

We are now in an exciting era of molecular bioenergetics. High-resolution x-ray structures have been determined for several of the key components of bioenergetics systems, including the respiratory enzymes cytochrome oxidase (1–4) and the bc1 complex (5, 6), the F1 component of the F1Fo-ATPase (7), the light-driven proton pump bacteriorhodopsin (8, 9), and the bacterial photosynthetic reaction center (10–12). Each of these membrane proteins generates (or can use) a protonmotive force across the membrane by using unique mechanisms. In cytochrome oxidase the complex oxygen chemistry is somehow linked at different stages to driving protons about 50 Å across the membrane against an electrochemical gradient. In this issue of the Proceedings, Hartmut Michel (45) offers a new model of how this process might work. He presents a challenge to a paradigm that has been generally accepted by researchers in the field for the past decade. To be sure, his proposal will spark healthy debate and a new round of experiments designed to test the predictions of the new model that distinguish it from other models that have been offered.
Background
Protein Structure and Overall Chemistry.
Cytochrome oxidase is a magnificent enzyme. A wide variety of biophysical techniques in combination with site-directed mutagenesis and the recent x-ray crystallography have resulted in a remarkable increase in our understanding of this enzyme in the past few years (for review see ref. 13). The x-ray structures have been determined for the 13-subunit bovine heart mitochondrial oxidase (3, 4) as well as the four-subunit oxidase from the soil bacterium Paracoccus denitrificans (1, 2). The x-ray structures show that subunits I, II, and III are very similar in both systems. The Paracoccus oxidase also has been isolated in a two-subunit form (subunits I and II) that is fully functional (2). The role of subunit II seems to be primarily to provide the site where cytochrome c is oxidized. Subunit I, which has 12 transmembrane helical spans, contains virtually everything else required for the redox chemistry and proton pumping functions.
Cytochrome oxidase is an integral membrane protein, and to understand its function it must be recognized that it has surfaces exposed to both inside and outside aqueous compartments. In the eukaryotic enzyme, the enzyme is located in the inner mitochondrial membrane, and the inside refers to the mitochondrial matrix. In the homologous prokaryotic oxidases, the enzyme is located in the cytoplasmic membrane and the inside corresponds to the bacterial cytoplasm.
The chemistry catalyzed by the enzyme is deceptively simple:
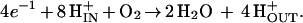 |
The four electrons are provided by the sequential reaction of four equivalents of cytochrome c that react with the enzyme at the outside surface. Eight protons per O2 are taken up from the inside, of which four are used in the chemistry and four are pumped to the outside. Because the protons and electrons used to make H2O come from opposite sides of the membrane, the chemistry by itself results in the net translocation of four charges (4 q) across the membrane per O2 and the generation of a transmembrane potential. In addition, the enzyme pumps four protons (per O2) electrogenically, i.e., one H+ per electron. The reaction, thus, results in eight charges (8 q) translocated across the membrane (per O2), providing an efficient source of energy for making ATP and driving active transport processes.
Electron Circuitry.
The electron transfer sequence is well defined and is linear (13). There are four metal redox centers in cytochrome oxidase: CuA (a bimetallic copper center), heme a, heme a3, and CuB. The heme a3 Fe and CuB are separated by only about 5 Å and comprise the binuclear center, which is the active site of the enzyme where oxygen is reduced to water. The binuclear center as well as heme a all are located at the same depth in the membrane, about 35% down (physically) from the outer surface. CuA oxidizes cytochrome c at the outside surface, and from there the electron is transferred first to heme a and then to the heme a3/CuB binuclear center. The only electron transfer that contributes directly to the transmembrane charge separation is from CuA to heme a. Most of the electrical work is caused by proton movements within the enzyme (14, 15).
Proton Circuitry.
Site-directed mutagenesis studies as well as the x-ray structures suggest two pathways for proton translocation (see ref. 16). A third “H-channel” recently has been proposed for the mammalian oxidase (3), but a functional role for this channel is not supported by mutagenesis studies on the bacterial enzymes (16, 17).
The D-channel starts at Asp-124 (Paracoccus numbering) near the inside surface. The channel entrance is within a region on the protein surface that could function as a proton-gathering antenna (18). This channel proceeds about 25 Å to Glu-278 deep within the protein interior, largely by a chain of hydrogen-bonded internal water molecules (19, 20). By changing side-chain conformation, Glu-278 has been proposed to shuttle the protons to the binuclear center that are required for the oxygen chemistry (19, 21). Glu-278 also has been proposed to shuttle pumped protons to the heme a3 ring D propionate (20), directed eventually to the outside surface. In either case, internal water molecules are likely to complete these proton pathways, at least transiently. Mutagenesis studies clearly implicate the D-channel in proton pumping.
