

Background: What are the essential roles of five tryptophans in the T7 primase?
Results: Replacement of Trp-42, -97, or -147 with the structurally similar tyrosine disturbs the conformation of the ZBD and reduces NTP binding and the catalysis step.
Conclusion: Trp-42, -97, and -147 contribute to template binding, NTP binding, and the catalysis step.
Significance: Trp-42, -97, and -147 play different but essential roles in T7 DNA primase.
Keywords: DNA Primase, DNA Replication, Enzyme Catalysis, Enzyme Mutation, RNA Synthesis, Bacteriophage T7, Tryptophan, Primer Synthesis
Abstract
DNA primases catalyze the synthesis of oligoribonucleotides required for the initiation of lagging strand DNA synthesis. Prokaryotic primases consist of a zinc-binding domain (ZBD) necessary for recognition of a specific template sequence and a catalytic RNA polymerase domain. Interactions of both domains with the DNA template and ribonucleotides are required for primer synthesis. Five tryptophan residues are dispersed in the primase of bacteriophage T7: Trp-42 in the ZBD and Trp-69, -97, -147, and -255 in the RNA polymerase domain. Previous studies showed that replacement of Trp-42 with alanine in the ZBD decreases primer synthesis, whereas substitution of non-aromatic residues for Trp-69 impairs both primer synthesis and delivery. However, the roles of tryptophan at position 97, 147, or 255 remain elusive. To investigate the essential roles of these residues, we replaced each tryptophan with the structurally similar tyrosine and examined the effect of this subtle alteration on primer synthesis. The substitution at position 42, 97, or 147 reduced primer synthesis, whereas substitution at position 69 or 255 did not. The functions of the tryptophans were further examined at each step of primer synthesis. Alteration of residue 42 disturbed the conformation of the ZBD and resulted in partial loss of the zinc ion, impairing binding to the ssDNA template. Replacement of Trp-97 with tyrosine reduced the binding affinity to NTP and the catalysis step. The replacement of Trp-147 with tyrosine also impaired the catalytic step. Therefore, Trp-42 is important in maintaining the conformation of the ZBD for template binding; Trp-97 contributes to NTP binding and the catalysis step; and Trp-147 maintains the catalysis step.
Introduction
The replisome of bacteriophage T7 consists of gene 5 DNA polymerase (gp5), the processivity factor, Escherichia coli thioredoxin (trx),3 gene 4 helicase-primase (gp4), and gene 2.5 ssDNA-binding protein (gp2.5) (1). The helicase and primase domains of gp4 are located in the carboxyl- and amino-terminal halves of gp4, respectively. In leading strand DNA synthesis, the helicase domain of the hexameric gp4 unwinds the parental dsDNA to produce an ssDNA template for the continuous DNA synthesis by the complex of gp5 and trx (gp5-trx). Due to the 5′ to 3′ polarity of synthesis by DNA polymerases and their requirement of a 3′-hydroxyl terminus to initiate synthesis, the lagging strand DNA is synthesized discontinuously, requiring a timely supply of oligoribonucleotides to serve as primers. The primase domain of gp4 recognizes specific sequences on the lagging strand template and catalyzes the synthesis of oligoribonucleotides for the initiation of synthesis of Okazaki fragments by the lagging strand gp5-trx (2). Formation of a replication loop containing a nascent Okazaki fragment allows both leading and lagging strand synthesis to progress in the same overall direction (3).
The primase domain of gp4 consists of an N-terminal Cys4 zinc-binding domain (ZBD; residues 1–56), a flexible linker (residues 57–67), and a C-terminal RNA polymerase domain (RPD; residues 68–255) (Fig. 1) (4). The ZBD plays a critical role in the recognition of primase recognition sequences (5) and in placing this sequence in juxtaposition to the RPD (4). The RPD consists of an N-terminal subdomain composed of four β strands and a C-terminal TOPRIM fold containing the active site for oligoribonucleotide synthesis. A shallow cleft is present between these two subdomains, a feature that is also found in E. coli DnaG primase (6, 7). The flexible linker allows the ZBD of one subunit in a hexameric gp4 to interact with the RPD of an adjacent subunit to reconstitute a functional primase and catalyze the primer synthesis in a trans mode (8, 9).
FIGURE 1.

Crystal structure of T7 DNA primase. T7 DNA primase fragment (residues 1–255) consists of a ZBD (residues 1–56, in red), a linker (residues 57–67), and an RPD (residues 68–255) (Protein Data Bank entry 1NUI (4)). The RPD consists of an N-terminal subdomain composed of four β strands (yellow) and a TOPRIM fold (blue). The Zn2+ in the ZBD and the two Mg2+ in the TOPRIM fold are shown as purple and green spheres, respectively. Five tryptophan residues (Trp-42, -69, -97, -147, and -255) in green are dispersed in the three subdomains. The structures of side chains of tryptophan and tyrosine are shown (inset). Tyrosine is similar in structure to tryptophan in that both have an aromatic plane and a hydrogen bond donor.
Oligoribonucleotide synthesis by the primase requires interactions of both the ZBD and RPD with the DNA template and ribonucleotides. The ZBD and RPD depicted in Fig. 2A undergo sequential contacts in each step during primer synthesis. In the first step, T7 primase binds to the 5′-GTC-3′ recognition sequence in ssDNA (10) (Fig. 2B). The ZBD plays an important role in the stabilization of the primase-DNA complex through an interaction with the RPD (11). Once bound to the recognition sequence, the primase catalyzes the synthesis of the diribonucleotide pppAC using the ATP and CTP bound at the active site (Fig. 2C). The 3′-cryptic C in the template is essential for recognition but not copied into the product (12). After diribonucleotide synthesis, the ssDNA template together with pppAC and the ZBD shifts relative to the RPD so that the next sequence on the template at the 5′-side of the primase recognition sequence moves into the active site of the RPD (Fig. 2D). The resulting pppAC is then extended to yield the functional tetraribonucleotide primers, pppACCC, pppACAC, or pppACCA, depending on the template sequence (2). NMR studies show that both the ZBD and RPD are associated with the ACCA-annealed template (4). After formation of the tetraribonucleotide, the primers are delivered to gp5-trx for the initiation of synthesis of Okazaki fragments. In addition to template-directed synthesis, the active site of the primase can also synthesize, albeit at a very low rate, random diribonucleotide in the absence of DNA (13).
FIGURE 2.

The mechanism of primer synthesis by T7 DNA primase. A, T7 DNA primase consists of a ZBD and a RPD. B, primase binds an ssDNA template with a primase recognition sequence (5′-GTC-3′) in the presence of ATP and CTP. C, after binding, primase catalyzes the synthesis of pppAC at the active site in the RPD. D, the ssDNA template together with the pppAC and the ZBD shifts relative to the RPD so that the 5′ template moves into the active site. E, the primase extends pppAC to the functional tetraribonucleotide. Finally, the primer is delivered to DNA polymerase.
Alterations in the ZBD lead to the loss of sequence-specific recognition, and changes in the RPD abolish the catalytic function, both defects resulting in failure to synthesize oligoribonucleotide (5, 14–16). Both the ZBD and RPD interact with the recognition sequence weakly during the initial synthesis of diribonucleotide (11). However, once a tetraribonucleotide is synthesized, a relatively stable complex involving the primase, the tetraribonucleotide, and the DNA template is formed. The linker connecting the ZBD and the RPD modulates interactions between the ZBD and RPD (9). A shorter linker connecting the ZBD and RPD facilitates extension of, for example, diribonucleotide to tetraribonucleotide.
Protein structure is stabilized by hydrogen binding, hydrophobic interactions, electrostatic interactions, and van der Waals forces. The hydrophobic interactions of non-polar side chains contribute significantly to the stabilization of the tertiary structures in proteins. Structurally, tryptophan has the largest accessible hydrophobic surface among the 20 natural amino acids, providing for the strongest stacking interactions with other aromatic residues (17–19). Tryptophan is also associated with a wide variety of protein functions. O-Acetylserine sulfhydrylase is a homodimeric enzyme catalyzing the last step of cysteine biosynthesis. Replacement of Trp-50 with tyrosine strongly affects its unfolding pathway (20). In another case, replacement of Trp-112 with phenylalanine in diacylglycerol kinase of E. coli reduces its expression level and its stability (17). Other studies also suggest that tryptophan plays a major role in the stabilization of proteins (21–23).
