

Abstract
Alzheimer's disease (AD) is a neurodegenerative disease, one of whose major pathological hallmarks is the accumulation of amyloid plaques comprised of aggregated β-amyloid (Aβ) peptides. It is now recognized that soluble Aβ oligomers may lead to synaptic dysfunctions early in AD pathology preceding plaque deposition. Aβ is produced by a sequential cleavage of amyloid precursor protein (APP) by the activity of β- and γ-secretases, which have been identified as major candidate therapeutic targets of AD. This paper focuses on how Aβ alters synaptic function and the functional consequences of inhibiting the activity of the two secretases responsible for Aβ generation. Abnormalities in synaptic function resulting from the absence or inhibition of the Aβ-producing enzymes suggest that Aβ itself may have normal physiological functions which are disrupted by abnormal accumulation of Aβ during AD pathology. This interpretation suggests that AD therapeutics targeting the β- and γ-secretases should be developed to restore normal levels of Aβ or combined with measures to circumvent the associated synaptic dysfunction(s) in order to have minimal impact on normal synaptic function.
1. Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disorder, causing loss of synaptic contacts and cognitive decline. It is widely believed that AD is initiated by synaptic dysfunction, which may be the basis for memory loss in early stages of the disease [1, 2]. Current theories implicate the production of amyloid beta (Aβ) as a key molecular event that ultimately leads to neuronal degeneration and the clinical pathology seen in AD [3]. Aβ is produced by sequential proteolytic cleavage of amyloid precursor protein (APP) by two endoproteolytic enzymes, β- and γ-secretase (Figure 1). Therefore, inhibiting the activity of these enzymes has surfaced as one of the major disease-modifying approaches for AD [4]. However, in order to develop effective therapeutics, a detailed molecular and cellular understanding of the role of both secretases in synaptic function is necessary. In addition, since accumulating evidence suggests that the initial pathology of AD is a result of synaptic dysfunction [1, 2], understanding how Aβ production alters normal synaptic function and what types of synaptic functions are differentially affected by Aβ becomes important in developing effective therapeutics for disease intervention. In this paper, we will summarize a number of experimental observations that address how Aβ affects synaptic function, and review data obtained from genetically altered mice developed to test the feasibility of blocking APP-processing enzymes which unveiled functional roles for these enzymes in normal synaptic transmission and plasticity. We will also discuss a body of work, which investigates how synaptic function is affected by currently available therapies that target APP-processing enzymes. Before that we will briefly introduce the topic and current understanding of synaptic plasticity, which are relevant for the later discussions.
Figure 1.
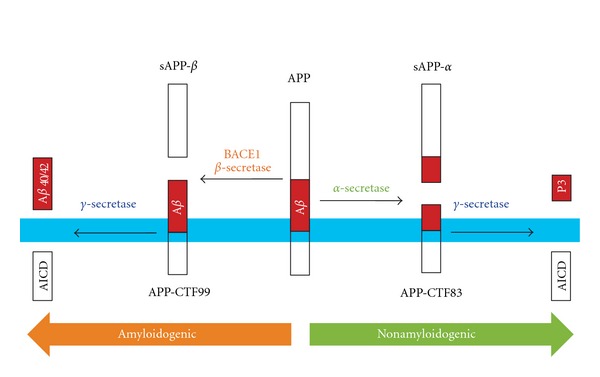
A diagram of amyloid precursor protein (APP) processing pathways. The transmembrane protein APP (membrane indicated in blue) can be processed by two pathways, the nonamyloidogenic α-secretase pathway and the amyloidogenic β-secretase pathway. In the nonamyloidogenic pathway, α-secretase cleaves in the middle of the β-amyloid (Aβ) region (red) to release the soluble APP-fragment sAPP-α. The APP C-terminal fragment 83 (APP-CTF83) is then cleaved by γ-secretase to release the APP intracellular domain (AICD) and P3 fragment. In the amyloidogenic pathway, β-secretase cleaves APP to produce the soluble fragment sAPP-β. APP-CTF99 is then cleaved by γ-secretase to produce Aβ 40, Aβ 42 and AICD.
2. Synaptic Plasticity and Memory Formation
It is widely believed that long-term changes in the strength of synaptic transmission underlie the formation of memories. Hebb is often recognized as the first person to crystallize this idea by proposing that coincident activity of pre- and postsynaptic neurons strengthens synaptic connections [5]. It was subsequently recognized that uncorrelated activity between two neurons should decrease the strength of synaptic transmission between them [6]. The strengthening of synaptic connections is termed long-term potentiation (LTP) and is experimentally produced by high-frequency stimulation [7], while the weakening of synaptic connections, produced by low-frequency stimulation [8, 9], is called long-term depression (LTD). Since their initial discovery, both LTP and LTD have been found to occur in a diverse set of synapses across many different brain areas (reviewed in [10]). These long lasting forms of synaptic plasticity share similar mechanisms of induction, expression, and maintenance with those of long-term consolidation of several forms of memory [11–19]. Moreover, long-term alterations in synaptic transmission, similar to characteristics of LTP and LTD, have been observed in vivo during various learning paradigms [20–24], which further suggests that LTP and LTD may be cellular substrates for memory formation.
While LTP and LTD are effective models for mediating synapse-specific changes required for memory formation, theoretical considerations indicate that maintaining the stability of the nervous system requires additional homeostatic plasticity mechanisms that operate at a slower time scale (hours to days) [25–29]. For example, without homeostatic regulation, the increase in postsynaptic activity after LTP might result in a vicious cycle of potentiation that not only degrades the capacity of neural circuits to store specific information but could also culminate in a run-away excitation of the neural network. There are several mechanisms of homeostasis that can stabilize the nervous system: adjusting excitatory synaptic transmission postsynaptically [26–30], modulating the excitability of neurons [31–33], changing inhibitory circuits [33–36], and altering presynaptic function [37–39]. While most studies of synaptic plasticity related to memory formation focus on LTP and LTD, it is prudent to understand that alterations in homeostatic plasticity can also affect learning and memory.
3. Molecular Mechanisms of Synaptic Plasticity: A Brief Overview
While LTP and LTD have been observed in many different brain areas, the majority of knowledge about their molecular mechanisms comes from studies in the hippocampus. This is partly because the hippocampus is an area of the brain that is critically involved in the formation of long-term memories (reviewed in [16]). In addition, the hippocampus is one of the areas highly susceptible to amyloid pathology in most AD brains (reviewed in [2]). Therefore, we will briefly review the mechanisms of synaptic plasticity in the hippocampus.
In the hippocampus, two major forms of LTP and LTD are observed: one that is dependent on NMDA receptor (NMDAR) activation and another that is independent of NMDARs [16, 40]. The most widely studied forms of LTP and LTD are those dependent on NMDARs in the CA1 region; hence, their mechanisms have been fairly well characterized. Therefore, most of our discussion will focus on the NMDAR-dependent forms of LTP and LTD. NMDARs, due to activity-dependent relief of their Mg2+ block [41], act as coincident detectors for pre- and postsynaptic activity. In addition, activation of NMDARs allows influx of Ca2+ [42–44], which can act as a second messenger to activate various downstream effectors in the postsynaptic neuron. It is thought that both the magnitude and temporal pattern of Ca2+ increase determine the expression of either LTP or LTD, by differentially regulating the activity of protein kinases and phosphatases [15]. One of the key downstream events of LTP and LTD is the regulation of synaptic AMPA receptors (AMPARs) (for review see [45, 46]). AMPARs are the major mediators of fast excitatory synaptic transmission in the central nervous system (CNS); therefore their function directly dictates synaptic strength. Several studies demonstrated that LTP increases the synaptic content of AMPARs, predominantly by an activity-dependent insertion of receptors containing the GluA1 subunit (GluR1) [47–49]. This requires concomitant activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII) and phosphorylation of the AMPAR subunit GluA1 at serine 818 (S818) [50] and serine 845 (S845) [51]. GluA1-S818 is a protein kinase C (PKC) phosphorylation site [50] while GluA1-S845 is a protein kinase A (PKA) phosphorylation site [52]. In addition to these two sites, phosphorylation of GluA1-S831, which can be phosphorylated by both PKC [52] and CaMKII [53, 54], has been shown to correlate with LTP [55, 56]. However, this site is not necessary for LTP [57] nor synaptic trafficking of AMPARs [47]. Many studies confirm that CaMKII, PKC, and PKA are involved in NMDAR-dependent LTP (reviewed in [46]). Consistent with a dominant role for GluA1 in mediating synaptic potentiation, GluA1 knockout mice [58], as well as mice lacking specific phosphorylation sites on GluA1 [59], display LTP deficits. On the other hand, NMDAR-dependent LTD is associated with an activity-dependent removal of synaptic AMPARs [60]. This process depends on endocytosis of GluA2-containing receptors [61–67] but also requires dephosphorylation at GluA1-S845 [56, 59, 68].
While regulation of synaptic AMPARs, through synaptic targeting and phosphorylation, is involved in the initial expression of LTP and LTD, maintenance of these forms of plasticity involves additional mechanisms. Collectively, data from many studies report that blocking new protein synthesis inhibits the late phase of long-term synaptic plasticity [69–74]. This parallels the requirement for new protein synthesis in the formation of long-term memory in intact animals [75, 76] (see review [77]). Transcriptional activation is also necessary for the maintenance of some forms of long-term synaptic plasticity [78]. So far, it is known that multiple transcription factors are activated immediately after induction of LTP. Increased transcription of several immediate early genes (IEG) is especially important [79] since they enhance new protein synthesis [12, 16]. Interestingly, some, if not all, of these transcriptional regulators are also required for long-term memory formation. Disruption of cAMP response element-binding protein (CREB) levels, a Ca2+-dependent transcription factor, in either the hippocampus or the amygdala has been found to impair specific long-term memory but not initial acquisition or short-term memory formation [80–82]. Inhibiting the expression of Arc/Arg 3.1 (activity-regulated cytoskeletal protein/activity-regulated gene 3.1), an IEG, in the hippocampus also impairs long-term memory consolidation [83].
4. Exogenous Aβ Application Alters Synaptic Function
Much of the molecular understanding of AD came from studying familial AD (FAD-) linked mutations, which have been found in genes encoding APP and presenilin 1 and 2 (PS1 and 2) in AD patients. These mutations are linked to elevated Aβ production [84, 85]. This is because many FAD-linked mutations make APP a more favorable substrate for the amyloidogenic cleavage pathway leading to increased Aβ production. Since FAD patients often harbor multiple mutations, many of the AD mouse models also carry several FAD mutations. However, depending on the combination of the mutations and their variants, distinct phenotypes are observed across age and brain regions studied (for an extensive recent review on electrophysiological studies of various AD transgenic (Tg) mouse models see [86]).
Although different AD mouse models show deficits in synaptic function, it cannot be taken for granted that these deficits are caused directly by the enhanced production of Aβ peptides (especially Aβ 42, which is the major component of extracellular senile plaques). In order to directly test the role of Aβ in altering synaptic function, many studies have investigated synaptic properties and synaptic plasticity following exogenous application of various Aβ peptides.
In vitro studies done in either the medial perforant path to dentate granule cells or the Schaffer collateral inputs to CA1 neurons reported that application of subneurotoxic concentrations of Aβ peptides (i.e., Aβ 42, Aβ 40, or Aβ 25–35) inhibit LTP induction without affecting basal synaptic transmission [87–89]. A similar result was found in an in vivo study, where naturally secreted Aβ collected from cells expressing mutated APP (V717F mutation in APP751) was injected into the CA1 region of hippocampus which prevented stable LTP maintenance [90]. This study further showed that soluble Aβ oligomers, not monomeric Aβ, or Aβ fibrils, are responsible for blocking LTP [90]. In addition, in vivo injection of Aβ peptides (i.e., Aβ 42 or the C-terminal of APP which contains the Aβ fragment) is reported to facilitate LTD and LTP reversal (called depotentiation) in the CA1 region [91]. A majority of studies suggest that while fibrillar Aβ accumulation is found in senile plaques that are a hallmark of AD, it is the soluble Aβ oligomers that disturb synaptic function and lead to neurodegeneration in AD [90, 92].
4.1. Postsynaptic Alterations by Aβ
Soluble Aβ oligomers in AD brains have been found to bind to neuronal surfaces [93], specifically to a subset of synapses where they colocalize with a postsynaptic density marker PSD95 [94], suggesting that Aβ may regulate postsynaptic function directly. One candidate target of Aβ is NMDARs. It was found that synthetic Aβ 40 peptides can selectively augment NMDAR current, without affecting AMPAR current, in the dentate gyrus of acute hippocampal slices [95]. Consistent with this, APPInd (V717F mutation) Tg mice show an enhancement in the ratio of NMDAR-to-AMPAR-mediated synaptic transmission in the CA1 region [96]. However, contradictory results are reported from later studies. A recent study showed that application of both synthetic Aβ 42 peptides and naturally secreted Aβ, from APPSwe (K670N/M671L mutation) Tg mice, promotes endocytosis of surface NMDARs and hence depresses NMDAR current in wildtype cultured cortical neurons [97]. Moreover, they also found reduced surface expression of NMDARs in cultured cortical neurons from APPSwe Tg mice [97]. Other studies found downregulation of surface AMPARs in neurons overexpressing either wildtype or APPSwe or when wildtype neurons were treated with exogenous Aβ 42 peptides [98, 99]. This is mediated not only by endocytosis of synaptic AMPARs via mechanisms shared by LTD [99] but also through a reduction in basal levels of GluA1-S845 phosphorylation by activating the calcium-dependent phosphatase, calcineurin, as well as interrupting extrasynaptic delivery of AMPARs [100]. Contradictory results on the effects of Aβ on AMPAR and NMDAR regulation may be due to several variables. First, there is evidence that Aβ 40 and Aβ 42 peptides may have distinct functions in AD pathology. For example, a majority of FAD-linked PS1 mutations cause a reduction in Aβ40 peptides and therefore an increase in the Aβ 42/Aβ 40 ratio [101, 102]. Second, there are differences in experimental preparations. Both Wu et al. [95] and Hsia et al. [96] were working with acute adult hippocampal slices, while Snyder et al. [97], Almeida et al. [98], Hsieh et al. [99], and Miñano-Molina et al. [100] were using either cultured neurons from embryonic mice or organotypic hippocampal slice cultures prepared from early postnatal mice. Third, the presence or absence of APP itself may have also affected the results. Indeed there is evidence that uncleaved full-length APP may promote synapse formation and enhance excitatory synaptic function (see [103] for a recent review).
In any case, Aβ-mediated alterations in NMDAR function suggest that Aβ will affect downstream Ca2+-dependent signaling pathways. Calcineurin, a Ca2+-activated protein phosphatase, may be one of the downstream signaling molecules affected by Aβ, since it is required for the inhibition of perforant pathway LTP [88], endocytosis of surface AMPARs [99], as well as dephosphorylation of GluA1-S845 [100]. In addition to activating calcineurin, Aβ prevents the activation of CaMKII, a Ca2+-dependent protein kinase necessary for LTP, and decreases the synaptic clustering of CaMKII, which correlates with a reduction in the phosphorylation of GluA1-S831, surface expression of GluA1, and AMPAR-mediated EPSCs [89, 104]. Together, these data are consistent with the idea that Aβ oligomers impair LTP and facilitate LTD [56, 105, 106].
Aβ has also been found to modify regulation of gene expression. Aβ peptides have been found to alter CREB signaling, causing synaptic dysfunction and memory deficits (reviewed in [107]). In addition, treating cultured hippocampal neurons with soluble Aβ oligomers induces rapid expression of the IEG Arc/Arg 3.1 [94], which is implicated in synaptic plasticity [83, 108, 109]. Because overexpression of Arc/Arg 3.1 causes learning dysfunction [110], possibly via reducing surface expression of GluA1-containing AMPARs [109], this would suggest that Aβ oligomer-induced Arc/Arg 3.1 expression may in fact interfere with normal synaptic plasticity. However, this study is seemingly at odds with the results of Echeverria and colleagues, which reported a strong inhibition of BDNF-induced increase in Arc expression in cultured cortical neurons treated with Aβ oligomers [111]. Similarly, there is also a report that synaptic plasticity-related genes, including Arc/Arg 3.1, are reduced in transgenic mice expressing FAD-linked mutations in APP and PS1 [112]. The apparent differences in Arc expression caused by Aβ could be due to different experimental systems or to the differential effects of different concentrations of Aβ oligomers.
