

Antisense oligonucleotides and short interfering RNAs (siRNAs) are nucleic acids-targeting reagents for gene expression modulation that are being developed as drugs for many applications. A number of useful synthetic nucleic acids analogues have been introduced recently to greatly improve their properties for use as therapeutics. However, their full effectiveness in cells and in vivo has often only been realized through development of suitable nonviral delivery systems. Among these, a range of natural and synthetic peptides have been found useful for enhancing cellular uptake and/or cell targeting of oligonucleotide analogues and siRNA. Such peptides are synthetically conjugated, used as noncovalent complexes, or used in combination with polymer, liposomal or exosome formulation techniques. This review begins by describing the modes of action of antisense reagents and siRNA and goes on to focus on recent advances in their peptide-mediated cell and in vivo delivery and how peptide use has influenced drug development. The review discusses the challenges associated with understanding the physiological and toxicological aspects of peptide-mediated delivery. Developments towards clinical use are also highlighted, with particular emphasis on peptide conjugates of oligonucleotide analogues used for treatment of neuromuscular diseases.
Introduction
Antisense oligonucleotides are used widely to interfere with biological processes in cells and are being developed in some cases as drugs.1,2 There are numerous types of oligonucleotide analogue of varying chemistries, as well as short interfering RNAs (siRNAs), and they encompass a range of intracellular targets and modes of action. Common to all oligonucleotide types is the need to be delivered into cells and tissues efficiently in order to carry out their targeting functions. Some types are able to enter certain cell classes in vivo (such as hepatocytes or kidney cells) reasonably efficiently in naked form (e.g., those containing both phosphorothioate (PS) linkages and certain sugar modifications),3 but bio-availability at the right cellular location may still be limited by poor cell trafficking and endosomal entrapment. In other cell types, cell targeting and entry is found to be poor for many naked oligonucleotide types, or their ability to reach the desired tissue is limited. Accordingly, there have been massive efforts to search for new delivery systems that may enhance tissue penetration, improve cell targeting and cell entry, as well as enhance intracellular bioavailability at the desired biological target. The most prevalent delivery systems for oligonucleotides and siRNA are cationic liposomes and other nanoparticle delivery systems, based initially on cell culture studies and later translated into sophisticated multicomponent in vivo vectors.4,5
Among the simpler methods that have been utilized to enhance cell delivery of oligonucleotides and siRNA are cationic peptide vectors, often referred to as cell-penetrating peptides (CPPs) or peptide transduction domains (PTDs). The first CPP was introduced in 19946 and since then there has been a continuous stream of new peptide delivery vectors where increased delivery and better pharmacological properties are claimed and a variety of applications demonstrated.7 The cellular entry and intracellular trafficking mechanisms behind peptide-mediated transduction are still not fully elucidated but are reviewed thoroughly elsewhere.8,9,10,11 In addition to CPPs, there are peptides designed to target to specific cells or tissues (homing peptides).12 This review concentrates on recent progress (since 2007) in all of the various peptide-mediated strategies for delivery of antisense oligonucleotides and siRNA into cells in culture and also in in vivo applications, and points to prospects for clinical development.
Modes of Action of Oligonucleotide and siRNA Therapeutics
There are several ways that short oligonucleotides and siRNA can affect cellular processes, schematically illustrated in Figure 1. The most obvious approach, originally proposed by Zamecnik and Stevenson,13 is antisense binding through Watson-Crick base pairing to messenger RNA (mRNA) to block translational initiation or elongation and thus decrease the levels of the corresponding protein (Figure 1a). Steric blocking of ribosomal elongation is known to be hard to achieve, but blocking of ribosomal initiation can be efficient by targeting the initiator AUG region, the 5′-cap region, or, where appropriate, an internal ribosome entry site. It is also possible to block polyadenylation.14 Such targeting requires delivery of oligonucleotides in stoichiometric amount to that of the targeted mRNA present in the cell at a given time. Steric blocking also requires the use of strongly binding oligonucleotide analogues to ensure good competition for the mRNA with the proteins involved in translation or other cellular processes.
Figure 1.

Schematic illustration of how synthetic oligonucleotides affect cellular processes. (a) Complementary oligonucleotide binds to mRNA and sterically blocks translation initiation or elongation. (b) Complementary oligonucleotide binds to mRNA and recruits RNase H. (c) siRNA cell internalization leads to one strand binding to mRNA and triggering RISC-dependent RNA cleavage. (d) A complementary oligonucleotide binds to the sense strand of a miRNA and blocks RISC activation. (e) An oligonucleotide masks the miRNA binding site on the mRNA and prevents RISC-mediated mRNA degradation or translation inhibition. (f) A splice site is masked on the mRNA, which results in exon exclusion. (g) A splice site is masked on the mRNA, which results in exon inclusion. mRNA, messenger RNA; miRNA, microRNA; RISC, RNA-induced silencing complex; siRNA, short interfering RNA.
Similarly to steric-blocking mRNA binding, some oligonucleotides are also capable of binding in triplex mode to certain specific DNA sequences and inhibiting transcription in the nucleus, leading to a decrease in mRNA production,15 but difficulties in obtaining sufficient potency when targeting chromosomal DNA selectively make gene expression inhibition through triplex formation currently less attractive as a therapeutic intervention mechanism. However, sequence-specific gene modification via triplex formation is a new subject that has recently shown great potential.16
A second mechanism involves antisense binding to RNA followed by recruitment and activation of RNase-H, which consequently leads to degradation of the mRNA target (Figure 1b). This mode of action is limited to those oligonucleotides containing a minimum stretch of 6–10 unmodified DNA or PS DNA units (known as gapmers), but outer nucleotides can be more heavily modified to enhance complementary RNA binding. In principle, catalytic turnover allows a lower concentration of oligonucleotides to be used compared to steric-blocking oligonucleotides. RNase-H–dependent antisense has been used very successfully to reduce expression levels of specific proteins, or to inhibit viral replication.17,18
In the case of RNA interference, one strand of an siRNA duplex (antisense or guide strand) becomes targeted to the mRNA as a result of recruitment by the RNA-induced silencing complex (RISC) cellular machinery and leads subsequently to RNA cleavage (Figure 1c). The siRNA can be targeted to any accessible part of the mRNA, similar to the RNase-H–dependent oligonucleotide targeting method. The sense (passenger) strand may include chemically modified nucleotides in any position, whereas the antisense strand only tolerates a limited range of modified nucleotides in specific locations.
Single-stranded oligonucleotide analogues have shown great promise as inhibitors of micro-RNA (miRNA) action by preventing miRNA binding to mRNA that would otherwise trigger the RNA interference machinery and result in mRNA downregulation or degradation (Figure 1d). Interestingly, good levels of intracellular activity have been achieved without requiring CPP-assisted delivery both in cell culture and in vivo (e.g., for peptide nucleic acid (PNA) and locked nucleic acid (LNA)),19,20 in contrast to naked oligonucleotides targeting mRNA, where high concentrations and extended periods are required in cell culture for RNase-H–dependent activity when delivered gymnotically (from Greek: gymnos, meaning naked).21 Oligonucleotides targeting miRNAs have been shown to rapidly enter cells and interfere with miRNA activity at submicromolar concentrations.19,22 Current thinking is that the oligonucleotide may meet the miRNA somewhere within the endosomal pathway,20 and therefore CPP-enhanced endosomal release may be unnecessary, although the generality of this conclusion for all cell types and miRNAs is yet to be established. Inhibition of miRNA is thought to occur by miRNA sequestration rather than by degradation.20 Therapeutic applications of anti-miRs have been recently reviewed.23 An alternative mechanism has also proven successful, where a short oligonucleotide binds the mRNA to cover the miRNA-binding site to again prevent RISC binding and subsequent mRNA downregulation (Figure 1e).24
Another important mode of action of antisense oligonucleotides involves splicing redirection of pre-mRNA in the cell nucleus. An oligonucleotide directed to regions at, or close to, a splice site can mask normal or aberrant splicing events leading either to exon exclusion (Figure 1f) or exon inclusion (Figure 1g). Splicing redirection model systems have been used very successfully to monitor peptide-mediated oligonucleotide delivery.25,26 In addition, the methodology is already being assessed in the clinic for treatment of neuromuscular and neurodegenerative diseases where targeting of specific exons in mutated dystrophin pre-mRNA with antisense phosphorodiamidate morpholino oligonucleotides (PMO) allows redirection of the splicing pattern to restore a correct reading frame (exon skipping), resulting in the regeneration of functional dystrophin protein expression.27 Stimulation of exon inclusion by antisense oligonucleotides is an alternative mechanism being examined for potential treatment for spinal muscular atrophy.28 In addition, exon skipping can be applied to wild-type genes to generate out-of-frame transcripts as a method of normal protein downregulation.29
Types of Oligonucleotide Analogue
In addition to low bioavailability, the poor stabilities of unmodified DNA and RNA and rapid clearance from the bloodstream have been major drawbacks for use as therapeutics. Thus, many different oligonucleotide analogues have been developed in order to increase stability, RNA binding, and other therapeutically useful properties. A selection of important oligonucleotide analogues that have been involved with peptide-mediated delivery is presented in Figure 2. Each analogue type has particular properties that make them suitable for different applications.2,30
Figure 2.

A selection of common oligonucleotide analogues delivered by peptide-mediated means. (a) Phosphorothioate (PS) linkages, (b) 2′-O-Methyl oligonucleotide (2′-OMe), (c) locked nucleic acid (LNA), (d) Phosphorodiamidate morpholino oligonucleotide (PMO), and (e) peptide nucleic acid (PNA).
Perhaps the most utilized and well-studied analogue is the PS linkage where a sulfur atom replaces one non-bridging oxygen atom in the phosphate backbone (Figure 2a). The PS linkage can be included in any DNA- or RNA-type oligonucleotide and increases nuclease resistance, whereas in an otherwise unmodified DNA it permits RNase-H activation. Introduction of the PS linkage reduces RNA-target affinity somewhat but leads to enhanced interaction with plasma proteins, thereby decreasing renal clearance rates.31 Despite some concerns as to possible toxic side effects at higher concentrations, PS linkages are included in all FDA-approved oligonucleotides to date, which unfortunately are currently very few.32,33
The 2′-O-methyl nucleotide (2′-OMe), in which a methyl group replaces a hydrogen atom in the 2′-hydroxyl group in the ribose ring of RNA (Figure 2b), also imparts nuclease resistance, but does not permit RNase-H activation. Such non-RNase-H inducing oligonucleotides are particularly valuable in certain applications where the target RNA needs to remain intact (e.g., splicing redirection). Although the 2′-OMe modification is insensitive to endonucleases, it is still partially susceptible to exonuclease degradation. By combining PS linkages and 2′-OMe nucleotides (PS-2′-OMe), much greater in vivo stability has been achieved resulting in several successful applications. Further enhancements of therapeutic activity have been achieved in some cases by including other 2′-O-alkylated nucleotides (such as 2′-O-methoxyethyl, MOE) or 2′-fluoro-2′-deoxynucleotides, resulting in “mixmers”. All these analogues are also used in the wings of gapmers, as well as in particular positions of siRNA.
A highly successful nucleotide analogue is LNA, which is a constrained RNA analogue having a methylene bridge between the 2′ and 4′ positions in the ribose ring (Figure 2c). LNA is also unable to activate RNase-H, but due to the constrained backbone LNA has a very high affinity for single-stranded DNA/RNA compared to other analogues. In addition to high affinity, LNAs display high in vivo stability and slower renal clearance, although in rare cases hepatotoxicity has been observed.34 The increased affinity allows LNA to be used in much shorter oligonucleotide than for many other analogue types (tiny LNAs,).35 However, nonspecific binding of longer sequences of LNAs, which could result in off-target effects, is alleviated by using LNA in combination with unmodified DNA or with other analogues, such as 2′-OMe, in steric-blocking applications. LNA is also used in RNase-H–dependent applications in the wings of all PS gapmers where the central section is PS-DNA.
A separate class of analogues are charge neutral. In PMO, the ribose is replaced by a morpholino moiety and the natural phosphodiester linkage is replaced by an uncharged phosphorodiamidate backbone (Figure 2d). The constrained structure and the lack of electrostatic repulsion between the oligonucleotide and its target both contribute to make PMO–RNA interactions more stable compared to DNA–RNA interactions. PMO does not activate RNase-H, but it has been used successfully for steric block antisense purposes, in many in vivo applications and in the clinic.36,37 Like PMO, PNA is an uncharged oligonucleotide analogue, where the sugar-phosphate backbone is replaced by repeating N-(2-aminoethyl)-glycine units linked together by amide bonds (Figure 2e). PNA has very high affinity for DNA/RNA that, like PMO, is due partly to the lack of charge repulsion of the peptide-like backbone. PNAs display high selectivity and mismatch discrimination towards its target strand and have been used effectively in many antisense applications and in vivo.16,38 Further, PNA containing a few additional cationic residues (e.g., lysine) have been used successfully to block miRNA activity without the use of a transfection agent or delivery peptide.19,20
The need for improved oligonucleotide therapies has accelerated advances in chemistry and many newer analogues have emerged recently. Unfortunately very few have been able to attain high stability, affinity, and selectivity, while maintaining low toxicity and off-target effects and which are available to researchers in sufficient quantities and reliability for in vivo studies. The analogues selected for mention in this review are those that not only have most of these above attributes but also have been most utilized in peptide-mediated delivery.
