

Abstract
We previously described an HIV-1-infected individual who developed resistance to vicriviroc (VCV), an investigational CCR5 antagonist, during 28 weeks of therapy (Tsibris AM et al., J. Virol. 82:8210–8214, 2008). To investigate the decay of VCV resistance mutations, a standard clonal analysis of full-length env (gp160) was performed on plasma HIV-1 samples obtained at week 28 (the time of VCV discontinuation) and at three subsequent time points (weeks 30, 42, and 48). During 132 days, VCV-resistant HIV-1 was replaced by VCV-sensitive viruses whose V3 loop sequences differed from the dominant pretreatment forms. A deep-sequencing analysis showed that the week 48 VCV-sensitive V3 loop form emerged from a preexisting viral variant. Enfuvirtide was added to the antiretroviral regimen at week 30; by week 48, enfuvirtide treatment selected for either the G36D or N43D HR-1 mutation. Growth competition experiments demonstrated that viruses incorporating the dominant week 28 VCV-resistant env were less fit than week 0 viruses in the absence of VCV but more fit than week 48 viruses. This week 48 fitness deficit persisted when G36D was corrected by either site-directed mutagenesis or week 48 gp41 domain swapping. The correction of N43D, in contrast, restored fitness relative to that of week 28, but not week 0, viruses. Virus entry kinetics correlated with observed fitness differences; the slower entry of enfuvirtide-resistant viruses corrected to wild-type rates in the presence of enfuvirtide. These findings suggest that while VCV and enfuvirtide select for resistance mutations in only one env subunit, gp120 and gp41 coevolve to maximize viral fitness under sequential drug selection pressures.
INTRODUCTION
HIV exists as a quasispecies, a collection of diverse but related RNA sequences that undergo mutation and selection (10, 12, 62). The likelihood that a given genome in the quasispecies contributes progeny to successive viral replication cycles is referred to as virus fitness (10). Antiretroviral therapy (ART) imposes a potent selection pressure that can modulate quasispecies composition. Viruses that incorporate resistance mutations demonstrate fitness advantages in the presence of drug; rapid population shifts toward these genomes are observed in clinical practice (26, 50, 53). Increased fitness in one environment may result in reduced fitness in another, and the removal of a drug selection pressure may lead to equally dramatic population shifts back toward viruses that do not contain resistance mutations (7, 31). The HIV-1 env gene plays a major role in determining viral fitness due to envelope-mediated differences in the efficiency of receptor binding and the kinetics of entry (27, 34, 46, 57).
Viruses that dominate after drug discontinuation are derived either from previously archived wild-type genomes or, if no such wild-type virus exists (e.g., in the case of transmitted drug resistance), from back mutation and selection (20, 23, 40). In vitro and in vivo studies have quantified the fitness costs of a range of specific resistance mutations to nucleoside reverse transcriptase inhibitors (NRTI), nonnucleoside reverse transcriptase inhibitors (NNRTI), protease inhibitors (PI), and entry inhibitors (enfuvirtide) (11). The fitness cost of the enfuvirtide-associated G36D mutation, a substitution in the first heptad repeat (HR-1) of gp41, results from reduced viral fusion kinetics (30, 49). The particular enfuvirtide mutation selected during therapy is a function of the total env genetic context, suggesting that sequences outside gp41 influence resistance development (25). Env context is also important in the development of CCR5 antagonist resistance mutations (24). The fitness cost of resistance to CCR5 antagonists remains unclear. Resistance to the CCR5 antagonist AD101 was not associated with a fitness cost in vitro, whereas we observed a loss of VCV resistance mutations in vivo after drug discontinuation, suggesting an in vivo fitness cost of those mutations (1, 59).
Clinically, the dominant mechanism of HIV-1 escape from a CCR5 antagonist is the emergence of CXCR4-using virus (13, 14, 44, 58, 60). Less commonly, resistance to CCR5 antagonists can emerge by the selection of mutations in the V3 loop of HIV gp120 that permit viral entry using the drug-bound CCR5 coreceptor (8, 17, 35, 36, 38, 39, 42, 43, 59); the particular mutations that emerge are strain specific. Resistance is associated with an increased reliance on the CCR5 amino terminus (NT) for entry, although in some cases viruses were highly reliant on CCR5 NT prior to the development of VCV resistance (5, 16, 38, 42, 51, 56). We previously characterized the emergence of VCV resistance in HIV-1 from a subtype C-infected individual and determined that resistance was conferred by a set of V3 mutations (59). The V3 changes accumulated during 28 weeks of therapy and resulted in more rapid entry rates in the presence of drug (45). The majority of these mutations were required for resistance (17). VCV was discontinued at study week 28, and the regimen was reoptimized with the addition of enfuvirtide. We now describe the decay of these VCV resistance-associated V3 mutations from the plasma viral population and characterize env evolution and viral fitness under the unique clinical circumstances of sequential drug selection pressures that act on different subunits of the envelope glycoprotein.
MATERIALS AND METHODS
Subject samples.
We identified one participant enrolled in ACTG 5211 (A5211; clinicaltrials.gov identifier NCT00082498), a phase IIb clinical trial of vicriviroc (VCV), an investigational CCR5 antagonist, who experienced protocol-defined virologic failure (VF) and developed VCV resistance (15, 59). Five time points were analyzed: study entry (study week 0; baseline), week 28 (maximal VCV resistance and drug discontinued), and three time points after VCV discontinuation (study weeks 30, 42, and 48). Per A5211 protocol, following virologic failure patients could reoptimize their ART regimens at the discretion of their local treating physician.
Calculation of maximal percent inhibition.
Subjects enrolled in A5211 had VCV susceptibility testing performed by Monogram Biosciences using the PhenoSense entry inhibitor assay (61).
Standard clonal analysis.
HIV-1 RNA was extracted from plasma, and full-length env was amplified. To minimize any potential founder effect, four independent reverse transcription (RT) reactions and PCR amplifications were performed and combined for each time point. These purified amplicons were then ligated into a TOPO-TA vector (Invitrogen) and electroporated into TOP10 cells. Subclones were isolated and sequenced by conventional (Sanger) methods as described previously (59). Between 12 and 28 clones were sequenced per time point.
Deep sequencing.
V3 amplicons were submitted in a blinded fashion to the Broad Institute for deep sequencing (454 Life Sciences, Roche) (33, 58).
Fitness determinations by mutation decay.
The relative fitness, 1 + s, was calculated using a two-time-point and a multiple-time-point measurement, where s is the selection coefficient (http://bis.urmc.rochester.edu/vFitness/FitnessTwo.aspx and http://bis.urmc.rochester.edu/vFitness/FitnessMulti.aspx) (32, 63). The time points available for analysis were designated days 1 (week 28), 12 (week 30), 91 (week 42), and 130 (week 48). Day 1 was defined as the first day after VCV was discontinued.
SDM.