4.2. Presynaptic Alterations by Aβ
Besides influencing postsynaptic function, Aβ is also implicated in presynaptic modifications. A recent study reported that 8 nM Aβ 42 globulomer (a highly stable globular oligomeric Aβ) could directly inhibit presynaptic P/Q type Ca2+ channels and decrease vesicle release [113]. Moreover, application of synthetic Aβ to cultured hippocampal neurons causes a downregulation of dynamin, a protein critical for synaptic vesicle endocytosis, and interrupts synaptic vesicle recycling [114, 115]. This result is consistent with the observed reduction in dynamin levels in human AD brains [116]. These findings may explain the observation that Aβ 42 globulomer causes a decrease in basal synaptic transmission at the Schaffer collateral to CA1 synapses in hippocampal slice culture [117]. Recently, Kelly et al. reported that the reduction in dynamin is dependent on Ca2+ influx through activated NMDARs as well as activation of a calcium-activated intracellular cysteine protease calpain [114, 118]. These results not only suggest that there may be retrograde signaling from postsynaptic to presynaptic terminals but also establish an interesting relationship between Aβ, NMDARs, and calpain. It has been found that Aβ 42 peptides can activate calpain-mediated cleavage of p35 to p25 [119], which then upregulates mRNA and protein expression of β-secretase (BACE1) [120, 121], a critical enzyme for Aβ formation (discussed in the following sections). This indicates that there is a positive feedback between Aβ production and calpain activation. Calpain inhibitors can fully prevent deficits in basal synaptic transmission caused by Aβ globulomer application in hippocampal slice culture to a comparable level as using an NMDAR antagonist memantine [117]. This suggests that Aβ acts through NMDARs and calpain: a potential signaling cascade being NMDAR-medicated Ca2+ influx activating intracellular calpain, which then promotes p25/cdk5-dependent transcription of downstream genes, including BACE1 [120].
4.3. Other Targets of Aβ That Affect Synaptic Plasticity
Recent studies suggest that the α7-nicotinic acetylcholine receptor (α7-nAChR), a Ca2+-permeable homopentameric ion channel highly expressed in the hippocampus and cerebral cortex [122], is another potential target of Aβ. High affinity binding between Aβ 42 peptides and α7-nAChRs [123, 124] either inhibits [125–128] or activates α7-nAChR signaling [129]. It is possible that Aβ 42 peptides may facilitate α7-nAChRs at low concentrations but may inhibit α7-nAChRs when the burden of Aβ increases [129, 130]. This concentration-dependent role of Aβ peptides is suggested from studies showing that at normal concentrations (picomolar range), Aβ peptides positively regulate presynaptic release at hippocampal synapses and facilitate CA1 LTP and learning by activating α7-nAChRs, whereas when the level of Aβ is low or high (nanomolar range), Aβ peptides cause either deficits in presynaptic function or abolish hippocampal LTP and learning via its interaction with α7-nAChRs [131–133].
Moreover, the concentration-dependent effect of Aβ is also reflected by its ability to regulate reactive oxygen species (ROS). ROS have been found to have physiological roles in maintaining normal synaptic plasticity. However, high levels of ROS have been found in both AD animal models and human patients, leading to oxidative damage related to AD pathology (reviewed in [134]). Recently, Ma and colleagues found that exogenous treatment of Aβ 42 (500 nM) increased mitochondria superoxide, which they reported is a cause of synaptic dysfunction induced by Aβ. In particular, decreasing mitochondrial superoxide levels reversed Aβ-induced CA1 LTP impairments [135]. Given the normal physiological role of Aβ and ROS at intermediate levels, this finding suggests that ROS imbalance, caused by Aβ toxicity, may lead to synaptic dysfunction in AD. It also implies that Aβ levels exceeding the normal range may initiate the abnormalities in synaptic function (Figure 2).
Figure 2.
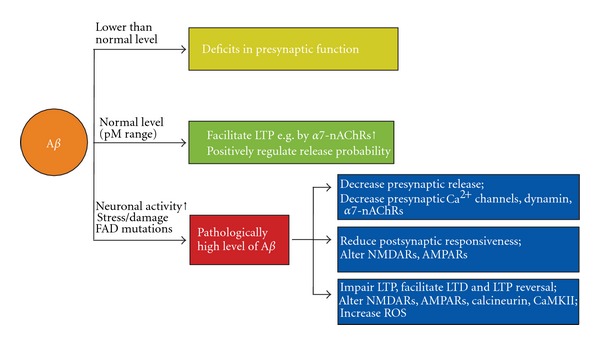
Concentration-dependent effects of Aβ on synaptic function. At normal physiological levels (picomolar range), Aβ peptides have positive effects on synaptic function: they can positively regulate presynaptic release probability and facilitate learning and LTP in CA1 by activating α7-nAChRs. However, when the concentration of Aβ peptides is lower than normal, presynaptic function is impaired. On the other hand, under pathological conditions, such as increased neuronal activity, stress, or the presence of familial Alzheimer's disease (FAD) mutations, the increase in Aβ peptide concentration produces pathological effects, including decreased presynaptic neurotransmitter release, reduced postsynaptic responsiveness, LTP impairment, and LTD facilitation. Therefore, maintaining the concentration of Aβ peptides within a normal physiological range is essential and should be the goal for developing effective treatments for Alzheimer's disease.
In summary, pathologically high levels of Aβ can disturb the ROS balance and interfere with both pre- and postsynaptic function, presumably by affecting NMDARs, presynaptic P/Q Ca2+ channels, and/or α7-nAChRs, thereby interrupting subsequent Ca2+ signaling leading to altered synaptic function.
5. Neuronal Activity Can Regulate APP Processing and Aβ Levels
Data from both transgenic mice and exogenous Aβ application studies suggest that alterations in Aβ levels change neuronal activity and synaptic function. In vivo two-photon Ca2+ imaging of APP23xPS45 mice showed that cortical neurons near amyloid plaques are hyperactive, while the percentage of hypoactive cortical neurons is enhanced at locations further away from a plaque [136]. The disparate change in neuronal activity relative to the location of a neuron to amyloid plaques may reflect differences in local Aβ concentration. It is now evident that neuronal activity itself can also regulate APP-processing leading to alterations in Aβ production. In 1993, a study reported that electrical stimulation not only increases neurotransmitter release in rat hippocampal slices but also enhances the release of APP cleavage products [137]. In agreement with this finding, ten years later, Kamenetz and colleagues [138] found that neuronal activity can bidirectionally control Aβ levels in organotypic hippocampal slice cultures from APPSwe Tg mice. Blocking neuronal activity in this preparation by tetrodotoxin (TTX) treatment reduced Aβ levels, while increasing neuronal activity with picrotoxin (PTX) enhanced Aβ secretion [138]. The experimental paradigm used by Kamenetz et al. to manipulate neuronal activity is reported to produce homeostatic synaptic plasticity termed “synaptic scaling” [28], which globally up- or downregulates all excitatory synapses following prolonged decrease or increase, respectively, in neuronal activity [29]. This suggests that Aβ may play a role in regulating homeostasis of excitatory synapses in normal brains. In addition, the cellular mechanism responsible for regulating APP-processing and Aβproduction in response to neuronal activity is possibly through enhancing the accessibility of APP to γ-secretase cleavage [138] and/or depressing γ-secretase function [139]. It has recently been shown that PS1, the catalytic subunit of the γ-secretase complex, is necessary to scale up excitatory synapses following reduced network activity and that PS1 knockout mice show deficits in synaptic scaling [140]. Moreover, Wu and colleagues have reported that the immediate early gene Arc is required for the activity-dependent increase in Aβ production [141]. They found that Arc directly binds the N terminus of PS1 and plays an important role in trafficking the γ-secretase complex to early endosomes where APP is processed through the amyloidogenic pathway to produce Aβ peptides. In addition, Arc contributes to Aβ levels and plaque load in APPSwe; PS1ΔE9 mice and Arc expression are elevated in medial frontal cortex of AD patients [141]. These results provide a cellular mechanism coupling Aβ generation to neuronal activity and may explain why people who suffer from hypoxia, which usually causes an abnormal enhancement in neuronal activity [142], have a higher risk for developing AD [143].
Consistent with the idea that Aβ induces homeostatic adaptation to increases in activity, in vivo studies have also shown that either electrical stimulation or endogenous whisker activity proportionally regulates interstitial fluid (ISF) Aβ levels in Tg2576 mice, which overexpress human APP carrying the Swedish (K670N/M671L) mutation [144–146]. However, there are also contradictory results. Tampellini et al. have shown that synaptic activity decreases intracellular Aβ in primary neuronal culture, as well as in the barrel cortex of 4-month-old Tg19959 mice, which overexpress human APP carrying the Swedish (K670N/M671L) and Indiana (V717F) mutations [147], likely by enhancing Aβ degradation [148]. Zhang et al. have reported that prolonged olfactory deprivation facilitates amyloid plaque deposition in the olfactory bulb and piriform cortex of 7–24-month-old Tg2576 mice [149]. These contradictions may be due to age, region, and paradigm differences. Another possibility is that normal neuronal activity regulates Aβ levels by balancing Aβ release and degradation and that either hyperactivity or hypoactivity may break this balance leading to Aβ accumulation.
6. Physiological Roles of APP and Aβ
Proteolytic processing of APP not only produces Aβ peptides but also other products. Some functions of these products have been identified (reviewed in [150]). For example, the cytoplasmic tail of APP, APP intracellular domain (AICD), is shown to participate in transcriptional regulation [151]. To evaluate other normal physiological roles of APP, mice lacking APP were generated. APP knockouts show enhanced excitatory synaptic activity and neurite growth [152], which is consistent with the finding that APP-deficient mice are more susceptible to glutamate-induced toxicity [153]. Similar to APP, Aβ peptides also have normal physiological functions. Normal levels (picomolar range) of Aβ peptides regulate synaptic function by positively increasing presynaptic release at hippocampal synapses and facilitating learning and LTP in CA1 [131–133]. Moreover, normal levels of Aβ may be essential for neurons, because preventing Aβ production by adding β- or γ-secretase inhibitors in cultured neurons causes cell death, which can be rescued by applying synthetic Aβ peptides to culture medium [154]. In addition, activity-dependent changes in Aβ may in fact play a role in maintaining homeostasis by acting as a negative feedback regulator of excitatory synaptic transmission [138].
Collectively, these data suggest that proteolytic processing of APP and the presence of a normal physiological dose of Aβ may be required for maintaining proper neuronal activity and brain function. While the therapeutic benefits of targeting APP-processing and Aβ production are still attractive, it should be noted that AD pathology is most likely triggered only when Aβ levels exceed the normal range and that the physiological processing of APP and Aβ production may be important in maintaining normal brain functions. Therefore, partial inhibition, but not complete blockade, of Aβ production might be a useful approach for AD therapeutics. A recent study supports this view. Immunizing APPInd Tg mice against Aβ, which lowered Aβ levels, decreased senile plaque formation and rescued loss of neuronal integrity seen previously in aged mice [155]. However, Aβ-immunotherapy in clinical trials reported severe complications, which must be overcome (for review articles on this topic please see [156–158]).
7. Role of BACE1 in Synaptic Function
As mentioned above, Aβ peptides are generated by sequential cleavage of APP by β- and γ-secretase (Figure 1). In the brain, beta-site APP cleaving enzyme (BACE1), a transmembrane aspartic protease, has been found to be the major neuronal β-secretase [159–162]. Mice lacking the BACE1 gene show no β-secretase activity and essentially no Aβ (Aβ 40 and Aβ 42) production in the brain compared to wildtype littermates. Initial characterization of BACE1 knockouts (BACE1−/−) showed that they are viable and fertile, with no gross differences in behavior or development [159–161, 163]. Furthermore, knocking out the BACE1 gene in mouse models of AD was able to rescue hippocampus-dependent memory deficits [163–165] and ameliorate impaired hippocampal cholinergic regulation of neuronal excitability [163]. These findings were quite encouraging and suggested that BACE1 may be a good therapeutic target for treating AD [4, 166, 167].
However, recent studies have found that BACE1 has normal physiological functions in synaptic transmission and plasticity in both CA1 and CA3 regions of the hippocampus (Table 1). Laird et al. found that BACE1−/− mice display deficits in both synaptic transmission and plasticity at the hippocampal Schaffer collateral to CA1 synapses [164]. While BACE1−/− mice display normal AMPAR- and NMDAR-mediated synaptic transmission, these synapses show a larger paired-pulse facilitation (PPF) ratio compared to wildtype littermates when tested with paired-pulse stimuli at a 50 ms interstimulus interval [164]. Changes in PPF ratio are linked to alterations in presynaptic function [168]. Therefore, the increase in PPF ratio observed in BACE1−/− mice indicates a reduction in presynaptic function, which is consistent with the high expression of BACE1 in presynaptic terminals [164]. In addition to reflecting presynaptic changes, recent data suggest that alterations in PPF ratio can also be caused by postsynaptic modifications, such as by varying the subunit composition of AMPARs [169]. Therefore, it is possible that knockout of BACE1 may also affect postsynaptic AMPAR function. Besides alterations in PPF ratio, BACE1−/− mice also showed a larger dedepression (reversal of LTD) induced by high frequency theta burst stimulation (TBS) at the Schaffer collateral inputs to CA1 [164]. In contrast, the same TBS protocol-induced LTP remained unchanged [164]. As LTP and dedepression have separate underlying mechanisms [56], these data suggest BACE1 may play a regulatory role in the dedepression pathway, while not affecting the mechanisms that lead to LTP. Laird and colleagues also found evidence that the enhanced dedepression is due to larger summation of responses during TBS, specifically following LTD induction. Enhanced summation of synaptic responses during the induction of de-depression despite normal basal synaptic transmission suggests that BACE1 may play a specific role in activity-dependent high-frequency information transfer across synapses. Also, the abnormal increase in the magnitude of de-depression reflects that LTD expression may be easily disrupted when knocking out BACE1, which could interfere with memory formation and storage. Consistent with this interpretation, detailed behavioral studies of BACE1−/− mice reported problems in both cognitive and emotional memory tests [164, 170, 171].
Table 1.
Summary of known synaptic effects of altering BACE1.
| Age | Aβ | Basal synaptic transmission | Presynaptic function | LTP | LTD | Reference | |
|---|---|---|---|---|---|---|---|
| BACE1 KO | 3–6 mo | No Aβ 40 or Aβ 42 | Normal (CA1 and CA3) | Increased PPF ratio (CA1 and CA3) | Normal LTP (4x TBS) but larger dedepression in CA1; no mossy fiber LTP (3x 100 Hz) and no dedepression in CA3 | Normal LTD (paired-pulse 1 Hz) in CA1, but slightly larger LTD (paired-pulse 1 Hz) in CA3 | [164, 174] |
|
| |||||||
| BACE1 KO + activation of α7-nAChRs | 3–6 mo | No Aβ 40 or Aβ 42 | Normal (CA3) | Restored PPF ratio (CA3) | Rescued mossy fiber LTP (CA3) (3x 100 Hz) | [175] | |
|
| |||||||
| BACE1+/−; 5XFAD APP/PS1 (Tg6799) | 6 mo | 66% decrease in Aβ 40 and 57% in Aβ 42 in brain; reduce amyloid plaque burden in hippocampus by 78% and anterior cingulate cortex by 44% | Remained significantly reduced (CA1) | Restored LTP to WT control levels (CA1) (3x TBS) | [176] | ||
|
| |||||||
| Adenoviral-Fbx2∗ in Tg2576 | 12–14 mo | 30% decrease in Aβ 42 | No change | Improved the impaired LTP (CA1) (3x TBS) 4 weeks after adenoviral injection | [177] | ||
∗Transfected into hippocampus. Fbx2 facilitates BACE1 degradation.