Strategies For Peptide-Mediated Delivery
There are two main ways of utilizing peptide vectors for the delivery of antisense oligonucleotides and siRNA. The first method involves chemical synthesis of peptides that are modified to enable covalent conjugation to oligonucleotides or other cargo types (Figure 3a). Such conjugation reactions may be carried out either on solid support or in solution, and often result in high yields. An advantage of covalent conjugation is that this results in a well-defined single entity that simplifies drug development. Linker types may be chemically stable, which ensures that the peptide–oligonucleotide conjugate remains intact throughout in vivo administration and the subsequent delivery process and that degradation now reflects only the proteolytic susceptibility of the particular peptide. There are numerous conjugation strategies, including thioether, thiol-maleimide, ester formation, and “click” chemistry (reviewed in ref. 39,40).
Figure 3.

Schematic illustration of formulation strategies for peptide-mediated oligonucleotide delivery. (a) Covalent conjugation between peptide vector and oligonucleotide via a stable or cleavable linker. (b) Complex formation between peptide and oligonucleotides through electrostatic and/or hydrophobic interactions. (c) Complex formation between lipid-conjugated peptide and oligonucleotides through electrostatic and/or hydrophobic interactions. (d) Oligonucleotide condensation by peptide-functionalized cationic polymers. (e) Lipid vesicle or exosome loaded with oligonucleotides and functionalized with a CPP. (f) Lipid vesicle or exosome loaded with oligonucleotides and functionalized with a targeting/homing peptide. CPP, cell-penetrating peptide; ON, oligonucleotide; siRNA, short interfering RNA.
Alternatively, a labile linker cleavable within the cell, such as a disulphide linkage, has proven very successful in studies of peptide-mediated delivery, both in cells and in vivo.41,42 Such disulphide bridges may remain sufficiently stable, if administration is rapid, but may be cleaved later when the conjugate reaches the reducing environment of the endosome/cytoplasm. This approach was initially favored in therapeutic design, because cleavage diminishes the risk of any detrimental effect of the peptide on the interaction of the oligonucleotide with its target. However, recent in vivo applications using stable linkages suggest that peptide-cargo cleavage is not essential. Recent studies have suggested also that thiol or disulfide groups might enhance cellular uptake, although the reasons for this are not as yet fully understood.43,44
Although covalent conjugation is very suitable for charge-neutral oligonucleotide analogues, there are technical difficulties in conjugation and purification of conjugates of highly cationic peptides with negatively charged oligonucleotides that have limited the type of peptide that can be conjugated.45 Sadly, meaningful comparative data on the effects of different peptide-oligonucleotide linkages in biological antisense assays are mostly lacking.
The second method of peptide-mediated oligonucleotide delivery exploits the complexing properties of CPPs and their derivatives (Figure 3b). For example, cationic CPPs can form complexes efficiently with negatively charged oligonucleotides.8 Further, some peptide vectors also contain hydrophobic elements (hydrophobic amino acids or addition of hydrophobic amino acids and/or other moieties such as fatty acids, lipids or cholesterol), which are designed for complex formation with both negatively charged and charge-neutral analogue types. Such complexing peptides form nanosized particles together with the oligonucleotides that are better able to translocate across the plasma membrane (in some cases perhaps avoiding the endosomal uptake system altogether)46 and can deliver the oligonucleotide cargo to the target with greater efficiency.
It is common to optimize empirically the ratio of the peptide over cargo to obtain the best cellular delivery. The noncovalent strategy does not require the oligonucleotide to be end-modified for conjugation, and thus complex formation is often achieved by simple mixing. Complex formation also results in protection of the oligonucleotide or siRNA from nuclease degradation. On the other hand, structures of varying and undefined size may be generated, which complicates drug characterization and subsequent in vivo use. In the cases of uncharged PMO or PNA, it has been common to first hybridize to a complementary DNA strand (leash) before complex formation with cationic peptide vectors.47
Recently, a new generation of chemically modified peptide vectors for noncovalent conjugation has been introduced (Figure 3c), based on the original work of Futaki et al. that involved stearylation of cationic poly-arginine peptides as delivery vectors for plasmid DNA.48 For example, a stearylated peptide showed noncovalent complex formation and enhanced cell delivery of a 2′-OMe splice-redirecting oligonucleotide.49 It seems that the peptide sequence needs to be carefully tuned to the type of cargo as well as to the application, since for example a stearylated poly-arginine peptide was found to be ineffective in cellular delivery of a splice-correcting oligonucleotide, despite being effective with a larger plasmid.
Other nanovectors involve peptides associated with cationic polymers (Figure 3d) or peptides complexed/cell surface displayed on liposomes or exosomes (Figure 3e). In addition to CPPs designed for general enhancement of cell entry, peptides can also be used to target specific cell surface receptors or tissues (homing peptides). These can be conjugated to an oligonucleotide by themselves (Figure 3a) or incorporated as a chimera with a CPP, or embedded on the surface of a nanovector (Figure 3f). This last concept combines the cell-penetrating or cell-targeting properties of a peptide with oligonucleotide or siRNA encapsulation technology (e.g., liposomes or exosomes) and this has recently opened up new possibilities for cell delivery.
Just as for any type of delivery agent, the advantages gained over a naked oligonucleotide or siRNA in use of a peptide vector (whether conjugate or complex) in enhancing cell and/or in vivo delivery must outweigh the disadvantages of the additional size of the molecule and the increased potential cost per gram. In the case of siRNA, for all except kidney and topical applications, it is clear that a complexation vector of some sort is required to obtain any significant in vivo activity. This is partly because the vector protects the siRNA from rapid degradation as well as helps the siRNA to passage into the cell and be released from endosomes. A simple peptide-based complexation vector is attractive compared to the better known but more complex lipid-based or polymeric nanoparticle vectors in terms of simplicity and cost of components. Here the question is more as to whether a peptide vector can be found to offer sufficient in vivo delivery power or tissue targeting compared to the other more sophisticated types of delivery vectors.
By contrast in the case of oligonucleotides, the advantages of using a peptide vector are fully dependent on the organ type and the application. For liver in vivo applications for example, there is already good delivery of naked PS oligonucleotides and a peptide vector seems unlikely to add more than a marginal benefit at best. However, delivery to muscle for example with naked oligonucleotide analogues, including PMO and PNA, requires very high dosages in animals to obtain any activity (see for example the muscular dystrophy applications described later) and here the substantial improvement in in vivo activity afforded by covalent peptide conjugation easily outweighs the added cost and effort of conjugation. In the examples that follow, readers should bear in mind that there are still relatively few peptide vectors currently being considered for therapeutic use and few if any publications have addressed toxicology, potential immune recognition, or demonstrated cost/benefit analysis. Most of the vectors describing enhanced cell delivery represent therefore research in progress.
Peptide Conjugates and Complexes For Antisense Oligonucleotide and siRNA Applications
Peptide-mediated delivery of RNase-H–dependent antisense oligonucleotides into cells in culture was first shown in 1994 through covalent CPP conjugation.50,51 The covalent CPP attachment concept was later used for steric-blocking PNA in downregulation of the galanin receptor in vivo.41 Since then many demonstrations of antisense and siRNA cell delivery by CPPs have been published. Tables 1–4 show some recent examples. The choice of antisense mechanism or siRNA is dependent on the particular application and this in turn influences the type of peptide delivery system available. In cases where the particular RNA target permits a choice of targeting methods available, the generally lower potency of steric-blocking oligonucleotides has to be balanced against the greater possibility of off-target effects in RNase-H–dependent or RISC-dependent activities. These effects may arise for example from binding of an oligonucleotide to incorrect target sequences that result in unwanted RNA cleavages, to miRNA-like effects of siRNA passenger strands, or to other cellular effects such as immune activation. Peptide-mediated delivery often results in raised levels of cellular activity but it could also lead to increases in off-target effects (compare use of cationic liposomes to enhance delivery which have suffered from the same problems). Whereas potency increases have been frequently demonstrated, off-target effects have rarely been measured systematically where peptide vectors have been used, and it is common to find only simple cell toxicity assays carried out. This limits the ability to compare methods effectively.
Table 1. Selection of peptide–oligonucleotide conjugates and complexes used for mRNA targeting.
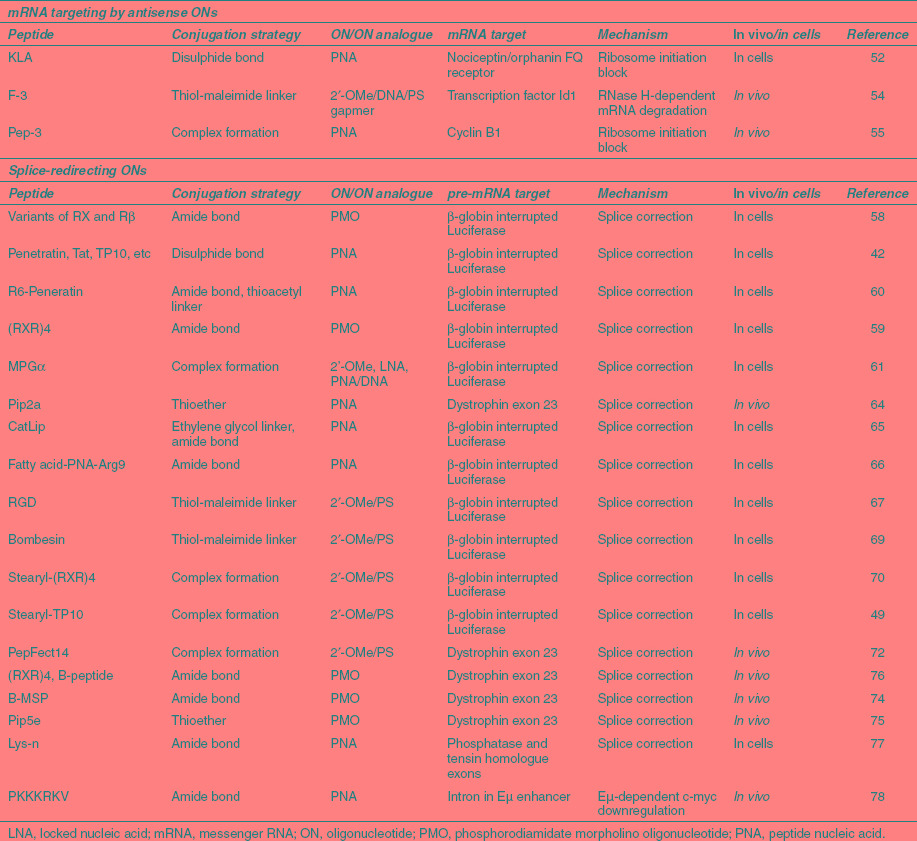
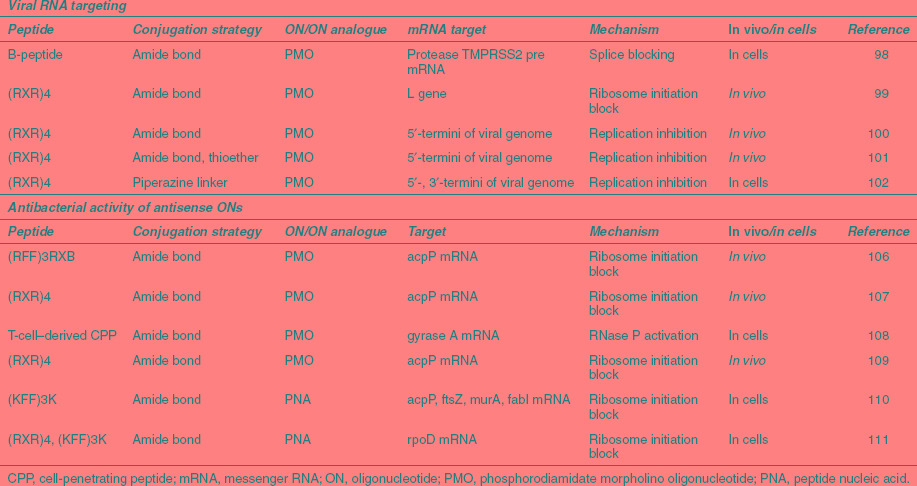
Table 4. Selection of peptide–oligonucleotide conjugates and complexes with antiviral and antibacterial activity.
mRNA targeting by antisense oligonucleotides
There are only a few recent examples of peptide conjugates of oligonucleotides used for targeting mRNA (Table 1). A variety of CPPs of cationic or amphipathic origin disulfide-conjugated to a 12-mer PNA targeting the nociceptin/orphanin FQ receptor mRNA were studied for receptor downregulation in cardiomyocytes.52 Among these CPPs, a repeating KLA amphipathic peptide was found to have the highest activity as a PNA conjugate in the absence of transfection agent, but no comparison was made with Arg-rich peptides. Interestingly, there was found no correlation of bioactivity with the level of fluorescence uptake of labeled conjugates, confirming previous studies with peptide-PNAs.53 Further, receptor downregulation was improved through CPP conjugation by a relatively modest amount. The significant phenotypic activity for naked PNA and lack of clear quantitative link between phenotypic and genotypic antisense activity calls into question as to the extent mRNA antisense behavior is truly observed in this model.
Perhaps the most promising example of mRNA targeting involves an RNase-H–dependent gapmer 2'-OMe/DNA/PS oligonucleotide that targets the transcription factor Id1 which was coupled to the F-3 31-mer peptide, a fragment of the high mobility group protein N2 which homes to neovessels in xenograft tumors.54 The conjugate showed substantial inhibition of tumor growth and metastasis in a mouse tumor xenograft model. The peptide has been shown to bind to nucleolin, which is overexpressed on the surface of cancer cells and is subsequently transported into the nucleus. It is likely that the cationic nature of the peptides may result in aggregation of this conjugate to form nanoparticles in solution that leads to partial protection against degradation in serum and which perhaps improves cell and in vivo activity. Although delivery of the oligonucleotide conjugate by the homing peptide into the tumor endothelium and dose-dependent Id1 protein knockdown was observed, both in cells and in vivo, surprisingly sequence- and dose-dependent downregulation of the corresponding Id1 mRNA was not reported, and thus there remains the question as to whether the biological activity observed was fully due to a sequence-specific antisense effect in this study. Nevertheless the strong in vivo activity observed is undeniable.