Full-length envelopes from plasma virus obtained at the week 48 visit were cloned into a pCR4-TOPO TA vector (Invitrogen, Carlsbad, CA). Back mutations at gp41 codons 36 (D→G) and 43 (D→N) were introduced into pTOPO-env using the QuickChange IIE site-directed mutagenesis (SDM) protocol (Stratagene, La Jolla, CA). The bidirectional full-length env sequencing of recombinant clones was performed to verify the presence of the desired substitutions and the absence of adventitious mutations. Recombinant marker vectors carrying hisD or PLAP fragments as sequence tags in nef were used, and recombinant viruses that incorporated participant-derived env genes were generated as previously described (30). env chimeras were constructed in an overlap PCR that combined the week 48 gp120 amplicon of p07Jenv48-1, p07Jenv48-3, or p07Jenv48-8 with the gp41 amplicon of p07Jenv0. The primer set used for gp120 amplification was 5′-AAGAAAGATAGACAGGTTAATTGATA-3′ (GC-env2A; forward primer) and 5′-TCTTTTTTCTCTCTCCACCACTC-3′ (gp120R; reverse primer). The primer set for gp41 amplification was 5′-CAGCAAAAAGGAGAGTGGTGG-3′ (gp41F; forward primer) and 5′-GGCCATCCAACTATACTACTTTTGGA-3′ (GC-env2B; reverse primer). Primers GC-env2A and GC-env2B were used for overlap PCR amplifications. The use of primer GC-env2B results in the amplification of approximately 40 nucleotides of the proximal portion of nef. Bidirectional full-length env sequencing was performed to confirm that chimeric env genes did not contain adventitious mutations, frameshifts, or deletions at the gp120-gp41 junction.
Enfuvirtide susceptibility assays.
Recombinant virus that incorporated HIV-1 envelopes were constructed by a modification of a previously described method (21, 28). Briefly, the cytomegalovirus (CMV) promoter was amplified and attached to a 265-bp segment of rev from pNL43 using overlap PCR. A second overlap PCR was then performed to link CMV-rev to the cloned or uncloned env amplicon of interest. These CMV-rev-env amplicons were then cotransfected into 293T cells with an NL4-3 envelope-deleted vector (30). Clonal susceptibility testing was performed as described previously (17, 59).
Viral replication kinetics assays.
Viral replication capacity was quantified by measuring HIV-1 p24 antigen production in U87-R5 cell-free culture supernatants in the absence of drug. Recombinant viruses carrying the specific env gene of interest were inoculated onto 12-well plates that contained 8 × 104 U87-R5 cells per well in 300 μl of Dulbecco's modified Eagle medium (DMEM) to yield a multiplicity of infection (MOI) of 0.001; each sample was tested in triplicate. After a 2-h incubation at 37°C, cells were washed twice with phosphate-buffered saline (PBS) and 1.0 ml of fresh medium was added. On days 2, 4, 6, 8, 10, and 12, 100 μl of supernatant was removed from each well for p24 antigen assay testing and replaced with 200 μl of fresh medium. Replication kinetics were determined twice, once with recombinants carrying the hisD marker and once with the PLAP marker, and the results were averaged. Replication rates were determined by plotting the change (increase) in p24 antigen over time.
HIV-1 entry kinetics assays.
We used a well-validated BlaM activity assay to quantify virus-cell fusion in real time as described previously (6, 37, 45). Fusion kinetics at 37°C were determined by real-time fluorimetry and monitored at 5-min intervals for 90 min. To normalize the data, the maximum entry of each recombinant virus was determined by the time point at which the maximum blue-to-green ratio occurred and was maintained (plateau). The correlation between time and percentage of fusion was calculated and curve fitted using GraphPad Prism 5 (GraphPad, La Jolla, CA).
Growth competition assays.
Pairwise growth competition assays were performed as described previously (29, 30). Briefly, recombinant marker viruses of interest carrying the hisD or PLAP sequence tags were mixed at ratios of 50:50 and inoculated into triplicate wells of a 12-well plate containing 8 × 104 U87-R5 cells per well in 300 μl of DMEM to yield an MOI of 0.001. After incubation at 37°C for 2 h, cells were washed twice with PBS, resuspended in 1 ml of DMEM, seeded into 12-well plates, and reincubated. The proportion of the two competing viral variants was estimated by quantifying PLAP and hisD sequences present in culture supernatants on days 1, 4, 7, 11, and 14 using real-time PCR with an ABI PRISM 7000 sequence detection system (Applied Biosystems, Inc.). Viral RNA was extracted from culture supernatants using the Qiagen kit and treated with RNase-free DNase (Qiagen, CA). Parameters for the real-time PCR were as described previously, except that the initial reverse transcription reaction was performed at 50°C for 30 min (30). Quantitative real-time reverse transcription-PCR was performed in triplicate for each sample. Experiments were performed twice and the results averaged.
Phylogenetic analyses.
Full-length gp160 sequences isolated during 48 weeks of observation were included in a phylogenetic analysis of gp120 and gp41 evolution. Nucleotide sequences were aligned by a Geneious alignment (cost matrix, 93%; gap open penalty, 12; gap extension penalty, 3; Geneious version 5.1.7) and separated into gp120 and gp41 coding region. A FindModel analysis (www.hiv.lanl.gov/content/sequence/findmodel/findmodel.html) demonstrated the near equivalence of the nucleotide substitution models Hasegawa-Kishino-Yano (HKY) plus gamma and general time reversible plus gamma (GTR). Bayesian phylogenies that used an HKY85-plus-gamma substitution model and consensus C env as an outgroup were constructed separately for gp120 and gp41 sequences using the MrBayes plugin of Geneious (18). The Monte Carlo Markov chain (MCMC) length was set to 1,100,000 iterations with trees sampled every 200th generation.
Nucleotide sequence accession numbers.
The nucleotide sequences from this study have been deposited under GenBank accession numbers EU663615 (p07Jenv0), EU663618 (p07Jenv28), and JQ855935 to JQ856014.
RESULTS
Clonal envelope analysis during sequential vicriviroc and enfuvirtide therapy.
We identified an individual enrolled in ACTG5211 who developed VCV resistance (59). The HIV-1 RNA load at study entry was 5.79 log10 copies/ml (Fig. 1). The genotypic susceptibility score (GSS) and phenotypic susceptibility score (PSS) of the background antiretroviral regimen after optimization at week 2 were 0.37 and 1.17, respectively (2, 55). VCV was discontinued due to a lack of virologic response 2 days after the week 28 blood draw (defined as 28 weeks from the start of VCV therapy), a time point notable for maximal VCV resistance and a maximal percent inhibition (MPI) of 0%. Antiretroviral therapy was reoptimized at week 30 by the addition of enfuvirtide and tipranavir to the regimen, but plasma HIV-1 RNA levels remained essentially unchanged. Plasma HIV-1 RNA measurements were not performed between week 30 and week 42, the first time point that enfuvirtide resistance mutations were detected.
Fig 1.
