Mitochondrial dysfunction is considered the central pathophysiological process contributing to progression of NALFD to NASH, and the quest to identify molecular mechanisms leads to the identification of lysosomal-mitochondrial axis in FFA-induced lipotoxicity and the potential role of lysosomal permeabilization in the progression of NASH [99]. In this study, liver cells were fed with high fat diet and observed in real time, lysosomal permeabilization and cathepsin B (a lysosomal protease) release in the cytoplasm occurred much earlier than mitochondrial dysfunction and cytochrome c release into the cytosol. Also inhibition of cathepsin B was protective against FFA-induced lipotoxicity [99]. Cathepsin B is also implicated in progression of liver fibrosis by its role in activation of hepatic stellate cells and aiding their differentiation into myofibroblasts [100].
5.5. Endoplasmic Reticulum Stress
Endoplasmic reticulum (ER) is an intracellular organelle with multiple important functions like protein synthesis, lipid synthesis, and so forth. When ER is put under stress (ER stress), it responds by a mechanism called unfolded protein response (UPR) [101]. UPR is designed to protect ER from the stress induced by various sources like viral infections, alcohol, or FFAs. But when the duration of ER stress is prolonged then UPR might not be able to cope and leads to apoptosis [102, 103]. Further information about the role of ER stress is addressed in this in vitro study where saturated fatty acid palmitic acid was able to induce ER stress and lead to apoptosis of hepatic cells [104].
Other mechanisms by which FFAs can lead to apoptosis include mitochondrial dysfunction via c-Jun N-terminal kinase (JNK) activation, pro-apoptotic protein Bax-induced mitochondrial permeabilization, free cholesterol-mediated ER stress, and ceramide-mediated apoptosis induced by death ligands like TNF/FAS [74, 105] (Figure 2).
Figure 2.
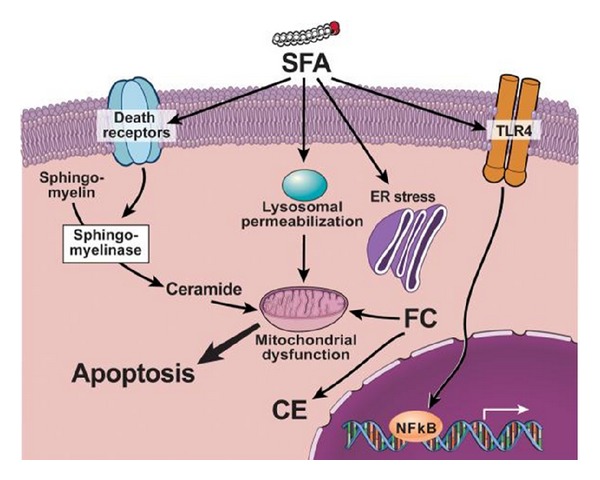
Reference [74]. FFA may activate several signaling pathways of apoptosis including upregulation and increased number of death receptors such as Fas and TRAIL receptor 5 (DR5), at the level of the plasma membrane, lysosomal permeabilization, and ER stress both coupled to mitochondrial dysfunction resulting in activation of the mitochondrial pathway of apoptosis. These toxic fatty acids may also activate TLR4 signaling resulting in up-regulation of several pro-inflammatory cytokines. Finally, other lipid types such as free cholesterol (FC) and ceramide may induce mitochondrial dysfunction and activate the mitochondrial pathway of apoptosis. Abbreviations: FFA: free fatty acids; SFA: saturated fatty acids. MUFA: monounsaturated fatty acids, FC: free cholesterol, CE. cholesteryl-ester; ER: endoplasmic reticulum.
Insummary, impaired central appetite regulation, genetic predisposition, dietary caloric excess, and lack of physical activity contribute to development of obesity. Obesity is a pro-inflammatory state and leads to insulin resistance via adipocytokines. Insulin resistance leads to increased lipolysis and exponentially high delivery of free fatty acids to liver. Accumulation of FFAs leads to hepatic steatosis and FFA-mediated lipotoxicity that eventually progresses to fibrosis/cirrhosis (Figure 3).
Figure 3.