Although the majority of studies characterizing synaptic function of BACE1−/− mice have been performed in the CA1 region of the hippocampus [163, 164, 171], the expression of BACE1 is most prominent in the mossy fiber terminals that synapse onto CA3 pyramidal neurons [164, 172]. Recently, we reported that BACE1−/− mice display severe deficits in presynaptic function at these synapses, including a reduction in presynaptic release and an absence of mossy fiber LTP, which is normally expressed by a long-term increase in presynaptic release [173]. Moreover, BACE1−/− mice exhibited a slightly larger mossy fiber LTD, which could not be reversed [174]. These results suggest that BACE1 function is crucial for normal synaptic transmission and activity-dependent presynaptic potentiation at these synapses. We further found evidence that the presynaptic dysfunction in BACE1−/− mice is likely at the level of presynaptic Ca2+ signaling, because the mossy fiber LTP deficit in BACE1−/− mice could be recovered by increasing the extracellular Ca2+ concentration. This suggests that the signaling downstream of Ca2+ is more or less intact in BACE1−/− mice, which was confirmed by the fact that the magnitude of presynaptic potentiation resulting from direct activation of the cAMP signaling pathway is normal in BACE1−/− mice [174]. Therefore, it is possible that manipulations that enhance presynaptic Ca2+ may overcome the synaptic deficits caused by inhibiting BACE1 activity. In line with this, we recently showed that activation of Ca2+-permeable α7-nAChRs, by nicotine or α7-nAChRs agonist, can restore PPF ratio and mossy fiber LTP in BACE1−/− mice [175]. The cellular mechanism of nicotine-induced rescue is dependent on the recruitment of Ca2+-induced Ca2+-release (CICR) from intracellular Ca2+ stores through ryanodine receptors [175]. These results suggest that nicotine and α7-nAChR agonists may be a potential pharmacological means to circumvent the synaptic dysfunctions caused by BACE1 inhibition.
Since synaptic deficits are seen in both the CA1 and CA3 regions of BACE1−/− mice, it indicates that BACE1 may play a general role in regulating presynaptic function. Reduced Aβ levels have been shown to produce deficits in presynaptic function [131], which may explain the synaptic phenotype seen in BACE1−/− mice. However, whether presynaptic deficits in BACE1−/− mice are solely due to a lack of APP-processing is unclear. An alternative possibility is that the synaptic dysfunction seen in BACE1−/− mice may arise from abnormal processing of substrates other than APP (Figure 3).
Figure 3.
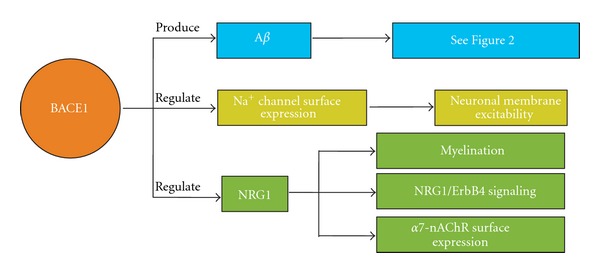
The roles of BACE1 in synaptic function. Besides cleaving APP to produce Aβ peptides, BACE1 has been found to have other substrates. It can process the β2 subunit of the voltage-gated sodium (Na+) channels, which can regulate Na+ channel surface expression and in turn modulate neuronal excitability. In addition, BACE1 can cleave NRG1, which plays a crucial role in myelination and NRG1/ErbB4 signaling. Recently, it has been showed that NRG1 can regulate cell surface expression of α7-nAChRs, which can also affect synaptic transmission.
It has been shown that the auxiliary β2 subunit of the voltage-gated sodium channel (Nav1) is a substrate of BACE1 [186, 187]. The β2 subunit of the Nav1 channel is important for plasma membrane expression of functional Na+ channels, which are critical for generating action potentials. Among the ten different types of Nav1 channels, Nav1.1, Nav1.2, Nav1.3, and Nav1.6 are expressed mainly in the central nervous system (CNS) [188]. BACE1 regulates the surface expression of these types of Nav1 channels by cleaving the β2 subunit. In transgenic mice overexpressing BACE1, there is an increase in the Nav1.1 α-subunit mRNA and protein levels, but a decrease in the surface expression of functional Nav1.1 channels due to cleavage of the β2 subunits [187, 189]. The interpretation is that the full-length β2 subunit promotes surface expression of Nav1.1 channels, but the β2-intracellular domain (ICD), which is produced by a sequential cleavage by BACE1 and γ-secretase, increases transcription of the Nav1.1 α-subunit gene. Consistent with this, BACE1−/− mice display a decrease in Nav1.1 α-subunit mRNA and protein [190]. However, there is a compensatory increase in the surface expression of Nav1.2 in BACE1−/− mice, which correlates with the hyperexcitability and seizure phenotypes seen in these mice [191]. These results suggest that the ability of BACE1 to regulate the Nav1 family of Na+ channels is rather complex but suggest a role for BACE1 in regulating neuronal excitability.
Another candidate substrate for BACE1 is neuregulin-1 (NRG1), which is an axonal signaling molecule critical for regulating myelination [192]. Willem and colleagues found that BACE1−/− mice show hypomyelination in the peripheral nerves [193], while another study detected loss of myelination in the central nerves [194]. Both of these studies showed an accumulation of unprocessed NRG1 and a reduction in its cleavage products, suggesting that NRG1 is a potential substrate for BACE1 cleavage and that this process is important for myelination of axons [193, 194]. Recently, it has been shown that the absence of NRG1 processing in BACE1−/− mice decreased postsynaptic function of ErbB4, a receptor for NRG1 [195]. NRG1/ErbB4 signaling has been suggested to regulate synaptic function and plasticity, mainly via regulation of postsynaptic glutamate receptors [196–198]. Additionally, abnormal processing of NRG1 may also affect presynaptic release by regulating the expression of α7-nAChRs [199, 200] which allows Ca2+ influx [122]. Indeed, presynaptic nAChRs can increase glutamate release [201–203], likely via the α7 containing nAChRs [204]. These results suggest that a lack of NRG1 cleavage resulting from BACE1 inhibition can alter synaptic function both pre- and postsynaptically.
Accumulating data on the biological roles of BACE1, particularly evidence that completes inhibition of BACE1 activity which is deleterious for normal neuronal function, suggests caution for using BACE1 inhibitors as a treatment for AD. In order to improve the development of effective therapeutics that target this enzyme, we need to identify ways to avoid the synaptic dysfunction associated with blocking BACE1, which may include partial inhibition strategies.
7.1. Partial Inhibition or Conditional Knockdown of BACE1
It has been shown that Aβ burden is dose dependent on BACE1 activity; therefore, partial inhibition or conditional knockdown of BACE1 may be beneficial for AD treatment. To test this, Kimura and colleagues crossed BACE1 heterozygous mice with a line of transgenic mice carrying a combination of 5 FAD-linked mutations in human APP and PS1 (5XFAD); they found that partial reduction of BACE1 improved remote and recent memory and restored CA1 LTP [176]. Researchers have also successfully suppressed BACE1 activity by using RNA interference (RNAi) in vitro [205, 206] and in vivo [164, 207]. Lentiviral BACE1 siRNA delivered into the hippocampus has been found to effectively reduce Aβ production, neurodegeneration, and behavioral deficits in APP transgenic mice [164, 207]. Characterizing synaptic function in the BACE1 siRNA knockdown models may provide information about acute effects of blocking BACE1 function. In addition, siRNA knockdown of BACE1 in APP transgenic lines will better approximate clinical situations, hence allowing us to better estimate the feasibility of developing an effective treatment for AD by BACE1 inhibition.
7.2. BACE1 Inhibitors
Since the identification of BACE1, the development of BACE1 inhibitors has been initiated. However, the progress was slow, probably due to the difficulty of identifying small molecules that can pass through the blood brain barrier and also have high stability and good pharmaceutical properties [208, 209]. So far, several BACE1 inhibitors have been discovered; among them only CTS-21166 has passed Phase I clinical trials (see review [208, 210]). Many BACE1 inhibitors have been shown to decrease soluble Aβ production, amyloid plaque deposition, as well as improve cognitive function in AD animal models [211–216]. Surprisingly, none of them have been tested to determine their ability to improve synaptic dysfunction, the cellular mechanism that correlates with cognitive decline. A critical question is whether these inhibitors can recover synaptic deficits seen in AD models or whether they may produce additional defects as seen in BACE1−/− mice.
7.3. Transcriptional and miRNA Regulation of BACE1
There are several reports of transcriptional regulation of BACE1. Nie et al. have shown that activation of α4β2 nAChR can decrease BACE1 transcription through the ERK1-NFκB pathway in vitro [217]; Wen and colleagues reported that overexpression of p25, an activator of cdk5, can increase BACE1 mRNA and protein levels likely through interactions of signal transducer and activator of transcription (STAT3) with the BACE1 promoter [120]. In addition, in the brains of sporadic AD patients, an increase in BACE1 levels is correlated with a decrease in a subset of microRNAs (miRNA), especially the miR-29a/b-1 miRNA cluster [218]. miRNAs regulate mRNA translation. Therefore, it is possible that an increase in specific miRNA levels can downregulate BACE1 protein expression and decrease Aβ burden. These findings provide various ways to regulate BACE1 expression.
7.4. Endogenous BACE1 Activity Modulators
Recently, studies have shown that during sporadic AD or in AD animal models, the activities of certain endogenous molecules are modified, causing an increase in BACE1 activity. For example, sphingosine-1-phosphate (S1P) phosphorylation of the translation initiation factor eIF2α and calpain activity are increased in AD, which can lead to an increase in BACE1 activity [117, 121, 219–221]. On the other hand, decreased activity in conjugated linoleic acid (CLA), acetylcholinesterase inhibitor galantamine (Gal), copper chaperone for superoxide dismutase (CCS), PPARγ coactivator-1α (PGC-1α), the trafficking molecule GGA3, as well as Fbx2-E3 ligase during AD can lead to increased BACE1 protein levels [177, 222–228]. So far, only the effect of Fbx2 on synaptic plasticity has been tested. Adenoviral-Fbx2 transfection significantly improves CA1 LTP in Tg2576 mice without affecting basal synaptic transmission [177]. While these molecules may be potential targets for controlling BACE1 activity, further studies need to verify whether synaptic function can be improved by manipulating the activity of these BACE1 modulators.
8. Presenilin: Its Physiological Roles and Relationship with Alzheimer's Disease
Presenilin 1 (PS1) is the catalytic component of the γ-secretase complex. Following BACE1 cleavage, γ-secretase cleaves the transmembrane domain of APP, releasing Aβ peptides (Figure 1). The active γ-secretase complex is composed of four different proteins, all of which are required for the protease to function (for a good review on the composition of γ-secretase, see [229]); however, PS1 receives the most attention stemming from its identification as the major locus for early onset FAD [230]. Since the accumulation and deposition of extracellular Aβ have been emphasized in the progression of AD [92], the identification of several FAD-linked mutations in PS1 led to many studies investigating how dysfunction of this protein contributes to AD. FAD-linked mutations in PS1 facilitate the production of the more pathogenic Aβ 42 peptide [85, 101], which is the major constituent of senile plaques found in the brains of AD patients. Here, we will briefly summarize the functions of presenilins and focus on how they play a role in normal synaptic regulation and also during AD. Key points are summarized in Table 2.
Table 2.
Summary of alterations in synaptic function by altering presenilin or γ-secretase activity.
| Age | Aβ | Basal synaptic transmission | Presynaptic function | LTP | LTD | Other | Reference | |
|---|---|---|---|---|---|---|---|---|
| PS1 cKO | 3–6 mo | Cortical Aβ 40 and Aβ 42 are decreased | Normal (CA1) | Normal PPF ratio (CA1) | Normal (CA1) (5x TBS) normal (CA1) (3x 100 Hz) | Normal (CA1) | [178] | |
|
| ||||||||
| 2 mo | Normal (CA1) | Decreased PPF ratio (CA1) | Decreased (CA1) (5x TBS) | Normal (CA1) | Reduced NMDAR function (CA1); decreased cortical synaptic levels of NR1, NR2A, αCaMKII, and CRE-dependent genes | [179] | ||
| 6 mo | Increased (CA1) | Decreased PPF ratio (CA1) | Decreased (CA1) (5x TBS) | Normal (CA1) | ||||
| PS cDKO | 2 mo | Increased expression of the NR2A subunit of NMDARs, specifically at postsynaptic density and presynaptic terminals of axo/dendritic synapses, trapped at synapses (CA1) | [180] | |||||
| 5 wk | Decreased Ca2+-dependent frequency facilitation and release probability (CA1) | Normal (CA1) (5x TBS) | Normal NMDAR-mediated synaptic response (CA1) | [181] | ||||
| 6 wk | Decreased (CA1) 5x TBS | Reduced NMDAR-mediated synaptic response (CA1) | ||||||
|
| ||||||||
| CA1-PS cDKO | 2 mo | Normal (CA1) | Normal (CA1) (5x TBS) | Normal NMDAR/AMPAR ratio | [182] | |||
|
| ||||||||
| CA3-PS cDKO | 2 mo | Decreased Ca2+-dependent frequency facilitation and release probability (CA1) | Decreased (CA1) (5x TBS) | Normal NMDAR/AMPAR ratio | [182] | |||
|
| ||||||||
| AppTg (Swe/Ind) | 3 mo | Normal (CA1) | Normal (CA1) | Increased (CA1) (5x TBS) | [183] | |||
| 6 mo | Age dependent increase in Aβ levels and plaque deposition | Decreased (CA1) | Normal (CA1) | Decreased (CA1) (5x TBS) | ||||
|
| ||||||||
| AppTg; PS1−/− | 3 mo | Normal (CA1) | Normal (CA1) | Increased (CA1) | [183] | |||
| 6 mo | Decreased cortical Aβ peptides and plaque formation | Decreased (CA1) | Normal (CA1) | Decreased (CA1) (5x TBS) | ||||
|
| ||||||||
| 3-4 mo | Normal (CA1) | Normal (CA1) | ||||||
| Tg2576 | 6-7 mo | 50% increase in soluble Aβ | Decreased (CA1)-stronger stimulus required to elicit similar sized postsynaptic responses | Decreased (CA1) | [184] | |||
| 14-15 mo | 1000% increase in soluble Aβ | Decreased (CA1) but similar to 6-7 months | Normal (CA1) | |||||
|
| ||||||||
| Tg2576 w/MRK-560 (γ-secretase inhibitor) | 6-7 mo | 1, 3, or 7 days of treatment reduced soluble Aβ levels | Partial recovery but not significant | Recovered (CA1) | [184] | |||
| 15 mo | 3 months of treatment reduced soluble Aβ levels | No improvement | ||||||
|
| ||||||||
| Tg2576 w/CHF5074 (γ-secretase modulator) | 6 mo | 4-week, subchronic, oral treatment reduced Aβ levels | Recovered (CA1) | [185] | ||||
To investigate the normal physiological functions of PS1, many genetic knockout experiments have been conducted. Knockout of PS1 causes abnormal development and perinatal death [231–235]. FAD-linked mutations have also been discovered in Presenilin 2 (PS2), which is highly similar to PS1 in both sequence and structure [236]; however, PS2 knockout mice are viable and fertile with only mild age-dependent pulmonary fibrosis and hemorrhage [237]. This suggests PS1 is sufficient to maintain the majority of regular physiological activities and that these two homologs share little overlapping function. Another study using PS1+/−; PS2−/− mice found that they could live normally until 6 months of age, after which most developed an autoimmune disease and benign skin hyperplasia [238]. The lethal effect of knocking out PS1 is not surprising considering that γ-secretase is involved in the processing of many other substrates beside APP [239–241], one of the most important being the Notch receptor, a protein that is critical in cell differentiation during embryonic development [231, 239, 240, 242].
γ-secretase still remains to be a promising candidate for AD drug targets because it is thought that the function of PS1 might not be as critical in the adult brain, unlike during embryonic development, and/or partial inhibition of the enzymatic activity may still be feasible. Encouragingly, mice with conditional knockout (cKO) of PS1, in which PS1 expression was eliminated in most neurons of the cerebral cortex in the postnatal brain, were viable and had nearly normal phenotypes, including normal basal synaptic transmission and plasticity, with only mild deficits in long-term spatial memory [178, 179]. Aβ 40 and Aβ 42 levels were also reduced in the cortex of PS1 cKO mice, providing evidence in support of targeting PS1 as a potential antiamyloid therapy in AD. Another promising finding was that regulation of Notch activity in the adult brain was unaffected and independent of PS1, contrasting the dependency of Notch signaling during embryonic brain development. This suggests PS2 may be able to compensate for the loss of PS1 in the adult brain and leads one to question whether knockout of both PS1 and PS2 will lead to more extreme deficits. To test this hypothesis, Saura and colleagues [179] generated forebrain-specific PS1/PS2 conditional double knockout (PS cDKO) mice. These mice exhibit cognitive impairments as well as deficits in hippocampal synaptic plasticity, which appear earlier than in the PS1 cKO mice. PS cDKO mice also developed age-dependent and progressive neurodegeneration, including loss of dendritic spines and presynaptic terminals [179]. Together, this suggests that in the adult brain the role of PS1 in regulating Notch signaling may not be as important but that presenilins are required for normal hippocampal synaptic plasticity, memory formation, and age-dependent neuronal survival.