Among peptide-complex delivery strategies, a PEGylated amphipathic peptide Pep-3 was shown to mediate in vivo delivery of a PNA analogue oligonucleotide targeting cyclin B1 mRNA following intravenous injection through the formation of nanoparticles, and this resulted in reduction in rate of PC3 tumor growth.55 Pep-3 is a short 15-mer peptide that combines lysine/arginine-rich hydrophilic domains and tryptophan/phenylalanine hydrophobic residues to form stable complexes with the antisense oligonucleotide. PEGylation was necessary for in vivo activity. In this case the effect is steric block, targeting close to the AUG start sequences, and sequence- and dose-dependence was well validated. A 20-fold excess of Pep-3 over oligomer was needed for optimal cell delivery, whereas in vivo a layer of PEGylated peptide was added after initial complexation with unmodified peptide to enhance delivery. Further Pep derivatives have been developed for siRNA (see below).
Splice-redirecting oligonucleotides
Splicing redirection has been used extensively for measuring the efficacy of peptide-mediated cell delivery of oligonucleotides. This is due to a very convenient readout system where HeLa cells are stably transfected with a luciferase gene that is interrupted by a mutated β-globin intron.56 The mutation causes aberrant splicing of luciferase pre-mRNA, thus preventing translation. Oligonucleotides that target particular sequences at or close to the aberrant splice site (e.g., the “705” site) induce correct splicing and thus restore luciferase activity. A similar construct involves use of green fluorescent protein (GFP) in place of luciferase.57 The reliable positive readout and large dynamic range of the method allows the efficacy of different peptide vectors, conjugation strategies, and analogue types to be compared.58 The assay is well validated for many analogue types and delivery methods and sequence-specificity is not in doubt for such studies.
Further, the HeLa 705 assay has served as a model system to help elucidate cellular uptake mechanisms.42 In general, CPP conjugates are delivered through endocytosis and the CPP is believed to help the release from endosomes.7,59,60 The uptake mechanisms of peptide-complexed oligonucleotides appear to be more diverse. Some peptide complexing agents such as MPGα appear to utilize endocytosis,61 whereas for other similar peptides a non-energy–dependent membrane-crossing pathway is proposed.62 To obtain splicing redirection, the oligonucleotide must be delivered into the cell nucleus. Debate continues as to whether endocytosis or energy-independent mechanisms of delivery are optimal in such applications in terms of bioavailability, which is known to be poorly correlated with cell uptake.42,61
In recent studies of conjugates of charge-neutral antisense PNA and PMO, earlier Penetratin and Tat transduction domains have now been replaced by Arg-rich CPPs (Table 1). In the paradigm peptides, Arg residues are spaced by aminohexanoyl (Ahx or X) and/or β-alanyl (β) moieties to enhance hydrophobicity and proteolytic resistance in serum, such as (R-Ahx-R)4-Ahx-βAla and (R-Ahx-R-R-βAla-R)2-Ahx-βAla called RXR4 and B-peptide respectively.36,63 The aminohexanoyl spacer in the R-Ahx-R motif was found to be optimal in the splicing-redirection assay among several other spacers tried.59 Based on an initial discovery, that the addition of six Arg residues to the Penetratin peptide (R6Pen) resulted in significantly enhanced splicing redirection of PNA in HeLa 705 cells,60 a series of peptides known as Pip (PNA/PMO internalizing peptides) was developed that include a hydrophobic core flanked by two Arg-rich regions, which when attached to PNA maintain good splicing redirection at submicromolar concentrations, yet are more serum stable and therefore usable in vivo.64
Another recent idea is to add a second chemical moiety such as a fatty acid to the end of an Arg-rich peptide to enhance delivery of the PNA conjugate.65 It was found that a decanoic acid was the optimal chain length to balance increased splicing redirection obtained with concurrent increases in cell toxicity as the chain length was extended. In a separate study, the addition of a C14 palmitoyl lipid chain to the free terminus of the PNA in a R9-PNA conjugate also increased splice-redirecting activity, and it was suggested that the increased activity was due to micelle formation.66
A conjugate between a 2′-OMe/PS antisense oligonucleotide and a bivalent RGD peptide, selectively targeting αvβ3 integrin, was found to enter cells via receptor-mediated endocytosis and was used for splicing redirection in melanoma cells.67 The RGD conjugate was found to enter cells by an endocytotic pathway distinct from that of naked oligonucleotide.68 Recently, a splice-redirecting antisense oligonucleotide was conjugated to bombesin peptide and its intracellular delivery was studied in gastrin-releasing peptide receptor expressing PC3 cells, which was transfected with a luciferase gene interrupted by an abnormally spliced intron. The results suggest that the conjugate enters cells via a process of gastrin-releasing peptide receptor-mediated endocytosis followed by trafficking to deep endomembrane compartments, leading to significantly higher luciferase expression compared to unmodified oligonucleotides.69
The principle of using a peptide targeted to a cell surface receptor, particularly an overexpressed marker on cancer cells, is an excellent one. The issue lies in whether there is sufficient molecular recognition of a peptide conjugated to an individual oligonucleotide to deliver enough through a specific receptor-mediated endocytotic pathway to provide a large boost in splicing redirection that has relevance in an in vivo setting. Demonstrations of quantitatively useful peptide-mediated delivery in cells and in vivo are awaited eagerly.
Various types of stearylated peptide have been used to aid redirection of splicing using the noncovalent strategy. Stearyl-RXR4 peptide at 5:1 ratio compared to 2′-OMe/PS oligonucleotide was much more effective at luciferase upregulation than stearyl-Arg9 in HeLa 705 cells.70 More effort has gone into developing stearyl derivatives of the peptide Transportan 10 (TP-10) as nucleic acid delivery agents.49 In general, this class of stearylated peptide delivery agent seems to utilize the endocytotic pathway.71 PF14, a peptide derivative of TP-10 containing pairs of ornithine as well as pairs of leucine residues, showed particularly good splicing redirection for 2′-OMe/PS oligonucleotides both in the HeLa 705 cell assay as well as in exon skipping in mouse mdx muscle cells.72
CPP-aided splicing redirection is also a promising mechanism for in vivo and therapeutic applications. The mdx mouse model of Duchenne muscular dystrophy (DMD), bearing a nonsense mutation in exon 23 of the dystrophin gene, is an important in vivo system for evaluation of peptide-delivery methods. Exon skipping with oligonucleotide analogues targeting exon 23 restores the transcript reading frame and hence production of a shorter but active dystrophin protein. RXR4-PMO and B-PMO conjugates showed dramatically improved dystrophin production, compared to naked PMO, following intravenous injection in the mdx mouse.27,63,73 PMO conjugated to a chimeric CPP comprising the B-peptide and a short muscle-specific peptide, known as B-MSP, showed higher activity in skeletal tissue in mdx mice than B-PMO.74 An improved Pip series Arg-rich peptide (Pip5e) conjugated to PMO showed enhanced dystrophin production in both skeletal muscles and heart using a single 25 mg/kg injection.75 This is a significant development, because, together with respiratory difficulties, cardiac deterioration is a major cause of death in DMD patients.
Peptide-PNA conjugates have also been used for in vivo antisense studies targeting DMD,76 as well as other targets. For example, it was reported recently that an amphipathic peptide covalently conjugated to a PNA was able to redirect splicing of the murine PTEN primary transcript in adipose tissue of male Balb/c mice after a low-dose intraperitoneal injection of 2.5 mg/kg.77 The study showed a clear dependence of the levels of PNA antisense activity in adipose and other tissues depending on the peptide carrier sequence.
Enhancement of splicing redirection by PNA or PMO is clearly a well-suited application of covalently attached CPPs and potentially other peptide types, and further peptide vector development here is likely. For negatively charged splice-directing oligonucleotides, good cell delivery enhancement has been observed in several cases by complexing with peptide vectors, but this has yet to translate into a sufficient advantage in vivo over naked oligonucleotides.
A cautionary tale serves as a reminder that not all intron-targeting leads to clear interpretation of observed biological activity. For example, a PNA targeted to an intronic sequence in a severe combined immunodeficiency mouse model of Burkitt's lymphoma conjugated to a Lys-rich nuclear localization signal peptide was shown to be effective in blocking tumor growth through multiple intravenous injections.78 This was a surprising result, because the Lys-rich peptide is very likely to be degraded rapidly in the circulation and thus insufficiently intact to act as an nuclear localization signal. Such peptide conjugation may instead alter the bioavailability of the PNA. No other peptide conjugates were examined. Further, the mechanism of action of the PNA in this case is obscure. The PNA was targeted to the Eμ enhancer sequence, which regulates c-myc overexpression. PNA binding may perhaps alter RNA secondary structure to downregulate c-myc expression, rather than altering splicing patterns.
Gene targeting PNA
Three recent examples of antigene PNA (AgPNA) targeting are described (Table 2). AgPNAs conjugated to Lys-rich or Arg-rich short peptides have shown some effectiveness in targeting the progesterone receptor promoter DNA in breast cancer cells resulting in downregulation of receptor protein production, but to achieve this activity, cells needed to be treated with µmol/l concentrations of AgPNA for several days.79 Further, it is not clear whether targeting was truly obtained at the DNA level or instead on short RNAs derived from transcripts at the promoter site. A perhaps more promising target for PNA conjugated to the peptide Lys8 is the repeated CAG sequences in the mRNA of huntingtin to block mutant expression selectively in cells, which would be a potential approach to treat Huntington's disease.80 However, there are alternative promising methods of targeting announced from the same laboratory involving use of other oligonucleotide analogues, siRNAs and miRNA mimics that may turn out to have higher allele-selectivity,81 and in vivo studies are awaited eagerly.
Table 2. Selection of peptide–-oligonucleotide conjugates and complexes used for anti gene targeting.

An interesting new application is the use of triplex-forming PNA conjugated with the classic CPP Penetratin to direct gene modification in the hematopoietic stem cells of mice, retaining unmodified differentiation capabilities.16 Sequence-specific gene modification was also observed in multiple somatic tissues following systemic administration, and also preserved in bone marrow and spleen of recipient mice following transplantation of bone marrow from Penetratin-PNA–treated mice. This suggests that peptide–PNA conjugates might have good potential application for treatment of monogenic hematological diseases such as thalassemia and sickle cell anemia.
miRNA-targeting oligonucleotides (anti-miRs)
Most of the anti-miR oligonucleotide types utilize 2′-OMe, LNA or 2′-fluoro analogues usually as mixmers of more than one analogue type or with DNA (Table 3). In vivo applications all utilize PS linkages. Currently, there are no papers describing peptide delivery of such anti-miRs. As discussed above, it seems that some naked oligonucleotide analogues may have the ability not only to enter cells through endocystosis but also can efficiently block miRNA activity without need for any enhancement of transfection by peptide or other vectors.19,20,35,82,83 Thus it seems unlikely that peptide-mediated delivery will be of value for such oligonucleotide types.
Table 3. Selection of oligonucleotides and peptide–oligonucleotide conjugates used for miRNA blocking.
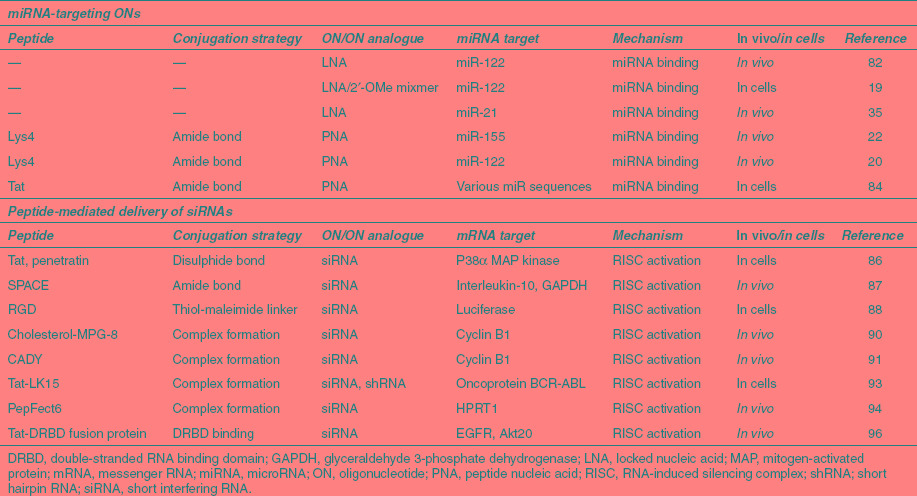
In the case of PNA anti-miRs, just a few added Lys residues enhances cell uptake significantly,19,22 and a terminal Cys residue also adds to the uptake potential and anti-miR activity.20 A longer peptide is not required for activity19 but conjugation of an Arg-rich Tat peptide to PNA anti-mIRs has also shown efficient cell delivery while maintaining miRNA regulation activity in cell culture.84,85 In addition to CPPs, it seems likely that CPPs would be worth investigating here for the future development of PNA anti-miRs targeted to specific tissue types or to cancer cells.
Peptide-mediated delivery of siRNAs
Covalent conjugation of siRNA with a CPP has met with limited success (Table 3). For example, reduction in mRNA expression was achieved only at high concentration of CPPs Tat or Penetratin conjugated to the 3′-end of the sense strand of siRNA,86 which is the most appropriate position for conjugation. An interesting peptide conjugated recently in this way is the cell and skin penetrating peptide called SPACE, identified by a phage display technique, which was evaluated for delivery of siRNA in vivo to two different targets, interleukin-10 and GAPDH mRNAs. Conjugation of siRNA with the SPACE peptide led to enhanced skin-absorptive properties and knockdown of corresponding protein targets.87 It is hoped to use such conjugates to treat atopic dermatitis. Unfortunately, there was no experiment described showing knockdown at the mRNA level, which is required to be sure that the effects observed in cells and mice on protein reduction are entirely due to an siRNA-directed RNA cleavage.