The K-channel contains Lys-254 as a key residue buried within the membrane and leads from the inside surface to Tyr-280, located at the enzyme active site. The x-ray structures reveal that Tyr-280 is crosslinked to His-276 (a ligand to CuB) (2, 3), and also that the hydroxyl moiety of this tyrosine is close enough to serve as a proton or hydrogen atom donor to help split the O—O bond after O2 binds to heme a3 (16, 22). Mutagenesis studies show that the primary function of the K-channel is to provide one or two protons necessary to accomplish the reduction of the heme a3/CuB binuclear center before the binding of O2.
Oxygenated Intermediates and the Catalytic Cycle.
Apart from any interest in proton pumping, cytochrome oxidase provides one of the best systems for studying the chemistry of oxygen activation and the splitting of the O—O bond (13). This experimental advantage is important because the mechanism of proton pumping is coupled mechanistically to the oxygen chemistry, and so it cannot be understood without a detailed knowledge of the oxygen chemistry. The catalytic mechanism (see refs. 23–27) can be simplified as the following sequence (Fig. 1), starting with the fully oxidized enzyme (Ox). It is noteworthy that electron transfer rates are mostly controlled by proton movements within the protein (28–30).
Figure 1.
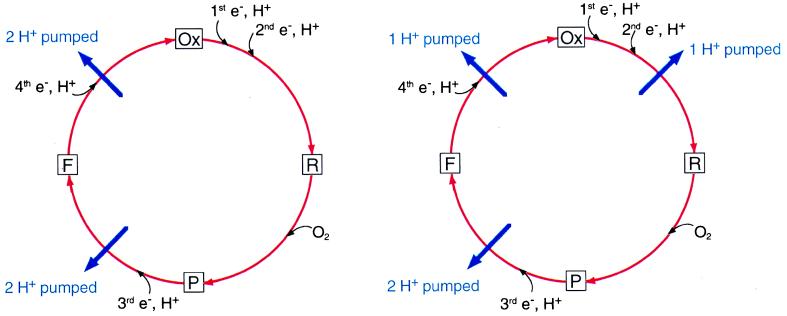
(Left) The catalytic cycle of the oxidase, schematically showing only the delivery of the last two electrons being coupled to the proton pump as proposed by Wikström (32). (Right) The suggested change by Michel (45) showing coupling during the reductive part of the catalytic cycle.
Step 1: Reduction of the binuclear center.
It generally is thought that O2 does not bind to the enzyme until at least the two-electron reduced state. The net uptake of two protons by the binuclear center accompanies the reduction of the binuclear center by two electrons, and the electron transfer appears to be kinetically limited by proton movements (28). This two-electron reduction often is called the Ox → R transition, and it can be divided into two one-electron steps, with the one-electron reduced intermediate (E) in between (Ox → E → R).
Step 2: Binding of oxygen, followed by formation of the P intermediate, or compound P.
No proton uptake from solution accompanies this step.
Step 3: Addition of the third electron, along with net uptake of another proton, resulting in the formation of the next intermediate, called compound F.
This step is the P → F transition.
Step 4: Addition of the fourth electron, accompanied by the net uptake of the fourth proton.
This step returns the enzyme to the Ox form and is called the F → Ox transition.
The Challenge to an Existing Paradigm: Which Steps Are Coupled to Proton Pumping?
It was first demonstrated in 1977 by Mårten Wikström at the University of Helsinki that cytochrome oxidase is a true proton pump (31). This finding was not at first widely accepted, and a spirited debate continued until about 1985. Wikström and his group since have gone on to establish much of the conceptual and an experimental framework for studying the oxidase mechanism, including important experimental evidence that the proton pump is coupled only to the P → F and F → Ox transitions, and that each of these one-electron redox steps results in the pumping of two protons (15, 32). This paradigm has been generally accepted for the past decade, though some problems have been pointed out and discussed (33). The “histidine cycle” models (1, 25, 34) of proton pumping have been designed around providing a reasonable mechanistic explanation for the pumping of two H+ each during the P → F and F → Ox transitions. In essence, one of the histidine ligands to CuB is postulated to cycle between an imidazolate (Im−) form that is ligated to the metal and an imidazolium (ImHH+) form that is dissociated from the metal and serves as a two-proton carrier. This elegant proposal explains the two-proton pumping steps and also contains the element of a gate built-in, providing proton flux in one direction (25). The driving force for the pump in this model is the uptake of chemical protons, as it also is in the new model.