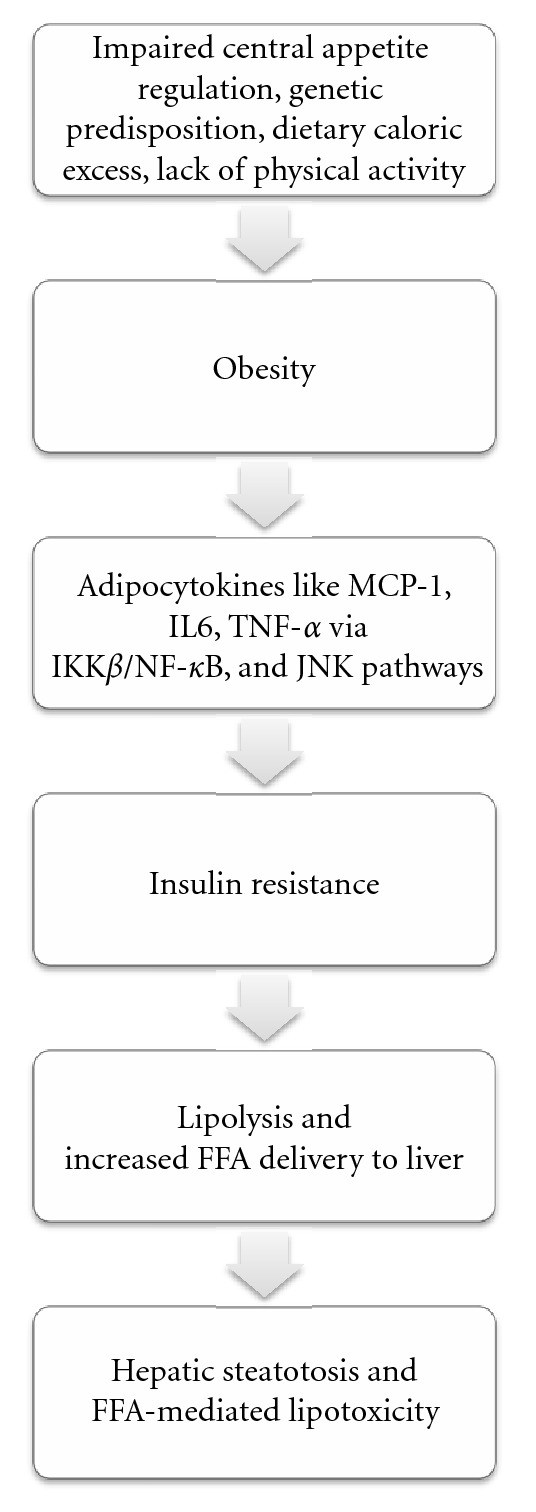
Development and progression of nonalcoholic fatty liver disease. Abbreviations: tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), monocyte chemoattractant protein-1 (MCP-1), I-kappa-B-kinase-β (IKKβ), nuclear-factor-kappa-B (NF-κB), Jun N-terminal kinase (JNK), and free fatty acids (FFAs).
In conclusion, NAFLD is increasing in prevalence and could become the most common cause of chronic liver disease in the near future in the Western world. It is very important to understand the complex molecular mechanisms and the mediator involved to develop new therapeutic targets for this disease.