Cyclic RGD peptides have been investigated as tri- and tetra-valent conjugates of siRNA and shown to bring about dose-dependent reduction of reporter luciferase expression in human melanoma cells, but not the monovalent or divalent conjugates.88 This result demonstrates that, as with oligonucleotides, multivalency is likely to be necessary for conjugates of siRNA for sufficient effectiveness as a cell-targeting approach.
In general, noncovalent complex formation has been a more fruitful strategy to siRNA delivery (Table 3).89 An amphipathic cholesterol-functionalized peptide carrier, Chol-MPG-8, was shown capable of complexing siRNA-targeting cyclin B1 and to lead to tumor growth reduction after intravenous injection in a PC-3 cancer model.90 The high stability of Chol-MPG-8–based nanoparticles and the slow release of the siRNA within cells allowed the use of low concentrations of siRNA. A peptide named CADY was also reported by the same research group to bring about inhibition of PC3 tumor growth when complexed with siRNA.91 CADY adopts a helical conformation within cell membranes, where charged amino acids are exposed on one side of the helix and tryptophan residues, found essential to good cellular uptake, are on the other side. Importantly, CADY–siRNA complexes were shown to bind to phospholipids on biological membranes via electrostatic interactions and enter cells by energy-independent translocation thereby bypassing the endosomal pathway.46,62,92 A fusion peptide of Tat (49-57) and a membrane lytic peptide LK15 was shown to form complexes with both short hairpin RNA and siRNA targeting BCR-ABL oncoprotein mRNA, but the short hairpin RNA induced a more stable silencing of the gene target in K562 chronic myeloid leukemia cells compared to siRNA complexes.93
Stearylated peptide vectors have also been used successfully to deliver siRNA. A TP10-derived lipopeptide (PF6) was designed to aid endosomal release through attachment of four pH titratable trifluoromethylquinoline moieties to a lysine side chain of TP10. PF6 was shown to form nanoparticles with siRNA and knockdown HPRT1 mRNA production in a range of cell types as well as in kidney, lung, and liver of mice upon tail vein infusion at 1 mg/kg.94
An alternative peptide–siRNA complexation approach utilizes a recombinant fusion of the HIV Tat protein PTD with a double-stranded RNA binding domain (DRBD) that binds to siRNA and neutralizes its negative charges. The PTD-DRBD peptide vector has shown excellent cellular delivery of siRNA into various primary and transformed cells.95 PTD-DRBD was used to package two siRNAs simultaneously (against EGFR and Akt20) to induce tumor-specific apoptosis in a glioblastoma model after intracerebral injection, and to also substantially increase mouse survival.96 The PTD-DRBD peptide is now being developed industrially, but its very large size makes it unsuitable for chemical synthesis. Further the general applicability for siRNA delivery in vivo, especially for systemic use, is in doubt in the absence of further examples of efficacy.
Viral RNA targeting
Although there are no examples of peptides being used for delivery of negatively charged oligonucleotides as antiviral agents, peptide conjugates of charge-neutral PNA and PMO have been investigated for some years. Peptide-PMO antiviral applications up to 2007 include targeting of Dengue virus, Cocksackievirus, Ebola, and Marburg viruses for example and are reviewed in reference 36. Despite reassuring results that PNA-peptides targeted to the HIV-1 trans-activation responsive element are nontoxic in mice at high (300 mg/kg) doses,97 antiviral development seems recently to have ceased for PNA peptides. By contrast, RXR4 and B-peptide conjugates of PMO oligonucleotides have recently shown high antiviral activities both in cell culture and in vivo in various systems (Table 4). For example, P-PMO conjugates interfered with splicing of hemagglutinin-activating protease TMPRSS2 pre-mRNA and suppressed viral titers by two to three log10 units in Influenza A cultures.98 Similarly, RXR4-PMOs blocked viral replication of human respiratory syncytial viruses both in cells and in BALB/c mice.99 Here, the PMO sequence was targeted to the start of the coding region. Interestingly, there was also reduction in lung viral titers even with treatment of mice 3 hours after infection, but not 8 hours after infection. This suggests the PMO needs to be present in the cell soon after infection if it is to have an impact on virus production.
Particularly promising results have been obtained in vivo in pre- and post-infection P-PMO treatment of pigs infected with porcine reproductive and respiratory syndrome virus both in reducing viremia and interstitial pneumonia.100 RXR4–PMO conjugates were also seen to substantially improve antisense activity in vivo over naked PMO in protection of mice upon infection with West Nile virus or coronavirus by targeting to specific viral RNA sites involved in translation or replication.101,102
Overall, the P-PMO approach is very promising, but it also must compete with other antiviral approaches including use of PMO analogues with cationic groups within the PMO as currently being exploited by AVI Biopharma (Bothell, WA).
Antibacterial activity of antisense PNA and PMO
Increasing multidrug resistance in bacteria and the difficulties in discovery of new small molecule antibiotics have prompted consideration of new strategies such as gene targeting by PNA and PMO for treating bacterial diseases. However, the delivery of PNAs into bacterial cells has proved to be problematic. Although some CPPs may themselves have intrinsic antimicrobial activity,103 peptide-mediated delivery of antisense PNA and PMO targeting specific essential bacterial genes was suggested more than 10 years using peptide–PNA conjugates104 and has shown recent promise as an antibacterial strategy (Table 4). In particular, Arg-rich peptides conjugated to PMO are able to penetrate the cell outer membrane and display gene-specific antibiotic activity in gram-negative bacteria.105 The peptide–PMO conjugates are able to reduce bacterial blood titers and increase survival in Escherichia coli infected mice.106,107 Very recently, a novel 22-residue Arg-rich peptide derived from human T cells, when conjugated to PMO targeting GyrA was able to induce broad range bacterial cell killing at concentrations much lower than required for activity by other Arg-rich peptide-PMOs.108 The higher than perhaps anticipated activity is attributed to non-gene–specific RNA targeting of the P-PMO in addition to a gene specific effect, suggesting partial bacteriocidal activity plays an important role here. Interestingly, PMO containing positive charges in the PMO backbone also have antisense activity but did not achieve the same efficiency as peptide-conjugated PMO.109
PNAs conjugated to a Lys-rich peptide have also been used successfully to validate essential gene targets in Escherichia coli by studying the relationship between decrease in mRNA expression and growth rate decline.110 Further, peptide-conjugated PNA targeting rpoD, a gene essential for bacterial growth, showed broad inhibition in multidrug-resistant Escherichia coli, Salmonella enterica, Klebsiella pneumoniae, and Shigella flexneri both in cells and in vivo.111
The variety of recent promising results shows that peptide-mediated delivery of PMO and PNA holds promise for further drug development against multidrug-resistant bacteria.
Polymer, Liposomal, and Exosome Delivery Methods Involving Peptides
Peptide-conjugated polymers
Synthetic and naturally occurring cationic polymers represent a large group of carriers for nucleic acids. Although initially developed for the purpose of gene delivery, numerous reports have affirmed their utility in oligonucleotide delivery. These linear or branched polymers range from the first discovered poly-L-lysine (PLL), to the most frequently used polyethyleneimine (PEI).112,113 They share the properties of condensing oligonucleotides into small particles (polyplexes) that facilitate cellular uptake via endocytosis. However, their delivery efficiencies and toxicities vary significantly. PEI has been produced in particular in an assortment of structural variants.114
A major drawback with use of PEI and many other cationic polymers is their non-biodegradable nature, which raises toxicity concerns.115 Furthermore, their efficiencies have been poor in vivo in many cases, due to unwanted interactions with serum components.115,116 Polyethylene glycol (PEG) linkers of different sizes have been included in PEI polymers in order to prolong the circulation time in blood (reviewed in ref. 117). Although such PEGylation significantly stabilizes polyplexes, it unfortunately often results in lower potency, due to inadequate cellular uptake and poor endosomal escape.117,118 Another strategy for stabilization of polyplexes involves use of hydrophobic modifications. For example, partial modifications of amines in PEI with tyrosine stabilize polyplexes significantly increases transfection of both siRNAs and splice-redirecting 2′-OMe oligonucleotides.119,120 Analogously, modification of polyamines with hydrophobic acrylates results in stabilization of cores for highly efficient siRNA and 2′-OMe anti-miR delivery in vivo.121,122
Peptide modifications of cationic polymers have been used successfully to either improve tissue targeting or overall delivery efficacy and to decrease the required dose. Pioneering work from the laboratory of Ernst Wagner showed that PEI-mediated gene transfer can be improved dramatically by incorporation of receptor-targeting peptide ligands (reviewed in ref. 117,123,124).
Many other groups have capitalized on this peptide/cationic polymer strategy to improve gene delivery profoundly.125,126 Surprisingly, examples of use of targeting peptides and CPPs to enhance polyplex delivery of oligonucleotides have been sparse. In one study. Tat peptide was conjugated to PEGylated PEI which, when complexed with 2′-OMe oligomers and injected intramuscularly into mdx mice, improved exon skipping of dystrophin pre-mRNA.127
Several successful studies on siRNA delivery have been reported. Schiffelers et al. utilized the integrin-binding peptide RGD attached to PEGylated PEI for delivery of siRNA targeting VEGF-R in tumor-bearing mice.128 Similarly, Wang et al. conjugated a bombesin peptide via a PEG spacer to a free thiol group in an amphiphilic polymer N-(1-aminoethyl)iminobis[N-(oleicyl-cysteinyl-histidinyl-1-aminoethyl)propionamide] (EHCO) to target bombesin receptors overexpressed on various cancer cells.129 The siRNA/functionalized polymer complex was shown to efficiently promote RNA interference responses in cells and systemic administration of anti-HIF-1α siRNA with bombesin-containing EHCO displayed significant advantage over nontargeted polymer in inducing target reduction in a xenograft model of human glioma.
PEGylated transferrin peptide combined with oligoethyleneimine was able to deliver siRNA highly efficiently into neuroblastoma-bearing mice.130 Further, an elegant demonstration of receptor-specific uptake of defined folate-PEG-siRNA conjugates together with a structurally defined polycation was shown in use of such complexes for specific gene silencing.131
Instead of targeting peptides, the Wagner group has obtained improved endosomal escape by inclusion of fusogenic peptides. In the initial study, PEGylated polylysine/PEI was covalently linked to the endosomolytic peptide, melittin. Since melittin possesses lytic properties at both neutral and low pH, amines within the peptide were masked by reaction with dimethylmaleic anhydride (DMMA). Dimethylmaleic groups are removed at low pH within endosomes, thus exposing the lytic peptide. Polycation-PEG-DMMA-melittin conjugates were mixed with siRNAs to form polyplexes that efficiently induced an RNA interferene response in cell culture.132 In a further study with the same components, a cysteine residue was incorporated into the PLL to allow disulfide formation with an siRNA containing a 5′-thiolated sense strand. Such conjugates were almost as efficient as polyplexes and displayed reduced cytotoxicity.133
Another important example of peptide/polymer combinations is a cyclodextrin containing polymer (CDP)-based system, which so far is the only platform reported for RNA interference delivery in humans.134 CDP is a short polycation that assembles with nucleic acids (e.g., siRNA) via electrostatic interactions. In order to facilitate endosomal escape, imidazole groups were incorporated on the ends of the polymer to act as proton sponges. For systemic delivery, the polymer was stabilized by inclusion of surface-functionalized PEG moieties. In contrast to other polymeric delivery systems, the CDP approach relies on noncovalent incorporation of PEG via conjugation to adamantane, a small molecule that forms a high affinity inclusion complex with cyclodextrin. Finally, in order to confer tissue targeting to solid tumors, the surface of CDP was coated with adamantane conjugated to the transferrin ligand for targeting of transferrin receptors that are expressed abundantly on the surface of tumors.135
Liposomal nanovectors with peptide ligands
Many common cationic liposomal vectors used for nucleic acid delivery in cell culture, such as lipofectin or lipofectamine, are not suited for in vivo use, because of their sensitivity to serum proteins and their toxicity.136,137 Surface functionalization with inert hydrophilic polymers such as PEG has improved in vivo use of such vectors (reviewed in ref. 138), but PEGylation decreases cell delivery due to poor membrane interactions and poor endosomal escape.118,139 New lipid formulations that include peptides have broadened the repertoire of liposomes used for oligonucleotide delivery. In particular, the inclusion of Arg-rich CPPs on such liposomes has proven as a potent strategy to enhance transfections.10,138 In a recent example of peptide-mediated siRNA delivery, liposomal nanoparticles that are octaarginine-modified was used to encapsulate siRNA and to silence an endogenous gene in liver cells as well as in vivo at low concentrations of siRNA (25 µg/mouse) using a single dose, without evidence for liver toxicity.140 In another example, cationic liposomes were modified by a chimeric peptide containing 16 lysine residues and a short multicell targeting peptide Y and used to deliver siRNA against luciferase and GAPDH targets.141
A further example of rational liposome design is the multifunctional envelope-type device (MEND) system.10 In the prototype MEND, oligonucleotides are condensed with polycations (protamine) and wrapped by a lipid envelope containing cationic, anionic, and helper lipids, such as dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) that improves endosomal escape. In addition to surface modification with PEG, peptide delivery vectors have been incorporated on to the surface of such liposomes using a stearylated peptide that spontaneously inserts into the lipid membrane. Novel versions of MEND contain cleavable PEG linkers as well as targeting peptides for specific receptors or fusogenic peptides, in order to confer targeting of specific tissues and controlled release of encapsulated cargoes (reviewed in ref. 118). MEND has been efficiently exploited for delivery of unmodified single-stranded antisense oligonucleotides in cells as well as siRNAs both in cells and ex vivo in dendritic cells.142,143,144 Very recently, an even more sophisticated MEND system was efficiently utilized for systemic, targeted delivery of siRNA to tumors.145
Similar liposomal delivery systems have been developed for drug delivery by Torchillin and colleagues, where the surface has been decorated with the CPP Tat.138 To incorporate the Tat peptide in a controlled manner, it was covalently linked to the anionic lipid phosphatidic acid that spontaneously inserts into liposomes. Such liposomes have been used primarily for targeted delivery of plasmid DNA to tumors, but have also been shown to be effective for siRNA delivery.146 CPPs have also been used in conjunction with conventional liposome-based transfection agents and synergistic effects reported for the delivery of splice redirecting 2′-OMe oligomers.147
Peptide-functionalized exosomes
Practically all nanoparticle vectors for oligonucleotide delivery have been developed for targeting liver or tumors. Development of efficient carriers for crossing of other biological barriers, such as the blood-brain barrier, is in its infancy. In addition to synthetic nonviral vectors, which still retain inherent risks of raising an immune response or causing systemic toxicity in vivo, an alternative biologically derived vector has recently been proposed that makes use of naturally occurring membrane vesicles secreted by most cells and found in all body fluids, which are thought to be key mediators of information transmission between cells in the body.148
A subgroup of such vesicles are termed exosomes, which have been shown recently to contain various RNA species. They exert their biological effects either by directly activating cell surface receptors on target cells or by transferring proteins from the cell of origin to the recipient.149 Furthermore, numerous studies suggest that exosomes, which are derived from the endolysosomal pathway and have a diameter of 40–120 nm, play a crucial role in the horizontal transfer of RNA to neighboring or distant recipient cells.150,151 Of particular interest is their ability to deliver miRNAs into recipient cells to induce gene silencing.152,153 Such properties indicate that they would be highly suitable candidates for oligonucleotide and siRNA delivery.