Five tryptophan residues are dispersed throughout the primase fragment (Fig. 1): Trp-42 in the ZBD, Trp-69 and Trp-97 in the N-terminal subdomain of the RPD, and Trp-147 and Trp-255 in the C-terminal TOPRIM fold of the RPD. A previous study showed that the replacement of Trp-42 with alanine in the ZBD greatly decreases tetraribonucleotide synthesis and partially reduces their utilization as primers by T7 DNA polymerase (24). In another study, the substitution of non-aromatic residues for Trp-69 significantly impairs the ability of gp4 to synthesize primers and deliver them to DNA polymerase (25). However, the substitution of alanine for tryptophan in these earlier studies is too drastic for meaningful conclusions about the essential roles of these tryptophans. Furthermore, the roles of tryptophan at positions 97, 147, or 255 in primer synthesis are not elucidated. Among 20 natural amino acids, tyrosine is structurally similar to tryptophan in that both residues have a hydrogen atom donor and a hydrophobic aromatic group (Fig. 1, inset). Nonetheless, the two residues are different with respect to the size of the aromatic group and the capability of stacking interaction to have different effects on primase function. Only replacement of tryptophan with the most similar residue, tyrosine, can disclose the essential roles of the five tryptophan residues in the T7 DNA primase. Our results show that such a subtle structural variation affects each step of primer synthesis, indicating the different and essential roles in primer synthesis.
EXPERIMENTAL PROCEDURES
Materials
Oligonucleotides used for site-directed mutagenesis and primer synthesis assays were purchased from Integrated DNA Technology. DNA purification kits and Ni-NTA minispin columns were purchased from Qiagen. Calf intestinal alkaline phosphatase (CIAP) was purchased from New England Biolabs. A non-tagged primase fragment plus the additional linkage between primase and helicase (residues 1–271) was purified as described previously (26).
Methods
Construction of Plasmids Containing Replacement of Codon for Tryptophan with Codon for Tyrosine or Alanine
Plasmid pET24-gp4 contains the sequence for phage T7 gene 4. Site-directed single point mutations in the codon for Trp-42, -69, -97, -147, or -255 were introduced into the gene 4 coding region of plasmid pET24-gp4 using either a standard megaprimer PCR procedure or the QuikChange® site-directed mutagenesis kit (Stratagene). The substitutions replaced the codon for tryptophan with that for tyrosine or alanine. Plasmids containing multiple substitutions for tryptophan residues were created by combination of mutagenic primers and template that already contained the replacement of tryptophan. For the overproduction of His-tagged primase, the primase coding region from the altered gene 4 in a plasmid was subcloned into pET28 at restriction sites NdeI and HindIII. Similarly, a DNA fragment encoding the substitution of alanine for Asp-237 was inserted into a plasmid coding the primase fragment. A DNA fragment encoding the substitution of tyrosine for Trp-42 was inserted into a plasmid coding the ZBD (pET24-ZBD) at restriction sites NdeI and HindIII. The presence of the expected substitutions in gene 4 was confirmed by DNA sequence analysis of the entire gene 4 coding region.
Complementation for Growth of T7 Phage
E. coli DH5α containing a plasmid encoding T7 gp4 with replacement of a single tryptophan with tyrosine or alanine at various positions was grown to an A600 of ∼1. About 0.3 ml of the bacterial culture and 0.1 ml of serially diluted phage solution were mixed with 3 ml of LB medium containing 0.7% agar and then plated on an LB plate containing kanamycin. After incubation at 37 °C overnight, the number of plaques was counted and normalized against the value obtained with wild-type gp4.
Purification of His-tagged Primase
The primase domain of gp4 mostly used in this study is located in the N-terminal 259 amino acids (residues 1–259) of gp4. The presence of six histidine residues at the N terminus of the primase facilitated the purification. Proteins were overproduced in E. coli BL21(DE3) at 37 °C. After the bacterial culture reached an A600 of ∼1, protein expression was induced by 1 mm isopropyl 1-thio-β-d-galactopyranoside at 25 °C for 5 h. Bacterial cells from 100–300 ml of culture were harvested, resuspended in 2–6 ml of buffer A (20 mm Tris-HCl, pH 7.5, and 0.3 m NaCl), and freeze-thawed three times in the presence of lysozyme (0.2 mg/ml) to rupture the cells. Clear bacterial lysate was collected by centrifugation and loaded onto a Ni-NTA column by gravity. Proteins were subsequently eluted from the column with buffer A containing 0, 50, 100, 250, or 500 mm imidazole. Small fractions (1 ml) were collected and analyzed by SDS-PAGE. Only the fractions containing pure primase fragment were collected, followed by dialysis against storage buffer (20 mm Tris-HCl, pH 7.5, 0.1 mm EDTA, and 50% glycerol). Most of the pure primase fragment was eluted with 250 mm imidazole. As judged on SDS-PAGE analysis, all purified primase fragments were >90% pure (supplemental Fig. S1).
Wild-type ZBD and the altered ZBD-W42Y were purified as described (4). Wild-type gp4, gp4-W97Y, gp4-W147Y, and gp4-W147A were purified as described (14, 15).
Template-directed Oligoribonucleotide Synthesis
The standard reaction contained the indicated amount of gp4 or primase fragment, template 1 containing 5′-TGGTC-3′ (a primase recognition sequence) or template 2 containing 5′-TGGTG-3′ (non-recognition sequence) (Table 1), 1 mm each ATP and [α-32P]CTP (0.1 mCi/ml) in reaction buffer B (40 mm Tris-HCl, pH 7.5, 50 mm potassium glutamate, 10 mm MgCl2, and 10 mm DTT) (15). After incubation at the indicated temperature for the indicated time, the reactions were terminated by the addition of EDTA to a final concentration of 40 mm, and the products were separated on a 25% denaturing polyacrylamide gel containing 3 m urea. After autoradiography of the gel, the amount of products was analyzed using a Fuji BAS 1000 bioimaging analyzer.
TABLE 1.
DNA templates used in oligoribonucleotide synthesis assay
The basic primase recognition site 5′-GTC-3′ at which the diribonucleotide pppAC is synthesized is indicated in boldface type. The complete primase recognition sites at which the tetraribonucleotides are synthesized are underlined. Template 2 lacks the cryptic cytosine necessary for primase recognition.
| Template | 5′-Sequence-3′ |
|---|---|
| 1 | T20ATTCG TAATC TGCAG GCA TGGTC AA TTTTT ATAGA GTTAT |
| 2 | T20ATTCG TAATC TGCAG GCA TGGTG AA TTTTT ATAGA GTTAT |
| 3 | CGTAA TCTGC AGGCA AAGTC AATTT |
| 4 | CGTAA TCTGC AGGCA GGGTC AATTT |
Template-independent Diribonucleotide Synthesis
T7 DNA primase can synthesize random diribonucleotide at low efficiency in the absence of DNA (13). The standard reaction contained a 2 μm concentration of the indicated primase fragment and 1 mm [α-32P]CTP (0.1 mCi/ml) in the absence of template in reaction buffer C (40 mm Tris-HCl, pH 7.5, 50 mm potassium glutamate, 10 mm MnCl2, and 10 mm DTT). The presence of Mn2+ instead of Mg2+ facilitates the diribonucleotide synthesis without template DNA (15). After reaction at 23 °C for 1 h, the product pppCC was dephosphorylated to CC by incubating with CIAP at 37 °C for 20 min. The reaction was terminated by the addition of EDTA, and the product CC was analyzed as described above.
Template-directed Diribonucleotide Synthesis
The standard reaction contained 0.4 μm primase fragment, 4 μm template 3 containing a basic primase recognition sequence 5′-AAGTC-3′ (Table 1), 1 mm each ATP and [α-32P]CTP (0.1 mCi/ml) in buffer B (15). Using this template, only pppAC was synthesized. After reaction at 23 °C for 30 min, the product pppAC was dephosphorylated to AC by incubation with CIAP at 37 °C for 20 min. The reaction was terminated by the addition of EDTA, and the AC was analyzed as described above.
Template-directed Oligoribonucleotide Extension
T7 DNA primase can extend the diribonucleotide AC to the tetraribonucleotide in the presence of template containing 5′-GGGTC-3′ (15). The standard extension reaction contained 0.4 μm primase fragment, 4 μm ssDNA template 4 containing a primase recognition sequence (5′-GGGTC-3′) (Table 1), 1 mm diribonucleotide 5′-AC-3′, and 1 mm [α-32P]CTP (0.1 mCi/ml) in buffer B (15). After incubation at 23 °C for 30 min, the reaction was terminated by the addition of EDTA, and the reaction products were analyzed as described above.