References
- 1.Ludwig J, Viggiano TR, McGill DB, Ott BJ. Nonalcoholic steatohepatitis. Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clinic Proceedings. 1980;55(7):434–438. [PubMed] [Google Scholar]
- 2.Preiss D, Sattar N. Non-alcoholic fatty liver disease: an overview of prevalence, diagnosis, pathogenesis and treatment considerations. Clinical Science. 2008;115(5-6):141–150. doi: 10.1042/CS20070402. [DOI] [PubMed] [Google Scholar]
- 3.Day CP. Natural history of NAFLD: remarkably benign in the absence of cirrhosis. Gastroenterology. 2005;129(1):375–378. doi: 10.1053/j.gastro.2005.05.041. [DOI] [PubMed] [Google Scholar]
- 4.Loria P, Lonardo A, Carulli L, et al. Review article: the metabolic syndrome and non-alcoholic fatty liver disease. Alimentary Pharmacology and Therapeutics, Supplement. 2005;22(supplement 2):31–36. doi: 10.1111/j.1365-2036.2005.02592.x. [DOI] [PubMed] [Google Scholar]
- 5.Sanyal AJ. AGA technical review on nonalcoholic fatty liver disease. Gastroenterology. 2002;123(5):1705–1725. doi: 10.1053/gast.2002.36572. [DOI] [PubMed] [Google Scholar]
- 6.Miele L, Forgione A, Hernandez AP, et al. The natural history and risk factors for progression of non-alcoholic fatty liver disease and steatohepatitis. European Review for Medical and Pharmacological Sciences. 2005;9(5):273–277. [PubMed] [Google Scholar]
- 7.Bhala N, Angulo P, van der Poorten D, et al. The natural history of nonalcoholic fatty liver disease with advanced fibrosis or cirrhosis: an international collaborative study. Hepatology. 2011;54(4):1208–1216. doi: 10.1002/hep.24491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clark JM. The epidemiology of nonalcoholic fatty liver disease in adults. Journal of Clinical Gastroenterology. 2006;40(supplement 1):S5–S10. doi: 10.1097/01.mcg.0000168638.84840.ff. [DOI] [PubMed] [Google Scholar]
- 9.Musso G, Gambino R, Cassader M. Non-alcoholic fatty liver disease from pathogenesis to management: an update. Obesity Reviews. 2010;11(6):430–445. doi: 10.1111/j.1467-789X.2009.00657.x. [DOI] [PubMed] [Google Scholar]
- 10.Day CP, James OFW. Steatohepatitis: a tale of two ‘Hits’? Gastroenterology. 1998;114(4):842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 11.Yamaguchi K, Yang L, McCall S, et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45(6):1366–1374. doi: 10.1002/hep.21655. [DOI] [PubMed] [Google Scholar]
- 12.Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43(2, supplement 1):S99–S112. doi: 10.1002/hep.20973. [DOI] [PubMed] [Google Scholar]
- 13.Larter CZ, Chitturi S, Heydet D, Farrell GC. A fresh look at NASH pathogenesis. Part 1: the metabolic movers. Journal of Gastroenterology and Hepatology. 2010;25(4):672–690. doi: 10.1111/j.1440-1746.2010.06253.x. [DOI] [PubMed] [Google Scholar]
- 14.Kennedy GC. The role of depot fat in the hypothalamic control of food intake in the rat. Proceedings of the Royal Society B. 1953;140(901):578–596. doi: 10.1098/rspb.1953.0009. [DOI] [PubMed] [Google Scholar]
- 15.Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443(7109):289–295. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- 16.Chitturi S, Farrell G, Frost L, et al. Serum leptin in NASH correlates with hepatic steatosis but not fibrosis: a manifestation of lipotoxicity? Hepatology. 2002;36(2):403–409. doi: 10.1053/jhep.2002.34738. [DOI] [PubMed] [Google Scholar]
- 17.Heymsfield SB, Greenberg AS, Fujioka K, et al. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. Journal of the American Medical Association. 1999;282(16):1568–1575. doi: 10.1001/jama.282.16.1568. [DOI] [PubMed] [Google Scholar]