Wood et al. recently provided the first proof-of-concept study for delivery of exogenous siRNA using such exosomes.154 Immature dendritic cells were chosen as source for exosomes, because they lack T-cell activators and are thus immunologically inert.155 In order to confer targeting of exosomes to the brain, dendritic cells were transfected with a plasmid expressing an exosomal protein Lamp2b fused to a brain-specific peptide, rabies virus glycoprotein-derived peptide (RVG),156 which becomes presented at the surface of exosomes. RVG-targeted exosomes were subsequently purified from cell cultures and loaded with exogenous siRNA by electroporation. Systemic delivery of GAPDH targeting siRNA with RVG-exosomes resulted in specific knockdown of GAPDH in several regions of mouse brain such as cortex and striatum. The potency of RVG-exosomes was tested by delivery of BACE-1 siRNA (targeting the enzyme β-secretase), which resulted in significant and specific target knockdown (60%) in the mouse brain cortex. This knockdown led to a reduction in the extracellular accumulation of Aβ1-42, the toxic amyloid species produced by BACE-1 that is implicated as a cause of Alzheimer's disease. No toxicity or immunogenicity was observed, even after repeated administration, a prerequisite for many disease applications.
Physiological Aspects of Peptide-Mediated Oligonucleotide and SiRNA in Vivo Delivery
Crucial to the clinical application of peptide-based oligonucleotide and siRNA therapies is the tailoring of such drugs for whole-body distribution, in order to maximize specific tissue targeting while minimizing induced toxic or immune responses. In vivo biodistribution data can be derived either by the assessment of an induced biological affect, by the detection of a directly labeled bio-cargo, or by use of a transgenic animal reporter system, yet each method has caveats in their use. Measurement of an induced biological affect is restricted to those tissues where the transcript targeted by the oligonucleotide or siRNA is expressed, for example, using dystrophin exon skipping and protein restoration as a read-out in the mdx mouse model.75 Fluorescent molecules or radioactive isotopes have been used successfully to label and follow the biodistribution of peptide conjugates of biocargoes such as PNA.157 However it is important to note that where the peptide part only is labeled, label detection may not always correlate with the location of the bioactive cargo. For example, a recent report of an N-terminally FITC-labeled B-PMO having crossed the blood-brain barrier and reaching Purkinje cells in wild-type mice should be viewed with caution until confirmed by other methods.158 An EGFP-654 transgenic mouse model that ubiquitously expresses an aberrantly spliced EGFP-654 pre-mRNA reporter gene has been used to assess functional biodistribution of B-PMO.159 However, such distribution assessments might not reflect the potential of a peptide-PMO to influence a disease pathology, whereas in an animal model of a disease such as DMD this becomes less of an issue. Nevertheless the use of all three of these techniques together can provide valuable information in building a view of how a peptide affects biodistribution of an oligonucleotide cargo.
An important parameter in systemic delivery is the route of administration (i.e., intravenous, intraperitoneal, subcutaneous, oral or nasal) in order to find the best biodistribution pattern for the particular application, but also to obtain optimal safety profiles.160 The efficacies of the delivery routes depend on the specific peptide ligand as well as the cell-targeting requirements of the studied disease, and must be balanced against the toxicology profile of the peptide-modified cargo. Very few direct comparisons of delivery routes have been published, but for B-PMO conjugates for example, higher efficiencies have been achieved via the intravenous delivery route compared to the subcutaneous route.159
Toxicity and unwanted immune activation by peptide conjugates of oligonucleotides and siRNA need to be carefully assessed in each case. For example, Penetratin–siRNA conjugates showed innate immune activation following intratracheal administration.161 By contrast, Penetratin–PNA conjugates did not induce an immunogenic response after intraperitoneal delivery and were well tolerated in mice at 100 mg/kg.97 Although CPPs do have potential to stimulate unwanted immunological responses, their small size and the use of analogues has reduced this risk significantly. For example, no immune response has been seen to date for B peptide-PMO162 and there are no reports of any peptide-PMO or peptide–PNA conjugate to date inducing a significant response.
The nature and levels of in vivo toxicity are not currently well understood for peptide conjugates of oligonucleotides or for peptide-based vector delivery routes, and there are very few published in vivo studies, probably because of commercial sensitivity of such data. However, peptide-PMO acts as a good case study. Here the PMO cargo has an amazingly good safety profile in animals and in many clinical trials.37,163,164 Thus the safety profile of a P-PMO conjugate will depend almost entirely upon the specific peptide sequence and is assessed in the context of the therapeutic dose required in a particular clinical setting. Amantana et al. investigated the safety profile in rats of an anti-c-myc PMO conjugated to the RXR4 CPP. Although lower doses of the CPP-PMO were well tolerated (15 mg/kg), adverse events such as lethargy, weight loss, and elevated blood urea nitrogen and serum creatinine levels were reported at much higher doses (150 mg/kg), with a lethal dose reported as 400 mg/kg.165 Toxicity has also been reported in non-human primates following a preclinical study with the B-PMO peptide conjugate developed by AVI BioPharma. Dose-dependent toxicity was observed in healthy Cynomolgus monkeys with four weekly intravenous injections of 9 mg/kg, causing tubular degeneration in the kidneys (AVI BioPharma website www.aviobio.com).63 No toxic effect was observed at higher doses in mdx mice, thus highlighting that the P-PMO toxicity threshold varies between species. Although lower toxic thresholds have been reported elsewhere, comparisons between studies assessing the use of CPPs to deliver non-oligomers are problematic, because the effect of the cargo cannot be distinguished from that of the CPP.166
In another useful study, Wancewicz et al. have reported acute toxicity in mice for a PNA containing nine additional Lys residues when dosed at 10.5 µmol/kg at three times a week for 2 weeks.77 Similarly a PNA conjugated to an amphipathic peptide containing Lys, Leu, and Ala residues showed severe nephrotoxocity, with profound proximal tubule necrosis at 40 mg/kg dose, whereas a PNA conjugate with mostly Arg and homoArg residues did not show such toxicity at this dose. Interestingly, the tissue distributions of these two peptide-PNAs differed widely, with the latter Arg-rich conjugate showing good splicing redirection in adipose tissue (the targeted organ) even at a low dose of 2.5 mg/kg.77
Thus, although immunogenicity seems unlikely to be an issue for most small peptides as oligonucleotide conjugates, toxicity profiles need to be evaluated carefully on a case by case basis to establish an effective yet safe dosing regimen in humans. There is clearly an important need to determine the exact nature of peptide-induced toxicity patterns, alongside the development of further peptide modifications, if such peptide applications are to reach the clinic.
Clinical Perspectives of Peptide-Mediated Delivery of Nucleic Acids
Efforts to translate synthetic peptides conjugates or complexes of oligonucleotides and siRNA to treat human disease have recently stepped up. Many of the polymeric, liposomal, and other nanovectors involving peptides have shown great promise for delivery in cell culture and in animal models. However, there are no examples yet of any of these reaching clinical trials apart from the application involving the human transferrin protein used in siRNA particle formation which was recently reported in the clinic.134 The application of short peptides in siRNA delivery applications needs to overcome remaining obstacles of reproducible and validated manufacture of the overall vectors. In addition, there needs to be better control of off-target effects that lead to toxicities. Peptide-based ligands displayed on such vectors are bound to feature more in clinical development in the near future and these are already helping to gain greater tissue and targeting specificity. It is hoped that further preclinical development studies here will be published soon to help move such therapies to the clinic as rapidly as possible.
Therapeutic approaches for neuromuscular disorders currently lead the way in clinical development of peptide-mediated delivery. Repeat systemic dose-escalation studies of naked PMO and 2′-OMe/PS oligonucleotides having been completed recently in DMD patients and further dose-escalations studies planned37,167 (current trials reviews in ref. 168), while recruitment for a clinical trial to treat spinal muscular atrophy is underway (clinicaltrials.gov identifier: NCT01494701), which is another neuromuscular disease where peptide-conjugated PMO is likely to be applied.
Recently there has been a concerted effort to evaluate the potential of CPPs and other peptides in some multi-systemic neuromuscular disorders. For example, our laboratories are currently involved in multi-center development of a peptide-PMO to treat DMD, based on initial evaluations in the mdx mouse as a model, and further preclinical therapeutic studies are underway to supplement data already published.75,159,165 Here, a lead Pip peptide conjugated to PMO has shown excellent dystrophin production in cardiac muscle, a tissue previously shown to take up naked antisense oligomers poorly, in addition to in skeletal muscles.75 However, evidence of induced renal toxicity in monkeys for the earlier B-PMO lead63 has highlighted the needs for caution and for obtaining a deeper understanding regarding possible toxic side effects of cationic CPPs.
It seems likely that the simplicity of the peptide conjugation approach and the ability to apply the principle of a steric-blocking approach across a broad range of neuromuscular and neurodegenerative diseases will likely drive the clinical development of peptide conjugates of oligonucleotides and their analogues forward for several years to come. New examples of preclinical development are expected soon in several of such diseases and there is great hope that one or more of these conjugates will make it to the clinic in the near future.
Acknowledgments
This work was supported by research grants to M.J.A.W. from the Muscular Dystrophy Campaign, Duchenne Ireland, and the Medical Research Council (G0900887), and joint research grants to M.J.A.W. and M.J.G. from Association Française contre les Myopathies (programme number 14784) and from the Wellcome Trust/Health Innovation Challenge Fund (HICF-1009-025). Work in the laboratory of M.J.G. was also supported by the Medical Research Council (MRC programme no. U105178803). S.E.L.A. is supported by a research fellowship from Swedish Society of Medical Research (SSMF). The authors declared no conflict of interest.