Determination of Zinc Content in Primase
Primase fragment stored in 40 μl of storage buffer (20 mm Tris-HCl, pH 7.5, 0.1 mm EDTA, and 50% glycerol) at a concentration of 16 μm, or the same volume of the storage buffer was dissolved in 0.5 ml of double-distilled concentrated nitric acid. After 1 h, the solution was diluted to 10 ml with Milli-Q water (18 megaohms). The zinc content in proteins or in buffer was analyzed using X series quadrupole inductively coupled plasma-mass spectrometry (ICP-MS) with collision cell technology using 72Ge, 103Rh, and 115In as the internal standards. The ICP-MS was calibrated using a series of zinc standard solutions prepared from single element stock solutions (High-Purity Standards, Charleston, SC). Quality assurance and quality control procedures were performed to ensure the accuracy of the zinc analysis. Method blanks were analyzed to monitor contamination. The limit of detection was determined as 3 times the S.D. value of nine replicate measurements of the calibration blanks. The zinc concentrations in all samples were above the limit of detection. The percentages of zinc contents of each sample were calculated by assigning a value of 100% for the wild-type primase fragment.
Circular Dichroism (CD) Spectroscopy
CD spectra of wild-type ZBD and ZBD-W42Y were recorded in a 1-mm cuvette using a JASCO J-710 instrument (JASCO, Easton, MD). A solution of the indicated ZBD (20 μm) in a buffer (50 mm Tris-HCl, pH 7.5, 1 mm DTT, 50 mm NaCl, and 1% glycerol) was placed in the cell, and spectra were recorded from 300 to 200 nm at 20 °C. Either buffer or sample was recorded four times. After subtracting the background signal from buffer, the final signal was normalized against the molar concentration of ZBD.
Surface Plasmon Resonance Analysis
The binding of gp4 to ATP was examined using surface plasmon resonance (SPR) on a Biacore 3000 instrument (15). The biotinylated ATP was immobilized on the surface of Sensor Chip SA by injecting 10 μl of 10 μm biotinylated ATP in a buffer (10 mm Tris-HCl, pH 7.5, 0.5 m NaCl, and 1 mm EDTA) at a flow rate of 20 μl/min. A lane in which biotin was immobilized instead of biotinylated ATP was used as control to remove background. Gp4 (0.7 μm) in 60 μl of flow buffer (10 mm Tris-HCl, pH 7.5, 10 mm MgCl2, 1 mm DTT, 50 mm potassium glutamate, and 1% glycerol) was injected at a flow rate of 50 μl/min. The binding was measured by subtracting a binding signal on biotin from a binding signal on biotinlyated ATP. After binding of protein, the chip was regenerated by injecting 100 μl of 1 m NaCl at a flow rate of 50 μl/min until the signal was restored to the level before injection of proteins.
Kinetic Analysis of Template-directed Diribonucleotide Synthesis
The template-dependent synthesis of diribonucleotide AC is dependent on the NTP concentration (10). The standard reaction contained a 0.4 μm concentration of the indicated primase, 4 μm template 3 containing 5′-AAGTC-3′ (Table 1), varying concentrations of ATP, and 2 mm [α-32P]CTP (0.1 mCi/ml) in buffer B. After incubation at 23 °C for 30 min, the product, pppAC, was dephosphorylated with CIAP to yield AC. After the reaction was terminated by the addition of EDTA, the product AC was separated from the other components by polyacrylamide gel electrophoresis. The amount of product was determined by comparing the intensity of the [α-32P]CMP in each band with the standards. The rates of product formation were plotted against ATP concentrations and fit to a Michaelis-Menten equation using GraphPad Prism (version 4.0c) to determine kcat and Km,ATP,
where v is the rate of synthesis of diribonucleotide at each concentration of ATP, kcat is the maximal rate of diribonucleotide synthesis, and Km,ATP is the concentration of ATP at half of the maximal rate.
The template-dependent synthesis of the diribonucleotide AC is also dependent on the DNA template concentration. The standard reactions were similar to the above except for varying concentrations of DNA template and a constant concentration of 2 mm ATP and 2 mm [α-32P]CTP (0.1 mCi/ml). After incubation at 23 °C for 10 min, the product was analyzed. The rates of product formation were plotted against template concentrations and fit to a Michaelis-Menten equation using GraphPad Prism (version 4.0c) to determine kcat and Km,DNA,
where v is the rate of synthesis of diribonucleotide at each concentration of DNA, kcat is the maximal rate of diribonucleotide synthesis, and Km,DNA is the concentration of DNA at half of the maximal rate.
RESULTS
Complementation of Phage Growth
Each of the five tryptophans in the primase domain (residues 1–259) of gp4 was replaced individually with tyrosine or alanine. The effect of the substitution on the in vivo function of gp4 was examined in a phage complementation assay. In this assay, the growth of phage T7Δ4 was dependent on gp4 exogenously expressed from a plasmid harbored in the T7-infected E. coli. Defects in the function of gp4 did not allow the phage to propagate, and no plaques were formed. In a control, a plasmid lacking gene 4 could not (less than 10−8-fold) support the growth of T7Δ4 phage compared with the one containing a wild-type gene 4 (Table 2). Substitution of tyrosine for tryptophan at each position did not result in a significant change in the ability of gp4 to support the growth of T7Δ4. Tryptophans at positions 97 and 147 were also replaced with alanine and examined for complementation of T7Δ4 growth. The gene 4 protein containing a substitution of alanine for Trp-97 could not support the phage growth, whereas the same alteration of Trp-147 decreased the supporting ability by about 20-fold. The alteration of Trp-147 also produced small plaques of T7Δ4, indicating the reduced ability for complementation. When cells expressing the altered gp4 were infected with wild-type T7 phage, no significant difference was observed between wild-type and the altered gp4. Thus, none of the altered proteins have a dominant negative effect on the function of the wild-type protein expressed by the phage.
TABLE 2.
Complementation of T7 phage growth by gp4 in which tryptophan in the primase domain is replaced with tyrosine or alanine
The ability of the full-length 63-kDa gp4 to support the growth of T7Δ4 phage was tested as described under “Experimental Procedures.” gp4 containing a single tryptophan substitution was expressed in E. coli DH5α. After infection with either T7Δ4 or wild-type T7 phage, the number of plaques was counted and normalized against the value obtained with wild-type gp4. Data were obtained from at least duplicated experiments.
| Alteration in gp4 | e.o.p.a |
|
|---|---|---|
| T7 Δ4 | T7 wild type | |
| No gp4 | <10−8 | 1.62 |
| Wild type | 1.00 | 1.00 |
| W42Y | 1.07 | 0.74 |
| W69Y | 1.00 | 0.56 |
| W97Y | 0.64 | 0.35 |
| W147Y | 0.71 | 0.44 |
| W255Y | 0.44 | 0.74 |
| W97A | <10−8 | 0.22 |
| W147A | 0.043b | 0.27 |
a Efficiency of plating; number of plaques relative to wild-type 63-kDa gp4.
b Small plaque.
To investigate the effect of the tryptophan substitutions on primase activity biochemically, primase fragments (residues 1–259) containing the replacement of tryptophan at various positions and an N-terminal His tag were purified using Ni-NTA affinity column chromatography. These primase fragments are designated as wild-type primase fragment, primase-W42Y, primase-W69Y, primase-W97Y, primase-W147Y, primase-W255Y, primase-W97A, and primase-W147A. A primase fragment containing the linker that connects the primase and helicase (residues 1–271) was also used for comparison, which has been characterized previously and shown to catalyze all of the reactions attributed to the primase domain (26).
Template-directed Oligoribonucleotide Synthesis
Wild-type primase fragment and the primase fragments with the replacement of a single tryptophan with tyrosine were first examined in the primer synthesis assay. The assay was performed at different temperatures using template 1 containing a primase recognition sequence 5′-TGGTC-3′ (Table 1), ATP, and radioactively labeled CTP (Fig. 3A). Generally, temperature affects the dynamic structure and conformation of a protein and therefore may influence the catalytic efficiency (27). The full-length 63-kDa gp4 and the primase fragment plus the linker between primase and helicase (residues 1–271) were also included in the assay. After separating the products (pppAC, pppACC, and pppACCA) of de novo synthesis (Fig. 3A), the amount of functional product pppACCA was measured (Fig. 3B). The 63-kDa gp4 exhibited the maximal activity for the production of pppACCA at 37 °C, whereas all of the primase fragments had optimal activity at 23 °C (Fig. 3B). At the optimal temperature of 23 °C, primase-W42Y catalyzed the synthesis of pppACCA at only 14% of the amount produced by the wild-type primase fragment (Fig. 3C). Similarly, primase-W147Y and primase-W97Y also exhibited reduced amounts compared with the wild-type primase fragment (32 and 64%, respectively). Primase-W69Y produced an amount of primer similar to wild-type, and primase-W255Y had slightly more than that of the wild-type primase fragment.