- 18.Morris DL, Rui L. Recent advances in understanding leptin signaling and leptin resistance. American Journal of Physiology-Endocrinology and Metabolism. 2009;297(6):E1247–E1259. doi: 10.1152/ajpendo.00274.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abdelmalek MF, Suzuki A, Guy C, et al. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology. 2010;51(6):1961–1971. doi: 10.1002/hep.23535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ouyang X, Cirillo P, Sautin Y, et al. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. Journal of Hepatology. 2008;48(6):993–999. doi: 10.1016/j.jhep.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abid A, Taha O, Nseir W, Farah R, Grosovski M, Assy N. Soft drink consumption is associated with fatty liver disease independent of metabolic syndrome. Journal of Hepatology. 2009;51(5):918–924. doi: 10.1016/j.jhep.2009.05.033. [DOI] [PubMed] [Google Scholar]
- 22.Assy N, Nasser G, Kamayse I, et al. Soft drink consumation linked with fatty liver in the absence of traditional risk factors. Canadian Journal of Gastroenterology. 2008;22(10):811–816. doi: 10.1155/2008/810961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sathiaraj E, Chutke M, Reddy MY, et al. A case-control study on nutritional risk factors in non-alcoholic fatty liver disease in Indian population. European Journal of Clinical Nutrition. 2011;65(4):533–537. doi: 10.1038/ejcn.2011.3. [DOI] [PubMed] [Google Scholar]
- 24.Kim CH, Kallman JB, Bai C, et al. Nutritional assessments of patients with non-alcoholic fatty liver disease. Obesity Surgery. 2010;20(2):154–160. doi: 10.1007/s11695-008-9549-0. [DOI] [PubMed] [Google Scholar]
- 25.Molendi-Coste O, Legry V, Leclercq IA. Dietary lipids and NAFLD: suggestions for improved nutrition. Acta Gastro-Enterologica Belgica. 2010;73(4):431–436. [PubMed] [Google Scholar]
- 26.Toshimitsu K, Matsuura B, Ohkubo I, et al. Dietary habits and nutrient intake in non-alcoholic steatohepatitis. Nutrition. 2007;23(1):46–52. doi: 10.1016/j.nut.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 27.Vilar L, Oliveira CPMS, Faintuch J, et al. High-fat diet: a trigger of non-alcoholic steatohepatitis? Preliminary findings in obese subjects. Nutrition. 2008;24(11-12):1097–1102. doi: 10.1016/j.nut.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 28.Fukunishi S, Nishio H, Fukuda A, et al. Long-term feeding of a synthetic diet rich in disaccharides induces hepatic fibrosis in nonalcoholic fatty liver disease in Zucker rats. International Journal of Molecular Medicine. 2010;25(2):187–193. [PubMed] [Google Scholar]
- 29.Diamant M, Blaak EE, de Vos WM. Do nutrient-gut-microbiota interactions play a role in human obesity, insulin resistance and type 2 diabetes? Obesity Reviews. 2011;12(4):272–281. doi: 10.1111/j.1467-789X.2010.00797.x. [DOI] [PubMed] [Google Scholar]
- 30.Hickman IJ, Jonsson JR, Prins JB, et al. Modest weight loss and physical activity in overweight patients with chronic liver disease results in sustained improvements in alanine aminotransferase, fasting insulin, and quality of life. Gut. 2004;53(3):413–419. doi: 10.1136/gut.2003.027581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nature Reviews Immunology. 2006;6(10):772–783. doi: 10.1038/nri1937. [DOI] [PubMed] [Google Scholar]
- 32.Krotkiewski M, Bjorntorp P, Sjostrom L, Smith U. Impact of obesity on metabolism in men and women. Importance of regional adipose tissue distribution. Journal of Clinical Investigation. 1983;72(3):1150–1162. doi: 10.1172/JCI111040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coon PJ, Rogus EM, Drinkwater D, Muller DC, Goldberg AP. Role of body fat distribution in the decline in insulin sensitivity and glucose tolerance with age. Journal of Clinical Endocrinology and Metabolism. 1992;75(4):1125–1132. doi: 10.1210/jcem.75.4.1400882. [DOI] [PubMed] [Google Scholar]
- 34.Shoelson SE, Herrero L, Naaz A. Obesity, Inflammation, and Insulin Resistance. Gastroenterology. 2007;132(6):2169–2180. doi: 10.1053/j.gastro.2007.03.059. [DOI] [PubMed] [Google Scholar]
- 35.Fried SK, Bunkin DA, Greenberg AS. Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: depot difference and regulation by glucocorticoid. Journal of Clinical Endocrinology and Metabolism. 1998;83(3):847–850. doi: 10.1210/jcem.83.3.4660. [DOI] [PubMed] [Google Scholar]