It is encouraging that conditional inactivation of PS1 is able to decrease Aβ levels in the adult brain without effecting Notch signaling [178]. In order to examine the possibility of using inactivation of PS1 as a therapy for AD, PS1 cKO mice have been crossed with transgenic mice expressing different FAD-linked mutations in APP. The first study developed postnatal neuron-specific inactivation of PS1 (PS1−/−) in transgenic mice overexpressing human APP with the London mutation (V717I), APPxPS1(−/−) [243]. This group had previously shown that APP(V717I) mice had increased levels of Aβ 42 peptides as early as 2 months, leading to plaque development at 13 months old [244], as well as cognitive impairment and reduced hippocampal LTP. APPxPS1(−/−) mice showed reduced Aβ and amyloid plaque formation, even at 18 months. While hippocampal CA1 LTP was rescued in APPxPS1(−/−) mice, they still showed impaired cognition. A second study used the forebrain-specific PS1 cKO mice, mentioned previously [178, 179], to inactivate PS1 in an APP transgenic that overexpressed human APP containing the Swedish (K670N/M671L) and Indiana (V717F) mutations to generate PS1 cKO;APP Tg [183]. Similar to the previous study, these mice also had reduced amyloid phenotypes compared to APP Tg mice, but there was still no long-term improvement in cognitive function. Conditional inactivation of PS1 was only able to rescue learning and memory deficits seen in young but not old mice [183]. Together, these data indicate a causative role for Aβ peptides in LTP deficits and demonstrate that inactivation of PS1 in Tg mice can decrease the amyloid pathology of AD and restore LTP impairments in young mice. One question elicited from the above studies is that, if conditional knockout of PS1 is able to reduce amyloid pathologies and rescue certain LTP impairments, why is it not able to rescue cognitive deficits seen in these AD mice and why is it not able to sustain LTP improvements in older mice? One possible explanation is the age-dependent accumulation of the APP C-terminal fragments (CTFs) caused by a lack of γ-secretase activity after conditional inactivation of PS1, leading to the buildup of γ-secretase substrates [183]. Another explanation is the non-γ-secretase functions of PS1 may be involved in aspects of memory formation, storage, or consolidation, by regulating intracellular calcium dynamics.
In addition to its proteolytic activity, PS1 is implicated in regulating neurotransmitter release [182, 245] and intracellular calcium dynamics [246–248] (Figure 4). It has been proposed that the full length PS1 can act as a passive endoplasmic reticulum (ER) Ca2+ leak channel [249] and that some FAD-linked PS1 mutations lack this property. However, it remains controversial [102, 250, 251] whether Ca2+ dysregulation that occurs during AD can be directly linked to alterations in ER Ca2+ leak channels formed by PS1. While the exact mechanism may be unknown, there is ample evidence that FAD-linked mutations in PS1 contribute to augmented cytosolic Ca2+ concentrations resulting from changes in intracellular ER Ca2+ dynamics [181, 246–248, 252–254]. FAD-linked mutations in PS1 appear to influence Ca2+ homeostasis by causing enhanced Ca2+ responses of ryanodine receptors (RyRs) [253–258] and inositol-1,4,5-triphosphate receptors (IP3Rs) [252, 259, 260] found in the ER [261], enhanced filling of ER Ca2+ stores [262, 263], and attenuation of capacitive Ca2+ entry (CCE) stores [264–267]. Presenilins have also been found to play a normal physiological role in regulating sarco-ER Ca2+-ATPase (SERCA) pumps that help maintain low cytosolic Ca2+ concentrations by pumping Ca2+ into ER stores [262]. SERCA activity also influences Aβ production, such that increased SERCA activity increases Aβ production [262].
Figure 4.
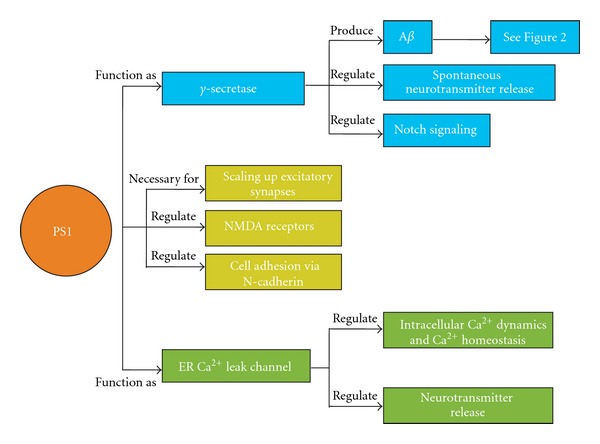
The roles of presenilins in synaptic function. Two main functions of presenilin 1 (PS1) focused on in this paper are its ability to function as part of the γ-secretase cleavage complex and also to function as an ER Ca2+ leak channel. The γ-secretase complex is responsible for the final cleavage of APP in the production of Aβ peptides. The γ-secretase complex has also been shown to regulate spontaneous neurotransmitter release and is crucial for the regulation of the notch signaling pathway especially during early development. The PS1 holoprotein has been proposed to function as an ER Ca2+ leak channel. It is responsible for regulating intracellular calcium dynamics and calcium homeostasis required for proper signaling and neurotransmitter release. In addition to these two main functions, knockout studies have shown that the PS1 protein is important for synaptic scaling, proper NMDAR-mediated responses, as well as cell adhesion mediated by N-cadherin. Through these studies it is clear that PS1 plays an important role in synaptic transmission and plasticity.
Synaptic transmission and plasticity are important cellular mechanisms underlying cognitive functions, and there is evidence that presenilins play a role in these mechanisms. Mice with PS1 cKO in the cortex showed normal basal synaptic transmission, LTP, and LTD in the hippocampal Schaffer collateral pathway [178], suggesting that in the adult brain, activity of PS2 is sufficient to maintain normal synaptic properties when PS1 is absent. In contrast to PS1 cKO mice, PS1/PS2 conditional double knockout (PS cDKO) shows reduced LTP and a decreased PPF ratio at these synapses as early as 2 months of age. By 6 months, PS cDKO mice showed even greater synaptic deficits, including loss of presynaptic inputs and enhanced basal synaptic transmission, in addition to reduced LTP and PPF ratio [179]. These synaptic impairments may explain the age-dependent deterioration in the cognition of the PS cDKO mice [179]. Collectively, these studies suggest that presenilins are essential for synaptic plasticity as well as learning and memory in the adult brain.
What is the cellular mechanism that mediates the effects of PS1 on synaptic plasticity? Saura et al. [179] found a reduction in the postsynaptic NMDAR-mediated response in PS cDKO mice, which correlated with a decrease in the cortical levels of synaptic NMDAR expression. Saura et al. also found that synaptic localization and delivery of NMDARs may depend on certain interactions with presenilins. Therefore, the downregulation of postsynaptic NMDARs is a reasonable explanation for why LTP and memory are impaired in PS cDKO mice. Loss of presenilin function also decreased the expression of both dendritic and synaptic αCaMKII levels as well as multiple CRE-dependent genes [179], which are all involved in the downstream signaling of NMDAR activation associated with LTP and memory formation (for a review on LTP and memory and the involved molecules, see [16]). This indicates that presenilins not only exert regulatory effects on NMDARs but also the signaling cascades that lead to LTP and memory formation. Surprisingly, later studies that looked specifically at CA1 neurons in the hippocampus revealed that, at 2 months, PS cDKO mice show an unexpected increase in the number of pre- and postsynaptic sites labeled for the NR2A subunits of NMDARs [180]. This increase is not accompanied by synapse loss or alterations in spine size, in agreement with previously documented morphology of PS cDKO mice at this age [179]. The authors [180] suggested that NMDARs become trapped at the synaptic membrane leading to excitotoxicity and eventual neurodegeneration that is present in PS cDKO mice at 6 months [179]. In addition, they suggested that LTP impairments are not due to a reduction in NMDAR number but may be more tightly linked to the reduced levels of αCaMKII present in the dendritic spines [180].
As previously mentioned, presynaptic function was also altered in PS cDKO mice: a reduced PPF ratio, which was attributed to abnormal presynaptic Ca2+ signaling, and a reduction in presynaptic release probability were observed [181, 182]. In addition, there was a loss of presynaptic inputs in older PS cDKO mice suggesting that certain signals necessary for maintaining axon terminals may be missing. PS1 has been found to localize at the synapse and regulate adhesive contact of pre- and postsynaptic compartments, mediated by N-cadherin [239], the major molecule that mediates Ca2+-dependent cell-cell interaction [268]. The diminution of N-cadherin-mediated cell-cell adhesion when presenilins are inhibited might cause the presynaptic defects in PS cDKO mice. One study sought to address the temporal progression of pre- and postsynaptic impairments in the Schaffer collateral pathway of PS cDKO mice [181]. They found that the decrease in presynaptic calcium-dependent facilitation and neurotransmitter release preceded postsynaptic impairments in NMDAR-mediated responses and LTP. Previous experiments in which presenilins were conditionally knocked out in either presynaptic, CA3 or postsynaptic, CA1 neurons [182] demonstrated that loss of presynaptic presenilin is sufficient to cause impaired glutamate neurotransmitter release and LTP, due to altered intracellular calcium signaling. However, loss of pre- or postsynaptic presenilin alone was not sufficient to cause impairments in NMDAR-mediated responses [182]. The authors propose a “trans-synaptic mechanism” to explain the alterations in postsynaptic NMDAR function [181]. In any case, presenilins are likely essential for regulating the intracellular calcium signals required for proper neurotransmitterrelease to insure normal short- and long-term plasticity. Indeed, several recent studies have found that PS1 function is important in regulating homeostatic plasticity [140] and neuronal ER Ca2+ homeostasis [246], as well as a novel function of the γ-secretase complex in regulating spontaneous neurotransmitters release [245]. Therefore, presynaptic dysfunction and altered calcium dynamics may be an early event leading to neuronal degeneration and pathogenesis in AD.
8.1. Gamma-Secretase Inhibitors and Modulators
The γ-secretase complex is critical in the formation of Aβ peptides; hence it is one of the key therapeutic targets for stopping the progression of AD. Although many classes of compounds exist that target the γ-secretase complex, not many have investigated their effects on synaptic transmission and plasticity. Numerous studies have documented the ability of different classes of γ-secretase inhibitors (GSI) and modulators (GSM) to reduce Aβ levels in the brain [269–278], as well as their effects on cognitive function in hippocampal-dependent memory task such as the Morris water maze or contextual fear conditioning [279–282]. There are two studies [184, 185] that looked at the effects of drug treatment on synaptic plasticity in a mouse model of AD. Both studies used Tg2576 mice to investigate the ability of the GSI, MRK-560 [184], or the GSM, CHF5074 [185], to restore hippocampal memory and synaptic plasticity. Each study used different initial starting times and durations of treatment. To understand the interaction between the age-dependent increase in Aβ and its effect on basal synaptic transmission and plasticity in the CA1 region of the hippocampus, Townsend et al. [184] compared synaptic activity across three different ages, young (3-4 months), middle (6-7 months), and old (14-15 months) mice. Basal synaptic transmission was assessed by measuring the input/output activity in CA1. Even though Aβ levels continue to increase with age, the greatest synaptic deficits in Tg2576 mice were seen at 6-7 months, and in particular LTP was impaired at this middle age but was normal in both young and old mice [184]. This suggests that soluble Aβ is inversely correlated with LTP, until plaque deposition occurs, when soluble Aβ can no longer predict LTP impairments [184]. Since 6-7-month-old mice showed the greatest deficits, they were given oral doses for 1, 3, or 7 days with the GSI, MRK-560. After 1 day, Aβ levels were significantly reduced and LTP began to improve. LTP improvements reached significance after 3 days of dosing. After 7 days of treatment, basal synaptic transmission began to recover but did not reach significance. This supports the theory that lowering Aβ levels can recover synaptic plasticity in 6-7-month-old Tg2576 mice, before plaque deposition. Balducci et al. [185] also focused on how the GSM, CHF5074 may be able to rescue synaptic deficits seen in plaque-free Tg2576 mice. After acute subcutaneous treatment with CHF5074, 5-month-old Tg2576 mice showed significantly reduced contextual memory impairments [185]. At 6 months old, after receiving a 4-week subchronic oral treatment, which reduced intraneuronal Aβ level, the impairments in recognition memory and hippocampal LTP were reversed. To determine if aged mice would also show improvements after treatment, daily doses of MRK-560 were given to Tg2576 mice from 12–15 months of age [184]. Since LTP was similar to wildtypes at this age, the focus was on basal synaptic transmission. Similar to middle-aged animals, treatment with MRK-560 significantly reduced Aβ levels; however, there was no improvement in basal synaptic transmission. The lack of functional recovery in older age group was also seen in APP Tg mice crossed with PS1 cKOs [183]. These results reveal that even though conditional inactivation of PS1 can successfully reduce Aβ production and the amyloid-associated neuropathological alterations, it does not prevent the impairments in both synaptic and cognitive functions [183]. Collectively, these studies suggest that the effects of Aβ on basal synaptic transmission and plasticity differ with age and that successful reduction of Aβ levels by targeting APP-processing enzymes may not recover synaptic dysfunctions.
9. Conclusion
It is clear that successful AD treatments will need to do more than just lower Aβ production; they will need to rescue cognitive as well as synaptic dysfunctions. Increasing evidence suggests the cognitive syndromes found in AD patients are preceded by changes in synaptic efficacy (reviewed in [1, 283]). Therefore, examining whether different strategies that target APP-processing enzymes rescue synaptic dysfunctions associated with AD is important. Several current reviews state why certain APP-processing drug therapies have failed in recent clinical trials and why current trials have not been able to generate more beneficial or significant results [284–286]. Testing the effects of potential AD therapeutics on synaptic function, in addition to behavioral analyses, will provide a better mechanistic understanding of the potential problems. It is also important to remember how different animal models may affect the outcome of results. For example, in mouse studies, genetic background has been shown to influence the effectiveness of certain γ-secretase targeting drugs [287–289]. In addition, many AD mouse models have been generated from FAD-linked mutations and may not fully recapitulate sporadic AD cases. In sum, mechanistic understanding of the normal synaptic functions of APP-processing enzymes will benefit the development of more effective treatments for AD.
Authors' Contribution
H. Wang and A. Megill contributed equally to this work.
Acknowledgment
This work was supported by R01-AG034606 to AK.