Now the paradigm that pumping is coupled only to the last two steps in the catalytic cycle is being challenged by Michel (45) on the basis of a re-evaluation of several key experiments described in the previous literature (going back to 1970). The conclusion of this re-evaluation (which is sure to be questioned in turn) is that the F → Ox transition is coupled to the pumping of only one proton and not two. Because the total proton pumping stoichiometry of four H+/O2 is not questioned, he postulates that one proton is pumped during the reduction of the binuclear center (Ox → R), before the binding of O2. The new mechanism that is proposed is tailored to these criteria. Indeed, a key experimental test of this model will be to determine whether reduction of the binuclear center results in proton pumping as predicted. It is this claim that the proton pump is coupled to the reduction of the binuclear center that is at the heart of Michel’s paper and is the most significant point.
The basis for Michel’s re-evaluation of the old data has several facets. Consideration of the structure of the enzyme caused him to estimate that heme a is located at an electrical depth in the membrane corresponding to 35%–40% of the membrane dielectric rather than the generally accepted value of 50% (35). He points out a possible source of error in the measurement of the value of 50% by Hinkle and Mitchell in 1970 (35), based on subsequent reports (36–38) that electron transfer from CuA to heme a is accompanied by the uptake of a fractional proton, 0.15 to 0.4, depending on the redox status of the binuclear center. The consequence of this new estimate of the dielectric depth of heme a influences the quantitation of the previous measurements of charge movements attributed to pumped protons (14, 32). In addition, Michel raises serious questions about the thermodynamic analysis of Wikström (32) in which it was concluded that the F → Ox transition pumps two protons. New experiments are not presented, but these assertions certainly will provide additional stimulus to do them. The experimental techniques available to examine these questions are much more sophisticated than they were in the 1980s, and the questions highlighted by the new model are the natural ones to address in any event.
Mechanistic Aspects of the New Model
The uptake of a proton from the inside accompanying the reduction of heme a at each step in the mechanism is a central component of the new model of proton pumping. There is strong evidence for proton uptake upon reduction of heme a when the binuclear site is oxidized (36–40), but it has yet to be demonstrated that this uptake is part of the pumping mechanism or whether it is peripheral. The general idea that the proton taken up upon reduction of heme a is central to proton pumping was explicit in some of the very first models of proton pumping (40). This concept also is built into the more recently proposed “glutamate trap” model (23), which proposes an electrostatic driving force for pumping similar to that in Michel’s model, but is not presented with molecular detail.
Michel provides a very detailed proposal, based essentially on charge compensation or electroneutrality. It is proposed that the proton taken up concomitant with the reduction of heme a comes through the D-channel, and goes to a cluster of protonatable groups (cluster A) above heme a, including the heme a propionates. Electron transfer from heme a to the binuclear center is accompanied by rapid redistribution of this proton to a second cluster of protonatable groups (cluster B), including the heme a3 propionates. This protonation provides electrostatic compensation for transfer of the electron to heme a3 and is the kinetically preferred pathway. However, this state is proposed to be transient, and the thermodynamically favored proton distribution for each intermediate is attained more slowly. This step involves the delivery of a proton from the inside, usually also via the D-channel, to protonate groups as required by the chemistry at the binuclear center. The delivery of the proton to the active site causes the electrostatic displacement of the proton temporarily stored near the outer surface in cluster B, and this proton exits to the outside. Indeed, experimental evidence has been presented that the oxidation and reduction of the enzyme is associated with the uptake and release of protons, respectively, from opposite sides of the membrane (41). The proton–proton electrostatic displacement at the heart of this new model is reminiscent of a recent proposal for the proton exit during the bacteriorhodopsin photocycle (42).
It is essential to postulate that the protons temporarily stored for pumping in the oxidase (e.g., bound to the heme propionates) not have access to the binuclear center, and this assertion is reasonable structurally. There is a natural segregation of the pumped protons and substrate protons beyond Glu-278 in this model.
The model is critically dependent on the relative rates of proton transit through different pathways. Proton transit rates are also of current interest in studies on the bacterial reaction centers in the context of the reduction and protonation of QB (43, 44), and clearly these rates are very dependent on numerous subtle factors and susceptible to large alterations by mutation or other small changes in protein structure/conformation.
The crucial point of why this proton pump should be unidirectional is not fully addressed. It is necessary to have a gating mechanism to prevent backflow of protons that would compromise the efficiency of the pump. Why should the D-channel be the preferred pathway to input protons to protonate a site or sites located most of the way (80%) across the dielectric barrier in cluster A or B, but not be a viable pathway for protons to leave when protonation of this site is disfavored? The answer may lie in simply electrostatic repulsion by the incoming proton, or possibly in subtle structural changes, for example in the orientation of Glu-278, which might serve to connect and disconnect this proton pathway in response to the state of the binuclear center.