- 36.Fukuhara A, Matsuda M, Nishizawa M, et al. Visfatin: a protein secreted by visceral fat that Mimics the effects of insulin. Science. 2005;307(5708):426–430. doi: 10.1126/science.1097243. [DOI] [PubMed] [Google Scholar]
- 37.Shimomura I, Funahashi T, Takahashi M, et al. Enhanced expression of PAI-1 in visceral fat: possible contributor to vascular disease in obesity. Nature Medicine. 1996;2(7):800–803. doi: 10.1038/nm0796-800. [DOI] [PubMed] [Google Scholar]
- 38.Steppan CM, Bailey ST, Bhat S, et al. The hormone resistin links obesity to diabetes. Nature. 2001;409(6818):307–312. doi: 10.1038/35053000. [DOI] [PubMed] [Google Scholar]
- 39.Kanda H, Tateya S, Tamori Y, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. Journal of Clinical Investigation. 2006;116(6):1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. Journal of Clinical Investigation. 2003;112(12):1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. Journal of Clinical Investigation. 2005;115(5):1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Freedland ES. Role of a critical visceral adipose tissue threshold (CVATT) in metabolic syndrome: implications for controlling dietary carbohydrates: A review. Nutrition and Metabolism. 2004;1(1):p. 12. doi: 10.1186/1743-7075-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fain JN, Madan AK, Hiler ML, Cheema P, Bahouth SW. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology. 2004;145(5):2273–2282. doi: 10.1210/en.2003-1336. [DOI] [PubMed] [Google Scholar]
- 44.Williamson RT. On the treatment of glycosuria and diabetes mellitus with sodium salicylate. British Medical Journal. 1901;1(2100):760–762. doi: 10.1136/bmj.1.2100.760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. Journal of the American Medical Association. 2001;286(3):327–334. doi: 10.1001/jama.286.3.327. [DOI] [PubMed] [Google Scholar]
- 46.Festa A, D’Agostino R, Williams K, et al. The relation of body fat mass and distribution to markers of chronic inflammation. International Journal of Obesity. 2001;25(10):1407–1415. doi: 10.1038/sj.ijo.0801792. [DOI] [PubMed] [Google Scholar]
- 47.Festa A, D’Agostino R, Tracy RP, Haffner SM. Elevated levels of acute-phase proteins and plasminogen activator inhibitor-1 predict the development of type 2 diabetes: the insulin resistance atherosclerosis study. Diabetes. 2002;51(4):1131–1137. doi: 10.2337/diabetes.51.4.1131. [DOI] [PubMed] [Google Scholar]
- 48.Freeman DJ, Norrie J, Caslake MJ, et al. C-reactive protein is an independent predictor of risk for the development of diabetes in the west of Scotland coronary prevention study. Diabetes. 2002;51(5):1596–1600. doi: 10.2337/diabetes.51.5.1596. [DOI] [PubMed] [Google Scholar]
- 49.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 50.Olson NC, Callas PW, Hanley AJG, et al. Circulating levels of TNF-α are associated with impaired glucose tolerance, increased insulin resistance, and ethnicity: the insulin resistance atherosclerosis study. Journal of Clinical Endocrinology and Metabolism. 2012;97(3):1032–1040. doi: 10.1210/jc.2011-2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zinman B, Hanley AJG, Harris SB, Kwan J, Fantus IG. Circulating tumor necrosis factor-α concentrations in a native canadian population with high rates of type 2 diabetes mellitus. Journal of Clinical Endocrinology and Metabolism. 1999;84(1):272–278. doi: 10.1210/jcem.84.1.5405. [DOI] [PubMed] [Google Scholar]
- 52.Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM. Tumor necrosis factor α inhibits signaling from the insulin receptor. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(11):4854–4858. doi: 10.1073/pnas.91.11.4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cai D, Yuan M, Frantz DF, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nature Medicine. 2005;11(2):183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hirosumi J, Tuncman G, Chang L, et al. A central, role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 55.Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH2-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser307. Journal of Biological Chemistry. 2000;275(12):9047–9054. doi: 10.1074/jbc.275.12.9047. [DOI] [PubMed] [Google Scholar]