References
- Kole R, Krainer AR., and, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov. 2012;11:125–140. doi: 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurreck J. Royal Society of Chemistry: Cambridge; 2008. Therapeutic Oligonucleotides. [Google Scholar]
- Bijsterbosch MK, Manoharan M, Rump ET, De Vrueh RL, van Veghel R, Tivel KL.et al. (1997In vivo fate of phosphorothioate antisense oligodeoxynucleotides: predominant uptake by scavenger receptors on endothelial liver cells Nucleic Acids Res 253290–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda Y. Update on non-viral delivery methods for cancer therapy: possibilities of a drug delivery system with anticancer activities beyond delivery as a new therapeutic tool. Expert Opin Drug Deliv. 2010;7:1079–1093. doi: 10.1517/17425247.2010.510511. [DOI] [PubMed] [Google Scholar]
- Xu L., and, Anchordoquy T. Drug delivery trends in clinical trials and translational medicine: challenges and opportunities in the delivery of nucleic acid-based therapeutics. J Pharm Sci. 2011;100:38–52. doi: 10.1002/jps.22243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derossi D, Joliot AH, Chassaing G., and, Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem. 1994;269:10444–10450. [PubMed] [Google Scholar]
- Said Hassane F, Saleh AF, Abes R, Gait MJ., and, Lebleu B. Cell penetrating peptides: overview and applications to the delivery of oligonucleotides. Cell Mol Life Sci. 2010;67:715–726. doi: 10.1007/s00018-009-0186-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margus H, Padari K., and, Pooga M. Cell-penetrating peptides as versatile vehicles for oligonucleotide delivery. Mol Ther. 2012;20:525–533. doi: 10.1038/mt.2011.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliano RL. Peptide-oligonucleotide conjugates for the delivery of antisense and siRNA. Curr Opin Mol Ther. 2005;7:132–136. [PubMed] [Google Scholar]
- Nakase I, Akita H, Kogure K, Gräslund A, Langel U, Harashima H.et al. (2011Efficient intracellular delivery of nucleic acid pharmaceuticals using cell-penetrating peptides Acc Chem Resepub ahead of print). [DOI] [PubMed]
- Juliano RL, Ming X., and, Nakagawa O. Cellular uptake and intracellular trafficking of antisense and siRNA oligonucleotides. Bioconjug Chem. 2012;23:147–157. doi: 10.1021/bc200377d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laakkonen P., and, Vuorinen K. Homing peptides as targeted delivery vehicles. Integr Biol (Camb) 2010;2:326–337. doi: 10.1039/c0ib00013b. [DOI] [PubMed] [Google Scholar]
- Zamecnik PC., and, Stephenson ML. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc Natl Acad Sci USA. 1978;75:280–284. doi: 10.1073/pnas.75.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett CF., and, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. 2010;50:259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- Mahato RI, Cheng K., and, Guntaka RV. Modulation of gene expression by antisense and antigene oligodeoxynucleotides and small interfering RNA. Expert Opin Drug Deliv. 2005;2:3–28. doi: 10.1517/17425247.2.1.3. [DOI] [PubMed] [Google Scholar]
- Rogers FA, Lin SS, Hegan DC, Krause DS., and, Glazer PM. Targeted gene modification of hematopoietic progenitor cells in mice following systemic administration of a PNA-peptide conjugate. Mol Ther. 2012;20:109–118. doi: 10.1038/mt.2011.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepsen JS., and, Wengel J. LNA-antisense rivals siRNA for gene silencing. Curr Opin Drug Discov Devel. 2004;7:188–194. [PubMed] [Google Scholar]
- Laxton C, Brady K, Moschos S, Turnpenny P, Rawal J, Pryde DC.et al. (2011Selection, optimization, and pharmacokinetic properties of a novel, potent antiviral locked nucleic acid-based antisense oligomer targeting hepatitis C virus internal ribosome entry site Antimicrob Agents Chemother 553105–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabani MM., and, Gait MJ. miR-122 targeting with LNA/2'-O-methyl oligonucleotide mixmers, peptide nucleic acids (PNA), and PNA-peptide conjugates. RNA. 2008;14:336–346. doi: 10.1261/rna.844108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres AG, Fabani MM, Vigorito E, Williams D, Al-Obaidi N, Wojciechowski F.et al. (2012Chemical structure requirements and cellular targeting of microRNA-122 by peptide nucleic acids anti-miRs Nucleic Acids Res 402152–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein CA, Hansen JB, Lai J, Wu S, Voskresenskiy A, Høg A.et al. (2010Efficient gene silencing by delivery of locked nucleic acid antisense oligonucleotides, unassisted by transfection reagents Nucleic Acids Res 38e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabani MM, Abreu-Goodger C, Williams D, Lyons PA, Torres AG, Smith KG.et al. (2010Efficient inhibition of miR-155 function in vivo by peptide nucleic acids Nucleic Acids Res 384466–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambari R, Fabbri E, Borgatti M, Lampronti I, Finotti A, Brognara E.et al. (2011Targeting microRNAs involved in human diseases: a novel approach for modification of gene expression and drug development Biochem Pharmacol 821416–1429. [DOI] [PubMed] [Google Scholar]
- Staton AA., and, Giraldez AJ. Use of target protector morpholinos to analyze the physiological roles of specific miRNA-mRNA pairs in vivo. Nat Protoc. 2011;6:2035–2049. doi: 10.1038/nprot.2011.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EL-Andaloussi S, Johansson HJ, Lundberg P., and, Langel Ü. Induction of splice correction by cell-penetrating peptide nucleic acids. J Gene Med. 2006;8:1262–1273. doi: 10.1002/jgm.950. [DOI] [PubMed] [Google Scholar]
- Abes S, Moulton H, Turner J, Clair P, Richard JP, Iversen P.et al. (2007Peptide-based delivery of nucleic acids: design, mechanism of uptake and applications to splice-correcting oligonucleotides Biochem Soc Trans 35Pt 153–55. [DOI] [PubMed] [Google Scholar]
- Wood MJ, Gait MJ., and, Yin H. RNA-targeted splice-correction therapy for neuromuscular disease. Brain. 2010;133 Pt 4:957–972. doi: 10.1093/brain/awq002. [DOI] [PubMed] [Google Scholar]
- Passini MA, Bu J, Richards AM, Kinnecom C, Sardi SP, Stanek LM.et al. (2011Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy Sci Transl Med 372ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JK, Malerba A, Popplewell L, Foster K., and, Dickson G. Antisense-induced myostatin exon skipping leads to muscle hypertrophy in mice following octa-guanidine morpholino oligomer treatment. Mol Ther. 2011;19:159–164. doi: 10.1038/mt.2010.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckstein F. The versatility of oligonucleotides as potential therapeutics. Expert Opin Biol Ther. 2007;7:1021–1034. doi: 10.1517/14712598.7.7.1021. [DOI] [PubMed] [Google Scholar]
- Levin AA. A review of the issues in the pharmacokinetics and toxicology of phosphorothioate antisense oligonucleotides. Biochim Biophys Acta. 1999;1489:69–84. doi: 10.1016/s0167-4781(99)00140-2. [DOI] [PubMed] [Google Scholar]
- Sanghvi YS. A status update of modified oligonucleotides for chemotherapeutics applications. Curr Protoc Nucleic Acid Chem. 2011;Chapter 4:Unit 4.1.1–Unit 4.122. doi: 10.1002/0471142700.nc0401s46. [DOI] [PubMed] [Google Scholar]
- Watts JK., and, Corey DR. Clinical status of duplex RNA. Bioorg Med Chem Lett. 2010;20:3203–3207. doi: 10.1016/j.bmcl.2010.03.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swayze EE, Siwkowski AM, Wancewicz EV, Migawa MT, Wyrzykiewicz TK, Hung G.et al. (2007Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals Nucleic Acids Res 35687–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obad S, dos Santos CO, Petri A, Heidenblad M, Broom O, Ruse C.et al. (2011Silencing of microRNA families by seed-targeting tiny LNAs Nat Genet 43371–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulton HM, Moulton JD.2008Antisense morpholino oligomers and their peptide conjugates Kurreck J.ed). Therapeutic Oligonucleotides Royal Society of Chemistry: Cambridge, UK. pp. 43–79 [Google Scholar]
- Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K.et al. (2011Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study Lancet 378595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey VN, Upadhyay A., and, Chaubey B. Prospects for antisense peptide nucleic acid (PNA) therapies for HIV. Expert Opin Biol Ther. 2009;9:975–989. doi: 10.1517/14712590903052877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh Y, Murat P., and, Defrancq E. Recent developments in oligonucleotide conjugation. Chem Soc Rev. 2010;39:2054–2070. doi: 10.1039/b911431a. [DOI] [PubMed] [Google Scholar]
- Lu K, Duan QP, Ma L., and, Zhao DX. Chemical strategies for the synthesis of peptide-oligonucleotide conjugates. Bioconjug Chem. 2010;21:187–202. doi: 10.1021/bc900158s. [DOI] [PubMed] [Google Scholar]
- Pooga M, Soomets U, Hällbrink M, Valkna A, Saar K, Rezaei K.et al. (1998Cell penetrating PNA constructs regulate galanin receptor levels and modify pain transmission in vivo Nat Biotechnol 16857–861. [DOI] [PubMed] [Google Scholar]
- Lundin P, Johansson H, Guterstam P, Holm T, Hansen M, Langel U.et al. (2008Distinct uptake routes of cell-penetrating peptide conjugates Bioconjug Chem 192535–2542. [DOI] [PubMed] [Google Scholar]
- Torres AG., and, Gait MJ. Exploiting cell surface thiols to enhance cellular uptake. Trends Biotechnol. 2012;30:185–190. doi: 10.1016/j.tibtech.2011.12.002. [DOI] [PubMed] [Google Scholar]
- Ezzat K, Helmfors H, Tudoran O, Juks C, Lindberg S, Padari K.et al. (2012Scavenger receptor-mediated uptake of cell-penetrating peptide nanocomplexes with oligonucleotides FASEB J 261172–1180. [DOI] [PubMed] [Google Scholar]
- Turner JJ, Williams D, Owen D., and, Gait MJ. Disulfide conjugation of peptides to oligonucleotides and their analogues. Curr Protocols Nucleic Acids Chemistry. 2005. pp. 4.28.21–24.28.21. [DOI] [PubMed]
- Konate K, Crombez L, Deshayes S, Decaffmeyer M, Thomas A, Brasseur R.et al. (2010Insight into the cellular uptake mechanism of a secondary amphipathic cell-penetrating peptide for siRNA delivery Biochemistry 493393–3402. [DOI] [PubMed] [Google Scholar]
- Rasmussen FW, Bendifallah N, Zachar V, Shiraishi T, Fink T, Ebbesen P.et al. (2006Evaluation of transfection protocols for unmodified and modified peptide nucleic acid (PNA) oligomers Oligonucleotides 1643–57. [DOI] [PubMed] [Google Scholar]
- Futaki S, Ohashi W, Suzuki T, Niwa M, Tanaka S, Ueda K.et al. (2001Stearylated arginine-rich peptides: a new class of transfection systems Bioconjug Chem 121005–1011. [DOI] [PubMed] [Google Scholar]
- Mäe M, El Andaloussi S, Lundin P, Oskolkov N, Johansson HJ, Guterstam P.et al. (2009A stearylated CPP for delivery of splice correcting oligonucleotides using a non-covalent co-incubation strategy J Control Release 134221–227. [DOI] [PubMed] [Google Scholar]
- Allinquant B, Hantraye P, Mailleux P, Moya K, Bouillot C., and, Prochiantz A. Downregulation of amyloid precursor protein inhibits neurite outgrowth in vitro. J Cell Biol. 1995;128:919–927. doi: 10.1083/jcb.128.5.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bongartz JP, Aubertin AM, Milhaud PG., and, Lebleu B. Improved biological activity of antisense oligonucleotides conjugated to a fusogenic peptide. Nucleic Acids Res. 1994;22:4681–4688. doi: 10.1093/nar/22.22.4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner Y, Wallukat G, Säälik P, Wiesner B, Pritz S., and, Oehlke J. Cellular uptake and biological activity of peptide nucleic acids conjugated with peptides with and without cell-penetrating ability. J Pept Sci. 2010;16:71–80. doi: 10.1002/psc.1198. [DOI] [PubMed] [Google Scholar]
- Turner JJ, Ivanova GD, Verbeure B, Williams D, Arzumanov AA, Abes S.et al. (2005Cell-penetrating peptide conjugates of peptide nucleic acids (PNA) as inhibitors of HIV-1 Tat-dependent trans-activation in cells Nucleic Acids Res 336837–6849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henke E, Perk J, Vider J, de Candia P, Chin Y, Solit DB.et al. (2008Peptide-conjugated antisense oligonucleotides for targeted inhibition of a transcriptional regulator in vivo Nat Biotechnol 2691–100. [DOI] [PubMed] [Google Scholar]
- Morris MC, Gros E, Aldrian-Herrada G, Choob M, Archdeacon J, Heitz F.et al. (2007A non-covalent peptide-based carrier for in vivo delivery of DNA mimics Nucleic Acids Res 35e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SH, Cho MJ., and, Kole R. Up-regulation of luciferase gene expression with antisense oligonucleotides: implications and applications in functional assay development. Biochemistry. 1998;37:6235–6239. doi: 10.1021/bi980300h. [DOI] [PubMed] [Google Scholar]
- Sazani P, Kang SH, Maier MA, Wei C, Dillman J, Summerton J.et al. (2001Nuclear antisense effects of neutral, anionic and cationic oligonucleotide analogs Nucleic Acids Res 293965–3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu RP, Youngblood DS, Hassinger JN, Lovejoy CE, Nelson MH, Iversen PL.et al. (2007Cell-penetrating peptides as transporters for morpholino oligomers: effects of amino acid composition on intracellular delivery and cytotoxicity Nucleic Acids Res 355182–5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abes R, Moulton HM, Clair P, Yang ST, Abes S, Melikov K.et al. (2008Delivery of steric block morpholino oligomers by (R-X-R)4 peptides: structure-activity studies Nucleic Acids Res 366343–6354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abes S, Turner JJ, Ivanova GD, Owen D, Williams D, Arzumanov A.et al. (2007Efficient splicing correction by PNA conjugation to an R6-Penetratin delivery peptide Nucleic Acids Res 354495–4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laufer SD, Recke AL, Veldhoen S, Trampe A., and, Restle T. Noncovalent peptide-mediated delivery of chemically modified steric block oligonucleotides promotes splice correction: quantitative analysis of uptake and biological effect. Oligonucleotides. 2009;19:63–80. doi: 10.1089/oli.2008.0160. [DOI] [PubMed] [Google Scholar]