Certainly, this proposal is not the last word on this topic but it provides stimulating ideas that can and will be rapidly tested experimentally.
Footnotes
A commentary on this article begins on page 12819.
References
- 1. Iwata S, Ostermeier C, Ludwig B, Michel H. Nature (London) 1995;376:660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 2.Ostermeier C, Harrenga A, Ermler U, Michel H. Proc Natl Acad Sci USA. 1997;94:10547–10553. doi: 10.1073/pnas.94.20.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoshikawa S, Shinzawa-Itoh K, Nakashima R, Yaono R, Yamashita E, Inoue N, Yao M, Fei M J, Libeu C P, Mizushima T, et al. Science. 1998;280:1723–1729. doi: 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]
- 4.Tsukihara T, Aoyama H, Yamashita E, Takashi T, Yamaguichi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 5.Hastings S F, Kaysser T M, Jiang F, Salerno J C, Gennis R B, Ingledew W J. Eur J Biochem. 1998;255:317–323. doi: 10.1046/j.1432-1327.1998.2550317.x. [DOI] [PubMed] [Google Scholar]
- 6.Xia D, Yu C-A, Kim H, Xia J-Z, Kachurin A M, Zhang L, Yu L, Deisenhofer J. Science. 1997;277:60–66. doi: 10.1126/science.277.5322.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abrahams J P, Leslie A G W, Lutter R, Walker J E. Nature (London) 1994;370:621–628. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- 8.Pebay-Peyroula E, Rummel G, Rosenbusch J P, Landau E M. Science. 1997;277:1676–1681. doi: 10.1126/science.277.5332.1676. [DOI] [PubMed] [Google Scholar]
- 9.Luecke H, Richter H-T, Lanyi J K. Science. 1998;280:1934–1937. doi: 10.1126/science.280.5371.1934. [DOI] [PubMed] [Google Scholar]
- 10.Deisenhofer J, Epp O, Miki K, Huber R, Michel H. Nature (London) 1985;318:619–624. doi: 10.1038/318618a0. [DOI] [PubMed] [Google Scholar]
- 11.Ermler U, Fritzsch G, Buchanan S K, Michel H. Curr Biol. 1994;2:925–936. doi: 10.1016/s0969-2126(94)00094-8. [DOI] [PubMed] [Google Scholar]
- 12.Allen J P, Feher G, Yeates T O, Komiya H, Rees D C. Proc Natl Acad Sci USA. 1988;85:8487–8491. doi: 10.1073/pnas.85.22.8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferguson-Miller S, Babcock G T. Chem Rev. 1996;7:2889–2907. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 14.Zaslavsky D L, Smirnova I A, Siletsky S A, Kaulen A D, Millett F, Konstantinov A A. FEBS Lett. 1995;359:27–30. doi: 10.1016/0014-5793(94)01443-5. [DOI] [PubMed] [Google Scholar]
- 15.Verkhovsky M I, Morgan J E, Verkhovskaya M L, Wikström M. Biochim Biophys Acta. 1997;1318:6–10. [Google Scholar]
- 16.Gennis R B. Biochim Biophys Acta. 1998;1365:241–248. doi: 10.1016/s0005-2728(98)00095-4. [DOI] [PubMed] [Google Scholar]
- 17.Pfitzner U, Odenwald A, Ostermann T, Weingard L, Ludwig B, Richter O-M H. J Biomemb Bioenerg. 1998;30:89–93. doi: 10.1023/a:1020515713103. [DOI] [PubMed] [Google Scholar]
- 18.Sacks V, Marantz Y, Aagaard A, Checover S, Nachliel E, Gutman M. Biochim Biophys Acta. 1998;1365:232–240. [Google Scholar]
- 19.Riistama S, Hummer G, Puustinen A, Dyer R B, Woodruff W H, Wikström M. FEBS Lett. 1997;414:275–280. doi: 10.1016/s0014-5793(97)01003-x. [DOI] [PubMed] [Google Scholar]
- 20.Hofacker I, Schulten K. Proteins. 1998;30:100–107. [PubMed] [Google Scholar]
- 21.Pomes R, Hummer G, Wikström M. Biochim Biophys Acta. 1998;1365:255–260. [Google Scholar]