- 56.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. Journal of Clinical Investigation. 2006;116(7):1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Musso G, Gambino R, Cassader M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD) Progress in Lipid Research. 2009;48(1):1–26. doi: 10.1016/j.plipres.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 58.Jou J, Choi SS, Diehl AM. Mechanisms of disease progression in nonalcoholic fatty liver disease. Seminars in Liver Disease. 2008;28(4):370–379. doi: 10.1055/s-0028-1091981. [DOI] [PubMed] [Google Scholar]
- 59.Diraison F, Moulin PH, Beylot M. Contribution of hepatic de novo lipogenesis and reesterification of plasma non esterified fatty acids to plasma triglyceride synthesis during non-alcoholic fatty liver disease. Diabetes and Metabolism. 2003;29(5):478–485. doi: 10.1016/s1262-3636(07)70061-7. [DOI] [PubMed] [Google Scholar]
- 60.Raman M, Allard J. Nonalcoholic fatty liver disease: a clinical approach and review. Canadian Journal of Gastroenterology. 2006;20(5):345–349. doi: 10.1155/2006/918262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wanless IR, Shiota K. The pathogenesis of nonalcoholic steatohepatitis and other fatty liver diseases: a four-step model including the role of lipid release and hepatic venular obstruction in the progression to cirrhosis. Seminars in Liver Disease. 2004;24(1):99–106. doi: 10.1055/s-2004-823104. [DOI] [PubMed] [Google Scholar]
- 62.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. Journal of Clinical Investigation. 2005;115(5):1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qureshi K, Abrams GA. Metabolic liver disease of obesity and role of adipose tissue in the pathogenesis of nonalcoholic fatty liver disease. World Journal of Gastroenterology. 2007;13(26):3540–3553. doi: 10.3748/wjg.v13.i26.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Choi SS, Diehl AM. Hepatic triglyceride synthesis and nonalcoholic fatty liver disease. Current Opinion in Lipidology. 2008;19(3):295–300. doi: 10.1097/MOL.0b013e3282ff5e55. [DOI] [PubMed] [Google Scholar]
- 65.Pohl J, Ring A, Korkmaz U, Ehehalt R, Stremmel W. FAT/CD36-mediated long-chain fatty acid uptake in adipocytes requires plasma membrane rafts. Molecular Biology of the Cell. 2005;16(1):24–31. doi: 10.1091/mbc.E04-07-0616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Koonen DPY, Jacobs RL, Febbraio M, et al. Increased hepatic CD36 expression contributes to dyslipidemia associated with diet-induced obesity. Diabetes. 2007;56(12):2863–2871. doi: 10.2337/db07-0907. [DOI] [PubMed] [Google Scholar]
- 67.Fujita K, Nozaki Y, Wada K, et al. Dysfunctional very-low-density lipoprotein synthesis and release is a key factor in nonalcoholic steatohepatitis pathogenesis. Hepatology. 2009;50(3):772–780. doi: 10.1002/hep.23094. [DOI] [PubMed] [Google Scholar]
- 68.Pettinelli P, Obregón AM, Videla LA. Molecular mechanisms of steatosis in nonalcoholic fatty liver disease. Nutricion Hospitalaria. 2011;26(3):441–450. doi: 10.1590/S0212-16112011000300003. [DOI] [PubMed] [Google Scholar]
- 69.Bugianesi E, Moscatiello S, Ciaravella MF, Marchesini G. Insulin resistance in nonalcoholic fatty liver disease. Current Pharmaceutical Design. 2010;16(17):1941–1951. doi: 10.2174/138161210791208875. [DOI] [PubMed] [Google Scholar]
- 70.Lafontan M, Berlan M. Do regional differences in adipocyte biology provide new pathophysiological insights? Trends in Pharmacological Sciences. 2003;24(6):276–283. doi: 10.1016/S0165-6147(03)00132-9. [DOI] [PubMed] [Google Scholar]
