

Abstract
The unfolded-protein response (UPR), activated by sensor molecules PERK, ATF6, and IRE1 to resolve endoplasmic reticulum (ER) stress, has emerged as a key target for host cells and viruses to control the infection outcomes. The UPR regulates ER protein folding, controls cell fate upon ER stress, and plays an important role in innate immunity. We and others have shown that human cytomegalovirus (HCMV) modulates the UPR. We show here that murine CMV (MCMV), the widely used CMV model for small animal infection, regulated the UPR in a manner similar to that of HCMV. This modulatory ability was triggered by virion entry and enhanced by viral immediate-early and early gene expression. Thus, while vulnerable at early times, MCMV became resistant to exogenous ER stress at late times of infection. MCMV activated the PERK-ATF4 pathway but only induced a subset of representative ATF4 targets at levels somewhat lower than those by the ER stress inducer tunicamycin. Moreover, MCMV induced ER chaperone Bip but actively blocked IRE1-mediated Xbp1(s) protein accumulation. ATF4 depletion severely attenuated viral growth at a low multiplicity of infection by modestly reducing viral DNA synthesis and more pronouncedly inhibiting late gene transcription. Collectively, we show that the UPR is a conserved target of CMVs and identify ATF4, a key UPR component, as a factor critical for MCMV infection. This work sets the stage for using the MCMV model to explore the role of this stress response in CMV biology, particularly during infection of the host, which is difficult to study in HCMV.
INTRODUCTION
Viruses are intracellular parasites that rely on hosts to provide resources to complete their life cycle. As a means to accomplish this, viruses hijack host processes and alter the cellular environment to subvert host cells for viral replication. During infection, many viruses induce endoplasmic reticulum (ER) stress and modulate subsequent cellular stress responses. The ER is the organelle where secretory and membrane proteins are modified and folded into their native conformations. One-third of the human proteome is modified in the ER (11, 43). In the ER lumen, newly synthesized polypeptides are modified by glycosylation and disulfide bond formation, and their folding is aided by ER-resident chaperones. Correctly folded proteins are transported in vesicles to the trans-Golgi network, where they are sorted to various locations to execute their functions. Meanwhile, any unfolded or misfolded proteins are eliminated by a process of ER-associated protein degradation (ERAD).
Unfolded or misfolded proteins, however, may accumulate in the ER lumen if ER function is compromised or if the ER is flooded by a surge of protein input, such as that resulting from virus infection. These misbehaved proteins not only lose their functionality, they also form aggregates that cause stress to the ER, becoming toxic to cells. To protect the central function of the ER, eukaryotic cells have evolved a conserved quality control system, termed the unfolded-protein response (UPR), to resolve ER stress and restore ER function (outlined in Fig. 1A). The UPR is initiated by three sensor molecules in the ER lumen, namely, PKR-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1). Activated PERK phosphorylates eukaryotic translation initiation factor 2α (eIF2α), leading to attenuation of global protein translation but selective upregulation of ATF4. ATF4 is a transcriptional factor that induces enzymes to overcome oxidative stress normally accompanying ER stress and stabilize redox homeostasis of the ER. Activated ATF6 translocates to the Golgi body, where its N-terminal fragment is cleaved and imported into the nucleus to induce the transcription of ER chaperone genes. The IRE1 pathway is the most ancient and evolutionally conserved UPR branch. Activated IRE1 mediates splicing of Xbp1 transcript to produce transcriptional factor Xbp1(s) that induces expression of ERAD enzymes. Collectively, these cytoprotective features of the UPR help cells eliminate ER stress and enhance ER function. However, if the damage to the ER is too great to overcome, the UPR induces molecular events, such as ATF4-mediated expression of C/EBP homologous protein (CHOP) or IRE1-mediated activation of c-Jun N terminus kinase (JNK), to commit damaged cells to apoptosis. Therefore, the UPR maintains a delicate balance among key ER activities to ensure a healthy ER environment under physiological conditions and relays stress signals into the regulatory circuit to control cell fate.
Fig 1.