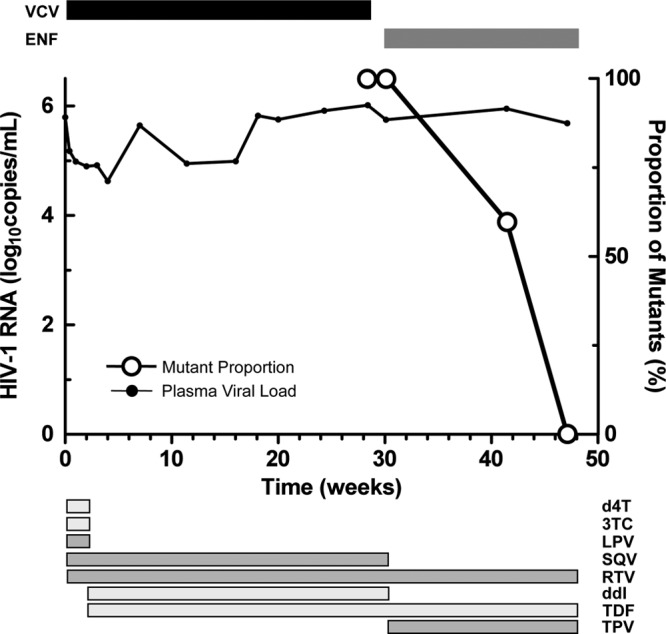
Vicriviroc resistance decay. Viral load is plotted along the left y axis and delineated by the black line. The proportion of VCV-resistant clones is plotted on the right y axis and shown in open circles. Treatment with VCV is shown above the graph. Additional antiretrovirals and their treatment durations are shown below the figure. d4T, stavudine; 3TC, lamivudine; LPV, lopinavir; ddI, didanosine; TDF, tenofovir; SQV, saquinavir; RTV, ritonavir (low-dose); ENF, enfuvirtide; TPV, tipranavir.
We isolated HIV-1 RNA from plasma samples obtained at weeks 30, 42, and 48, generated full-length env amplicons, and performed a clonal sequence analysis (Table 1). V3 sequences from the week 0 and 28 plasma samples have been previously reported and are shown for comparison; VCV treatment selected for the K305R, S306P, T307I, F318I, T320R, and G321E mutations (numbering according to the HXB2 reference) (59). For week 30 and 42 sequences, a V3 loop was classified as VCV resistant or VCV sensitive by the presence or absence, respectively, of the full complement of these VCV resistance mutations. At week 30, 24 of 24 (100%) clonal V3 loop sequences carried VCV resistance-associated mutations (17, 59). These resistant forms constituted 15 of 25 (60%) sequences at week 42 and 0 of 25 sequences (0%) at week 48. VCV-sensitive V3 loop forms were detected in 10 of 25 (40%) and 28 of 28 (100%) sequences at weeks 42 and 48, respectively. The VCV-sensitive sequences emerging at weeks 42 and 48 differed from the dominant week 0 V3 loop sequence by a K305R substitution. The targeted deep sequencing of V3 amplicons from week 0 plasma to a depth of 112,818× coverage demonstrated that a V3 loop form with the single K305R substitution was present at 0.2% (239 of 112,818 filtered, aligned V3 loop sequences) of the week 0 plasma sample (58).
Table 1.
Clonal changes in V3 loop and HR-1 sequences during sequential vicriviroc and enfuvirtide therapya
| Study wk | MPI | Changes in: |
|||||
|---|---|---|---|---|---|---|---|
| V3 loop |
HR-1 gp41 |
||||||
| No. of clones | Sequence | VCV susceptibility | No. of clones | Sequence | ENF susceptibility | ||
| 0 | 97 | 17 | CTRPGNNTRKSTRIGPGQTFFATGDIIGDIRQAHC | S | 19 | GIVQQQSN | S |
| 2 | -----------I----------R------------ | S | |||||
| 28 | 0 | 12 | ---------RPI--------I-RE---------Y- | R | 20 | -------- | S |
| 30 | NP | 17 | ---------RPI--------I-RE---------Y- | R | 24 | -------- | S |
| 5 | ---------RPI--------I-RE----------- | R | |||||
| 2 | ---------RPI----------RE---------Y- | R | |||||
| 42 | NP | 12 | ---------RPI--------I-RE---------Y- | R | 15 | D------- | R |
| 10 | ---------R------------------------- | S | 9 | -------D | R | ||
| 2 | ---------RPI--------I-RE----------- | R | 1 | D------D | R | ||
| 1 | ---------R---T------I-RE---------Y- | R | |||||
| 48 | 90 | 27 | ---------R------------------------- | S | 15 | -------D | R |
| 1 | ---L-----R------------------------- | S | 12 | D------- | R | ||
| 1 | --A----D | R | |||||
MPI, maximal percent inhibition in the Monogram PhenoSense entry inhibitor assay, which was performed on plasma samples; ENF, enfuvirtide; S, sensitive; R, resistant; NP, not performed.
An analysis of gp41 sequences showed no consistent changes during the initial 28-week period of vicriviroc therapy (data not shown). HR-1 sequences remained wild type at week 30 (Table 1). At week 42, all clones contained the enfuvirtide resistance mutation G36D or N43D in HR-1; one G36D/N43D double mutant was also detected. By week 48, N43D became the dominant enfuvirtide resistance mutation (16 of 28 clones [57%] versus 12 of 28 clones [43%] for G36D); an N43D-containing mutant also carried a V38A mutation. Week 48 G36D- or N43D-containing env demonstrated enfuvirtide 50% inhibitory concentrations (IC50s) of 8.5 μM (95% confidence intervals [CI], 4.4 to 16.7 μM) and 15.5 μM (1.8 to 134.3 μM), respectively, compared to the previously observed week 0 and week 28 env IC50s of 116 nM (86 to 157 nM) and 74 nM (57 to 96 nM), respectively (59). Mutations in HR-2 at gp41 positions 125 and 138 have been described in some enfuvirtide-resistant viruses but were not detected in plasma samples at any time point (48, 64). We observed HR-2 changes at position 128 (T-to-S substitution) in week 28 and week 48 envelopes and at position 145 (N-to-S substitution) in some week 48 envelopes (see Fig. S1 in the supplemental material). The G36D and N43D mutations were found in week 42 and 48 full-length envelopes in association with VCV-resistant and VCV-sensitive V3 loop forms; no consistent pattern of gp41 and V3 loop gp120 mutation segregation was evident.
Relative fitness calculations of VCV-resistant virus.
We next used two web-based fitness calculators to determine the relative fitness of viruses containing VCV resistance mutations in the absence of drug (32, 63). A calculation using viral proportions at only two time points, weeks 28 and 48, resulted in a fitness (1 + s) estimate of 89% for VCV-resistant virus relative to wild-type virus. That is, VCV-resistant virus demonstrated an 11% fitness disadvantage, relative to VCV-sensitive viruses, in the absence of VCV. A multipoint estimator based on the method of least squares that considered all viral proportion measurements from weeks 28, 30, 42, and 48 produced a similar result with a relative fitness estimate of 89% for VCV-resistant viruses or a fitness disadvantage of 11%.