FIGURE 3.

Template-directed oligoribonucleotide synthesis by primases at different temperatures. A, the standard reaction for primer synthesis contained 0.2 μm gp4 or the indicated primase fragment, 4 μm template 1 containing 5′-TGGTC-3′ (Table 1), 1 mm ATP, and 1 mm [α-32P]CTP (0.1 mCi/ml) in buffer B. Wild-type primase fragment, primase fragments containing one tryptophan replaced with tyrosine, and primase fragment containing the linker between helicase and primase domains were used in these experiments. After incubation at 16, 23, 30, 37, or 44 °C for 30 min, the di-, tri-, and tetraribonucleotide products (indicated to the left) were separated on a 25% denaturing polyacrylamide gel. Lane 1, gp4; lane 2, primase fragment with a linker; lane 3, wild-type primase fragment (wt); lane 4, primase-W42Y; lane 5, primase-W69Y; lane 6, primase-W97Y; lane 7, primase-W147Y; lane 8, primase-W255Y. B, the amount of pppACCA formed in the reaction was measured and plotted against the reaction temperature for each protein. C, the amount of pppACCA synthesized at 23 °C by each protein is compared. The percentage was calculated by assigning a value of 100% for the wild-type primase fragment. Representative data from multiple experiments are shown.
The primase fragment containing the linker region (residues 1–271) showed a pattern similar to the other primase fragments in the template-dependent oligoribonucleotide synthesis except for somewhat better activity at higher temperatures. Thus, the additional 12 residues in the linker appear to increase the thermal stability. In contrast to the primase fragment, the primase domain within gp4 is attached to the helicase domain that oligomerizes to form hexamer. Consequently, the primase domain within gp4 is located in a more crowded environment than is the primase fragment. Therefore, the primase fragment and the primase domain in gp4 may function optimally at different temperatures (27).
Identification of Defects in Primase-W42Y, -W97Y, and -W147Y
Replacement of tryptophan at position 42, 147, or 97 with tyrosine resulted in a significant reduction in primer synthesis (Fig. 3). This defect in primer synthesis could reflect alterations in any one of a number of steps. Initially, the ZBD and RPD together must recognize the basic primase recognition sequence, 5′-GTC-3′. Then the RPD must catalyze the formation of a phosphodiester bond to create pppAC, a step that requires the binding of ATP and CTP. Finally the diribonucleotide is extended to the tetraribonucleotide, provided the appropriate sequences are present in the template. To analyze the role of tryptophan in each step of tetraribonucleotide synthesis, we examined primase activity in several different steps.
Requirement for Primase Recognition Site
The cryptic cytosine in the primase recognition sequence is critical for efficient primer synthesis by T7 primase. To determine if the altered primases recognize the specific template sequence in a way similar to that of the wild-type primase fragment, an oligonucleotide either containing (template 1, 5′-TGGTC-3′) or lacking (template 2, 5′-TGGTG-3′) the cryptic cytosine was used to examine primer synthesis (supplemental Fig. S2). Neither the wild-type nor the altered primase fragments efficiently catalyzed the synthesis of oligoribonucleotide in the absence of the cryptic cytosine. Only the full-length gp4 synthesized appreciable amounts of tetraribonucleotide with template lacking the cryptic cytosine, and the amount of synthesis was only 17% of that observed when the cryptic cytosine was present.
Template-independent Diribonucleotide Synthesis
The active site within the RPD of the primase catalyzes the synthesis, albeit at a greatly reduced rate, of random diribonucleotide in the absence of a DNA template (5). Thus, in the presence of CTP alone, T7 DNA primase catalyzed the synthesis of pppCC. The pppCC was incubated with CIAP to convert it to CC that was more readily identified on a polyacrylamide gel. Primase-W42Y, primase-W69Y, and primase-W255Y all synthesized diribonucleotide approximately equal to that observed with the wild-type primase fragment (Fig. 4A). Primase-W97Y and primase-W147Y catalyzed ∼50% of the pppCC observed with the wild-type primase fragment. As a control, primase-D237A, in which the catalytic residue Asp-237 within the active site of the RPD was replaced with alanine, did not catalyze the synthesis of the diribonucleotide, in confirmation of earlier studies (15). These results suggest that Trp-97 and Trp-149 contribute to the catalytic activity of the primase.
FIGURE 4.
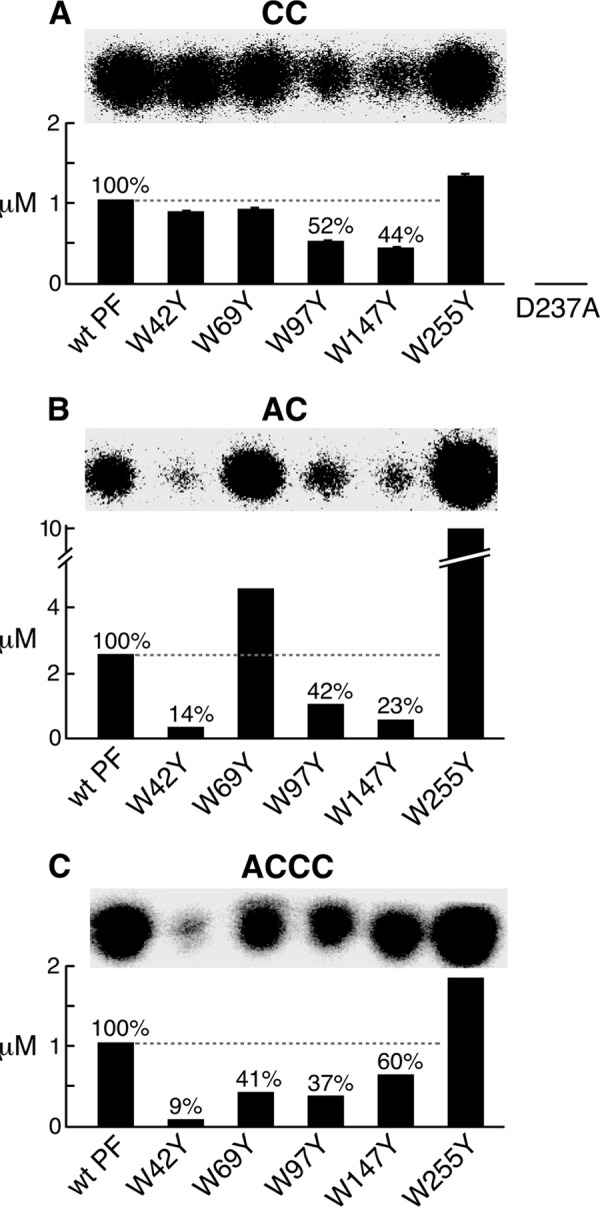
Catalytic activity, diribonucleotide synthesis, and diribonucleotide extension. A, template-independent diribonucleotide synthesis. The standard reaction contained a 2 μm concentration of the indicated primase and 1 mm [α-32P]CTP (0.1 mCi/ml) in buffer C. Wild-type primase fragment, primase fragments containing one tryptophan replaced with tyrosine, and primase-D237A were used in these experiments. After incubation at 23 °C for 1 h, the product pppCC was dephosphorylated with CIAP to yield CC. The product CC was separated, and its amount was determined. The percentage was calculated by assigning a value of 100% for the wild-type primase fragment. The S.E. values (error bars) were obtained from two independent experiments. B, template-directed diribonucleotide synthesis. Reactions contained a 0.4 μm concentration of the indicated primase, 4 μm template 3 containing 5′-AAGTC-3′ (Table 1), 1 mm ATP, and 1 mm [α-32P]CTP (0.1 mCi/ml) in buffer B. After incubation at 23 °C for 30 min, the product pppAC was dephosphorylated to AC as described in A. The amount of AC synthesized by each primase was determined as in A. C, template-dependent extension of diribonucleotide. The reaction contained 0.4 μm indicated primase, 4 μm template 4 containing 5′-GGGTC-3′ (Table 1), 1 mm 5′-AC-3′, and 1 mm [α-32P]CTP (0.1 mCi/ml) in the same buffer B. After incubation at 23 °C for 30 min, the products were separated, and the amount of ACCC was measured as in A.