MCMV modulates the UPR during infection. (A) Outline of the UPR and MCMV-mediated regulation of this cellular response reported in the present study. Only ER function-enhancing features of the UPR are shown. UPR-induced apoptosis in response to excessive ER stress is described in the text. Despite the induction of eIF2α phosphorylation, CMVs have developed a means to prevent attenuation of global protein translation. (B) MCMV modulates the UPR during infection. MEF10.1 cells were mock infected (MCMV, −) or infected with MCMV (MCMV, +) at an MOI of 2. Cells were then treated with 2 μg of tunicamycin/ml (TM, 2) or solvent DMSO only (TM, 0) for 1 h at 1, 24, or 48 hpi. Drug-containing medium was removed, and the cells were washed twice, cultured in drug-free medium for another 4 h, and collected at the indicated times for analysis. The cells were fractionated before being analyzed for the key components of the UPR signaling pathways by immunoblotting, since Xbp1(s) could only be detected in the nuclear fraction but not in the total lysate. Bip was determined in the cytoplasmic fraction. All other UPR components were determined in the nuclear fraction. Viral protein IE1 was used as the infection control. Actin and histone H3 were used as the loading control for the cytoplasmic and nuclear fractions, respectively. (C) Experiment design to test the effect of exogenous ER stress on MCMV infection. MEF10.1 cells were infected with MCMV at an MOI of 2 and 2 μg of tunicamycin/ml (TM), 2 μM thapsigargin (TG), or DMSO was added to the culture at the indicated times (indicated by a downward arrow). Drug-containing medium was then removed 1 h later (indicated by an upward arrow), and the cells were washed twice and maintained in drug-free medium for the remainder of the experiment. Cell-free virus was collected at either 24 h after drug treatment or 72 hpi (depicted by black triangle), and the virus titer was determined by TCID50 assay. (D) Virus production under various conditions as described in panel C. The upper panel shows the virus titer produced within 24 h after drug treatment. The lower panel shows the virus titer produced between the time of drug withdrawal and 72 hpi. The time points when cells were treated with the drugs are indicated.
This balance is often disturbed, however, under pathological conditions, particularly during virus infection. For example, most viruses produce a large amount of proteins that need to be folded and processed in the ER to support infection. In the ongoing battle between host and pathogen, host cells utilize the UPR to limit viral replication by restricting biosynthesis or inducing apoptosis, whereas viruses have developed means to hijack the UPR to enhance ER function for a successful infection. Increasing evidence suggests that this intricate host-virus interaction bears a profound consequence on viral disease. For example, the members of the Flaviviridae family, such as West Nile virus, dengue virus, and hepatitis C virus, activate the beneficial components of the UPR in certain cell types to maintain their replication but induce UPR-mediated apoptosis in other cell types central to underlying viral diseases (1, 22, 24, 40). The members of the Herpesviridae family have also been reported to modulate the UPR, and each virus appears to accomplish this via distinct mechanisms with different consequences (25). Herpes simplex virus 1, an alphaherpesvirus, actively inhibits the PERK pathway by the viral glycoprotein gB (32), whereas varicella-zoster virus, another alphaherpesvirus, activates both PERK and IRE1 pathways (10). Viral protein LMP1 encoded by Epstein-Barr virus (EBV), a gammaherpesvirus, activates the UPR during latent infection (26). The UPR in turn sustains LMP1 expression and its oncogenic activity in an ATF4-dependent manner. In addition, extrinsic ER stress reactivates EBV from latent infection and Xbp1, the key component of the IRE1 pathway, induces EBV lytic gene expression (4). These diverse examples underscore a critical role of the UPR regulation in viral infection and pathogenesis.