Replication kinetics of HIV-1 recombinants incorporating env genes isolated during VCV and ENF therapy.
Emerging enfuvirtide resistance during the period of VCV resistance mutation decay could confound relative fitness calculations for VCV-resistant virus. To empirically determine the replication kinetics of viruses that contain VCV resistance mutations, we generated recombinant viruses that incorporated participant-derived envelopes isolated at week 0 (baseline), week 28 (maximal VCV resistance), and week 48 (20 weeks off VCV therapy, 18 weeks on enfuvirtide therapy) into an NL4-3 backbone (Fig. 2). A sequence comparison demonstrated numerous amino acid substitutions across the length of gp120 in these isolates (see Fig. S1 in the supplemental material). Full-length envelope clones were chosen that contained the dominant gp120 sequence for each time point; viral replication kinetics were studied for 10 to 16 days (59). VCV-sensitive week 0 viruses demonstrated greater p24 antigen yields than VCV-resistant week 28 viruses in replicate experiments (Fig. 3). The replication kinetics of recombinant viruses that incorporated either of two different week 48 envelopes, p07Jenv48-3 and p07Jenv48-8, demonstrated very poor p24 yield. This may be due to the presence of the HR-1 mutation G36D or N43D, respectively, in these clones.
Fig 2.

env construct schematic. The dominant week 0 and week 48 subject-derived full-length env constructs were used to construct chimeric envelopes and generate site-directed mutants at gp41 positions 36 and 43. Amino acid numbering is based on the HXB2 reference sequence. Plasmids are named after the subject identifier (07J) and the treatment week from which each env was isolated. Chimeric env genes are denoted by the Greek character χ; env genes with site-directed reversion mutations are denoted by the abbreviation SDM followed by the gp41 amino acid position of the reversion. Arrows denote the location of site-directed reversion mutations.
Fig 3.

Replication kinetics. Recombinant viruses that incorporated patient-derived env constructs were grown in U87-R5 cell cultures during approximately 2 weeks. (A) Native week 0, 28, and 48 dominant envelopes. (B) Native week 0 and 28 envelopes with G36D-corrected week 48 chimeras. (C) Native week 0 and 28 envelopes with week 48 envelopes that had G36D corrected by chimeric domain swapping or site-directed mutagenesis. (D) Week 0 and 28 envelopes with an N43D-corrected week 48 env. The data for p07Jenv0 and p07Jenv28 replication kinetics shown in each of the four panels were generated independently for each experiment.
To evaluate the contribution of sequence changes in gp41 to the observed replication kinetics of week 48 viruses, we constructed chimeric envelopes that combined week 48 gp120 with week 0 gp41 and, additionally, corrected the G36D ENF resistance mutation by site-directed mutagenesis while maintaining an intact full-length week 48 env. Two G36D-containing week 48 envelopes were used as backbones for domain swapping, clone 748-1 (p07Jenv48-1) with an MPI for VCV of 100% and clone 748-3 (p07Jenv48-3) with an MPI of 90%. We further cloned a full-length env that contained an N43D HR-1 mutation and used site-directed mutagenesis to revert position 43 to the wild-type asparagine. The removal of the G36D mutation by either SDM or chimeric domain swapping in the 748-1 and 748-3 backbones improved p24 antigen yield, but not to the levels seen with week 0 virus. Recombinant viruses with full-length week 48 env corrected by SDM at position 36 showed more p24 antigen production than viruses that expressed the chimeric week 48 gp120/week 0 gp41 env but less than week 28 viruses. In contrast, the correction of the N43D mutation in week 48 env by SDM or domain swapping resulted in greater p24 production than did week 28 virus; the replication of the chimeric env p07J0χ48-8 approached that of week 0 virus.
Relative replicative fitness of HIV-1 recombinants carrying VCV resistance mutations selected in vivo.
The replicative fitness of recombinant viruses carrying envelopes isolated during VCV and ENF therapy were tested in a series of growth competition assays. Viruses that incorporated the full-length env of the dominant week 28 virus carrying VCV but not ENF resistance mutations were less fit than the week 0 viruses lacking VCV or ENF resistance mutations in the absence of VCV (Fig. 4A). We were unable to use native week 48 env-containing virus carrying ENF but not VCV resistance mutations for fitness experiments due to their poor replication (as described above). To circumvent this problem, we tested our previously constructed chimeric week 48 env and native week 48 env with site-directed corrections at gp41 position G36 or N43 in growth competition assays. Virus that contained a chimeric week 48 gp120/week 0 gp41 env (no VCV or ENF resistance mutations) were less fit than week 0 and week 28 viruses, respectively (Fig. 4B to F). Recombinant viruses that incorporated a native week 48 env with the correction of the 36G mutation were more fit than the chimeric week 48 construct, but a fitness deficit relative to week 28 virus persisted. To demonstrate the generalizability of these findings, we repeated this series of growth competition assays using N43D-corrected week 48 envelopes (Fig. 5). The correction of N43D by domain swapping (p07Jenv0χ48-8) resulted in a virus that was less fit than week 0 virus (Fig. 5A) but more fit than week 28 virus (Fig. 5B), whereas the SDM correction of the point mutant (p07Jenv48SDM43) resulted in a virus that had essentially the same fitness as week 0 virus (Fig. 5C) and was more fit than week 28 virus (Fig. 5D). The SDM-corrected virus p07Jenv48SDM43 likewise demonstrated greater fitness than the gp41 domain-swapped p07Jenv0χ48-8 (Fig. 5E).
Fig 4.

Growth competition assays between (A) native week 0 and 28 env genes, (B) native week 0 and chimeric week 48 (48-1) env, (C) native week 28 env and chimeric week 48-1 env, (D) native week 0 and G36D site-corrected week 48-1 env, (E) native week 28 env and G36D site-corrected week 48-1 env, and (F) chimeric week 48-1 env and G36D site-corrected week 48-1 env.
Fig 5.

Growth competition assays between (A) native week 0 and chimeric week 48 (48-8) env, (B) native week 28 env and chimeric week 48-8 env, (C) native week 0 env and an N43D site-corrected week 48-8 env, (D) native week 28 env and an N43D site-corrected week 48-8 env, and (E) chimeric week 48-8 env and an N43D site-corrected week 48-8 env.
Entry rates of virus constructs.
To investigate the mechanisms responsible for these fitness differences, we assessed the entry kinetics of native and laboratory-generated week 48 env-containing viruses (Fig. 6). When tested on TZM-bl cells, the G36D-containing p07Jenv48-1 and the N43D-containing p07Jenv48-8 demonstrated longer times to half-maximal fusion than p07Jenv0; p07Jenv48-8 did not reach maximal fusion even after 90 min of observation (Table 2). Statistically significant differences in fusion rates between native week 0 and week 48 envelopes and between p07Jenv48-1 and p07Jenv48-8 were observed (nonoverlapping 95% confidence intervals). The addition of 1 μM enfuvirtide improved the fusion rates of p07Jenv48-1 and p07Jenv48-8 to a half-maximal fusion that was not statistically different from that of p07Jenv0 in the absence of drug (Fig. 6A). Entry rates of the week 48 constructs p07Jenv48SDM36, p07Jenv48SDM43, p07Jenv0χ48-1, and p07Jenv0χ48-8 were all slower than that of week 0 virus, but due to wide confidence intervals they could not be distinguished from the entry rates of virus that incorporated native week 48 env genes or from each other (Fig. 6B).