Template-directed Diribonucleotide Synthesis
The first step of de novo oligoribonucleotide synthesis is recognition of the basic recognition sequence, 5′-GTC-3′, in the template and synthesis of the diribonucleotide pppAC (28). In order to examine the initial diribonucleotide synthesis, we have used a template (template 3, Table 1) containing only the basic recognition sequence and lacking the 5′ guanosines or thymines, allowing the synthesis of only pppAC. After removal of the terminal triphosphate from the pppAC with CIAP, the product AC can be identified on a polyacrylamide gel (Fig. 4B). Primase-W42Y, primase-W147Y, and primase-W97Y had significantly less activity, with ∼14, 23, and 42% of the product produced by wild-type primase fragment, respectively. Interestingly, primase-W69Y and primase-W255Y displayed more product compared with the wild-type primase fragment.
Extension to Tetraribonucleotide
Extension of diribonucleotide to the functional tetraribonucleotide by T7 DNA primase was examined using an oligonucleotide template containing the full primase recognition site 5′-GGGTC-3′ (template 4, Table 1). The diribonucleotide, 5′-AC-3′, was supplied so that the first step of primer synthesis could be bypassed using the preformed diribonucleotide. The tetraribonucleotide 5′-ACCC-3′ formed by the incorporation of [α-32P]CMP was measured after separation on a polyacrylamide gel (Fig. 4C). Primase-W42Y had only 9% of the product observed with the wild-type primase fragment. Primase-W69Y, primase-W97Y, and primase-W147Y had decreased amount (40–60%) relative to the wild-type primase fragment.
Zinc Content and Conformation of Primase-W42Y
The catalytic site of primase-W42Y appeared normal, as determined by the template-independent synthesis of diribonucleotide. However, it was severely defective in template-dependent synthesis of the diribonucleotide as well as the extension of the diribonucleotide to tetraribonucleotide. Trp-42 resides within the ZBD of the primase, and its location thus suggests a role in recognition and binding of the specific template sequence. However, this function requires a specific conformation of ZBD and proper coordination with zinc ion. In earlier studies, we have shown that the cysteines of the ZBD are essential for binding zinc, as is His-33 (5, 29). Substitution of other amino acids for these residues leads to a loss of zinc (30). We measured the zinc contents of primase-W42Y and wild-type primase fragment by ICP-MS. Primase-W42Y has ∼50% of the zinc content of the wild-type primase fragment (Fig. 5A). The replacement of Trp-42 with tyrosine obviously reduced the binding affinity of the primase fragment to zinc ion.
FIGURE 5.

Zinc content of primase-W42Y and conformation of ZBD-W42Y. A, the zinc content of the wild-type (wt) primase fragment (PF) and primase-W42Y was determined by ICP-MS using 40 μl of a 16 μm concentration of the indicated primase fragment. The percentage of zinc in primase-W42Y or in the buffer was calculated by assigning a value of 100% for the wild-type primase fragment. B, circular dichroism spectra of the wild-type ZBD and ZBD-W42Y were recorded from 300 to 200 nm using a 20 μm concentration of the indicated ZBD in 50 mm Tris-HCl, pH 7.5, 1 mm DTT, 50 mm NaCl, and 1% glycerol. The spectrum for each sample was determined four times at 20 °C, and the background signal obtained from buffer was subtracted. The final signal was normalized against protein molar concentration.
The binding of zinc within the ZBD is the major determinant in the conformation of this subdomain of the primase fragment. In view of the reduced binding affinity of zinc in primase-W42Y, we compared the CD spectra of the wild-type ZBD and the ZBD-W42Y (Fig. 5B). We used only the ZBD instead of the primase fragment in order to exclude the signal from the RPD. A UV CD spectrum provides information about the α-helices, β-sheets, and random coils of a protein (31, 32). The wild-type ZBD had a CD spectrum similar to that reported previously (30). The negative trough at about 230 nm corresponds to the ordered protein structure consisting of β-sheet and α-helix shown in the crystal structure (Fig. 1). The positive peak at 220 nm for ZBD-W42Y indicates the formation of a random coil conformation in the protein structure. Together with the loss of zinc shown in Fig. 5A, the results suggest that the replacement of Trp-42 with tyrosine reduces the zinc binding in the ZBD and, as a result, disturbs the conformation of ZBD. The disturbed conformation of ZBD may impair the effective binding of the primase to ssDNA template with the recognition sequence.
Oligoribonucleotide Synthesis by Primase Fragments with Alanine Replacing Trp-97 or Trp-147
Other than the replacement of tryptophan with tyrosine at position 42, replacements at position 147 or 97 also decreased activity in primer synthesis (Fig. 3). To further address the roles of Trp-97 and Trp-147, we replaced Trp-97 or Trp-147 with alanine and examined oligoribonucleotide synthesis by the altered primase fragments.
The primase fragment containing alanine at position 97 (primase-W97A) had a more severe defect than did primase-W97Y in de novo oligoribonucleotide synthesis (Fig. 6A). Detailed analysis of each step in primer synthesis confirmed that the alteration in primase-W97A almost abrogated catalytic activity (Fig. 6, B–D). These results are in agreement with the inability of gp4-W97A to support the growth of T7Δ4 (Table 2). In contrast, replacement of Trp-147 with alanine (primase-W147A) did not show a significant difference from primase-W147Y except for the ability to extend diribonucleotide to tetraribonucleotide (Fig. 7D).
FIGURE 6.

Oligoribonucleotide synthesis by primase fragments with alanine replacing tryptophan. A, the standard reaction for primer synthesis contained a 0.2 μm concentration of the indicated primase fragment, 4 μm template 1 containing 5′-TGGTC-3′ (Table 1), 1 mm ATP, and 1 mm [α-32P]CTP (0.1 mCi/ml) in buffer B. Wild-type primase fragment and primase fragments containing substitution of tyrosine or alanine for tryptophan at position 97 or 147 were used. After incubation at 23 °C for 30 min, the di-, tri-, and tetraribonucleotide products (indicated to the left) were separated, and the amount of pppACCA was measured. The percentage of activity of each protein was calculated by assigning a value of 100% for the wild-type primase fragment. B, template-independent diribonucleotide synthesis. The standard reaction contained a 2 μm concentration of the indicated primase and 1 mm [α-32P]CTP (0.1 mCi/ml) in buffer C. After incubation at 23 °C for 1 h, the product pppCC was dephosphorylated to yield CC. The product CC was separated, and the amount was determined. The percentage of activity of each protein was calculated as described in A. C, template-directed diribonucleotide synthesis. Reactions contained 0.4 μm indicated primase, 4 μm template 3 containing 5′-AAGTC-3′ (Table 1), 1 mm ATP, and 1 mm [α-32P]CTP (0.1 mCi/ml) in the same buffer B. After incubation at 23 °C for 30 min, the product pppAC was dephosphorylated to AC as described in B. The amount of AC synthesized by each primase fragment was determined as in B. D, template-dependent extension of diribonucleotide. The reaction contained 0.4 μm indicated primase, 4 μm template 4 containing 5′-GGGTC-3′ (Table 1), 1 mm 5′-AC-3′, and 1 mm [α-32P]CTP (0.1 mCi/ml) in the same buffer B. After incubation at 23 °C for 30 min, the products were separated, and the amount of pppACCC was measured as in A. Representative data from multiple experiments are presented in A–D.
FIGURE 7.

Binding of gp4 to ATP determined by surface plasmon resonance. Biotinylated ATP (300 RU) was immobilized on the surface of the chip via biotin-streptavidin interaction. The wild-type gp4, gp4-W97Y, gp4-W147Y, and gp4-W147A (0.7 μm) in 60 μl of flow buffer were injected at a flow rate of 50 μl/min. Binding of gp4 to ATP is represented by the difference in binding signal obtained by flowing gp4 over the lane with either biotinylated ATP or biotin, as shown in response units on the y axis. The arrows indicate the start and end of injection of protein in each panel.