We and others have previously shown that human cytomegalovirus (HCMV), the prototypical betaherpesvirus, also deals with ER stress and modulates the UPR during infection. HCMV is the leading viral cause of birth defects in newborns and a common source of infectious complications in immunocompromised individuals. The ability to deal with defensive host stress responses, such as the UPR, is particularly important for HCMV, which has a protracted replication cycle. HCMV uses multiple strategies to modulate the UPR. Viral protein pUL38 activates the PERK pathway and prevents elevated JNK activation during its infection (42). HCMV also induces ATF6-independent expression of the ER chaperone protein Bip to facilitate virion assembly (8, 9). However, the role and associated mechanisms of other UPR components during virus infection remain elusive. Evidence is emerging in recent years that the UPR also plays an important role in regulating innate immunity and inflammation (30, 43), and the involvement of this activity in viral disease has started to gain appreciation (2, 28, 29). To facilitate our effort to dissect the role and regulation of the UPR during CMV infection, we began to analyze the UPR in murine CMV (MCMV) infection. MCMV and HCMV are conserved in their colinear genomes, biology, and viral pathogenesis, so MCMV provides a tractable small-animal model to study CMV infection, particularly in the host (35, 39).
In the present study, we report that MCMV induces ER stress and modulates the UPR similarly to HCMV. Moreover, our results suggest that MCMV hijacks this cellular stress response by desensitizing host cells from extrinsic ER stress and protecting viral replication from deleterious consequences of ER stress at late times of infection. Importantly, we provide evidence that ATF4 plays a critical role to promote MCMV DNA replication and late gene expression in a multiplicity of infection (MOI)-dependent manner. Our work sets the stage for using the MCMV model to explore this important cellular stress response in CMV biology in future studies.
MATERIALS AND METHODS
Plasmids and reagents.
The following retroviral expression vectors were derived from pRetro-EBNA (23). pYD-C453 carried the ATF4 coding sequence (42). pYD-C443 carried the coding sequence of an ATF4 dominant-negative variant (ATF4ΔRK) (18). pYD-C245 carried the red fluorescent protein (DsRed) coding sequence (3). The lentiviral expression constructs pYD-sh066 and pYD-sh068 or the lentiviral expression constructs pYD-sh070 and pYD-sh073 carried short hairpin RNAs (shRNAs) targeting the PERK or ATF4 coding sequence, respectively (acquired from the Children's Discovery Institute, RNAi Consortium, and Genome Institute at Washington University). shRNA targeting sequences are available upon request.
Primary antibodies against the following proteins were used in the present study: ATF4, CHOP, Xbp1(s), Bip, and eIF2α (Santa Cruz); p-eIF2α (BioSource International); MCMV IE1 and E1 (generous gifts from Stipan Jonjic, University of Rijeka, Croatia); MCMV gB (a generous gift from Anthony Scalzo, University of Western Australia); actin (Abcam); and histone H3 (BioLegend). Tunicamycin, thapsigargin, and phosphonoacetic acid (PAA) were purchased from Sigma-Aldrich.
Cells and viruses.
Mouse embryonic fibroblast 10.1 cells (MEF10.1) (16) were propagated in Dulbecco modified Eagle medium supplemented with 10% fetal calf serum plus nonessential amino acid and 1 mM sodium pyruvate. To create expressing cells, MEF10.1 cells were transduced three times with retroviruses reconstituted from the aforementioned retroviral vectors. Control cells expressing empty vector were made by transduction with retrovirus reconstituted from pRetro-EBNA. Similarly, MEF10.1 cells stably expressing shRNAs targeting ATF4 or PERK were generated by transduction once with lentivirus reconstituted from lentiviral expressing constructs. At 24 h after transduction, cells were selected with 2 μg of puromycin/ml for 3 days before being used for the experiments described here.
Recombinant MCMV BAC clones used in the present study were derived from the self-excisable parental MCMV BAC clone, pSM3fr, which carried a full-length genome of the MCMV Smith strain (41). All recombinant MCMV bacterial artificial chromosome (BAC) clones in used here were created according to a BAC recombineering protocol that we have previously established (34, 42). These BAC clones were then used to reconstitute corresponding recombinant MCMV viruses. The BAC clone pSMgfp carried the green fluorescent protein (GFP) expression cassette inserted at the MCMV IE2 intergenic locus (A. R. Fehr and D. Yu, unpublished data) and was used to produce wild-type virus SMgfp. pSMsubM86 carried a kanamycin mutagenic cassette in place of M86. pSMinM79 carried a 88-bp insert at nucleotide 403 of the M79 coding sequence, resulting in a frameshift mutation, and was used to reconstitute mutant MCMV virus SMinM79 (T. J. Chapa and D. Yu, unpublished data).