Fig 6.

Entry kinetics of viral isolates and constructs. (A) The entry of week 0 and week 48 enfuvirtide-resistant isolates was assessed in TZM-bl cells in the presence and absence of 1 μM enfuvirtide. (B) Chimeric or site-corrected week 48-1 and 48-8 env genes. Error bars represent the standard errors of the means of results from at least two experiments, each performed in triplicate. Nonlinear regression was used to estimate a fitted curve.
Table 2.
Entry rates of HIV-1 constructsa
| Construct | t1/2 max, in min (95% CI) | r2 |
|---|---|---|
| p07Jenv0 | 15.0 (12.2–19.2) | 0.948 |
| p07Jenv48-1 | 37.3 (27.8–56.3) | 0.892 |
| p07Jenv48-8 | 91.0 (60.0–183.5) | 0.943 |
| p07Jenv48-1 + ENF | 11.3 (8.8–16.0) | 0.921 |
| p07Jenv48-8 + ENF | 8.2 (6.2–11.9) | 0.969 |
| p07Jenv48SDM36 | 32.6 (24.4–49.0) | 0.968 |
| p07Jenv48SDM43 | 58.1 (39.8–107.8) | 0.971 |
| p07Jenv0χ48-1 | 55.2 (32.3–189.1) | 0.893 |
| p07Jenv0χ48-8 | 51.2 (33.0–113.8) | 0.909 |
t1/2 max, time to half-maximal fusion; 95% CI; 95% confidence intervals; ENF, enfuvirtide.
Evolutionary effect of VCV- and ENF-containing antiretroviral therapy.
Our growth competition data demonstrated that the pairing of gp120 and gp41 env domains can affect viral fitness. To determine the evolutionary relationship between env segments during the 48-week period of observation, we used Bayesian phylogenetic inference with gp120 sequences and the isolated gp41 domain (Fig. 7). Only clones with complete full-length sequences were included in this analysis; all VCV- and enfuvirtide-sensitive and -resistant env forms are represented. Preresistant week 16 (containing K305R, T307I, T320R, and G321E mutations) and week 19 (week 16 V3 changes plus F318I) gp120 sequences that contained some, but not all, V3 VCV mutations and the majority of VCV-resistant week 28 gp120 sequences were intermingled with a week 0 subpopulation (59) (Fig. 7A). VCV-resistant gp120 forms persisted at weeks 30 and 42 and continued to explore sequence space around the week 28 sequences, even after losing VCV mutations. These populations, however, were lost by week 48 and replaced by gp120 sequences most closely related to a cluster that contained week 42 sequences without VCV resistance mutations and the dominant pretreatment week 0 forms. The gp41 tree demonstrated a simpler topology (Fig. 7B); week 16 and 19 sequences clustered in a subclade that was not found at later time points. ENF-sensitive gp41 forms from weeks 28 and 30 and ENF-resistant forms from weeks 42 and 48 were interspersed, as were pre-VCV and later gp41 sequences. No segregation of G36D- and N43D-containing sequences was noted.
Fig 7.

Genotypic evolution of gp120 (A) and gp41 (B) during sequential VCV and ENF therapy. Bayesian phylogenetic inferences are shown for both env domains and contain sequences identified by time point and the presence of VCV or ENF resistance mutations. The time points are color coded identically for both domains; sequences that contain the full complement of V3 VCV resistance mutations are indicated by triangles. gp41 forms with ENF resistance mutations and are similarly depicted by triangles. A double-mutant G36D/N43D gp41 sequence is indicated by a star and a V38A/N43D-containing gp41 is shown as a square. Black, week 0; yellow, week 16; green, week 19; red, week 28; purple, week 30; blue, week 42; light blue, week 48. C, subtype C consensus gp120 and gp41, respectively.
DISCUSSION
We measured the decay of VCV resistance in an individual in whom VCV therapy failed and subsequently was discontinued. The later addition of ENF to the antiretroviral regimen provided a unique opportunity to study env sequence evolution under sequential drug selection pressures. HIV-1 V3 loop sequences that conferred VCV resistance were lost from the plasma within 20 weeks of VCV discontinuation, a time course similar to that observed for the decay of the M184V reverse transcriptase mutation after lamivudine discontinuation (7). The quantification of VCV-resistant and -susceptible variants by clonal sequence analysis of V3 from serial plasma samples showed an estimated fitness disadvantage of 10 to 11% for VCV-resistant viruses in the absence of VCV. In comparison, reported fitness disadvantages associated with enfuvirtide, didanosine, or lamivudine resistance range from 4 to 65% (9, 29, 31, 40, 54). No decay in VCV resistance mutations was observed in the first 3 weeks after VCV discontinuation. This plateau phase could not be accounted for in the two-time-point fitness estimator. The sampling of more time points during these weeks would have permitted an exact quantification of the total plateau time, as well as a precise determination of the start of resistance decay.
The persistence of resistant viruses in plasma for several weeks after drug discontinuation has been observed in patients infected with NRTI- and enfuvirtide-resistant viruses (31, 40). The maintenance of a less fit viral variant in plasma, followed by the rapid extinction of this variant, may reflect the time interval between drug discontinuation and the stochastic activation of a cell or cellular population that harbors the archived wild-type viral variant. Alternatively, this plateau time period may result from prolonged drug-receptor occupancy after vicriviroc discontinuation (52). Although we have demonstrated a fitness disadvantage of VCV resistance, the effect of resistance on viral population size, or viral load remains unclear.
We previously reported that viruses incorporating these VCV-resistant V3 loop forms demonstrate enhanced replication in vitro in the presence of CCR5 antagonists (59). In this patient, there was very little change in plasma HIV-1 RNA levels during 48 weeks. Changes to the background antiretroviral regimen during this time period make it difficult to correlate changes in viral fitness with any changes in viral load. It is possible that a transient viral load reduction accompanied the addition of enfuvirtide and tipranavir in week 30. HIV-1 RNA loads were similar in weeks 30 and 42 and suggest that enfuvirtide resistance mutations restored virus fitness in this new fitness landscape.