- 22.Proshlyakov D A, Pressler M A, Babcock G T. Proc Natl Acad Sci USA. 1998;95:8020–8025. doi: 10.1073/pnas.95.14.8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rich P R, Jünemann S, Meunier B. J Bioenerg Biomemb. 1997;30:131–137. doi: 10.1023/a:1020524014920. [DOI] [PubMed] [Google Scholar]
- 24.Konstantinov A A. J Bioenerg Biomemb. 1998;30:121–130. doi: 10.1023/a:1020571930850. [DOI] [PubMed] [Google Scholar]
- 25.Wikström M, Morgan J E, Verkhovsky M I. J Bioenerg Biomemb. 1998;30:139–145. doi: 10.1023/a:1020576031758. [DOI] [PubMed] [Google Scholar]
- 26.Karpefors M, Ädelroth P, Aagaard A, Sigurdson H, Ek M S, Brzezinski P. Biochim Biophys Acta. 1998;1365:159–169. doi: 10.1016/s0005-2728(98)00058-9. [DOI] [PubMed] [Google Scholar]
- 27.Brzezinski P, Ädelroth P. J Bioenerg Biomemb. 1998;30:99–107. doi: 10.1023/a:1020567729941. [DOI] [PubMed] [Google Scholar]
- 28.Verkhovsky M I, Morgan J E, Wikström M. Biochemistry. 1995;34:7483–7491. doi: 10.1021/bi00022a023. [DOI] [PubMed] [Google Scholar]
- 29.Ädelroth P, Ek M S, Mitchell D M, Gennis R B, Brzezinski P. Biochemistry. 1997;36:13824–13829. doi: 10.1021/bi9629079. [DOI] [PubMed] [Google Scholar]
- 30.Hallén S, Nilsson T. Biochemistry. 1992;31:11853–11859. doi: 10.1021/bi00162a025. [DOI] [PubMed] [Google Scholar]
- 31.Wikström M. Nature (London) 1977;266:271–273. doi: 10.1038/266271a0. [DOI] [PubMed] [Google Scholar]
- 32.Wikström M. Nature (London) 1989;338:776–778. doi: 10.1038/338776a0. [DOI] [PubMed] [Google Scholar]
- 33.Wikström M, Morgan J E, Verkhovsky M I. Biochim Biophys Acta. 1997;1318:299–306. [Google Scholar]
- 34.Morgan J E, Verkhovsky M I, Wikström M. J Bioenerg Biomembr. 1994;26:599–608. doi: 10.1007/BF00831534. [DOI] [PubMed] [Google Scholar]
- 35.Hinkle P, Mitchell P. Bioenergetics. 1970;1:45–60. doi: 10.1007/BF01516088. [DOI] [PubMed] [Google Scholar]
- 36.Mitchell R, Rich P R. Biochim Biophys Acta. 1994;1186:19–26. doi: 10.1016/0005-2728(94)90130-9. [DOI] [PubMed] [Google Scholar]
- 37.Capitanio N, Vygodina T V, Capitanio G, Konstantinov A A, Nicholls P, Papa S. Biochim Biophys Acta. 1997;1318:255–265. doi: 10.1016/s0005-2728(96)00143-0. [DOI] [PubMed] [Google Scholar]
- 38.Blair D F, Ellis J, Walther R, Wang H, Gray H B, Chan S I. J Biol Chem. 1986;261:11524–11537. [PubMed] [Google Scholar]
- 39.Carithers R P, Palmer G. J Biol Chem. 1981;256:7967–7976. [PubMed] [Google Scholar]
- 40.Artzatbanov V Y, Konstantinov A A, Skulachev V P. FEBS Lett. 1978;87:180–185. doi: 10.1016/0014-5793(78)80327-5. [DOI] [PubMed] [Google Scholar]
- 41.Capitanio N, Capitanio G, De Nitto E, Papa S. FEBS Lett. 1997;414:414–418. doi: 10.1016/s0014-5793(97)01043-0. [DOI] [PubMed] [Google Scholar]
- 42.Lanyi J K. Biochim Biophys Acta. 1998;1365:17–22. [Google Scholar]
- 43.Paddock M L, Senft M E, Graige M S, Rongey S H, Turanchik T, Feher G, Okamura M Y. Photosyn Res. 1998;55:281–291. [Google Scholar]
- 44.Abresch E C, Paddock M L, Stowell M H B, McPhillips T M, Axelrod H L, Soltis S M, Rees D C, Okamura M Y, Feher G. Photosyn Res. 1998;55:119–125. [Google Scholar]
- 45.Michel H. Proc Natl Acad Sci USA. 1998;95:12819–12824. doi: 10.1073/pnas.95.22.12819. [DOI] [PMC free article] [PubMed] [Google Scholar]