- 71.Cusi K. Role of insulin resistance and lipotoxicity in non-alcoholic steatohepatitis. Clinics in Liver Disease. 2009;13(4):545–563. doi: 10.1016/j.cld.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 72.Listenberger LL, Han X, Lewis SE, et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(6):3077–3082. doi: 10.1073/pnas.0630588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mantzaris MD, Tsianos EV, Galaris D. Interruption of triacylglycerol synthesis in the endoplasmic reticulum is the initiating event for saturated fatty acid-induced lipotoxicity in liver cells. FEBS Journal. 2011;278(3):519–530. doi: 10.1111/j.1742-4658.2010.07972.x. [DOI] [PubMed] [Google Scholar]
- 74.Alkhouri N, Dixon LJ, Feldstein AE. Lipotoxicity in nonalcoholic fatty liver disease: not all lipids are created equal. Expert Review of Gastroenterology and Hepatology. 2009;3(4):445–451. doi: 10.1586/egh.09.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Puri P, Baillie RA, Wiest MM, et al. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology. 2007;46(4):1081–1090. doi: 10.1002/hep.21763. [DOI] [PubMed] [Google Scholar]
- 76.de Almeida IT, Cortez-Pinto H, Fidalgo G, Rodrigues D, Camilo ME. Plasma total and free fatty acids composition in human non-alcoholic steatohepatitis. Clinical Nutrition. 2002;21(3):219–223. doi: 10.1054/clnu.2001.0529. [DOI] [PubMed] [Google Scholar]
- 77.Delarue J, LeFoll C, Corporeau C, Lucas D. n-3 long chain polyunsaturated fatty acids: a nutritional tool to prevent insulin resistance associated to type 2 diabetes and obesity? Reproduction Nutrition Development. 2004;44(3):289–299. doi: 10.1051/rnd:2004033. [DOI] [PubMed] [Google Scholar]
- 78.Li ZZ, Berk M, McIntyre TM, Feldstein AE. Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: role of stearoyl-Coa desaturase. Journal of Biological Chemistry. 2009;284(9):5637–5644. doi: 10.1074/jbc.M807616200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McClain CJ, Barve S, Deaciuc I. Good fat/bad fat. Hepatology. 2007;45(6):1343–1346. doi: 10.1002/hep.21788. [DOI] [PubMed] [Google Scholar]
- 80.Feldstein AE, Canbay A, Angulo P, et al. Hepatocyte apoptosis and Fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. 2003;125(2):437–443. doi: 10.1016/s0016-5085(03)00907-7. [DOI] [PubMed] [Google Scholar]
- 81.Ramalho RM, Cortez-Pinto H, Castro RE, et al. Apoptosis and Bcl-2 expression in the livers of patients with steatohepatitis. European Journal of Gastroenterology and Hepatology. 2006;18(1):21–29. doi: 10.1097/00042737-200601000-00005. [DOI] [PubMed] [Google Scholar]
- 82.Syn WK, Teaberry V, Choi SS, Diehl AM. Similarities and differences in the pathogenesis of alcoholic and nonalcoholic steatohepatitis. Seminars in Liver Disease. 2009;29(2):200–210. doi: 10.1055/s-0029-1214375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Syn WK, Choi SS, Diehl AM. Apoptosis and cytokines in non-alcoholic steatohepatitis. Clinics in Liver Disease. 2009;13(4):565–580. doi: 10.1016/j.cld.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wieckowska A, Zein NN, Yerian LM, Lopez AR, McCullough AJ, Feldstein AE. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology. 2006;44(1):27–33. doi: 10.1002/hep.21223. [DOI] [PubMed] [Google Scholar]
- 85.Ibrahim SH, Kohli R, Gores GJ. Mechanisms of lipotoxicity in NAFLD and clinical implications. Journal of Pediatric Gastroenterology and Nutrition. 2011;53(2):131–140. doi: 10.1097/MPG.0b013e31822578db. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Delhalle S, Duvoix A, Schnekenburger M, Morceau F, Dicato M, Diederich M. An introduction to the molecular mechanisms of apoptosis. Annals of the New York Academy of Sciences. 2003;1010:1–8. doi: 10.1196/annals.1299.001. [DOI] [PubMed] [Google Scholar]