To reconstitute virus, 2 to 5 μg of MCMV BAC DNA was electroporated into normal MEF10.1 cells or M79-expressing MEF10.1 cells (Chapa and Yu, unpublished) (for reconstituted SMinM79). The culture medium was changed 24 h later, and virus stock was prepared by collecting cell-free culture supernatant when the entire monolayer of cells was lysed. Alternatively, virus stocks were produced by collecting cell-free supernatant from infected culture at an MOI of 0.01. Virus titers were determined in duplicate by using a 50% tissue culture infectious dose (TCID50) assay in normal or M79-expressing (for SMinM79) MEF10.1 cells.
Viral growth analysis.
MEF10.1 cells were seeded in 12-well dishes overnight to produce a subconfluent monolayer. The cells were inoculated with recombinant MCMV virus for 1 h, the inoculum was removed, the monolayers were rinsed with phosphate-buffered saline (PBS), and fresh medium was replenished. At various times postinfection, cell-free media from infected cultures were collected, and virus titers in the media were determined by TCID50 assay in MEF10.1 cells.
Protein analysis.
Immunoblot analysis was performed as previously described (34). The cells were collected, washed, and lysed in the sodium dodecyl sulfate (SDS)-containing sample buffer. Proteins from equal cell numbers were resolved by electrophoresis on an SDS-containing polyacrylamide gel, transferred to a polyvinylidene difluoride membrane, hybridized with primary antibodies, reacted with horseradish peroxidase-conjugated secondary antibodies, and visualized by using SuperSignal West Pico chemiluminescent substrate (Thermo Scientific). In some experiments, cytoplasmic and nuclear fractionation of cell lysates was also performed at 4°C using a protocol from the David Ron lab (http://ron.medschl.cam.ac.uk/protocols/NucCyto.pdf) for analysis. The cells were collected by scraping into ice-cold PBS containing 1 mM EDTA, pelleted, and lysed in harvest buffer (10 mM HEPES [pH 7.9], 50 mM NaCl, 0.5 M sucrose, 0.1 mM EDTA, 0.5% Triton X-100, and a freshly added cocktail of protease and phosphatase inhibitors and 1 mM dithiothreitol [DTT]) by a 5-min incubation on ice. Nuclei were pelleted by centrifugation at 200 × g for 10 min. The supernatants were further cleared by centrifugation at 16,100 × g for 15 min and used as cytoplasmic fractions. Nuclei pellets were washed with buffer A (10 mM HEPES [pH 7.9], 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, and a freshly added cocktail of protease and phosphatase inhibitors and 1 mM DTT), resuspended, and extracted in 4 volumes of nuclear extraction buffer (10 mM HEPES [pH 7.9], 500 mM NaCl, 0.1 mM EDTA, 0.1 mM EGTA, 0.1% NP-40, and a freshly added cocktail of protease and phosphatase inhibitors and 1 mM DTT) by vortexing for 15 min. The lysates were cleared by centrifugation at 16,100 × g for 10 min and used as nuclear extracts.
DNA and RNA analysis.
Intracellular viral DNA in MCMV-infected cells was measured by quantitative PCR (qPCR) as previously described (34). Viral or cellular DNA was quantified by real-time PCR analysis with SYBR green PCR Master Mix (Clontech) and a primer pair specific for the MCMV IE1 or mouse actin gene, respectively (Table 1). The amount of viral DNA was normalized by dividing viral IE1 gene equivalents by actin gene equivalents.
Table 1.