ATP Binding Affinity of gp4 Containing Substitution at Position 97 or 147
Replacement of Trp-97 or Trp-147 with tyrosine or alanine reduced activity in the template-independent diribonucleotide synthesis, with the most severe defect in primase-W97A. One possible explanation for this result is that the binding affinity of the altered primase fragment to NTP is reduced. To examine this possibility, we measured the binding affinity of wild-type and altered gp4 to ATP using surface plasmon resonance (15). In these assays, biotinylated ATP was immobilized on the surface of a SA chip (300 response units (RU)). Each gp4 was flowed over the chip, and the binding of protein was measured (Fig. 7). Wild-type gp4 rapidly associated with the immobilized ATP, resulting in an increased signal of 700 RU. Replacement of Trp-97 with tyrosine greatly reduced the association rate and binding signal (∼150 RU). The difficulty in purifying gp4-W97A prevented a measurement of its affinity for ATP. However, the difficulty in purification suggests that it has a different conformation from wild-type gp4. Elution of gp4-W97A from phosphocellulose required much higher salt concentration than that for the other gp4. In addition, gp4-W97A did not bind to ATP-agarose resin, indicating that gp4-W97A has lost binding affinity to ATP. gp4-W147Y showed similar binding affinity with ATP compared with wild-type gp4, whereas replacement of Trp-147 with alanine resulted in an obvious reduction in binding affinity. Comparison of gp4-W97Y and gp4-W147Y with wild-type gp4 demonstrates that replacement of tryptophan with tyrosine at position 97 more drastically decreased the binding affinity to NTP, whereas the alteration at position 147 gave similar binding affinity to NTP, suggesting that Trp-97 plays the major role in NTP binding.
Kinetic Analysis of Diribonucleotide Synthesis
The de novo synthesis of oligoribonucleotides requires the binding of NTP and the subsequent formation of a phosphodiester bond at the active site. The reduced activity observed with proteins having tyrosine replacements in template-independent diribonucleotide synthesis can be partially attributed to a reduced binding affinity to NTP. However, these alterations could also affect the catalytic step. To determine if the substitutions at position 97 or 147 affect the catalytic rate as well as the binding of the protein to NTP, diribonucleotide synthesis catalyzed by the wild-type primase fragment, primase-W97Y, primase-W97A, primase-W147Y, and primase-W147A was examined. In the experiment shown in Fig. 8, diribonucleotide synthesis was measured at varying concentrations of ATP ranging from 0.05 to 2 mm and excess [α-32P]CTP (2 mm, 0.1 mCi/ml). The product formation was linearly increased with the reaction time under these conditions. The rate of diribonucleotide synthesis was dependent on ATP concentration (Fig. 8A). Under condition of excess CTP (2 mm) over its Km value (0.85 mm) (10), the rates of product formation were plotted against ATP concentrations and fit to a Michaelis-Menten equation. Wild-type primase fragment yielded an apparent Km,ATP of 0.24 mm and a kcat for diribonucleotide synthesis of 0.31 min−1 (Fig. 8B). Replacement of Trp-97 with tyrosine reduced the kcat value by 2-fold, whereas replacement with alanine reduced the kcat 22-fold. The substitution of tyrosine and alanine for Trp-147 reduced the values of kcat by 5- and 3-fold, respectively. Replacement of Trp-97 with tyrosine increased the Km,ATP by 1.5-fold, and replacement with alanine increased it by 5-fold. However, the same alteration at position 147 exhibited a Km,ATP value similar to that of wild-type primase.
FIGURE 8.

Kinetic analysis of ATP-dependent diribonucleotide synthesis. A, reactions contained a 0.4 μm concentration of the indicated primase, 4 μm template 3 containing 5′-AAGTC-3′ (Table 1), varying concentrations of ATP, and 2 mm [α-32P]CTP (0.1 mCi/ml) in buffer B. After incubation at 23 °C for 30 min, the product pppAC was dephosphorylated to yield AC. The product AC was separated, and the amount of AC was measured. Plots of the rates of the product formation against ATP concentrations were fit to a Michaelis-Menten equation using GraphPad Prism (version 4.0c) to determine kcat and Km,ATP. B, the kinetic parameters of kcat and Km,ATP obtained from each protein are summarized.
Physical binding of ATP measured by SPR indicates that alterations at position 97 give rise to a more severe defect than alterations at position 147. Because Km,ATP was determined in a biochemical assay that involves both NTP binding and the catalytic formation of diribonucleotides, the Km,ATP might reflect more than the physical binding of NTP. The kinetic analysis and SPR data clearly show that replacement of tryptophan with tyrosine or alanine at position 97 reduces the maximal catalytic rate and decreases the binding affinity to ATP, whereas alteration at position 147 reduces only the maximal catalytic rate. Therefore, Trp-97 plays roles in maintaining both the catalytic rate and NTP binding; Trp-147 plays a role in the catalytic step but has little effect on binding of NTP.
Diribonucleotide synthesis requires the primase to bind two NTPs and the DNA template. The binding affinities of these primases to DNA template were also measured using similar kinetic methods except for varying the template concentrations at saturated NTP concentrations (Fig. 9). All of the primase fragments except for primase-W97A showed similar binding affinities to DNA template. However, the maximal catalytic rates, kcat, varied among the primases.
FIGURE 9.
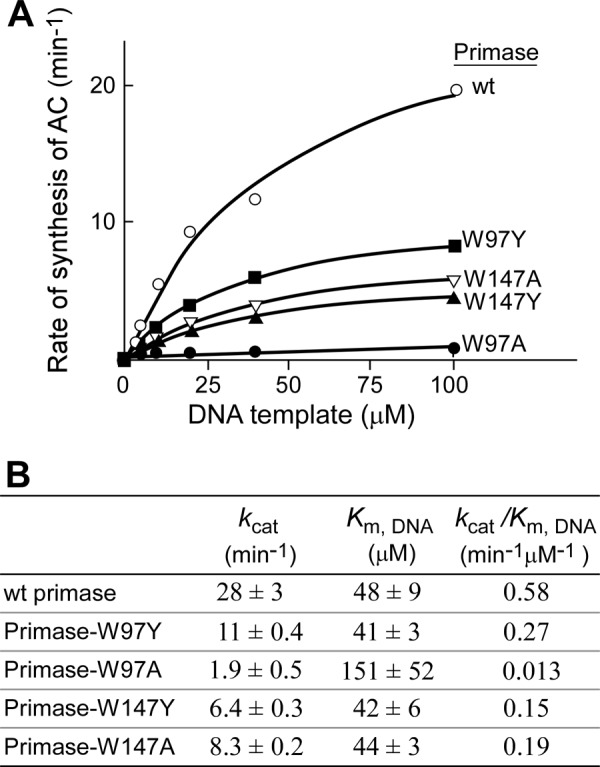
Kinetic analysis of template-dependent diribonucleotide synthesis. A, reactions contained a 0.4 μm concentration of the indicated primase, varying concentrations of DNA template 3 containing 5′-AAGTC-3′ (Table 1), 2 mm ATP, and 2 mm [α-32P]CTP (0.1 mCi/ml) in buffer B. After incubation at 23 °C for 10 min, the product pppAC was dephosphorylated to yield AC. The product AC was separated, and the amount of AC was measured. Plots of the rates of the product formation against DNA template concentrations were fit to a Michaelis-Menten equation using GraphPad Prism (version 4.0c) to determine kcat and Km,DNA. B, the kinetic parameters of kcat and Km,DNA obtained from each protein are summarized.
Single Tryptophans in the Primase Domain
The results derived from the biochemical analysis of the altered primase fragments show that the tryptophans at positions 42, 147, and 97 play important roles in template-directed oligoribonucleotide synthesis. Of these residues, Trp-42 is clearly important in the maintenance of ZBD conformation for binding of DNA template, and Trp-97 and Trp-147 are important for NTP binding and/or the catalysis step. Another approach to address the importance of individual tryptophans in the primase is to replace all tryptophans with tyrosine except for a single tryptophan and to examine its function in vivo and in vitro.
Gp4 in which all of the tryptophans were replaced with tyrosine was unable to support the growth of T7Δ4 phage (supplemental Table S1). The ability of each of the five gp4 in which only one tryptophan remained in the primase domain (residues 1–259) was examined to complement growth of T7Δ4 phage (supplemental Table S1). Only gp4 in which Trp-97 remained could support the growth of T7Δ4 phage with a plating efficiency of ∼8% of that observed with wild-type gp4.
The primase fragments lacking tryptophan or containing a single tryptophan were purified, and they were designated as primase-noW, -W42only, -W69only, -W97only, -W147only, and -W255only. They were examined for synthesis of oligoribonucleotide. Of the six altered primases, only primase-W97only exhibited some oligoribonucleotide synthesis, but the observed synthesis was less than 1% of that observed for the wild-type primase fragment (supplemental Fig. S3A). Primase-W97only catalyzed the synthesis of the tetraribonucleotide in a concentration-dependent manner (supplemental Fig. S3, B and C), where the activity ranged from 0.1 to 0.6% of that observed with the wild-type primase fragment.