The V3 loop sequences of VCV-sensitive viruses that emerged after VCV discontinuation were not identical to pretreatment V3 sequence forms. The relatively rapid decay of HIV-1 VCV resistance in this individual was most consistent with the reemergence of sensitive virus from a preexisting variant rather than the back mutation of multiple V3 loop amino acid substitutions (23, 40). Deep sequencing confirmed that the dominant week 48 V3 loop sequence was present as a minor variant in the week 0 plasma sample. We previously demonstrated that two V3 sequences differing by a single nucleotide can be differentiated with a sensitivity threshold of 0.10 to 0.12% when sequencing to a depth of approximately 100,000× coverage (58). Thus, the detection of K305R at baseline is unlikely to represent an artifact introduced by amplification or sequencing error. We have previously shown that an isolated K305R mutation in this env backbone does not affect CCR5 antagonist susceptibility (17). Additional investigation is required, however, to elucidate the factors responsible for the emergence of one V3 form over another.
Calculating the fitness cost of VCV resistance by observing the decay of V3 loop mutations in this subject is confounded by the simultaneous appearance of HR-1 mutations that have their own effects on viral fitness. The weak activity of this participant's nonsuppressive background antiretroviral regimen likely explains the emergence of the dominant N43D and subdominant G36D HR-1 mutations 12 weeks into ENF therapy. For the direct measurement of the fitness effects of VCV resistance and to explore the contribution of gp41 sequence changes, we conducted pairwise growth competition experiments. VCV-sensitive (week 0) virus was more fit than VCV-resistant (week 28) virus in the absence of drug, a finding that was suggested by the in vivo loss of these V3 loop forms after drug discontinuation. We surmised that HR-1 mutations were responsible for the poor replication kinetics of week 48 viral constructs, but our methods of correcting these mutations led to different findings. The back mutation to wild-type N43 corrected the fitness deficits of this week 48 env and suggests that the posttreatment appearance of a V3 loop sequence that incorporated the K305R mutation was permissible due to similar effects of either V3 form on overall viral fitness.
Different effects of chimeric domain swapping on virus fitness were observed and depended on the specific env backbone that was used. The pairing of the majority week 0 gp41 region with a week 48 gp120 domain originally isolated from a G36D-containing env did not restore viral fitness and produced viruses that were less fit than native week 48 env genes with site-directed HR-1 corrections. The pairing of this same week 0 gp41 with a previously N43D-associated week 48 gp120 domain restored fitness relative to VCV-resistant virus and the week 48 env with a site-directed correction at position 43, but it did not restore fitness to wild-type week 0 levels. Wild-type virus fitness could not be restored to week 0 levels by either molecular technique. Taken together, these findings suggest that gp120 and gp41 segments in this subject coevolved to maximize viral fitness, in contrast to a previous report of ENF resistance decay, and highlight the complexities of envelope subdomain interactions (22). A phylogenetic analysis of gp120 and gp41 sequences did reveal differences in env domain evolution at earlier time points during VCV therapy, but by week 48, 18 weeks after ENF therapy began, both gp120 and gp41 forms most closely resembled pretreatment sequences. Divergent evolutionary pathways were not suggested and corroborate our finding in growth competition experiments that gp120 and gp41 must coevolve through drug selection pressures, even when these pressures act mainly on one sequence domain. We speculate that this coevolution maximizes some key feature of viral entry, most likely entry kinetics, but also could be more directly related to receptor or coreceptor affinity.
The fitness of an N43D-containing dominant week 48 env was restored by reverting this mutation to its wild-type form. It is not clear why the correction of the G36D mutation by site-directed mutagenesis or domain exchange only partially restored viral fitness in this subdominant viral population. This poor env fitness was demonstrated in three different week 48 env backbones and suggests that this finding was not related to some unique feature of a particular clone we selected. The N43D-containing env genes became the majority clones from week 42 to 48, and it seems likely that regions outside the gp120 V3 loop or the HR-1 gp41 domain contributed to these observed fitness differences. The presence of compensatory changes in env has been previously described for both V3 loop and HR-1 mutations (19, 25, 41, 47, 64).
We used a virus-cell fusion assay to confirm the slower entry rate of a G36D-containing env mutant and extended this observation to virus that contained the N43D gp41 HR-1 mutation (49). The slower entry rates we previously reported for VCV-resistant isolates correlated with reduced fitness in this study (45). The association between entry rates and the reduced fitness of week 48 env constructs, however, was less clear. While chimeric and SDM constructs displayed delayed entry kinetics, they could not be statistically differentiated from the entry rates of native week 48 env. A trend toward intermediate entry rates was noted that could explain some fitness differences, but an alternate mechanism (e.g., changes in binding affinity) may explain changes in viral fitness for these envelopes. The finding that enfuvirtide increased the entry rates of enfuvirtide-resistant clonal isolates to wild-type levels is novel and unexpected. Enfuvirtide resistance mutations in HR-1 reduce enfuvirtide binding while still permitting association with HR2. A model has been proposed for entry in the presence of enfuvirtide, although our virus isolates are admittedly not enfuvirtide dependent for entry (3, 4). In this model, enfuvirtide stabilizes the HR-1–HR-2 interaction and prevents premature prefusion conformational changes that can inactivate virus entry in the absence of drug. Further work is required to clarify whether our observation can be generalized to other enfuvirtide-resistant clinical isolates.
Several limitations of our approach deserve mention. Our analysis is based on observations from a single subject infected with subtype C virus and is restricted to the effect of envelope changes on virus fitness. Selection pressures during the time interval we studied shifted due to the changing optimized background regimen, and we could not explicitly calculate this contribution to fitness. The interval between time points was variable and likely affected the slope of the decay curve. We could not experimentally model the effect of HR-1 mutation emergence on VCV resistance decay. We therefore consider our measurements to represent a minimum decay rate, and thus the fitness differences we observed may represent a minimum fitness difference. Growth competition and kinetics assays were performed in U87 and TZM cell lines that have greater CD4 and CCR5 expression levels than resting peripheral blood mononuclear cells and may not recapitulate the in vivo environment encountered by these viruses. We have previously noted, however, that while the choice of cell type affects the absolute rates of virus entry, the relative differences in virus entry rates are generalizable across a range of receptor and coreceptor expression levels (45).
The withdrawal of VCV was associated with the rapid decay of VCV resistance in this highly treatment-experienced individual with multidrug-resistant HIV-1 and contrasts with a previous in vitro report that used laboratory-adapted virus (1). We did not observe an association between VCV resistance, its decay, and plasma viral load. The limited number of HIV-infected patients in whom CCR5 antagonist resistance develops and the dearth of published data on the decay of these mutations make it difficult to draw general conclusions about the viral fitness costs associated with this resistance. Fitness measurements in HIV-1 isolates from additional patients failing and discontinuing CCR5 antagonist therapy will be needed to assess the generalizability of these results to the overall estimated fitness costs of CCR5 antagonist resistance-associated mutations.
Supplementary Material
ACKNOWLEDGMENTS
A.M.N.T. was supported by K08 AI081547. R.P. was supported in part by CHAIN, funded through the European Community's Seventh Framework Programme FP7/2007-2013 under grant agreement no. 22313. This work was also supported in part by the AIDS Clinical Trials Group, which is funded by the National Institute of Allergy and Infectious Diseases, National Institutes of Health (AI068636 [AIDS Clinical Trials Group], AI069419 [Cornell CTU], AI051966 [to R.M.G.], and AI055357 [to D.R.K.]) and the National Center for Research Resources (RR024996 [Cornell CTSC] and RR016482 [to D.R.K.]). The Schering-Plough Research Institute provided the study drug, funds to support patient screening, and specialized laboratory assays.