- Rydström A, Deshayes S, Konate K, Crombez L, Padari K, Boukhaddaoui H.et al. (2011Direct translocation as major cellular uptake for CADY self-assembling peptide-based nanoparticles PLoS ONE 6e25924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulton HM., and, Moulton JD. Morpholinos and their peptide conjugates: therapeutic promise and challenge for Duchenne muscular dystrophy. Biochim Biophys Acta. 2010;1798:2296–2303. doi: 10.1016/j.bbamem.2010.02.012. [DOI] [PubMed] [Google Scholar]
- Ivanova GD, Arzumanov A, Abes R, Yin H, Wood MJ, Lebleu B.et al. (2008Improved cell-penetrating peptide-PNA conjugates for splicing redirection in HeLa cells and exon skipping in mdx mouse muscle Nucleic Acids Res 366418–6428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppelhus U, Shiraishi T, Zachar V, Pankratova S., and, Nielsen PE. Improved cellular activity of antisense peptide nucleic acids by conjugation to a cationic peptide-lipid (CatLip) domain. Bioconjug Chem. 2008;19:1526–1534. doi: 10.1021/bc800068h. [DOI] [PubMed] [Google Scholar]
- Shen G, Fang H, Song Y, Bielska AA, Wang Z., and, Taylor JS. Phospholipid conjugate for intracellular delivery of peptide nucleic acids. Bioconjug Chem. 2009;20:1729–1736. doi: 10.1021/bc900048y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam MR, Dixit V, Kang H, Li ZB, Chen X, Trejo J.et al. (2008Intracellular delivery of an anionic antisense oligonucleotide via receptor-mediated endocytosis Nucleic Acids Res 362764–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam MR, Ming X, Dixit V, Fisher M, Chen X., and, Juliano RL. The biological effect of an antisense oligonucleotide depends on its route of endocytosis and trafficking. Oligonucleotides. 2010;20:103–109. doi: 10.1089/oli.2009.0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming X, Alam MR, Fisher M, Yan Y, Chen X., and, Juliano RL. Intracellular delivery of an antisense oligonucleotide via endocytosis of a G protein-coupled receptor. Nucleic Acids Res. 2010;38:6567–6576. doi: 10.1093/nar/gkq534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehto T, Abes R, Oskolkov N, Suhorutsenko J, Copolovici DM, Mäger I.et al. (2010Delivery of nucleic acids with a stearylated (RxR)4 peptide using a non-covalent co-incubation strategy J Control Release 14142–51. [DOI] [PubMed] [Google Scholar]
- Hassane FS, Abes R, El Andaloussi S, Lehto T, Sillard R, Langel U.et al. (2011Insights into the cellular trafficking of splice redirecting oligonucleotides complexed with chemically modified cell-penetrating peptides J Control Release 153163–172. [DOI] [PubMed] [Google Scholar]
- Ezzat K, Andaloussi SE, Zaghloul EM, Lehto T, Lindberg S, Moreno PM.et al. (2011PepFect 14, a novel cell-penetrating peptide for oligonucleotide delivery in solution and as solid formulation Nucleic Acids Res 395284–5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MJA. Towards an oligonucleotide therapy for Duchenne muscular dystrophy: a complex development challenge. Sci Transl Med. 2010;2:25ps15. doi: 10.1126/scitranslmed.3000512. [DOI] [PubMed] [Google Scholar]
- Yin H, Moulton HM, Betts C, Seow Y, Boutilier J, Iverson PL.et al. (2009A fusion peptide directs enhanced systemic dystrophin exon skipping and functional restoration in dystrophin-deficient mdx mice Hum Mol Genet 184405–4414. [DOI] [PubMed] [Google Scholar]
- Yin H, Saleh AF, Betts C, Camelliti P, Seow Y, Ashraf S.et al. (2011Pip5 transduction peptides direct high efficiency oligonucleotide-mediated dystrophin exon skipping in heart and phenotypic correction in mdx mice Mol Ther 191295–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Lu Q., and, Wood M. Effective exon skipping and restoration of dystrophin expression by peptide nucleic acid antisense oligonucleotides in mdx mice. Mol Ther. 2008;16:38–45. doi: 10.1038/sj.mt.6300329. [DOI] [PubMed] [Google Scholar]
- Wancewicz EV, Maier MA, Siwkowski AM, Albertshofer K, Winger TM, Berdeja A.et al. (2010Peptide nucleic acids conjugated to short basic peptides show improved pharmacokinetics and antisense activity in adipose tissue J Med Chem 533919–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boffa LC, Cutrona G, Cilli M, Matis S, Damonte G, Mariani MR.et al. (2007Inhibition of Burkitt's lymphoma cells growth in SCID mice by a PNA specific for a regulatory sequence of the translocated c-myc Cancer Gene Ther 14220–226. [DOI] [PubMed] [Google Scholar]
- Hu J., and, Corey DR. Inhibiting gene expression with peptide nucleic acid (PNA)–peptide conjugates that target chromosomal DNA. Biochemistry. 2007;46:7581–7589. doi: 10.1021/bi700230a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Matsui M., and, Corey DR. Allele-selective inhibition of mutant huntingtin by peptide nucleic acid-peptide conjugates, locked nucleic acid, and small interfering RNA. Ann N Y Acad Sci. 2009;1175:24–31. doi: 10.1111/j.1749-6632.2009.04975.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui M., and, Corey DR. Allele-selective inhibition of trinucleotide repeat genes. Drug Discov Today. 2012;17:443–450. doi: 10.1016/j.drudis.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmén J, Lindow M, Schütz S, Lawrence M, Petri A, Obad S.et al. (2008LNA-mediated microRNA silencing in non-human primates Nature 452896–899. [DOI] [PubMed] [Google Scholar]
- Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME.et al. (2010Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection Science 327198–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SY, Ju Y., and, Park H. A highly effective and long-lasting inhibition of miRNAs with PNA-based antisense oligonucleotides. Mol Cells. 2009;28:341–345. doi: 10.1007/s10059-009-0134-8. [DOI] [PubMed] [Google Scholar]
- Oh SY, Ju Y, Kim S., and, Park H. PNA-based antisense oligonucleotides for micrornas inhibition in the absence of a transfection reagent. Oligonucleotides. 2010;20:225–230. doi: 10.1089/oli.2010.0238. [DOI] [PubMed] [Google Scholar]
- Turner JJ, Jones S, Fabani MM, Ivanova G, Arzumanov AA., and, Gait MJ. RNA targeting with peptide conjugates of oligonucleotides, siRNA and PNA. Blood Cells Mol Dis. 2007;38:1–7. doi: 10.1016/j.bcmd.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Hsu T., and, Mitragotri S. Delivery of siRNA and other macromolecules into skin and cells using a peptide enhancer. Proc Natl Acad Sci USA. 2011;108:15816–15821. doi: 10.1073/pnas.1016152108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam MR, Ming X, Fisher M, Lackey JG, Rajeev KG, Manoharan M.et al. (2011Multivalent cyclic RGD conjugates for targeted delivery of small interfering RNA Bioconjug Chem 221673–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meade BR., and, Dowdy SF. Enhancing the cellular uptake of siRNA duplexes following noncovalent packaging with protein transduction domain peptides. Adv Drug Deliv Rev. 2008;60:530–536. doi: 10.1016/j.addr.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crombez L, Morris MC, Dufort S, Aldrian-Herrada G, Nguyen Q, Mc Master G.et al. (2009Targeting cyclin B1 through peptide-based delivery of siRNA prevents tumour growth Nucleic Acids Res 374559–4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divita G, Heitz F, Morris MC., and, Aldrian-Herrada G. Cell-penetrating peptides for intracellular delivery of molecules, US Patent 2009/015603 (ed). 2009.
- Crombez L, Aldrian-Herrada G, Konate K, Nguyen QN, McMaster GK, Brasseur R.et al. (2009A new potent secondary amphipathic cell-penetrating peptide for siRNA delivery into mammalian cells Mol Ther 1795–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthanari Y, Pluen A, Rajendran R, Aojula H., and, Demonacos C. Delivery of therapeutic shRNA and siRNA by Tat fusion peptide targeting BCR-ABL fusion gene in Chronic Myeloid Leukemia cells. J Control Release. 2010;145:272–280. doi: 10.1016/j.jconrel.2010.04.011. [DOI] [PubMed] [Google Scholar]
- EL-Andaloussi S, Lehto T, Mäger I, Rosenthal-Aizman K, Oprea II, Simonson OE.et al. (2011Design of a peptide-based vector, PepFect6, for efficient delivery of siRNA in cell culture and systemically in vivo Nucleic Acids Res 393972–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi A, Meade BR, Chang YC, Fredrickson CT, Willert K, Puri N.et al. (2009Efficient siRNA delivery into primary cells by a peptide transduction domain-dsRNA binding domain fusion protein Nat Biotechnol 27567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michiue H, Eguchi A, Scadeng M., and, Dowdy SF. Induction of in vivo synthetic lethal RNAi responses to treat glioblastoma. Cancer Biol Ther. 2009;8:2306–2313. doi: 10.4161/cbt.8.23.10271. [DOI] [PubMed] [Google Scholar]
- Chaubey B, Tripathi S., and, Pandey VN. Single acute-dose and repeat-doses toxicity of anti-HIV-1 PNA TAR-penetratin conjugate after intraperitoneal administration to mice. Oligonucleotides. 2008;18:9–20. doi: 10.1089/oli.2007.0088. [DOI] [PubMed] [Google Scholar]
- Böttcher-Friebertshäuser E, Stein DA, Klenk HD., and, Garten W. Inhibition of influenza virus infection in human airway cell cultures by an antisense peptide-conjugated morpholino oligomer targeting the hemagglutinin-activating protease TMPRSS2. J Virol. 2011;85:1554–1562. doi: 10.1128/JVI.01294-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai SH, Stein DA, Guerrero-Plata A, Liao SL, Ivanciuc T, Hong C.et al. (2008Inhibition of respiratory syncytial virus infections with morpholino oligomers in cell cultures and in mice Mol Ther 161120–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opriessnig T, Patel D, Wang R, Halbur PG, Meng XJ, Stein DA.et al. (2011Inhibition of porcine reproductive and respiratory syndrome virus infection in piglets by a peptide-conjugated morpholino oligomer Antiviral Res 9136–42. [DOI] [PubMed] [Google Scholar]
- Burrer R, Neuman BW, Ting JP, Stein DA, Moulton HM, Iversen PL.et al. (2007Antiviral effects of antisense morpholino oligomers in murine coronavirus infection models J Virol 815637–5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deas TS, Bennett CJ, Jones SA, Tilgner M, Ren P, Behr MJ.et al. (2007In vitro resistance selection and in vivo efficacy of morpholino oligomers against West Nile virus Antimicrob Agents Chemother 512470–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splith K., and, Neundorf I. Antimicrobial peptides with cell-penetrating peptide properties and vice versa. Eur Biophys J. 2011;40:387–397. doi: 10.1007/s00249-011-0682-7. [DOI] [PubMed] [Google Scholar]
- Good L., and, Nielsen PE. Peptide nucleic acid (PNA) antisense effects in Escherichia coli. Curr Issues Mol Biol. 1999;1:111–116. [PubMed] [Google Scholar]
- Tilley LD, Hine OS, Kellogg JA, Hassinger JN, Weller DD, Iversen PL.et al. (2006Gene-specific effects of antisense phosphorodiamidate morpholino oligomer-peptide conjugates on Escherichia coli and Salmonella enterica serovar typhimurium in pure culture and in tissue culture Antimicrob Agents Chemother 502789–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilley LD, Mellbye BL, Puckett SE, Iversen PL., and, Geller BL. Antisense peptide-phosphorodiamidate morpholino oligomer conjugate: dose-response in mice infected with Escherichia coli. J Antimicrob Chemother. 2007;59:66–73. doi: 10.1093/jac/dkl444. [DOI] [PubMed] [Google Scholar]
- Mellbye BL, Puckett SE, Tilley LD, Iversen PL., and, Geller BL. Variations in amino acid composition of antisense peptide-phosphorodiamidate morpholino oligomer affect potency against Escherichia coli in vitro and in vivo. Antimicrob Agents Chemother. 2009;53:525–530. doi: 10.1128/AAC.00917-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesolowski D, Tae HS, Gandotra N, Llopis P, Shen N., and, Altman S. Basic peptide-morpholino oligomer conjugate that is very effective in killing bacteria by gene-specific and nonspecific modes. Proc Natl Acad Sci USA. 2011;108:16582–16587. doi: 10.1073/pnas.1112561108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellbye BL, Weller DD, Hassinger JN, Reeves MD, Lovejoy CE, Iversen PL.et al. (2010Cationic phosphorodiamidate morpholino oligomers efficiently prevent growth of Escherichia coli in vitro and in vivo J Antimicrob Chemother 6598–106. [DOI] [PubMed] [Google Scholar]
- Goh S, Boberek JM, Nakashima N, Stach J., and, Good L. Concurrent growth rate and transcript analyses reveal essential gene stringency in Escherichia coli. PLoS ONE. 2009;4:e6061. doi: 10.1371/journal.pone.0006061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai H, You Y, Yan H, Meng J, Xue X, Hou Z.et al. (2012Antisense inhibition of gene expression and growth in gram-negative bacteria by cell-penetrating peptide conjugates of peptide nucleic acids targeted to rpoD gene Biomaterials 33659–667. [DOI] [PubMed] [Google Scholar]
- Inaki Y, Ishikawa T., and, Takemoto K. Synthesis and interactions of poly-L-lysines containing nucleic acid bases. Nucleic Acids Symp Ser. 1980. pp. s137–s140. [PubMed]