References
- 1.Shankar GM, Walsh DM. Alzheimer’s disease: synaptic dysfunction and Abeta. Molecular Neurodegeneration. 2009;4, article 48 doi: 10.1186/1750-1326-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walsh DM, Selkoe DJ. Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron. 2004;44(1):181–193. doi: 10.1016/j.neuron.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 3.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 4.Citron M. Strategies for disease modification in Alzheimer’s disease. Nature Reviews Neuroscience. 2004;5(9):677–685. doi: 10.1038/nrn1495. [DOI] [PubMed] [Google Scholar]
- 5.Hebb DO. Organization of Behavior. New York, NY, USA: John Wiley & Sons; 1949. [Google Scholar]
- 6.Stent GS. A physiological mechanism for Hebb’s postulate of learning. Proceedings of the National Academy of Sciences of the United States of America. 1973;70(4):997–1001. doi: 10.1073/pnas.70.4.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. Journal of Physiology. 1973;232(2):331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dudek SM, Bear MF. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(10):4363–4367. doi: 10.1073/pnas.89.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron. 1992;9(5):967–975. doi: 10.1016/0896-6273(92)90248-c. [DOI] [PubMed] [Google Scholar]
- 10.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44(1):5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 11.Bailey CH, Bartsch D, Kandel ER. Toward a molecular definition of long-term memory storage. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(24):13445–13452. doi: 10.1073/pnas.93.24.13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barco A, Bailey CH, Kandel ER. Common molecular mechanisms in explicit and implicit memory. Journal of Neurochemistry. 2006;97(6):1520–1533. doi: 10.1111/j.1471-4159.2006.03870.x. [DOI] [PubMed] [Google Scholar]
- 13.Bear MF. A synaptic basis for memory storage in the cerebral cortex. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(24):13453–13459. doi: 10.1073/pnas.93.24.13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bliss TV, Collingridge GL, Morris RG. Introduction. Long-term potentiation and structure of the issue. Philosophical Transactions of the Royal Society B. 2003;358(1432):607–611. doi: 10.1098/rstb.2003.1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lisman J. A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(23):9574–9578. doi: 10.1073/pnas.86.23.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lynch MA. Long-term potentiation and memory. Physiological Reviews. 2004;84(1):87–136. doi: 10.1152/physrev.00014.2003. [DOI] [PubMed] [Google Scholar]
- 17.Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annual Review of Neuroscience. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- 18.Morris RG. Elements of a neurobiological theory of hippocampal function: the role of synaptic plasticity, synaptic tagging and schemas. European Journal of Neuroscience. 2006;23(11):2829–2846. doi: 10.1111/j.1460-9568.2006.04888.x. [DOI] [PubMed] [Google Scholar]
- 19.Paulsen O, Sejnowski TJ. Natural patterns of activity and long-term synaptic plasticity. Current Opinion in Neurobiology. 2000;10(2):172–179. doi: 10.1016/s0959-4388(00)00076-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rodrigues SM, Schafe GE, LeDoux JE. Molecular mechanisms underlying emotional learning and memory in the lateral amygdala. Neuron. 2004;44(1):75–91. doi: 10.1016/j.neuron.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 21.Schafe GE, Doyère V, LeDoux JE. Tracking the fear engram: the lateral amygdala is an essential locus of fear memory storage. The Journal of Neuroscience. 2005;25(43):10010–10014. doi: 10.1523/JNEUROSCI.3307-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313(5790):1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- 23.Rioult-Pedotti MS, Friedman D, Hess G, Donoghue JP. Strengthening of horizontal cortical connections following skill learning. Nature Neuroscience. 1998;1(3):230–234. doi: 10.1038/678. [DOI] [PubMed] [Google Scholar]
- 24.Stefan K, Wycislo M, Gentner R, et al. Temporary occlusion of associative motor cortical plasticity by prior dynamic motor training. Cerebral Cortex. 2006;16(3):376–385. doi: 10.1093/cercor/bhi116. [DOI] [PubMed] [Google Scholar]
- 25.Abraham WC, Bear MF. Metaplasticity: the plasticity of synaptic plasticity. Trends in Neurosciences. 1996;19(4):126–130. doi: 10.1016/s0166-2236(96)80018-x. [DOI] [PubMed] [Google Scholar]
- 26.Bear MF, Cooper LN, Ebner FF. A physiological basis for a theory of synapse modification. Science. 1987;237(4810):42–48. doi: 10.1126/science.3037696. [DOI] [PubMed] [Google Scholar]
- 27.Bienenstock EL, Cooper LN, Munro PW. Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. The Journal of Neuroscience. 1982;2(1):32–48. doi: 10.1523/JNEUROSCI.02-01-00032.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391(6670):892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- 29.Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nature Reviews Neuroscience. 2004;5(2):97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- 30.Abbott LF, Nelson SB. Synaptic plasticity: taming the beast. Nature Neuroscience. 2000;3(supplement):1178–1183. doi: 10.1038/81453. [DOI] [PubMed] [Google Scholar]
- 31.Aizenman CD, Akerman CJ, Jensen KR, Cline HT. Visually driven regulation of intrinsic neuronal excitability improves stimulus detection in vivo. Neuron. 2003;39(5):831–842. doi: 10.1016/s0896-6273(03)00527-0. [DOI] [PubMed] [Google Scholar]
- 32.Desai NS, Rutherford LC, Turrigiano GG. BDNF regulates the intrinsic excitability of cortical neurons. Learning & Memory. 1999;6(3):284–291. [PMC free article] [PubMed] [Google Scholar]
- 33.Maffei A, Nelson SB, Turrigiano GG. Selective reconfiguration of layer 4 visual cortical circuitry by visual deprivation. Nature Neuroscience. 2004;7(12):1353–1359. doi: 10.1038/nn1351. [DOI] [PubMed] [Google Scholar]
- 34.Kilman V, van Rossum MC, Turrigiano GG. Activity deprivation reduces miniature IPSC amplitude by decreasing the number of postsynaptic GABAA receptors clustered at neocortical synapses. The Journal of Neuroscience. 2002;22(4):1328–1337. doi: 10.1523/JNEUROSCI.22-04-01328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maffei A, Nataraj K, Nelson SB, Turrigiano GG. Potentiation of cortical inhibition by visual deprivation. Nature. 2006;443(7107):81–84. doi: 10.1038/nature05079. [DOI] [PubMed] [Google Scholar]
- 36.Morales B, Choi SY, Kirkwood A. Dark rearing alters the development of GABAergic transmission in visual cortex. The Journal of Neuroscience. 2002;22(18):8084–8090. doi: 10.1523/JNEUROSCI.22-18-08084.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burrone J, O’Byrne M, Murthy VN. Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature. 2002;420(6914):414–418. doi: 10.1038/nature01242. [DOI] [PubMed] [Google Scholar]
- 38.Thiagarajan TC, Lindskog M, Tsien RW. Adaptation to synaptic inactivity in hippocampal neurons. Neuron. 2005;47(5):725–737. doi: 10.1016/j.neuron.2005.06.037. [DOI] [PubMed] [Google Scholar]
- 39.Thiagarajan TC, Piedras-Renteria ES, Tsien RW. α- and βCaMKII. Inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron. 2002;36(6):1103–1114. doi: 10.1016/s0896-6273(02)01049-8. [DOI] [PubMed] [Google Scholar]
- 40.Nicoll RA, Malenka RC. Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature. 1995;377(6545):115–118. doi: 10.1038/377115a0. [DOI] [PubMed] [Google Scholar]
- 41.Malenka RC, Nicoll RA. Long-term potentiation—a decade of progress? Science. 1999;285(5435):1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- 42.Kovalchuk Y, Eilers J, Lisman J, Konnerth A. NMDA receptor-mediated subthreshold Ca2+ signals in spines of hippocampal neurons. The Journal of Neuroscience. 2000;20(5):1791–1799. doi: 10.1523/JNEUROSCI.20-05-01791.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yuste R, Majewska A, Cash SS, Denk W. Mechanisms of calcium influx into hippocampal spines: heterogeneity among spines, coincidence detection by NMDA receptors, and optical quantal analysis. The Journal of Neuroscience. 1999;19(6):1976–1987. doi: 10.1523/JNEUROSCI.19-06-01976.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Connor JA, Petrozzino J, Pozzo-Miller LD, Otani S. Calcium signals in long-term potentiation and long-term depression. Canadian Journal of Physiology and Pharmacology. 1999;77(9):722–734. [PubMed] [Google Scholar]
- 45.Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annual Review of Neuroscience. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- 46.Lee H-K. Synaptic plasticity and phosphorylation. Pharmacology & Therapeutics. 2006;112(3):810–832. doi: 10.1016/j.pharmthera.2006.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287(5461):2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- 48.Shi S, Hayashi Y, Esteban JA, Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 2001;105(3):331–343. doi: 10.1016/s0092-8674(01)00321-x. [DOI] [PubMed] [Google Scholar]
- 49.Shi SH, Hayashi Y, Petralia RS, et al. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284(5421):1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- 50.Boehm J, Kang MG, Johnson RC, Esteban J, Huganir RL, Malinow R. Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron. 2006;51(2):213–225. doi: 10.1016/j.neuron.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 51.Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nature Neuroscience. 2003;6(2):136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- 52.Roche KW, O’Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16(6):1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- 53.Barria A, Derkach V, Soderling T. Identification of the Ca2+/calmodulin-dependent protein kinase II regulatory phosphorylation site in the α-amino-3-hydroxyl-5-methyl-4- isoxazole-propionate-type glutamate receptor. The Journal of Biological Chemistry. 1997;272(52):32727–32730. doi: 10.1074/jbc.272.52.32727. [DOI] [PubMed] [Google Scholar]
- 54.Mammen AL, Kameyama K, Roche KW, Huganir RL. Phosphorylation of the α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor GluR1 subunit by calcium/calmodulin-dependent kinase II. The Journal of Biological Chemistry. 1997;272(51):32528–32533. doi: 10.1074/jbc.272.51.32528. [DOI] [PubMed] [Google Scholar]
- 55.Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276(5321):2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- 56.Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405(6789):955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- 57.Lee HK, Takamiya K, He K, Song L, Huganir RL. Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. Journal of Neurophysiology. 2010;103(1):479–489. doi: 10.1152/jn.00835.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zamanillo D, Sprengel R, Hvalby O, et al. Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science. 1999;284(5421):1805–1811. doi: 10.1126/science.284.5421.1805. [DOI] [PubMed] [Google Scholar]
- 59.Lee HK, Takamiya K, Han JS, et al. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003;112(5):631–643. doi: 10.1016/s0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- 60.Carroll RC, Beattie EC, von Zastrow M, Malenka RC. Role of AMPA receptor endocytosis in synaptic plasticity. Nature Reviews Neuroscience. 2001;2(5):315–324. doi: 10.1038/35072500. [DOI] [PubMed] [Google Scholar]
- 61.Daw MI, Chittajallu R, Bortolotto ZA, et al. PDZ proteins interacting with C-terminal GluR2/3 are involved in a PKC-dependent regulation of AMPA receptors at hippocampal synapses. Neuron. 2000;28(3):873–886. doi: 10.1016/s0896-6273(00)00160-4. [DOI] [PubMed] [Google Scholar]
- 62.Kim CH, Chung HJ, Lee HK, Huganir RL. Interaction of the AMPA receptor subunit GluR2/3 with PDZ domains regulates hippocampal long-term depression. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(20):11725–11730. doi: 10.1073/pnas.211132798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee SH, Simonetta A, Sheng M. Subunit rules governing the sorting of internalized AMPA receptors in hippocampal neurons. Neuron. 2004;43(2):221–236. doi: 10.1016/j.neuron.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 64.Lin JW, Ju W, Foster K, et al. Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nature Neuroscience. 2000;3(12):1282–1290. doi: 10.1038/81814. [DOI] [PubMed] [Google Scholar]
- 65.Lüthi A, Chittajallu R, Duprat F, et al. Hippocampal LTD expression involves a pool of AMPARs regulated by the NSF-GluR2 interaction. Neuron. 1999;24(2):389–399. doi: 10.1016/s0896-6273(00)80852-1. [DOI] [PubMed] [Google Scholar]
- 66.Man HY, Lin JW, Ju WH, et al. Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron. 2000;25(3):649–662. doi: 10.1016/s0896-6273(00)81067-3. [DOI] [PubMed] [Google Scholar]
- 67.Osten P, Khatri L, Perez JL, et al. Mutagenesis reveals a role for ABP/GRIP binding to GluR2 in synaptic surface accumulation of the AMPA receptor. Neuron. 2000;27(2):313–325. doi: 10.1016/s0896-6273(00)00039-8. [DOI] [PubMed] [Google Scholar]
- 68.Lee HK, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21(5):1151–1162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- 69.Frey U, Krug M, Reymann KG, Matthies H. Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Research. 1988;452(1-2):57–65. doi: 10.1016/0006-8993(88)90008-x. [DOI] [PubMed] [Google Scholar]
- 70.Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288(5469):1254–1257. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- 71.Kelleher RJ, III, Govindarajan A, Jung HY, Kang H, Tonegawa S. Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell. 2004;116(3):467–479. doi: 10.1016/s0092-8674(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 72.Krug M, Lossner B, Ott T. Anisomycin blocks the late phase of long-term potentiation in the dentate gyrus of freely moving rats. Brain Research Bulletin. 1984;13(1):39–42. doi: 10.1016/0361-9230(84)90005-4. [DOI] [PubMed] [Google Scholar]
- 73.Manahan-Vaughan D, Kulla A, Frey JU. Requirement of translation but not transcription for the maintenance of long-term depression in the CA1 region of freely moving rats. The Journal of Neuroscience. 2000;20(22):8572–8576. doi: 10.1523/JNEUROSCI.20-22-08572.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stanton PK, Sarvey JM. Blockade of long-term potentiation in rat hippocampal CA1 region by inhibitors of protein synthesis. The Journal of Neuroscience. 1984;4(12):3080–3088. doi: 10.1523/JNEUROSCI.04-12-03080.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Davis HP, Squire LR. Protein synthesis and memory: a review. Psychological Bulletin. 1984;96(3):518–559. [PubMed] [Google Scholar]
- 76.Flexner JB, Flexner LB, Stellar E. Memory in mice as affected by intracerebral puromycin. Science. 1963;141(3575):57–59. doi: 10.1126/science.141.3575.57. [DOI] [PubMed] [Google Scholar]
- 77.Sutton MA, Schuman EM. Dendritic protein synthesis, synaptic plasticity, and memory. Cell. 2006;127(1):49–58. doi: 10.1016/j.cell.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 78.Nguyen PV, Abel T, Kandel ER. Requirement of a critical period of transcription for induction of a late phase of LTP. Science. 1994;265(5175):1104–1107. doi: 10.1126/science.8066450. [DOI] [PubMed] [Google Scholar]
- 79.Tischmeyer W, Grimm R. Activation of immediate early genes and memory formation. Cellular and Molecular Life Sciences. 1999;55(4):564–574. doi: 10.1007/s000180050315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guzowski JF, Mcgaugh JL. Antisense oligodeoxynucleotide-mediated disruption of hippocampal cAMP response element binding protein levels impairs consolidation of memory for water maze training. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(6):2693–2698. doi: 10.1073/pnas.94.6.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lamprecht R, Hazvi S, Dudai Y. cAMP response element-binding protein in the amygdala is required for long- but not short-term conditioned taste aversion memory. The Journal of Neuroscience. 1997;17(21):8443–8450. doi: 10.1523/JNEUROSCI.17-21-08443.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annual Review of Neuroscience. 1998;21:127–148. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- 83.Guzowski JF, Lyford GL, Stevenson GD, et al. Inhibition of activity-dependent arc protein expression in the rat hippocampus impairs the maintenance of long-term potentiation and the consolidation of long-term memory. The Journal of Neuroscience. 2000;20(11):3993–4001. doi: 10.1523/JNEUROSCI.20-11-03993.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lendon CL, Ashall F, Goate AM. Exploring the etiology of Alzheimer disease using molecular genetics. Journal of the American Medical Association. 1997;277(10):825–831. [PubMed] [Google Scholar]