Because Trp-97 is indispensable for oligoribonucleotide synthesis, we examined the additional contribution of tryptophans at the other positions to the activity. Primase fragments containing two tryptophans (primase-W42/W97only, -W69/W97only, -W97/W147only, and -W97/W255only) were purified and assayed for oligoribonucleotide synthesis (supplemental Fig. S4A). All of these altered primases had lower activity than the wild-type primase fragment but higher activity than primase-W97only. Restoration of the second tryptophan obviously improves primase activity with activity increasing in the following order: Trp-42 > Trp-147 > Trp-69 > Trp-255 (supplemental Fig. S4B). This trend is in good agreement with the results shown in Fig. 3, confirming that tryptophans at positions 42, 97, and 147 play crucial roles in the template-dependent synthesis of oligoribonucleotide.
DISCUSSION
T7 DNA primase catalyzes the synthesis of short oligoribonucleotides for use as primers to initiate the synthesis of Okazaki fragments (2). As shown in Fig. 2, primer synthesis includes several steps. The ZBD and RPD recognize the basic primase recognition sequence, 5′-GTC-3′ (10). After binding of ATP and CTP, the RPD catalyzes the formation of a phosphodiester bond to create pppAC. Finally, the diribonucleotide is extended to the tetraribonucleotide, provided the appropriate sequences are present in the template (2). Interactions of both the ZBD and RPD with the DNA template and ribonucleotides are necessary for primer synthesis (4). These steps require a sequence of conformational changes of the T7 DNA primase.
The hydrophobic interaction among amino acid residues within a protein is important to maintain its stability and conformation during the catalytic process. Among the 20 natural amino acids, tryptophan has the largest accessible hydrophobic surface and consequently the strongest hydrophobic interaction with other amino acids in a local environment. Different from hydrogen bond interactions, these hydrophobic interactions can be effective in a longer range, up to 10 nm (33). Tryptophan has shown to play a major role in the folding of a protein (20) and its stabilization (17, 21–23).
Five tryptophans are dispersed in the primase domain of gp4: Trp-42 in the ZBD, Trp-69 and Trp-97 in the N-terminal portion of the RPD, and Trp-147 and Trp-255 in the TOPRIM fold of the RPD (4). Except for Trp-255, located near the linker between the primase and helicase domains, all tryptophan residues are surrounded by other aromatic residues (4). In the present study, we replaced each tryptophan with tyrosine, the amino acid with the closest structure to tryptophan among the 20 natural amino acids. We have observed that these replacements identify roles of each tryptophan in several steps of primer synthesis.
In a previous study, the replacement of Trp-42 with alanine in the ZBD decreased de novo synthesis of tetraribonucleotide and partially reduced its utilization as primers by DNA polymerase (24). However, replacement of tryptophan with alanine is too drastic to meaningfully deduce the structural roles of the tryptophan from the properties of the altered ZBD. In the present study, Trp-42 was replaced with tyrosine. Even with such a subtle alteration in structure, primase-W42Y exhibits defects in the de novo synthesis of oligoribonucleotides, template-directed diribonucleotide synthesis, and the extension of the oligoribonucleotides. However, the catalytic activity of primase-W42Y is not affected. Biophysical analysis shows that the replacement of Trp-42 with tyrosine partially disturbs the conformation of ZBD and reduces the binding affinity to zinc and consequently weakens the binding of primase to the primase recognition sequence, a step prior to the catalytic step.
The ZBD plays a crucial role in the recognition of the primase recognition sequence and the binding of this sequence to the primase (4, 12). In crystal structures, Trp-42 is located in a group of β-sheets containing aromatic groups. The zinc ion is coordinated with four cysteines at the loops connecting these β-sheets. Replacement of tryptophan with tyrosine may weaken the interaction in the β-sheets and therefore disturb the conformation of the ZBD, reducing the binding affinity to the zinc ion. In our previous study, replacement of the aromatic His-33 with alanine also disturbs the conformation of the ZBD, although His-33 does not directly interact with the zinc binding site (30). Collectively, Trp-42 plays a critical role in maintaining the conformation of ZBD.
Trp-69 is located in the N-terminal portion of the RPD near the linker between the RPD and the ZBD. As shown in earlier studies, gp4-W69A cannot support the growth of T7Δ4 phage (25). Wild-type gp4, gp4-W69Y, and gp4-W69K catalyze the synthesis of oligoribonucleotide at similar efficiency (25), showing that the tryptophan at this position is not crucial for primer synthesis. However, only wild-type gp4 and gp4-W69Y can effectively deliver primers to DNA polymerase for the initiation of the synthesis of Okazaki fragment. We conclude that the presence of an aromatic group at position 69 is necessary for primer delivery. In this work, we have examined the function of primase-W69Y at each step of primer synthesis. Although the wild-type primase fragment and primase-W69Y show similar activity for primer synthesis, primase-W69Y has a higher efficiency in diribonucleotide synthesis but a lower efficiency for extension of the diribonucleotide.
Trp-97 is located at the N-terminal portion of the RPD. Amino acid sequence analysis shows that this tryptophan is conserved at the comparable position in the primases of E. coli, Salmonella typhimurium, Bacillus subtilis, phage T4, and phage T3 (34). Gp4 in which Trp-97 is replaced with alanine does not support the growth of phage T7Δ4. Replacement of Trp-97 with tyrosine in the primase fragment decreases oligoribonucleotide synthesis and extension of the diribonucleotide, presumably due to the reduction in the catalytic activity of the primase. SPR and kinetic analysis further reveal that replacement of Trp-97 with tyrosine reduces not only ATP binding affinity but also the catalysis rate. Replacement of Trp-97 with alanine results in a more drastic decrease in binding affinity for ATP and catalytic rate, further supporting the importance of a bulkier aromatic residue at position 97 in primer synthesis. The importance of Trp-97 is also supported by other experiments. Trp-97 is the only one of the five tryptophans that can allow for the growth of T7Δ4 phage, albeit at a reduced efficiency, when the other four are replaced with tyrosine. Primase with only Trp-97 left is the only primase that retains some activity when the other four tryptophans are replaced with tyrosine.
Trp-147 is located in the TOPRIM fold of the RPD. Replacement of Trp-147 with tyrosine decreases the formation of phosphodiester bond at the active site, indicating this reaction is sensitive to an environment composed of this aromatic residue. However, replacement of Trp-147 with alanine results in higher activity than replacement with tyrosine, mostly due to better extension of diribonucleotides. Further analysis shows that replacement of Trp-147 with tyrosine does not reduce the binding affinity of primase to NTP but decreases the catalysis rate. In the SPR results (Fig. 7), the binding affinities of primase to ATP are similar for wild-type gp4 and gp4-W147Y. In kinetic analysis (Figs. 8 and 9), the binding affinities for NTP and template are also similar for wild-type and altered primases containing an alteration at position 147, indicating that the alteration at position 147 does not affect ATP binding or template binding. The reduced activity results from the defect in the catalysis step, suggesting an essential role of Trp-147 in the catalytic step.
The catalytic domain consists of an N-terminal subdomain of RPD and TOPRIM fold (Fig. 1). Two Mg2+ ions are located in the TOPRIM fold and also near the shallow cleft between the two subdomains. The Mg2+ ions mediate the catalysis of diphosphodiester bond formation between the bound NTPs. The substitution at position 97 reduces NTP binding affinity and the catalysis step; the substitution at position 147 only affects the catalysis step. These results suggest that Trp-97 might have more extensive contact with NTP than Trp-147, possibly involving the hydrophobic interaction. Alternatively, Trp-97 might have more contribution to the conformation of primase that is optimal for NTP binding than Trp-147.
In summary, the five tryptophans discussed above are located in different subdomains of the primase and clearly show different roles in the steps of primer synthesis. Trp-42 is not involved in the catalytic step but is required to maintain the conformation of the ZBD that is essential to bind primase to its recognition sequence. Trp-97 is involved in both catalysis rate and NTP binding, whereas Trp-147 is involved in only catalysis. Trp-42, -97, and -147 play different but essential roles in primer synthesis by T7 DNA primase.
Acknowledgments
We greatly thank Susan J. Chai for technical support, Steve Moskowitz (Advanced Medical Graphics) for preparing the figures, Charles Langmuir and Zhongxing Chen (Department of Earth and Planetary Sciences, Harvard University) for the ICP-MS measurements, and Gregory Verdine and Tom Grossmann (Departments of Chemistry and Chemical Biology, Harvard University) for use of the circular dichroism facility.
This work was supported, in whole or in part, by National Institutes of Health Grant GM 54937 (to C. C. R.).