- Boussif O, Lezoualc'h F, Zanta MA, Mergny MD, Scherman D, Demeneix B.et al. (1995A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine Proc Natl Acad Sci USA 927297–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wightman L, Kircheis R, Rössler V, Carotta S, Ruzicka R, Kursa M.et al. (2001Different behavior of branched and linear polyethylenimine for gene delivery in vitro and in vivo J Gene Med 3362–372. [DOI] [PubMed] [Google Scholar]
- Plank C, Oberhauser B, Mechtler K, Koch C., and, Wagner E. The influence of endosome-disruptive peptides on gene transfer using synthetic virus-like gene transfer systems. J Biol Chem. 1994;269:12918–12924. [PubMed] [Google Scholar]
- Merkel OM, Librizzi D, Pfestroff A, Schurrat T, Buyens K, Sanders NN.et al. (2009Stability of siRNA polyplexes from poly(ethylenimine) and poly(ethylenimine)-g-poly(ethylene glycol) under in vivo conditions: effects on pharmacokinetics and biodistribution measured by Fluorescence Fluctuation Spectroscopy and Single Photon Emission Computed Tomography (SPECT) imaging J Control Release 138148–159. [DOI] [PubMed] [Google Scholar]
- Wagner E.2011Polymers for siRNA delivery: inspired by viruses to be targeted, dynamic, and precise Acc Chem Resepub ahead of print). [DOI] [PubMed]
- Hatakeyama H, Akita H., and, Harashima H. A multifunctional envelope type nano device (MEND) for gene delivery to tumours based on the EPR effect: a strategy for overcoming the PEG dilemma. Adv Drug Deliv Rev. 2011;63:152–160. doi: 10.1016/j.addr.2010.09.001. [DOI] [PubMed] [Google Scholar]
- Creusat G, Rinaldi AS, Weiss E, Elbaghdadi R, Remy JS, Mulherkar R.et al. (2010Proton sponge trick for pH-sensitive disassembly of polyethylenimine-based siRNA delivery systems Bioconjug Chem 21994–1002. [DOI] [PubMed] [Google Scholar]
- Zaghloul EM, Viola JR, Zuber G, Smith CI., and, Lundin KE. Formulation and delivery of splice-correction antisense oligonucleotides by amino acid modified polyethylenimine. Mol Pharm. 2010;7:652–663. doi: 10.1021/mp900220p. [DOI] [PubMed] [Google Scholar]
- Akinc A, Zumbuehl A, Goldberg M, Leshchiner ES, Busini V, Hossain N.et al. (2008A combinatorial library of lipid-like materials for delivery of RNAi therapeutics Nat Biotechnol 26561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love KT, Mahon KP, Levins CG, Whitehead KA, Querbes W, Dorkin JR.et al. (2010Lipid-like materials for low-dose, in vivo gene silencing Proc Natl Acad Sci USA 1071864–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogris M. Nucleic acid based therapeutics for tumor therapy. Anticancer Agents Med Chem. 2006;6:563–570. doi: 10.2174/187152006778699158. [DOI] [PubMed] [Google Scholar]
- Ogris M., and, Wagner E. To be targeted: is the magic bullet concept a viable option for synthetic nucleic acid therapeutics. Hum Gene Ther. 2011;22:799–807. doi: 10.1089/hum.2011.065. [DOI] [PubMed] [Google Scholar]
- Kilk K, El-Andaloussi S, Järver P, Meikas A, Valkna A, Bartfai T.et al. (2005Evaluation of transportan 10 in PEI mediated plasmid delivery assay J Control Release 103511–523. [DOI] [PubMed] [Google Scholar]
- Kleemann E, Dailey LA, Abdelhady HG, Gessler T, Schmehl T, Roberts CJ.et al. (2004Modified polyethylenimines as non-viral gene delivery systems for aerosol gene therapy: investigations of the complex structure and stability during air-jet and ultrasonic nebulization J Control Release 100437–450. [DOI] [PubMed] [Google Scholar]
- Sirsi SR, Schray RC, Guan X, Lykens NM, Williams JH, Erney ML.et al. (2008Functionalized PEG-PEI copolymers complexed to exon-skipping oligonucleotides improve dystrophin expression in mdx mice Hum Gene Ther 19795–806. [DOI] [PubMed] [Google Scholar]
- Schiffelers RM, Ansari A, Xu J, Zhou Q, Tang Q, Storm G.et al. (2004Cancer siRNA therapy by tumor selective delivery with ligand-targeted sterically stabilized nanoparticle Nucleic Acids Res 32e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XL, Xu R., and, Lu ZR. A peptide-targeted delivery system with pH-sensitive amphiphilic cell membrane disruption for efficient receptor-mediated siRNA delivery. J Control Release. 2009;134:207–213. doi: 10.1016/j.jconrel.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Tietze N, Pelisek J, Philipp A, Roedl W, Merdan T, Tarcha P.et al. (2008Induction of apoptosis in murine neuroblastoma by systemic delivery of transferrin-shielded siRNA polyplexes for downregulation of Ran Oligonucleotides 18161–174. [DOI] [PubMed] [Google Scholar]
- Dohmen C., and, Wagner E. Multifunctional CPP polymer system for tumor-targeted pDNA and siRNA delivery. Methods Mol Biol. 2011;683:453–463. doi: 10.1007/978-1-60761-919-2_32. [DOI] [PubMed] [Google Scholar]
- Meyer M, Philipp A, Oskuee R, Schmidt C., and, Wagner E. Breathing life into polycations: functionalization with pH-responsive endosomolytic peptides and polyethylene glycol enables siRNA delivery. J Am Chem Soc. 2008;130:3272–3273. doi: 10.1021/ja710344v. [DOI] [PubMed] [Google Scholar]
- Meyer M, Dohmen C, Philipp A, Kiener D, Maiwald G, Scheu C.et al. (2009Synthesis and biological evaluation of a bioresponsive and endosomolytic siRNA-polymer conjugate Mol Pharm 6752–762. [DOI] [PubMed] [Google Scholar]
- Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA.et al. (2010Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles Nature 4641067–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis ME. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: from concept to clinic. Mol Pharm. 2009;6:659–668. doi: 10.1021/mp900015y. [DOI] [PubMed] [Google Scholar]
- Scheule RK, St George JA, Bagley RG, Marshall J, Kaplan JM, Akita GY.et al. (1997Basis of pulmonary toxicity associated with cationic lipid-mediated gene transfer to the mammalian lung Hum Gene Ther 8689–707. [DOI] [PubMed] [Google Scholar]
- Filion MC., and, Phillips NC. Toxicity and immunomodulatory activity of liposomal vectors formulated with cationic lipids toward immune effector cells. Biochim Biophys Acta. 1997;1329:345–356. doi: 10.1016/s0005-2736(97)00126-0. [DOI] [PubMed] [Google Scholar]
- Torchilin VP. Tat peptide-mediated intracellular delivery of pharmaceutical nanocarriers. Adv Drug Deliv Rev. 2008;60:548–558. doi: 10.1016/j.addr.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Ambegia E, Ansell S, Cullis P, Heyes J, Palmer L., and, MacLachlan I. Stabilized plasmid-lipid particles containing PEG-diacylglycerols exhibit extended circulation lifetimes and tumor selective gene expression. Biochim Biophys Acta. 2005;1669:155–163. doi: 10.1016/j.bbamem.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Yamauchi J, Khalil IA, Kajimoto K, Akita H., and, Harashima H. Cell penetrating peptide-mediated systemic siRNA delivery to the liver. Int J Pharm. 2011;419:308–313. doi: 10.1016/j.ijpharm.2011.07.038. [DOI] [PubMed] [Google Scholar]
- Tagalakis AD, He L, Saraiva L, Gustafsson KT., and, Hart SL. Receptor-targeted liposome-peptide nanocomplexes for siRNA delivery. Biomaterials. 2011;32:6302–6315. doi: 10.1016/j.biomaterials.2011.05.022. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Kogure K, Yamada Y, Futaki S., and, Harashima H. Significant and prolonged antisense effect of a multifunctional envelope-type nano device encapsulating antisense oligodeoxynucleotide. J Pharm Pharmacol. 2006;58:431–437. doi: 10.1211/jpp.58.4.0002. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Kogure K, Futaki S., and, Harashima H. Octaarginine-modified multifunctional envelope-type nano device for siRNA. J Control Release. 2007;119:360–367. doi: 10.1016/j.jconrel.2007.03.010. [DOI] [PubMed] [Google Scholar]
- Akita H, Kogure K, Moriguchi R, Nakamura Y, Higashi T, Nakamura T.et al. (2010Nanoparticles for ex vivo siRNA delivery to dendritic cells for cancer vaccines: programmed endosomal escape and dissociation J Control Release 143311–317. [DOI] [PubMed] [Google Scholar]
- Hatakeyama H, Akita H, Ito E, Hayashi Y, Oishi M, Nagasaki Y.et al. (2011Systemic delivery of siRNA to tumors using a lipid nanoparticle containing a tumor-specific cleavable PEG-lipid Biomaterials 324306–4316. [DOI] [PubMed] [Google Scholar]
- Zhang C, Tang N, Liu X, Liang W, Xu W., and, Torchilin VP. siRNA-containing liposomes modified with polyarginine effectively silence the targeted gene. J Control Release. 2006;112:229–239. doi: 10.1016/j.jconrel.2006.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabulo S, Resina S, Simões S, Lebleu B., and, Pedroso de Lima MC. A non-covalent strategy combining cationic lipids and CPPs to enhance the delivery of splice correcting oligonucleotides. J Control Release. 2010;145:149–158. doi: 10.1016/j.jconrel.2010.03.021. [DOI] [PubMed] [Google Scholar]
- Ratajczak J, Wysoczynski M, Hayek F, Janowska-Wieczorek A., and, Ratajczak MZ. Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia. 2006;20:1487–1495. doi: 10.1038/sj.leu.2404296. [DOI] [PubMed] [Google Scholar]
- Camussi G, Deregibus MC, Bruno S, Grange C, Fonsato V., and, Tetta C. Exosome/microvesicle-mediated epigenetic reprogramming of cells. Am J Cancer Res. 2011;1:98–110. [PMC free article] [PubMed] [Google Scholar]
- Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ., and, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- Skog J, Würdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M.et al. (2008Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers Nat Cell Biol 101470–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegtel DM, Cosmopoulos K, Thorley-Lawson DA, van Eijndhoven MA, Hopmans ES, Lindenberg JL.et al. (2010Functional delivery of viral miRNAs via exosomes Proc Natl Acad Sci USA 1076328–6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecalvo A, Larregina AT, Shufesky WJ, Stolz DB, Sullivan ML, Karlsson JM.et al. (2012Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes Blood 119756–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S., and, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29:341–345. doi: 10.1038/nbt.1807. [DOI] [PubMed] [Google Scholar]
- Quah BJ., and, O'Neill HC. The immunogenicity of dendritic cell-derived exosomes. Blood Cells Mol Dis. 2005;35:94–110. doi: 10.1016/j.bcmd.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Lentz TL. Rabies virus binding to an acetylcholine receptor alpha-subunit peptide. J Mol Recognit. 1990;3:82–88. doi: 10.1002/jmr.300030205. [DOI] [PubMed] [Google Scholar]
- Maier MA, Esau CC, Siwkowski AM, Wancewicz EV, Albertshofer K, Kinberger GA.et al. (2006Evaluation of basic amphipathic peptides for cellular delivery of antisense peptide nucleic acids J Med Chem 492534–2542. [DOI] [PubMed] [Google Scholar]
- Du L, Kayali R, Bertoni C, Fike F, Hu H, Iversen PL.et al. (2011Arginine-rich cell-penetrating peptide dramatically enhances AMO-mediated ATM aberrant splicing correction and enables delivery to brain and cerebellum Hum Mol Genet 203151–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jearawiriyapaisarn N, Moulton HM, Buckley B, Roberts J, Sazani P, Fucharoen S.et al. (2008Sustained dystrophin expression induced by peptide-conjugated morpholino oligomers in the muscles of mdx mice Mol Ther 161624–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemskerk H, de Winter C, van Kuik P, Heuvelmans N, Sabatelli P, Rimessi P.et al. (2010Preclinical PK and PD studies on 2'-O-methyl-phosphorothioate RNA antisense oligonucleotides in the mdx mouse model Mol Ther 181210–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moschos SA, Jones SW, Perry MM, Williams AE, Erjefalt JS, Turner JJ.et al. (2007Lung delivery studies using siRNA conjugated to TAT(48-60) and penetratin reveal peptide induced reduction in gene expression and induction of innate immunity Bioconjug Chem 181450–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Moulton HM, Iversen PL, Juang J, Li J, Spurney CF.et al. (2008Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modifies morpholino oligomer Proc Natl Acad Sci USA 10514814–14819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sazani P, Ness KP, Weller DL, Poage DW, Palyada K., and, Shrewsbury SB. Repeat-dose toxicology evaluation in cynomolgus monkeys of AVI-4658, a phosphorodiamidate morpholino oligomer (PMO) drug for the treatment of duchenne muscular dystrophy. Int J Toxicol. 2011;30:313–321. doi: 10.1177/1091581811403505. [DOI] [PubMed] [Google Scholar]
- Sazani P, Ness KP, Weller DL, Poage D, Nelson K., and, Shrewsbury AS. Chemical and mechanistic toxicology evaluation of exon skipping phosphorodiamidate morpholino oligomers in mdx mice. Int J Toxicol. 2011;30:322–333. doi: 10.1177/1091581811403504. [DOI] [PubMed] [Google Scholar]
- Amantana A, Moulton HM, Cate ML, Reddy MT, Whitehead T, Hassinger JN.et al. (2007Pharmacokinetics, biodistribution, stability and toxicity of a cell-penetrating peptide-morpholino oligomer conjugate Bioconjug Chem 181325–1331. [DOI] [PubMed] [Google Scholar]
- Aguilera TA, Olson ES, Timmers MM, Jiang T., and, Tsien RY. Systemic in vivo distribution of activatable cell penetrating peptides is superior to that of cell penetrating peptides. Integr Biol (Camb) 2009;1:371–381. doi: 10.1039/b904878b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goemans NM, Tulinius M, van den Akker JT, Burm BE, Ekhart PF, Heuvelmans N.et al. (2011Systemic administration of PRO051 in Duchenne's muscular dystrophy N Engl J Med 3641513–1522. [DOI] [PubMed] [Google Scholar]
- Muntoni F., and, Wood MJ. Targeting RNA to treat neuromuscular disease. Nat Rev Drug Discov. 2011;10:621–637. doi: 10.1038/nrd3459. [DOI] [PubMed] [Google Scholar]