- 85.Scheuner D, Eckman C, Jensen M, et al. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nature Medicine. 1996;2(8):864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 86.Marchetti C, Marie H. Hippocampal synaptic plasticity in Alzheimer's disease: what have we learned so far from transgenic models? Reviews in the Neurosciences. 2011;22(4):373–402. doi: 10.1515/RNS.2011.035. [DOI] [PubMed] [Google Scholar]
- 87.Chen QS, Kagan BL, Hirakura Y, Xie CW. Impairment of hippocampal long-term potentiation by Alzheimer amyloid β-peptides. Journal of Neuroscience Research. 2000;60(1):65–72. doi: 10.1002/(SICI)1097-4547(20000401)60:1<65::AID-JNR7>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 88.Chen QS, Wei WZ, Shimahara T, Xie CW. Alzheimer amyloid β-peptide inhibits the late phase of long-term potentiation through calcineurin-dependent mechanisms in the hippocampal dentate gyrus. Neurobiology of Learning and Memory. 2002;77(3):354–371. doi: 10.1006/nlme.2001.4034. [DOI] [PubMed] [Google Scholar]
- 89.Zhao D, Watson JB, Xie CW. Amyloid β prevents activation of calcium/calmodulin-dependent protein kinase II and AMPA receptor phosphorylation during hippocampal long-term potentiation. Journal of Neurophysiology. 2004;92(5):2853–2858. doi: 10.1152/jn.00485.2004. [DOI] [PubMed] [Google Scholar]
- 90.Walsh DM, Klyubin I, Fadeeva JV, et al. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416(6880):535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 91.Kim JH, Anwyl R, Suh YH, Djamgoz MB, Rowan MJ. Use-dependent effects of amyloidogenic fragments of β-amyloid precursor protein on synaptic plasticity in rat hippocampus in vivo. The Journal of Neuroscience. 2001;21(4):1327–1333. doi: 10.1523/JNEUROSCI.21-04-01327.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tanzi RE. The synaptic Abeta hypothesis of Alzheimer disease. Nature Neuroscience. 2005;8(8):977–979. doi: 10.1038/nn0805-977. [DOI] [PubMed] [Google Scholar]
- 93.Gong Y, Chang L, Viola KL, et al. Alzheimer’s disease-affected brain: presence of oligomeric Abeta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(18):10417–10422. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lacor PN, Buniel MC, Chang L, et al. Synaptic targeting by Alzheimer’s-related amyloid β oligomers. The Journal of Neuroscience. 2004;24(45):10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wu J, Anwyl R, Rowan MJ. β-amyloid selectively augments NMDA receptor-mediated synaptic transmission in rat hippocampus. Neuroreport. 1995;6(17):2409–2413. doi: 10.1097/00001756-199511270-00031. [DOI] [PubMed] [Google Scholar]
- 96.Hsia AY, Masliah E, Mcconlogue L, et al. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(6):3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Snyder EM, Nong Y, Almeida CG, et al. Regulation of NMDA receptor trafficking by amyloid-β . Nature Neuroscience. 2005;8(8):1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- 98.Almeida CG, Tampellini D, Takahashi RH, et al. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiology of Disease. 2005;20(2):187–198. doi: 10.1016/j.nbd.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 99.Hsieh H, Boehm J, Sato C, et al. AMPAR removal underlies Aβ-induced synaptic depression and dendritic spine loss. Neuron. 2006;52(5):831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Miñano-Molina AJ, España J, Martín E, et al. Soluble oligomers of amyloid-β peptide disrupt membrane trafficking of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor contributing to early synapse dysfunction. The Journal of Biological Chemistry. 2011;286(31):27311–27321. doi: 10.1074/jbc.M111.227504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Borchelt DR, Thinakaran G, Eckman CB, et al. Familial Alzheimer’s disease-linked presenilin I variants elevate Aβ1- 42/1-40 ratio in vitro and in vivo. Neuron. 1996;17(5):1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 102.Thinakaran G, Sisodia SS. Presenilins and Alzheimer disease: the calcium conspiracy. Nature Neuroscience. 2006;9(11):1354–1355. doi: 10.1038/nn1106-1354. [DOI] [PubMed] [Google Scholar]
- 103.Hoe HS, Lee HK, Pak DT. The upside of APP at synapses. CNS Neuroscience & Therapeutics. 2012;18(1):47–56. doi: 10.1111/j.1755-5949.2010.00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gu Z, Liu W, Yan Z. β-amyloid impairs AMPA receptor trafficking and function by reducing Ca2+/calmodulin-dependent protein kinase II synaptic distribution. The Journal of Biological Chemistry. 2009;284(16):10639–10649. doi: 10.1074/jbc.M806508200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Knobloch M, Farinelli M, Konietzko U, Nitsch RM, Mansuy IM. Aβ oligomer-mediated long-term potentiation impairment involves protein phosphatase 1-dependent mechanisms. The Journal of Neuroscience. 2007;27(29):7648–7653. doi: 10.1523/JNEUROSCI.0395-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid β protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62(6):788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Saura CA, Valero J. The role of CREB signaling in Alzheimer’s disease and other cognitive disorders. Reviews in the Neurosciences. 2011;22(2):153–169. doi: 10.1515/RNS.2011.018. [DOI] [PubMed] [Google Scholar]
- 108.Steward O, Worley PF. A cellular mechanism for targeting newly synthesized mRNAs to synaptic sites on dendrites. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(13):7062–7068. doi: 10.1073/pnas.131146398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shepherd JD, Rumbaugh G, Wu J, et al. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52(3):475–484. doi: 10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Guzowski JF. Insights into immediate-early gene function in hippocampal memory consolidation using antisense oligonucleotide and fluorescent imaging approaches. Hippocampus. 2002;12(1):86–104. doi: 10.1002/hipo.10010. [DOI] [PubMed] [Google Scholar]
- 111.Echeverria V, Berman DE, Arancio O. Oligomers of β-amyloid peptide inhibit BDNF-induced arc expression in cultured cortical neurons. Current Alzheimer Research. 2007;4(5):518–521. doi: 10.2174/156720507783018190. [DOI] [PubMed] [Google Scholar]
- 112.Dickey CA, Loring JF, Montgomery J, Gordon MN, Eastman PS, Morgan D. Selectively reduced expression of synaptic plasticity-related genes in amyloid precursor protein + presenilin-1 transgenic mice. The Journal of Neuroscience. 2003;23(12):5219–5226. doi: 10.1523/JNEUROSCI.23-12-05219.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nimmrich V, Grimm C, Draguhn A, et al. Amyloid β oligomers (Aβ(1-42) globulomer) suppress spontaneous synaptic activity by inhibition of P/Q-type calcium currents. The Journal of Neuroscience. 2008;28(4):788–797. doi: 10.1523/JNEUROSCI.4771-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kelly BL, Vassar R, Ferreira A. β-amyloid-induced dynamin 1 depletion in hippocampal neurons: a potential mechanism for early cognitive decline in Alzheimer disease. The Journal of Biological Chemistry. 2005;280(36):31746–31753. doi: 10.1074/jbc.M503259200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kelly BL, Ferreira A. Beta-amyloid disrupted synaptic vesicle endocytosis in cultured hippocampal neurons. Neuroscience. 2007;147(1):60–70. doi: 10.1016/j.neuroscience.2007.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yao PJ, Zhu M, Pyun EI, et al. Defects in expression of genes related to synaptic vesicle trafficking in frontal cortex of Alzheimer’s disease. Neurobiology of Disease. 2003;12(2):97–109. doi: 10.1016/s0969-9961(02)00009-8. [DOI] [PubMed] [Google Scholar]
- 117.Nimmrich V, Reymann KG, Strassburger M, et al. Inhibition of calpain prevents NMDA-induced cell death and β-amyloid-induced synaptic dysfunction in hippocampal slice cultures. British Journal of Pharmacology. 2010;159(7):1523–1531. doi: 10.1111/j.1476-5381.2010.00652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kelly BL, Ferreira A. β-amyloid-induced dynamin 1 degradation is mediated by N-methyl-D-aspartate receptors in hippocampal neurons. The Journal of Biological Chemistry. 2006;281(38):28079–28089. doi: 10.1074/jbc.M605081200. [DOI] [PubMed] [Google Scholar]
- 119.Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405(6784):360–364. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- 120.Wen Y, Yu WH, Maloney B, et al. Transcriptional regulation of β-secretase by p25/cdk5 leads to enhanced amyloidogenic processing. Neuron. 2008;57(5):680–690. doi: 10.1016/j.neuron.2008.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liang B, Duan BY, Zhou XP, Gong JX, Luo ZG. Calpain activation promotes BACE1 expression, amyloid precursor protein processing, and amyloid plaque formation in a transgenic mouse model of alzheimer disease. The Journal of Biological Chemistry. 2010;285(36):27737–27744. doi: 10.1074/jbc.M110.117960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Seguela P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain α7: a nicotinic cation channel highly permeable to calcium. The Journal of Neuroscience. 1993;13(2):596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wang HY, Lee DH, D’Andrea MR, Peterson PA, Shank RP, Reitz AB. β-amyloid1-42 binds to α7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. The Journal of Biological Chemistry. 2000;275(8):5626–5632. doi: 10.1074/jbc.275.8.5626. [DOI] [PubMed] [Google Scholar]
- 124.Wang HY, Lee DH, Davis CB, Shank RP. Amyloid peptide Aβ1-42 binds selectively and with picomolar affinity to α7 nicotinic acetylcholine receptors. Journal of Neurochemistry. 2000;75(3):1155–1161. doi: 10.1046/j.1471-4159.2000.0751155.x. [DOI] [PubMed] [Google Scholar]
- 125.Liu Q, Kawai H, Berg DK. β-amyloid peptide blocks the response of α7-containing nicotinic receptors on hippocampal neurons. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(8):4734–4739. doi: 10.1073/pnas.081553598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Guan ZZ, Miao H, Tian JY, Unger C, Nordberg A, Zhang X. Suppressed expression of nicotinic acetylcholine receptors by nanomolar β-amyloid peptides in PC12 cells. Journal of Neural Transmission. 2001;108(12):1417–1433. doi: 10.1007/s007020100017. [DOI] [PubMed] [Google Scholar]
- 127.Pettit DL, Shao Z, Yakel JL. Beta-amyloid(1-42) peptide directly modulates nicotinic receptors in the rat hippocampal slice. The Journal of Neuroscience. 2001;21(1):p. RC120. doi: 10.1523/JNEUROSCI.21-01-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chen L, Yamada K, Nabeshima T, Sokabe M. α7 nicotinic acetylcholine receptor as a target to rescue deficit in hippocampal LTP induction in β-amyloid infused rats. Neuropharmacology. 2006;50(2):254–268. doi: 10.1016/j.neuropharm.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 129.Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD. β-amyloid activates the mitogen-activated protein kinase cascade via hippocampal α7 nicotinic acetylcholine receptors: in vitro and in vivo mechanisms related to Alzheimer’s disease. The Journal of Neuroscience. 2001;21(12):4125–4133. doi: 10.1523/JNEUROSCI.21-12-04125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dougherty JJ, Wu J, Nichols RA. β-amyloid regulation of presynaptic nicotinic receptors in rat hippocampus and neocortex. The Journal of Neuroscience. 2003;23(17):6740–6747. doi: 10.1523/JNEUROSCI.23-17-06740.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I. Amyloid-β as a positive endogenous regulator of release probability at hippocampal synapses. Nature Neuroscience. 2009;12(12):1567–1576. doi: 10.1038/nn.2433. [DOI] [PubMed] [Google Scholar]
- 132.Palop JJ, Mucke L. Amyloid-β-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nature Neuroscience. 2010;13(7):812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Puzzo D, Privitera L, Leznik E, et al. Picomolar amyloid-β positively modulates synaptic plasticity and memory in hippocampus. The Journal of Neuroscience. 2008;28(53):14537–14545. doi: 10.1523/JNEUROSCI.2692-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Massaad CA, Klann E. Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxidants and Redox Signaling. 2011;14(10):2013–2054. doi: 10.1089/ars.2010.3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ma T, Hoeffer CA, Wong H, et al. Amyloid β-induced impairments in hippocampal synaptic plasticity are rescued by decreasing mitochondrial superoxide. The Journal of Neuroscience. 2011;31(15):5589–5595. doi: 10.1523/JNEUROSCI.6566-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Busche MA, Eichhoff G, Adelsberger H, et al. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science. 2008;321(5896):1686–1689. doi: 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- 137.Nitsch RM, Farber SA, Growdon JH, Wurtman RJ. Release of amyloid β-protein precursor derivatives by electrical depolarization of rat hippocampal slices. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(11):5191–5193. doi: 10.1073/pnas.90.11.5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kamenetz F, Tomita T, Hsieh H, et al. APP processing and synaptic function. Neuron. 2003;37(6):925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- 139.Lesné S, Ali C, Gabriel C, et al. NMDA receptor activation inhibits α-secretase and promotes neuronal amyloid-β production. The Journal of Neuroscience. 2005;25(41):9367–9377. doi: 10.1523/JNEUROSCI.0849-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pratt KG, Zimmerman EC, Cook DG, Sullivan JM. Presenilin 1 regulates homeostatic synaptic scaling through Akt signaling. Nature Neuroscience. 2011;14(9):1112–1114. doi: 10.1038/nn.2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wu J, Petralia RS, Kurushima H, et al. Arc/Arg3.1 regulates an endosomal pathway essential for activity-dependent beta-amyloid generation. Cell. 2011;147(3):615–628. doi: 10.1016/j.cell.2011.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Talos DM, Follett PL, Folkerth RD, et al. Developmental regulation of α-amino-3-hydroxy-5-methyl-4-isoxazole- propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury. II. Human cerebral white matter and cortex. Journal of Comparative Neurology. 2006;497(1):61–77. doi: 10.1002/cne.20978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Desmond DW, Moroney JT, Sano M, Stern Y. Incidence of dementia after ischemic stroke: results of a longitudinal study. Stroke. 2002;33(9):2254–2260. doi: 10.1161/01.str.0000028235.91778.95. [DOI] [PubMed] [Google Scholar]
- 144.Cirrito JR, Yamada KA, Finn MB, et al. Synaptic activity regulates interstitial fluid amyloid-β levels in vivo. Neuron. 2005;48(6):913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 145.Bero AW, Yan P, Roh JH, et al. Neuronal activity regulates the regional vulnerability to amyloid-β 2 deposition. Nature Neuroscience. 2011;14(6):750–756. doi: 10.1038/nn.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Cirrito JR, Kang JE, Lee J, et al. Endocytosis is required for synaptic activity-dependent release of amyloid-β in vivo. Neuron. 2008;58(1):42–51. doi: 10.1016/j.neuron.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Li F, Calingasan NY, Yu F, et al. Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. Journal of Neurochemistry. 2004;89(5):1308–1312. doi: 10.1111/j.1471-4159.2004.02455.x. [DOI] [PubMed] [Google Scholar]
- 148.Tampellini D, Rahman N, Gallo EF, et al. Synaptic activity reduces intraneuronal Aβ, promotes APP transport to synapses, and protects against Aβ-related synaptic alterations. The Journal of Neuroscience. 2009;29(31):9704–9713. doi: 10.1523/JNEUROSCI.2292-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhang XM, Xiong K, Cai Y, et al. Functional deprivation promotes amyloid plaque pathogenesis in Tg2576 mouse olfactory bulb and piriform cortex. European Journal of Neuroscience. 2010;31(4):710–721. doi: 10.1111/j.1460-9568.2010.07103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Pearson HA, Peers C. Physiological roles for amyloid β peptides. The Journal of Physiology. 2006;575, part 1:5–10. doi: 10.1113/jphysiol.2006.111203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Cao X, Sudhof TC. A transcriptively active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293(5527):115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- 152.Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J. Synapse formation and function is modulated by the amyloid precursor protein. The Journal of Neuroscience. 2006;26(27):7212–7221. doi: 10.1523/JNEUROSCI.1450-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Steinbach JP, Müller U, Leist M, Li ZW, Nicotera P, Aguzzi A. Hypersensitivity to seizures in β-amyloid precursor protein deficient mice. Cell Death and Differentiation. 1998;5(10):858–866. doi: 10.1038/sj.cdd.4400391. [DOI] [PubMed] [Google Scholar]