This article contains supplemental Table S1 and Figs. S1–S4.
- trx
- thioredoxin
- ICP-MS
- inductively coupled plasma-mass spectrometry
- nt
- nucleotide
- Ni-NTA
- nickel-nitrilotriacetic acid
- CIAP
- calf intestinal alkaline phosphatase
- SPR
- surface plasmon resonance
- ZBD
- zinc-binding domain
- RPD
- RNA polymerase domain
- RU
- response units.
REFERENCES
- 1. Richardson C. C. (1983) Bacteriophage T7. Minimal requirements for the replication of a duplex DNA molecule. Cell 33, 315–317 [DOI] [PubMed] [Google Scholar]
- 2. Frick D. N., Richardson C. C. (2001) DNA primases. Annu. Rev. Biochem. 70, 39–80 [DOI] [PubMed] [Google Scholar]
- 3. Lee J., Chastain P. D., 2nd, Griffith J. D., Richardson C. C. (2002) Lagging strand synthesis in coordinated DNA synthesis by bacteriophage T7 replication proteins. J. Mol. Biol. 316, 19–34 [DOI] [PubMed] [Google Scholar]
- 4. Kato M., Ito T., Wagner G., Richardson C. C., Ellenberger T. (2003) Modular architecture of the bacteriophage T7 primase couples RNA primer synthesis to DNA synthesis. Mol. Cell 11, 1349–1360 [DOI] [PubMed] [Google Scholar]
- 5. Kusakabe T., Hine A. V., Hyberts S. G., Richardson C. C. (1999) The Cys4 zinc finger of bacteriophage T7 primase in sequence-specific single-stranded DNA recognition. Proc. Natl. Acad. Sci. U.S.A. 96, 4295–4300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Keck J. L., Roche D. D., Lynch A. S., Berger J. M. (2000) Structure of the RNA polymerase domain of E. coli primase. Science 287, 2482–2486 [DOI] [PubMed] [Google Scholar]
- 7. Podobnik M., McInerney P., O'Donnell M., Kuriyan J. (2000) A TOPRIM domain in the crystal structure of the catalytic core of Escherichia coli primase confirms a structural link to DNA topoisomerases. J. Mol. Biol. 300, 353–362 [DOI] [PubMed] [Google Scholar]
- 8. Lee S. J., Richardson C. C. (2002) Interaction of adjacent primase domains within the hexameric gene 4 helicase-primase of bacteriophage T7. Proc. Natl. Acad. Sci. U.S.A. 99, 12703–12708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Qimron U., Lee S. J., Hamdan S. M., Richardson C. C. (2006) Primer initiation and extension by T7 DNA primase. EMBO J. 25, 2199–2208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Frick D. N., Kumar S., Richardson C. C. (1999) Interaction of ribonucleoside triphosphates with the gene 4 primase of bacteriophage T7. J. Biol. Chem. 274, 35899–35907 [DOI] [PubMed] [Google Scholar]
- 11. Lee S. J., Zhu B., Hamdan S. M., Richardson C. C. (2010) Mechanism of sequence-specific template binding by the DNA primase of bacteriophage T7. Nucleic Acids Res. 38, 4372–4383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mendelman L. V., Richardson C. C. (1991) Requirements for primer synthesis by bacteriophage T7 63-kDa gene 4 protein. Roles of template sequence and T7 56-kDa gene 4 protein. J. Biol. Chem., 266, 23240–23250 [PubMed] [Google Scholar]
- 13. Bernstein J. A., Richardson C. C. (1988) A 7-kDa region of the bacteriophage T7 gene 4 protein is required for primase but not for helicase activity. Proc. Natl. Acad. Sci. U.S.A. 85, 396–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee S. J., Richardson C. C. (2001) Essential lysine residues in the RNA polymerase domain of the gene 4 primase-helicase of bacteriophage T7. J. Biol. Chem. 276, 49419–49426 [DOI] [PubMed] [Google Scholar]
- 15. Lee S. J., Richardson C. C. (2005) Acidic residues in the nucleotide-binding site of the bacteriophage T7 DNA primase. J. Biol. Chem. 280, 26984–26991 [DOI] [PubMed] [Google Scholar]
- 16. Kusakabe T., Richardson C. C. (1996) The role of the zinc motif in sequence recognition by DNA primases. J. Biol. Chem. 271, 19563–19570 [DOI] [PubMed] [Google Scholar]
- 17. Clark E. H., East J. M., Lee A. G. (2003) The role of tryptophan residues in an integral membrane protein. Diacylglycerol kinase. Biochemistry, 42, 11065–11073 [DOI] [PubMed] [Google Scholar]
- 18. Chothia C. (1976) Nature of accessible and buried surfaces in proteins. J. Mol. Biol. 105, 1–12 [DOI] [PubMed] [Google Scholar]
- 19. Burley S. K., Petsko G. A. (1985) Aromatic-aromatic interaction. A mechanism of protein structure stabilization. Science 229, 23–28 [DOI] [PubMed] [Google Scholar]
- 20. Campanini B., Raboni S., Vaccari S., Zhang L., Cook P. F., Hazlett T. L., Mozzarelli A., Bettati S. (2003) Surface-exposed tryptophan residues are essential for O-acetylserine sulfhydrylase structure, function, and stability. J. Biol. Chem. 278, 37511–37519 [DOI] [PubMed] [Google Scholar]
- 21. Subramaniam V., Jovin T. M., Rivera-Pomar R. V. (2001) Aromatic amino acids are critical for stability of the bicoid homeodomain. J. Biol. Chem. 276, 21506–21511 [DOI] [PubMed] [Google Scholar]
- 22. Davidson W. S., Arnvig-McGuire K., Kennedy A., Kosman J., Hazlett T. L., Jonas A. (1999) Structural organization of the N-terminal domain of apolipoprotein A-I. Studies of tryptophan mutants. Biochemistry 38, 14387–14395 [DOI] [PubMed] [Google Scholar]
- 23. Liu J., Yong W., Deng Y., Kallenbach N. R., Lu M. (2004) Atomic structure of a tryptophan-zipper pentamer. Proc. Natl. Acad. Sci. U.S.A. 101, 16156–16161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kato M., Ito T., Wagner G., Ellenberger T. (2004) A molecular handoff between bacteriophage T7 DNA primase and T7 DNA polymerase initiates DNA synthesis. J. Biol. Chem. 279, 30554–30562 [DOI] [PubMed] [Google Scholar]
- 25. Zhu B., Lee S. J., Richardson C. C. (2010) Direct role for the RNA polymerase domain of T7 primase in primer delivery. Proc. Natl. Acad. Sci. U.S.A. 107, 9099–9104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Frick D. N., Baradaran K., Richardson C. C. (1998) An N-terminal fragment of the gene 4 helicase/primase of bacteriophage T7 retains primase activity in the absence of helicase activity. Proc. Natl. Acad. Sci. U.S.A. 95, 7957–7962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Daniel R. M., Dunn R. V., Finney J. L., Smith J. C. (2003) The role of dynamics in enzyme activity. Annu. Rev. Biophys. Biomol. Struct. 32, 69–92 [DOI] [PubMed] [Google Scholar]
- 28. Frick D. N., Richardson C. C. (1999) Interaction of bacteriophage T7 gene 4 primase with its template recognition site. J. Biol. Chem. 274, 35889–35898 [DOI] [PubMed] [Google Scholar]
- 29. Mendelman L. V., Beauchamp B. B., Richardson C. C. (1994) Requirement for a zinc motif for template recognition by the bacteriophage T7 primase. EMBO J. 13, 3909–3916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Akabayov B., Lee S. J., Akabayov S. R., Rekhi S., Zhu B., Richardson C. C. (2009) DNA recognition by the DNA primase of bacteriophage T7. A structure-function study of the zinc-binding domain. Biochemistry 48, 1763–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Whitmore L., Wallace B. A. (2008) Protein secondary structure analyses from circular dichroism spectroscopy. Methods and reference databases. Biopolymers 89, 392–400 [DOI] [PubMed] [Google Scholar]
- 32. Greenfield N. J. (2006) Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 1, 2876–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Israelachvili J., Pashley R. (1982) The hydrophobic interaction is long-range, decaying exponentially with distance. Nature 300, 341–342 [DOI] [PubMed] [Google Scholar]
- 34. Ilyina T. V., Gorbalenya A. E., Koonin E. V. (1992) Organization and evolution of bacterial and bacteriophage primase helicase systems. J. Mol. Evol. 34, 351–357 [DOI] [PubMed] [Google Scholar]