- 87.Aoyama T, Paik YH, Seki E. Toll-like receptor signaling and liver fibrosis. Gastroenterology Research and Practice. 2010;2010:8 pages. doi: 10.1155/2010/192543.192543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schaeffler A, Gross P, Buettner R, et al. Fatty acid-induced induction of Toll-like receptor-4/nuclear factor-κB pathway in adipocytes links nutritional signalling with innate immunity. Immunology. 2009;126(2):233–245. doi: 10.1111/j.1365-2567.2008.02892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Igolnikov AC, Spatz K, Green RM. C3H/HeJ mice with mutations of the toll-like receptor 4 (TLR-4) are resistant to methionine-choline deficient (MCD) diet induced non-alcoholic steatohepatitis (NASH) Hepatology. 2002;36(4):p. 404A-A. [Google Scholar]
- 90.Gabele E, Dostert K, Hofmann C, et al. DSS induced colitis increases portal LPS levels and enhances hepatic inflammation and fibrogenesis in experimental NASH. Journal of Hepatology. 2011;55(6):1391–1399. doi: 10.1016/j.jhep.2011.02.035. [DOI] [PubMed] [Google Scholar]
- 91.Faubion WA, Gores GJ. Death receptors in liver biology and pathobiology. Hepatology. 1999;29(1):1–4. doi: 10.1002/hep.510290101. [DOI] [PubMed] [Google Scholar]
- 92.Guicciardi ME, Gores GJ. Apoptosis as a mechanism for liver disease progression. Seminars in Liver Disease. 2010;30(4):402–410. doi: 10.1055/s-0030-1267540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rolo AP, Teodoro JS, Palmeira CM. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radical Biology and Medicine. 2012;52(1):59–69. doi: 10.1016/j.freeradbiomed.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 94.Brand MD. The sites and topology of mitochondrial superoxide production. Experimental Gerontology. 2010;45(7-8):466–472. doi: 10.1016/j.exger.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Murphy MP. How mitochondria produce reactive oxygen species. Biochemical Journal. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tessari P, Coracina A, Cosma A, Tiengo A. Hepatic lipid metabolism and non-alcoholic fatty liver disease. Nutrition, Metabolism and Cardiovascular Diseases. 2009;19(4):291–302. doi: 10.1016/j.numecd.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 97.Ricci C, Pastukh V, Leonard J, et al. Mitochondrial DNA damage triggers mitochondrial-superoxide generation and apoptosis. American Journal of Physiology-Cell Physiology. 2008;294(2):C413–C422. doi: 10.1152/ajpcell.00362.2007. [DOI] [PubMed] [Google Scholar]
- 98.Feldstein AE, Gores GJ. Apoptosis in alcoholic and nonalcoholic steatohepatitis. Frontiers in Bioscience. 2005;10(3):3093–3099. doi: 10.2741/1765. [DOI] [PubMed] [Google Scholar]
- 99.Li Z, Berk M, McIntyre TM, Gores GJ, Feldstein AE. The lysosomal-mitochondrial axis in free fatty acid-induced hepatic lipotoxicity. Hepatology. 2008;47(5):1495–1503. doi: 10.1002/hep.22183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Moles A, Tarrats N, Fernández-Checa JC, Marí M. Cathepsins B and D drive hepatic stellate cell proliferation and promote their fibrogenic potential. Hepatology. 2009;49(4):1297–1307. doi: 10.1002/hep.22753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nature Reviews Molecular Cell Biology. 2007;8(7):519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 102.Ji C, Kaplowitz N. ER stress: can the liver cope? Journal of Hepatology. 2006;45(2):321–333. doi: 10.1016/j.jhep.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 103.Kaplowitz N, Tin AT, Shinohara M, Ji C. Endoplasmic reticulum stress and liver injury. Seminars in Liver Disease. 2007;27(4):367–377. doi: 10.1055/s-2007-991513. [DOI] [PubMed] [Google Scholar]
- 104.Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. American Journal of Physiology-Endocrinology and Metabolism. 2006;291(2):E275–E281. doi: 10.1152/ajpendo.00644.2005. [DOI] [PubMed] [Google Scholar]
- 105.Malhi H, Gores GJ. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Seminars in Liver Disease. 2008;28(4):360–369. doi: 10.1055/s-0028-1091980. [DOI] [PMC free article] [PubMed] [Google Scholar]