- 154.Plant LD, Boyle JP, Smith IF, Peers C, Pearson HA. The production of amyloid β peptide is a critical requirement for the viability of central neurons. The Journal of Neuroscience. 2003;23(13):5531–5535. doi: 10.1523/JNEUROSCI.23-13-05531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Buttini M, Masliah E, Barbour R, et al. β-amyloid immunotherapy prevents synaptic degeneration in a mouse model of Alzheimer’s disease. The Journal of Neuroscience. 2005;25(40):9096–9101. doi: 10.1523/JNEUROSCI.1697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Schenk D, Games D, Seubert P. Potential treatment opportunities for Alzheimer’s disease through inhibition of secretases and Aβ immunization. Journal of Molecular Neuroscience. 2001;17(2):259–267. doi: 10.1385/JMN:17:2:259. [DOI] [PubMed] [Google Scholar]
- 157.Lemere CA. Developing novel immunogens for a safe and effective Alzheimer’s disease vaccine. Progress in Brain Research. 2009;175:83–93. doi: 10.1016/S0079-6123(09)17506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Town T. Alternative Aβ immunotherapy approaches for Alzheimer’s disease. CNS & Neurological Disorders—Drug Targets. 2009;8(2):114–127. doi: 10.2174/187152709787847306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Cai H, Wang Y, McCarthy D, et al. BACE1 is the major β-secretase for generation of Aβ peptides by neurons. Nature Neuroscience. 2001;4(3):233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- 160.Luo Y, Bolon B, Kahn S, et al. Mice deficient in BACE1, the Alzheimer’s β-secretase, have normal phenotype and abolished β-amyloid generation. Nature Neuroscience. 2001;4(3):231–232. doi: 10.1038/85059. [DOI] [PubMed] [Google Scholar]
- 161.Roberds SL, Anderson J, Basi G, et al. BACE knockout mice are healthy despite lacking the primary β-secretase activity in brain: implications for Alzheimer’s disease therapeutics. Human Molecular Genetics. 2001;10(12):1317–1324. doi: 10.1093/hmg/10.12.1317. [DOI] [PubMed] [Google Scholar]
- 162.Vassar R, Bennett BD, Babu-Khan S, et al. β-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286(5440):735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 163.Ohno M, Sametsky EA, Younkin LH, et al. BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer’s disease. Neuron. 2004;41(1):27–33. doi: 10.1016/s0896-6273(03)00810-9. [DOI] [PubMed] [Google Scholar]
- 164.Laird FM, Cai H, Savonenko AV, et al. BACE1, a major determinant of selective vulnerability of the brain to amyloid-β amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. The Journal of Neuroscience. 2005;25(50):11693–11709. doi: 10.1523/JNEUROSCI.2766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ohno M, Chang L, Tseng W, et al. Temporal memory deficits in Alzheimer’s mouse models: rescue by genetic deletion of BACE1. European Journal of Neuroscience. 2006;23(1):251–260. doi: 10.1111/j.1460-9568.2005.04551.x. [DOI] [PubMed] [Google Scholar]
- 166.Citron M. Alzheimer’s disease: treatments in discovery and development. Nature Neuroscience. 2002;5(supplement):1055–1057. doi: 10.1038/nn940. [DOI] [PubMed] [Google Scholar]
- 167.Vassar R. β-secretase (BACE) as a drug target for Alzheimer’s disease. Advanced Drug Delivery Reviews. 2002;54(12):1589–1602. doi: 10.1016/s0169-409x(02)00157-6. [DOI] [PubMed] [Google Scholar]
- 168.Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. Journal of Neurophysiology. 1993;70(4):1451–1459. doi: 10.1152/jn.1993.70.4.1451. [DOI] [PubMed] [Google Scholar]
- 169.Rozov A, Zilberter Y, Wollmuth LP, Burnashev N. Facilitation of currents through rat Ca2+-permeable AMPA receptor channels by activity-dependent relief from polyamine block. Journal of Physiology. 1998;511(2):361–377. doi: 10.1111/j.1469-7793.1998.361bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Harrison SM, Harper AJ, Hawkins J, et al. BACE1 (β-secretase) transgenic and knockout mice: identification of neurochemical deficits and behavioral changes. Molecular and Cellular Neuroscience. 2003;24(3):646–655. doi: 10.1016/s1044-7431(03)00227-6. [DOI] [PubMed] [Google Scholar]
- 171.Ma H, Lesné S, Kotilinek L, et al. Involvement of β-site APP cleaving enzyme 1 (BACE1) in amyloid precursor protein-mediated enhancement of memory and activity-dependent synaptic plasticity. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(19):8167–8172. doi: 10.1073/pnas.0609521104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Zhao J, Fu Y, Yasvoina M, et al. β-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer’s disease pathogenesis. The Journal of Neuroscience. 2007;27(14):3639–3649. doi: 10.1523/JNEUROSCI.4396-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Nicoll RA, Schmitz D. Synaptic plasticity at hippocampal mossy fibre synapses. Nature Reviews Neuroscience. 2005;6(11):863–876. doi: 10.1038/nrn1786. [DOI] [PubMed] [Google Scholar]
- 174.Wang H, Song L, Laird F, Wong PC, Lee HK. BACE1 knock-outs display deficits in activity-dependent potentiation of synaptic transmission at mossy fiber to CA3 synapses in the hippocampus. The Journal of Neuroscience. 2008;28(35):8677–8681. doi: 10.1523/JNEUROSCI.2440-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wang H, Song L, Lee A, Laird F, Wong PC, Lee HK. Mossy fiber long-term potentiation deficits in BACE1 knock-outs can be rescued by activation of α7 nicotinic acetylcholine receptors. The Journal of Neuroscience. 2010;30(41):13808–13813. doi: 10.1523/JNEUROSCI.1070-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kimura R, Devi L, Ohno M. Partial reduction of BACE1 improves synaptic plasticity, recent and remote memories in Alzheimer’s disease transgenic mice. Journal of Neurochemistry. 2010;113(1):248–261. doi: 10.1111/j.1471-4159.2010.06608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Gong B, Chen F, Pan Y, et al. SCFFbx2-E3-ligase-mediated degradation of BACE1 attenuates Alzheimer’s disease amyloidosis and improves synaptic function. Aging Cell. 2010;9(6):1018–1031. doi: 10.1111/j.1474-9726.2010.00632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Yu H, Saura CA, Choi SY, et al. APP processing and synaptic plasticity in presenilin-1 conditional knockout mice. Neuron. 2001;31(5):713–726. doi: 10.1016/s0896-6273(01)00417-2. [DOI] [PubMed] [Google Scholar]
- 179.Saura CA, Choi SY, Beglopoulos V, et al. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42(1):23–36. doi: 10.1016/s0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- 180.Aoki C, Lee J, Nedelescu H, Ahmed T, Ho A, Shen J. Increased levels of NMDA receptor NR2A subunits at pre- and postsynaptic sites of the hippocampal CA1: an early response to conditional double knockout of presenilin 1 and 2. Journal of Comparative Neurology. 2009;517(4):512–523. doi: 10.1002/cne.22151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zhang D, Zhang C, Ho A, Kirkwood A, Sudhof TC, Shen J. Inactivation of presenilins causes pre-synaptic impairment prior to post-synaptic dysfunction. Journal of Neurochemistry. 2010;115(5):1215–1221. doi: 10.1111/j.1471-4159.2010.07011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zhang C, Wu B, Beglopoulos V, et al. Presenilins are essential for regulating neurotransmitter release. Nature. 2009;460(7255):632–636. doi: 10.1038/nature08177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Saura CA, Chen G, Malkani S, et al. Conditional inactivation of presenilin 1 prevents amyloid accumulation and temporarily rescues contextual and spatial working memory impairments in amyloid precursor protein transgenic mice. The Journal of Neuroscience. 2005;25(29):6755–6764. doi: 10.1523/JNEUROSCI.1247-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Townsend M, Qu Y, Gray A, et al. Oral treatment with a γ-secretase inhibitor improves long-term potentiation in a mouse model of Alzheimer’s disease. Journal of Pharmacology and Experimental Therapeutics. 2010;333(1):110–119. doi: 10.1124/jpet.109.163691. [DOI] [PubMed] [Google Scholar]
- 185.Balducci C, Mehdawy B, Mare L, et al. The γ-secretase modulator CHF5074 restores memory and hippocampal synaptic plasticity in plaque-free Tg2576 mice. Journal of Alzheimer’s Disease. 2011;24(4):799–816. doi: 10.3233/JAD-2011-101839. [DOI] [PubMed] [Google Scholar]
- 186.Wong HK, Sakurai T, Oyama F, et al. β subunits of voltage-gated sodium channels are novel substrates of β-site amyloid precursor protein-cleaving enzyme (BACE1) and γ-secretase. The Journal of Biological Chemistry. 2005;280(24):23009–23017. doi: 10.1074/jbc.M414648200. [DOI] [PubMed] [Google Scholar]
- 187.Kim DY, Carey BW, Wang H, et al. BACE1 regulates voltage-gated sodium channels and neuronal activity. Nature Cell Biology. 2007;9(7):755–764. doi: 10.1038/ncb1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lai HC, Jan LY. The distribution and targeting of neuronal voltage-gated ion channels. Nature Reviews Neuroscience. 2006;7(7):548–562. doi: 10.1038/nrn1938. [DOI] [PubMed] [Google Scholar]
- 189.Kim DY, Kovacs DM. Surface trafficking of sodium channels in cells and in hippocampal slices. Methods in Molecular Biology. 2011;793:351–361. doi: 10.1007/978-1-61779-328-8_23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kim DY, Gersbacher MT, Inquimbert P, Kovacs DM. Reduced sodium channel Na(v)1.1 levels in BACE1-null mice. The Journal of Biological Chemistry. 2011;286(10):8106–8116. doi: 10.1074/jbc.M110.134692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Hu X, Zhou X, He W, et al. BACE1 deficiency causes altered neuronal activity and neurodegeneration. The Journal of Neuroscience. 2010;30(26):8819–8829. doi: 10.1523/JNEUROSCI.1334-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Lemke G. Neuregulin-1 and myelination. Science’s STKE. 2006;2006(325):p. pe11. doi: 10.1126/stke.3252006pe11. [DOI] [PubMed] [Google Scholar]
- 193.Willem M, Garratt AN, Novak B, et al. Control of peripheral nerve myelination by the β-secretase BACE1. Science. 2006;314(5799):664–666. doi: 10.1126/science.1132341. [DOI] [PubMed] [Google Scholar]
- 194.Hu X, Hicks CW, He W, et al. Bace1 modulates myelination in the central and peripheral nervous system. Nature Neuroscience. 2006;9(12):1520–1525. doi: 10.1038/nn1797. [DOI] [PubMed] [Google Scholar]
- 195.Savonenko AV, Melnikova T, Laird FM, Stewart KA, Price DL, Wong PC. Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(14):5585–5590. doi: 10.1073/pnas.0710373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Li B, Woo RS, Mei L, Malinow R. The neuregulin-1 receptor erbB4 controls glutamatergic synapse maturation and plasticity. Neuron. 2007;54(4):583–597. doi: 10.1016/j.neuron.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Huang YZ, Won S, Ali DW, et al. Regulation of neuregulin signaling by PSD-95 interacting with ErbB4 at CNS synapses. Neuron. 2000;26(2):443–455. doi: 10.1016/s0896-6273(00)81176-9. [DOI] [PubMed] [Google Scholar]
- 198.Gu Z, Jiang Q, Fu AK, Ip NY, Yan Z. Regulation of NMDA receptors by neuregulin signaling in prefrontal cortex. The Journal of Neuroscience. 2005;25(20):4974–4984. doi: 10.1523/JNEUROSCI.1086-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Liu Y, Ford B, Mann MA, Fischbach GD. Neuregulins increase α7 nicotinic acetylcholine receptors and enhance excitatory synaptic transmission in GABAergic interneurons of the hippocampus. The Journal of Neuroscience. 2001;21(15):5660–5669. doi: 10.1523/JNEUROSCI.21-15-05660.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Zhong C, Du C, Hancock M, Mertz M, Talmage DA, Role LW. Presynaptic type III neuregulin 1 is required for sustained enhancement of hippocampal transmission by nicotine and for axonal targeting of α7 nicotinic acetylcholine receptors. The Journal of Neuroscience. 2008;28(37):9111–9116. doi: 10.1523/JNEUROSCI.0381-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.McGehee DS, Heath MJ, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 1995;269(5231):1692–1696. doi: 10.1126/science.7569895. [DOI] [PubMed] [Google Scholar]
- 202.Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383(6602):713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- 203.Maggi L, Le Magueresse C, Changeux JP, Cherubini E. Nicotine activates immature “silent” connections in the developing hippocampus. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(4):2059–2064. doi: 10.1073/pnas.0437947100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Le Magueresse C, Safiulina V, Changeux JP, Cherubini E. Nicotinic modulation of network and synaptic transmission in the immature hippocampus investigated with genetically modified mice. Journal of Physiology. 2006;576, part 2:533–546. doi: 10.1113/jphysiol.2006.117572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Dong WJ, Feng GF, Gong HL, Liu SH, Hu HT. Specific suppression of beta-secretase gene expression by short interfering RNA in mammalian cells. Sichuan Da Xue Xue Bao Yi Xue Ban. 2006;37(6):821–824. [PubMed] [Google Scholar]
- 206.Kao SC, Krichevsky AM, Kosik KS, Tsai LH. BACE1 suppression by RNA interference in primary cortical neurons. The Journal of Biological Chemistry. 2004;279(3):1942–1949. doi: 10.1074/jbc.M309219200. [DOI] [PubMed] [Google Scholar]
- 207.Singer O, Marr RA, Rockenstein E, et al. Targeting BACE1 with siRNAs ameliorates Alzheimer disease neuropathology in a transgenic model. Nature Neuroscience. 2005;8(10):1343–1349. doi: 10.1038/nn1531. [DOI] [PubMed] [Google Scholar]
- 208.Ghosh AK, Gemma S, Tang J. β-secretase as a therapeutic target for Alzheimer’s disease. Neurotherapeutics. 2008;5(3):399–408. doi: 10.1016/j.nurt.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Citron M. β-secretase inhibition for the treatment of Alzheimer’s disease—promise and challenge. Trends in Pharmacological Sciences. 2004;25(2):92–97. doi: 10.1016/j.tips.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 210.Luo X, Yan R. Inhibition of BACE1 for therapeutic use in Alzheimer’s disease. International Journal of Clinical and Experimental Pathology. 2010;3(6):618–628. [PMC free article] [PubMed] [Google Scholar]
- 211.Takahashi H, Fukumoto H, Maeda R, Terauchi J, Kato K, Miyamoto M. Ameliorative effects of a non-competitive BACE1 inhibitor TAK-070 on Aβ peptide levels and impaired learning behavior in aged rats. Brain Research. 2010;1361:146–156. doi: 10.1016/j.brainres.2010.09.032. [DOI] [PubMed] [Google Scholar]
- 212.Fukumoto H, Takahashi H, Tarui N, et al. A noncompetitive BACE1 inhibitor TAK-070 ameliorates Aβ pathology and behavioral deficits in a mouse model of Alzheimer’s disease. The Journal of Neuroscience. 2010;30(33):11157–11166. doi: 10.1523/JNEUROSCI.2884-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Zhu Z, Li C, Wang X, et al. 2,2′,4′-trihydroxychalcone from Glycyrrhiza glabra as a new specific BACE1 inhibitor efficiently ameliorates memory impairment in mice. Journal of Neurochemistry. 2010;114(2):374–385. doi: 10.1111/j.1471-4159.2010.06751.x. [DOI] [PubMed] [Google Scholar]
- 214.Chang WP, Huang X, Downs D, et al. β-secretase inhibitor GRL-8234 rescues age-related cognitive decline in APP transgenic mice. FASEB Journal. 2011;25(2):775–784. doi: 10.1096/fj.10-167213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Hussain I, Hawkins J, Harrison D, et al. Oral administration of a potent and selective non-peptidic BACE-1 inhibitor decreases β-cleavage of amyloid precursor protein and amyloid-β production in vivo. Journal of Neurochemistry. 2007;100(3):802–809. doi: 10.1111/j.1471-4159.2006.04260.x. [DOI] [PubMed] [Google Scholar]
- 216.Ghosh AK, Kumaragurubaran N, Hong L, et al. Potent memapsin 2 (β-secretase) inhibitors: design, synthesis, protein-ligand X-ray structure, and in vivo evaluation. Bioorganic & Medicinal Chemistry Letters. 2008;18(3):1031–1036. doi: 10.1016/j.bmcl.2007.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Nie HZ, Li ZQ, Yan QX, et al. Nicotine decreases beta-amyloid through regulating BACE1 transcription in SH-EP1-α4β2 nAChR-APP695 cells. Neurochemical Research. 2011;36(5):904–912. doi: 10.1007/s11064-011-0420-7. [DOI] [PubMed] [Google Scholar]
- 218.Hébert SS, Horré K, Nicolaï L, et al. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/β-secretase expression. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(17):6415–6420. doi: 10.1073/pnas.0710263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Takasugi N, Sasaki T, Suzuki K, et al. BACE1 activity is modulated by cell-associated sphingosine-1-phosphate. The Journal of Neuroscience. 2011;31(18):6850–6857. doi: 10.1523/JNEUROSCI.6467-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.O’Connor T, Sadleir KR, Maus E, et al. Phosphorylation of the translation initiation factor eIF2α increases BACE1 levels and promotes amyloidogenesis. Neuron. 2008;60(6):988–1009. doi: 10.1016/j.neuron.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.ILL-Raga G, Palomer E, Wozniak MA, et al. Activation of PKR causes amyloid ß-peptide accumulation via de-repression of bace1 expression. PLoS ONE. 2011;6(6) doi: 10.1371/journal.pone.0021456.e21456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Li YC, Chen Q, Wan XZ, Yang XL, Liu X, Zhong L. Effects of conjugated linoleic acid on cleavage of amyloid precursor protein via PPARγ . Neurological Sciences. 2011;32(6):1095–1101. doi: 10.1007/s10072-011-0711-4. [DOI] [PubMed] [Google Scholar]