We thank the A5211 protocol team members, the participating ACTG sites, and the study participants for their contributions to this work.
Footnotes
Published ahead of print 4 April 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Anastassopoulou CG, et al. 2007. Escape of HIV-1 from a small molecule CCR5 inhibitor is not associated with a fitness loss. PLoS Pathog. 3:e79 doi:10.1371/journal.ppat.0030079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anderson JA, et al. 2008. Genotypic susceptibility scores and HIV type 1 RNA responses in treatment-experienced subjects with HIV type 1 infection. AIDS Res. Hum. Retrovir. 24:685–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baldwin C, Berkhout B. 2008. Mechanistic studies of a T20-dependent human immunodeficiency virus type 1 variant. J. Virol. 82:7735–7740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baldwin CE, et al. 2004. Emergence of a drug-dependent human immunodeficiency virus type 1 variant during therapy with the T20 fusion inhibitor. J. Virol. 78:12428–12437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Berro R, Sanders RW, Lu M, Klasse PJ, Moore JP. 2009. Two HIV-1 variants resistant to small molecule CCR5 inhibitors differ in how they use CCR5 for entry. PLoS Pathog. 5:e1000548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cavrois M, De Noronha C, Greene WC. 2002. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat. Biotechnol. 20:1151–1154 [DOI] [PubMed] [Google Scholar]
- 7. Deeks SG, et al. 2005. Interruption of treatment with individual therapeutic drug classes in adults with multidrug-resistant HIV-1 infection. J. Infect. Dis. 192:1537–1544 [DOI] [PubMed] [Google Scholar]
- 8. Delobel P, et al. 2010. Shift in phenotypic susceptibility suggests a competition mechanism in a case of acquired resistance to maraviroc. AIDS 24:1382–1384 [DOI] [PubMed] [Google Scholar]
- 9. Devereux HL, Emery VC, Johnson MA, Loveday C. 2001. Replicative fitness in vivo of HIV-1 variants with multiple drug resistance-associated mutations. J. Med. Virol. 65:218–224 [DOI] [PubMed] [Google Scholar]
- 10. Domingo E, Holland JJ. 1997. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 51:151–178 [DOI] [PubMed] [Google Scholar]
- 11. Dykes C, Demeter LM. 2007. Clinical significance of human immunodeficiency virus type 1 replication fitness. Clin. Microbiol. Rev. 20:550–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eigen M. 1993. Viral quasispecies. Sci. Am. 269:42–49 [DOI] [PubMed] [Google Scholar]
- 13. Fatkenheuer G, et al. 2008. Subgroup analyses of maraviroc in previously treated R5 HIV-1 infection. N. Engl. J. Med. 359:1442–1455 [DOI] [PubMed] [Google Scholar]
- 14. Gulick RM, et al. 2008. Maraviroc for previously treated patients with R5 HIV-1 infection. N. Engl. J. Med. 359:1429–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gulick RM, et al. 2007. Phase 2 study of the safety and efficacy of vicriviroc, a CCR5 inhibitor, in HIV-1-Infected, treatment-experienced patients: AIDS clinical trials group 5211. J. Infect. Dis. 196:304–312 [DOI] [PubMed] [Google Scholar]
- 16. Henrich TJ, et al. 2011. Differential use of CCR5 by HIV-1 clinical isolates resistant to small molecule CCR5 antagonists. Antimicrob. Agents Chemother. 56:1931–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Henrich TJ, et al. 2010. Evolution of CCR5 antagonist resistance in an HIV-1 subtype C clinical isolate. J. Acquir. Immune Defic. Syndr. 55:420–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huelsenbeck JP, Ronquist F. 2001. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755 [DOI] [PubMed] [Google Scholar]
- 19. Jenwitheesuk E, Samudrala R. 2005. Heptad-repeat-2 mutations enhance the stability of the enfuvirtide-resistant HIV-1 gp41 hairpin structure. Antivir. Ther. 10:893–900 [PubMed] [Google Scholar]
- 20. Kijak GH, et al. 2002. Origin of human immunodeficiency virus type 1 quasispecies emerging after antiretroviral treatment interruption in patients with therapeutic failure. J. Virol. 76:7000–7009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kirchherr JL, et al. 2007. High throughput functional analysis of HIV-1 env genes without cloning. J. Virol. Methods 143:104–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kitchen CM, et al. 2009. Two-way Bayesian hierarchical phylogenetic models: an application to the co-evolution of gp120 and gp41 during and after enfuvirtide treatment. Comput. Stat. Data Anal. 53:766–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kitchen CM, et al. 2006. Continued evolution in gp41 after interruption of enfuvirtide in subjects with advanced HIV type 1 disease. AIDS Res. Hum. Retrovir. 22:1260–1266 [DOI] [PubMed] [Google Scholar]
- 24. Kuhmann SE, et al. 2004. Genetic and phenotypic analyses of human immunodeficiency virus type 1 escape from a small-molecule CCR5 inhibitor. J. Virol. 78:2790–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Labrosse B, et al. 2006. Role of the envelope genetic context in the development of enfuvirtide resistance in human immunodeficiency virus type 1-infected patients. J. Virol. 80:8807–8819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Larder BA, Darby G, Richman DD. 1989. HIV with reduced sensitivity to zidovudine (AZT) isolated during prolonged therapy. Science 243:1731–1734 [DOI] [PubMed] [Google Scholar]
- 27. Lassen KG, et al. 2009. Elite suppressor-derived HIV-1 envelope glycoproteins exhibit reduced entry efficiency and kinetics. PLoS Pathog. 5:e1000377 doi:10.1371/journal.ppat.1000377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lin NH, et al. 2010. The design and validation of a novel phenotypic assay to determine HIV-1 coreceptor usage of clinical isolates. J. Virol. Methods 169:39–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lu J, Kuritzkes DR. 2001. A novel recombinant marker virus assay for comparing the relative fitness of HIV-1 reverse transcriptase variants. J. Acquir. Immune Defic. Syndr. 27:7–13 [DOI] [PubMed] [Google Scholar]
- 30. Lu J, Sista P, Giguel F, Greenberg M, Kuritzkes DR. 2004. Relative replicative fitness of human immunodeficiency virus type 1 mutants resistant to enfuvirtide (T-20). J. Virol. 78:4628–4637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marconi V, et al. 2008. Viral dynamics and in vivo fitness of HIV-1 in the presence and absence of enfuvirtide. J. Acquir. Immune Defic. Syndr. 48:572–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maree AF, Keulen W, Boucher CA, De Boer RJ. 2000. Estimating relative fitness in viral competition experiments. J. Virol. 74:11067–11072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Margulies M, et al. 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Marozsan AJ, et al. 2005. Differences in the fitness of two diverse wild-type human immunodeficiency virus type 1 isolates are related to the efficiency of cell binding and entry. J. Virol. 79:7121–7134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McNicholas P, et al. 2010. Characterization of emergent HIV resistance in treatment-naive subjects enrolled in a vicriviroc phase 2 trial. J. Infect. Dis. 201:1470–1480 [DOI] [PubMed] [Google Scholar]