- 223.Li Q, Wu D, Zhang L, Zhang Y. Effects of galantamine on β-amyloid release and β-site cleaving enzyme 1 expression in differentiated human neuroblastoma SH-SY5Y cells. Experimental Gerontology. 2010;45(11):842–847. doi: 10.1016/j.exger.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 224.Gray EH, De Vos KJ, Dingwall C, Perkinton MS, Miller CC. Deficiency of the copper chaperone for superoxide dismutase increases amyloid-β production. Journal of Alzheimer’s Disease. 2010;21(4):1101–1105. doi: 10.3233/JAD-2010-100717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Katsouri L, Parr C, Bogdanovic N, Willem M, Sastre M. PPARγ co-activator-1α (PGC-1α) reduces amyloid-β generation through a PPARγ-dependent mechanism. Journal of Alzheimer’s Disease. 2011;25(1):151–162. doi: 10.3233/JAD-2011-101356. [DOI] [PubMed] [Google Scholar]
- 226.Kang EL, Cameron AN, Piazza F, Walker KR, Tesco G. Ubiquitin regulates GGA3-mediated degradation of BACE1. The Journal of Biological Chemistry. 2010;285(31):24108–24119. doi: 10.1074/jbc.M109.092742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Tesco G, Koh YH, Kang EL, et al. Depletion of GGA3 stabilizes BACE and enhances β-secretase activity. Neuron. 2007;54(5):721–737. doi: 10.1016/j.neuron.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Sarajärvi T, Haapasalo A, Viswanathan J, et al. Down-regulation of seladin-1 increases BACE1 levels and activity through enhanced GGA3 depletion during apoptosis. The Journal of Biological Chemistry. 2009;284(49):34433–34443. doi: 10.1074/jbc.M109.036202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.De Strooper B. Aph-1, Pen-2, and nicastrin with presenilin generate an active γ-secretase complex. Neuron. 2003;38(1):9–12. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- 230.Sherrington R, Rogaev EI, Liang Y, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375(6534):754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 231.Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. Skeletal and CNS defects in presenilin-1-deficient mice. Cell. 1997;89(4):629–639. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- 232.Wong PC, Zheng H, Chen H, et al. Presenilin 1 is required for Notch 1 and Dll1 expression in the paraxial mesoderm. Nature. 1997;387(6630):288–292. doi: 10.1038/387288a0. [DOI] [PubMed] [Google Scholar]
- 233.De Strooper B, Saftig P, Craessaerts K, et al. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391(6665):387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- 234.Donoviel DB, Hadjantonakis AK, Ikeda M, Zheng H, Hyslop PS, Bernstein A. Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes & Development. 1999;13(21):2801–2810. doi: 10.1101/gad.13.21.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Marjaux E, Hartmann D, De Strooper B. Presenilins in memory, Alzheimer’s disease, and therapy. Neuron. 2004;42(2):189–192. doi: 10.1016/s0896-6273(04)00218-1. [DOI] [PubMed] [Google Scholar]
- 236.Levy-Lahad E, Wasco W, Poorkaj P, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269(5226):973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 237.Herreman A, Hartmann D, Annaert W, et al. Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(21):11872–11877. doi: 10.1073/pnas.96.21.11872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Tournoy J, Bossuyt X, Snellinx A, et al. Partial loss of presenilins causes seborrheic keratosis and autoimmune disease in mice. Human Molecular Genetics. 2004;13(13):1321–1331. doi: 10.1093/hmg/ddh151. [DOI] [PubMed] [Google Scholar]
- 239.Uemura K, Kitagawa N, Kohno R, et al. Presenilin 1 is involved in maturation and trafficking of N-cadherin to the plasma membrane. Journal of Neuroscience Research. 2003;74(2):184–191. doi: 10.1002/jnr.10753. [DOI] [PubMed] [Google Scholar]
- 240.Kopan R, Ilagan MX. γ-secretase: proteasome of the membrane? Nature Reviews Molecular Cell Biology. 2004;5(6):499–504. doi: 10.1038/nrm1406. [DOI] [PubMed] [Google Scholar]
- 241.Lleó A. Activity of γ-secretase on substrates other than APP. Current Topics in Medicinal Chemistry. 2008;8(1):9–16. doi: 10.2174/156802608783334060. [DOI] [PubMed] [Google Scholar]
- 242.Handler M, Yang X, Shen J. Presenilin-1 regulates neuronal differentiation during neurogenesis. Development. 2000;127(12):2593–2606. doi: 10.1242/dev.127.12.2593. [DOI] [PubMed] [Google Scholar]
- 243.Dewachter I, Reversé D, Caluwaerts N, et al. Neuronal deficiency of presenilin 1 inhibits amyloid plaque formation and corrects hippocampal long-term potentiation but not a cognitive defect of amyloid precursor protein [V7171] transgenic mice. The Journal of Neuroscience. 2002;22(9):3445–3453. doi: 10.1523/JNEUROSCI.22-09-03445.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Moechars D, Dewachter I, Lorent K, et al. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. The Journal of Biological Chemistry. 1999;274(10):6483–6492. doi: 10.1074/jbc.274.10.6483. [DOI] [PubMed] [Google Scholar]
- 245.Pratt KG, Zhu P, Watari H, Cook DG, Sullivan JM. A novel role for γ-secretase: selective regulation of spontaneous neurotransmitter release from hippocampal neurons. The Journal of Neuroscience. 2011;31(3):899–906. doi: 10.1523/JNEUROSCI.4625-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Zhang H, Sun S, Herreman A, De Strooper B, Bezprozvanny I. Role of presenilins in neuronal calcium homeostasis. The Journal of Neuroscience. 2010;30(25):8566–8580. doi: 10.1523/JNEUROSCI.1554-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends in Neurosciences. 2008;31(9):454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Supnet C, Bezprozvanny I. The dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium. 2010;47(2):183–189. doi: 10.1016/j.ceca.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Tu H, Nelson O, Bezprozvanny A, et al. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell. 2006;126(5):981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Gandy S, Doeven MK, Poolman B. Alzheimer disease: presenilin springs a leak. Nature Medicine. 2006;12(10):1121–1123. doi: 10.1038/nm1006-1121. [DOI] [PubMed] [Google Scholar]
- 251.Shilling D, Mak DO, Kang DE, Foskett JK. Lack of evidence for presenilins as endoplasmic reticulum Ca2+ leak channels. The Journal of Biological Chemistry. 2012;287:10933–10944. doi: 10.1074/jbc.M111.300491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Cheung KH, Mei L, Mak DO, et al. Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer’s disease-linked presenilin mutants in human cells and mouse neurons. Science Signaling. 2010;3(114):p. ra22. doi: 10.1126/scisignal.2000818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Smith IF, Hitt B, Green KN, Oddo S, LaFerla FM. Enhanced caffeine-induced Ca2+ release in the 3xTg-AD mouse model of Alzheimer’s disease. Journal of Neurochemistry. 2005;94(6):1711–1718. doi: 10.1111/j.1471-4159.2005.03332.x. [DOI] [PubMed] [Google Scholar]
- 254.Stutzmann GE, Smith I, Caccamo A, Oddo S, LaFerla FM, Parker I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. The Journal of Neuroscience. 2006;26(19):5180–5189. doi: 10.1523/JNEUROSCI.0739-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Chakroborty S, Goussakov I, Miller MB, Stutzmann GE. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. The Journal of Neuroscience. 2009;29(30):9458–9470. doi: 10.1523/JNEUROSCI.2047-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Goussakov I, Miller MB, Stutzmann GE. NMDA-mediated Ca2+ influx drives aberrant ryanodine receptor activation in dendrites of young Alzheimer’s disease mice. The Journal of Neuroscience. 2010;30(36):12128–12137. doi: 10.1523/JNEUROSCI.2474-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Stutzmann GE, Smith I, Caccamo A, Oddo S, Parker I, Laferla F. Enhanced ryanodine-mediated calcium release in mutant PS1-expressing Alzheimer’s mouse models. Annals of the New York Academy of Sciences. 2007;1097:265–277. doi: 10.1196/annals.1379.025. [DOI] [PubMed] [Google Scholar]
- 258.Supnet C, Grant J, Kong H, Westaway D, Mayne M. Amyloid-β-(1- 42) increases ryanodine receptor-3 expression and function in neurons of TgCRND8 mice. The Journal of Biological Chemistry. 2006;281(50):38440–38447. doi: 10.1074/jbc.M606736200. [DOI] [PubMed] [Google Scholar]
- 259.Cheung KH, Shineman D, Müller M, et al. Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of insP3 receptor channel gating. Neuron. 2008;58(6):871–883. doi: 10.1016/j.neuron.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Stutzmann GE, Caccamo A, LaFerla FM, Parker I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer’s-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. The Journal of Neuroscience. 2004;24(2):508–513. doi: 10.1523/JNEUROSCI.4386-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21(1):13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- 262.Green KN, Demuro A, Akbari Y, et al. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid β production. Journal of Cell Biology. 2008;181(7):1107–1116. doi: 10.1083/jcb.200706171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Leissring MA, Paul BA, Parker I, Cotman CW, LaFerla FM. Alzheimer’s presenilin-1 mutation potentiates inositol 1,4,5- trisphosphate-mediated calcium signaling in Xenopus oocytes . Journal of Neurochemistry. 1999;72(3):1061–1068. doi: 10.1046/j.1471-4159.1999.0721061.x. [DOI] [PubMed] [Google Scholar]
- 264.Herms J, Schneider I, Dewachter I, Caluwaerts N, Kretzschmar H, Van Leuven F. Capacitive calcium entry is directly attenuated by mutant presenilin-1, independent of the expression of the amyloid precursor protein. The Journal of Biological Chemistry. 2003;278(4):2484–2489. doi: 10.1074/jbc.M206769200. [DOI] [PubMed] [Google Scholar]
- 265.Leissring MA, Akbari Y, Fanger CM, Cahalan MD, Mattson MP, LaFerla FM. Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. Journal of Cell Biology. 2000;149(4):793–798. doi: 10.1083/jcb.149.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Smith IF, Boyle JP, Vaughan PF, Pearson HA, Cowburn RF, Peers CS. Ca2+ stores and capacitative Ca2+ entry in human neuroblastoma (SH-SY5Y) cells expressing a familial Alzheimer’s disease presenilin-1 mutation. Brain Research. 2002;949(1-2):105–111. doi: 10.1016/s0006-8993(02)02970-0. [DOI] [PubMed] [Google Scholar]
- 267.Yoo AS, Cheng I, Chung S, et al. Presenilin-mediated modulation of capacitative calcium entry. Neuron. 2000;27(3):561–572. doi: 10.1016/s0896-6273(00)00066-0. [DOI] [PubMed] [Google Scholar]
- 268.Tepass U, Truong K, Godt D, Ikura M, Peifer M. Cadherins in embryonic and neural morphogenesis. Nature Reviews Molecular Cell Biology. 2000;1(2):91–100. doi: 10.1038/35040042. [DOI] [PubMed] [Google Scholar]
- 269.Basi GS, Hemphill S, Brigham EF, et al. Amyloid precursor protein selective gamma-secretase inhibitors for treatment of Alzheimer’s disease. Alzheimer’s Research & Therapy. 2010;2(6, article 36) doi: 10.1186/alzrt60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Dovey HF, John V, Anderson JP, et al. Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. Journal of Neurochemistry. 2001;76(1):173–181. doi: 10.1046/j.1471-4159.2001.00012.x. [DOI] [PubMed] [Google Scholar]
- 271.Eriksen JL, Sagi SA, Smith TE, et al. NSAIDs and enantiomers of flurbiprofen target γ-secretase and lower Aβ 42 in vivo. The Journal of Clinical Investigation. 2003;112(3):440–449. doi: 10.1172/JCI18162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Imbimbo BP, Del Giudice E, Colavito D, et al. 1-(3′,4′-Dichloro-2-fluoro[1,1′-biphenyl]-4-yl) -cyclopropanecarboxylic acid (CHF5074), a novel γ-secretase modulator, reduces brain β-amyloid pathology in a transgenic mouse model of Alzheimer’s disease without causing peripheral toxicity. Journal of Pharmacology and Experimental Therapeutics. 2007;323(3):822–830. doi: 10.1124/jpet.107.129007. [DOI] [PubMed] [Google Scholar]
- 273.Pettersson M, Kauffman GW, Am Ende CW, et al. Novel γ-secretase modulators: a review of patents from 2008 to 2010. Expert Opinion on Therapeutic Patents. 2011;21(2):205–226. doi: 10.1517/13543776.2011.547479. [DOI] [PubMed] [Google Scholar]
- 274.Yan Q, Zhang J, Liu H, et al. Anti-inflammatory drug therapy alters β-amyloid processing and deposition in an animal model of Alzheimer’s disease. The Journal of Neuroscience. 2003;23(20):7504–7509. doi: 10.1523/JNEUROSCI.23-20-07504.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Imbimbo BP, Panza F, Frisardi V, et al. Therapeutic intervention for Alzheimer’s disease with γ-secretase inhibitors: still a viable option? Expert Opinion on Investigational Drugs. 2011;20(3):325–341. doi: 10.1517/13543784.2011.550572. [DOI] [PubMed] [Google Scholar]
- 276.Martone RL, Zhou H, Atchison K, et al. Begacestat (GSI-953): a novel, selective thiophene sulfonamide inhibitor of amyloid precursor protein γ-secretase for the treatment of Alzheimer’s disease. Journal of Pharmacology and Experimental Therapeutics. 2009;331(2):598–608. doi: 10.1124/jpet.109.152975. [DOI] [PubMed] [Google Scholar]
- 277.Kounnas MZ, Danks AM, Cheng S, et al. Modulation of γ-secretase reduces β-amyloid deposition in a transgenic mouse model of Alzheimer’s disease. Neuron. 2010;67(5):769–780. doi: 10.1016/j.neuron.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Wolfe MS. Inhibition and modulation of γ-secretase for Alzheimer’s disease. Neurotherapeutics. 2008;5(3):391–398. doi: 10.1016/j.nurt.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Comery TA, Martone RL, Aschmies S, et al. Acute γ-secretase inhibition improves contextual fear conditioning in the Tg2576 mouse model of Alzheimer’s disease. The Journal of Neuroscience. 2005;25(39):8898–8902. doi: 10.1523/JNEUROSCI.2693-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Dash PK, Moore AN, Orsi SA. Blockade of γ-secretase activity within the hippocampus enhances long-term memory. Biochemical and Biophysical Research Communications. 2005;338(2):777–782. doi: 10.1016/j.bbrc.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 281.Imbimbo BP, Hutter-Paier B, Villetti G, et al. CHF5074, a novel γ-secretase modulator, attenuates brain β-amyloid pathology and learning deficit in a mouse model of Alzheimer’s disease. British Journal of Pharmacology. 2009;156(6):982–993. doi: 10.1111/j.1476-5381.2008.00097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Kukar T, Prescott S, Eriksen JL, et al. Chronic administration of R-flurbiprofen attenuates learning impairments in transgenic amyloid precursor protein mice. BMC Neuroscience. 2007;8, article 54 doi: 10.1186/1471-2202-8-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298(5594):789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 284.Sastre M, Gentleman SM. NSAIDs: how they work and their prospects as therapeutics in Alzheimer's disease. Frontiers in Aging Neuroscience. 2010;2, article 20 doi: 10.3389/fnagi.2010.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Imbimbo BP, Solfrizzi V, Panza F. Are NSAIDs useful to treat Alzheimer’s disease or mild cognitive impairment? Frontiers in Aging Neuroscience. 2010;2, article 19 doi: 10.3389/fnagi.2010.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Galimberti D, Scarpini E. Alzheimer’s disease: from pathogenesis to disease-modifying approaches. CNS & Neurological Disorders—Drug Targets. 2011;10(2):163–174. doi: 10.2174/187152711794480438. [DOI] [PubMed] [Google Scholar]
- 287.Czirr E, Leuchtenberger S, Dorner-Ciossek C, et al. Insensitivity to Aβ42-lowering nonsteroidal anti-inflammatory drugs and γ-secretase inhibitors is common among aggressive presenilin-1 mutations. The Journal of Biological Chemistry. 2007;282(34):24504–24513. doi: 10.1074/jbc.M700618200. [DOI] [PubMed] [Google Scholar]
- 288.Page RM, Baumann K, Tomioka M, et al. Generation of Aβ38 and Aβ42 is independently and differentially affected by familial Alzheimer disease-associated presenilin mutations and γ-secretase modulation. The Journal of Biological Chemistry. 2008;283(2):677–683. doi: 10.1074/jbc.M708754200. [DOI] [PubMed] [Google Scholar]
- 289.Hahn S, Brüning T, Ness J, et al. Presenilin-1 but not amyloid precursor protein mutations present in mouse models of Alzheimer’s disease attenuate the response of cultured cells to γ-secretase modulators regardless of their potency and structure. Journal of Neurochemistry. 2011;116(3):385–395. doi: 10.1111/j.1471-4159.2010.07118.x. [DOI] [PubMed] [Google Scholar]