- 36. McNicholas PM, et al. 2011. Mapping and characterization of vicriviroc resistance mutations from human immunodeficiency virus type-1 isolated from treatment-experienced subjects enrolled in a phase II study (VICTOR-E1). J. Acquir. Immune Defic. Syndr. 56:222–229 [DOI] [PubMed] [Google Scholar]
- 37. Miyauchi K, Kim Y, Latinovic O, Morozov V, Melikyan GB. 2009. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 137:433–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ogert RA, et al. 2010. Clinical resistance to vicriviroc through adaptive V3 loop mutations in HIV-1 subtype D gp120 that alter interactions with the N-terminus and ECL2 of CCR5. Virology 400:145–155 [DOI] [PubMed] [Google Scholar]
- 39. Ogert RA, et al. 2008. Mapping resistance to the CCR5 co-receptor antagonist vicriviroc using heterologous chimeric HIV-1 envelope genes reveals key determinants in the C2–V5 domain of gp120. Virology 373:387–399 [DOI] [PubMed] [Google Scholar]
- 40. Paredes R, et al. 2009. In vivo fitness cost of the M184V mutation in multidrug-resistant human immunodeficiency virus type 1 in the absence of lamivudine. J. Virol. 83:2038–2043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pastore C, et al. 2006. Human immunodeficiency virus type 1 coreceptor switching: V1/V2 gain-of-fitness mutations compensate for V3 loss-of-fitness mutations. J. Virol. 80:750–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pfaff JM, et al. 2010. HIV-1 resistance to CCR5 antagonists associated with highly efficient use of CCR5 and altered tropism on primary CD4+ T cells. J. Virol. 84:6505–6514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pfizer 2007. Maraviroc tablets NDA 22–128. Antiviral Drugs Advisory Committee (AVDAC) briefing document, p 103 www.fda.gov/OHRMS/DOCKETS/AC/07/briefing/2007-4283b1-01-Pfizer.pdf
- 44. Pfizer 2007. Maraviroc tablets NDA 22–128. Antiviral Drugs Advisory Committee (AVDAC) briefing document, p 104 www.fda.gov/OHRMS/DOCKETS/AC/07/briefing/2007-4283b1-01-Pfizer.pdf
- 45. Putcharoen O, et al. 2012. HIV-1 Clinical isolates resistant to CCR5 antagonists exhibit delayed entry kinetics that are corrected in the presence of drug. J. Virol. 86:1119–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rangel HR, et al. 2003. Role of the human immunodeficiency virus type 1 envelope gene in viral fitness. J. Virol. 77:9069–9073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ray N, Blackburn LA, Doms RW. 2009. HR-2 mutations in HIV-1 gp41 restore fusion kinetics delayed by HR-1 mutations that cause clinical resistance to enfuvirtide. J. Virol. 83:2989–2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ray N, et al. 2007. Clinical resistance to enfuvirtide does not affect susceptibility of human immunodeficiency virus type 1 to other classes of entry inhibitors. J. Virol. 81:3240–3250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Reeves JD, et al. 2005. Enfuvirtide resistance mutations: impact on human immunodeficiency virus envelope function, entry inhibitor sensitivity, and virus neutralization. J. Virol. 79:4991–4999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Richman DD, et al. 1994. Nevirapine resistance mutations of human immunodeficiency virus type 1 selected during therapy. J. Virol. 68:1660–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Roche M, et al. 2011. HIV-1 escape from the CCR5 antagonist maraviroc associated with an altered and less-efficient mechanism of gp120-CCR5 engagement that attenuates macrophage tropism. J. Virol. 85:4330–4342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schurmann D, et al. 2007. Antiviral activity, pharmacokinetics and safety of vicriviroc, an oral CCR5 antagonist, during 14-day monotherapy in HIV-infected adults. AIDS 21:1293–1299 [DOI] [PubMed] [Google Scholar]
- 53. Schuurman R, et al. 1995. Rapid changes in human immunodeficiency virus type 1 RNA load and appearance of drug-resistant virus populations in persons treated with lamivudine (3TC). J. Infect. Dis. 171:1411–1419 [DOI] [PubMed] [Google Scholar]
- 54. Sharma PL, Crumpacker CS. 1997. Attenuated replication of human immunodeficiency virus type 1 with a didanosine-selected reverse transcriptase mutation. J. Virol. 71:8846–8851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Swanstrom R, et al. 2004. Weighted phenotypic susceptibility scores are predictive of the HIV-1 RNA response in protease inhibitor-experienced HIV-1-infected subjects. J. Infect. Dis. 190:886–893 [DOI] [PubMed] [Google Scholar]
- 56. Tilton JC, et al. 2010. A maraviroc-resistant HIV-1 with narrow cross-resistance to other CCR5 antagonists depends on both N-terminal and extracellular loop domains of drug-bound CCR5. J. Virol. 84:10863–10876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Troyer RM, et al. 2009. Variable fitness impact of HIV-1 escape mutations to cytotoxic T lymphocyte (CTL) response. PLoS Pathog. 5:e1000365 doi:10.1371/journal.ppat.1000365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tsibris AM, et al. 2009. Quantitative deep sequencing reveals dynamic HIV-1 escape and large population shifts during CCR5 antagonist therapy in vivo. PLoS One 4:e5683 doi:10.1371/journal.pone.0005683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tsibris AM, et al. 2008. In vivo emergence of vicriviroc resistance in a human immunodeficiency virus type 1 subtype C-infected subject. J. Virol. 82:8210–8214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Westby M, et al. 2006. Emergence of CXCR4-using human immunodeficiency virus type 1 (HIV-1) variants in a minority of HIV-1-infected patients following treatment with the CCR5 antagonist maraviroc is from a pretreatment CXCR4-using virus reservoir. J. Virol. 80:4909–4920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Westby M, et al. 2007. Reduced maximal inhibition in phenotypic susceptibility assays indicates that viral strains resistant to the CCR5 antagonist maraviroc utilize inhibitor-bound receptor for entry. J. Virol. 81:2359–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wilke CO. 2005. Quasispecies theory in the context of population genetics. BMC Evol. Biol. 5:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wu H, et al. 2006. Modeling and estimation of replication fitness of human immunodeficiency virus type 1 in vitro experiments by using a growth competition assay. J. Virol. 80:2380–2389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Xu L, et al. 2005. Emergence and evolution of enfuvirtide resistance following long-term therapy involves heptad repeat 2 mutations within gp41. Antimicrob. Agents Chemother. 49:1113–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.