Primers used in qPCR analysis
| Primer | Orientationa | Sequence (5′–3′) |
|---|---|---|
| MCMV IE1 | F | CAGGGTGGATCATGAAGCCT |
| R | AGCGCATCGAAAGACAACG | |
| MCMV E1 | F | GAATCCGAGGAGGAAGACGAT |
| R | GGTGAACGTTTGCTCGATCTC | |
| MCMV gB (M55) | F | GCGATGTCCGAGTGTGTCAAG |
| R | CGACCAGCGGTCTCGAATAAC | |
| Mouse PERK | F | TACCCATTCAGCACCCAGATGGAA |
| R | TCCCGAGTCCAACTGATCAAAGCA | |
| Mouse actin | F | GCTGTATTCCCCTCCATCGTG |
| R | CACGGTTGGCCTTAGGGTTCA | |
| Mouse ASNS | F | TTCCGTGCAGTGTCTGAGTG |
| R | GCAGCAGTTGGTGTATCCATTG | |
| Mouse Slc3a2 | F | TGTTAGGCCCAATTCACAAGAAC |
| R | TGGGAGCCCAAAGTGGGATT | |
| Mouse ERO1L | F | AGCATGATGATTCGTCAGACAG |
| R | AGGGTTAAGGAGTAAGTCCACAT | |
| Mouse C/EBPβ | F | GACTGACGCAACACACGTGTAA |
| R | ATCAACAACCCCGCAGGAA |
F, forward; R, reverse.
RNA was measured by reverse transcription-coupled qPCR as previously described (42). To analyze cellular gene transcripts, RNA was reversed transcribed, and the amount of mouse ASNS, Slc3a2, C/EBPβ, ERO1L, or actin transcript was quantified by real-time PCR using the SYBR Advantage qPCR Premix and the primer pairs listed in Table 1. To analyze viral transcripts, RNA was reversed transcribed, and the amount of viral IE1, E1, or gB (M55) transcript was quantified by real-time PCR using the SYBR Advantage qPCR Premix and the primer pairs listed in Table 1. The amount of viral transcript was normalized using actin as the internal control.
To analyze PERK transcript, total RNA was extracted as described above, and PERK or actin transcript was reversed transcribed and then analyzed by semiquantitative PCR using the primer pairs listed in Table 1. PCR products were resolved on an agarose gel by electrophoresis, and the product from actin transcript was used as the internal control.
Cell viability and proliferation analysis.
To measure cell proliferation, MEF10.1 cells overexpressing a targeted gene were seeded onto a 12-well dish at the density of 105 cells/well. At various days after seeding, the cells were fixed with 1% paraformaldehyde and stained with DAPI (4′,6′-diamidino-2-phenylindole). Three fields of each well (i.e., the left, center, and right side of the well) were captured by inverted fluorescence tissue culture microscope. The number of DAPI-stained nuclei in each field was automatically scored by using CellProfiler software (http://www.cellprofiler.org/). Three wells were counted for each cell type at each time point, and the average was used to represent the cell number at any given time point. To quantify cell viability, the cells were seeded and treated as described above, except that they were costained with propidium iodide and DAPI. Propidium iodide only stained dead or dying cells with compromised plasma membrane, whereas DAPI stained every nucleus. The numbers of propidium iodide- or DAPI-stained nuclei were automatically counted by CellProfiler. The ratio of propidium iodide-positive nuclei to DAPI-positive cells for each field was calculated, and the average was presented as the percentage of cell death for each tested cell type. Cells that were starved in PBS for 6 h were included as the positive control for cell death.
Viral infectivity analysis.
MCMV-infected cells were collected by trypsinization and low-speed centrifugation at 4 h postinfection (hpi), fixed in 1% paraformaldehyde, and permeabilized with 0.1% Triton X-100. The cells were stained with mouse antibody against the MCMV IE1 protein and subsequently with an Alexa Fluor 555-conjugated goat anti-mouse secondary antibody (Invitrogen). The cells were analyzed by flow cytometry, and the percentage of IE1-positive cells represented viral infectivity.
RESULTS
MCMV modulates the unfolded protein response during infection.
To test the hypothesis that modulation of the UPR is conserved in MCMV, we first examined major components of three UPR signaling pathways during MCMV infection. In this experiment, MEF10.1 cells were mock infected or infected with BAC pSM3fr-derived MCMV, and the key components of the UPR in these cells were examined at different times postinfection (hpi). Activation of the PERK pathway induces ATF4 and CHOP expression. We found that, like HCMV (21, 42), MCMV induced ATF4 protein accumulation, as early as 6 hpi (Fig. 1B). Interestingly, despite elevated ATF4 accumulation, CHOP was hardly detected, even though it could be robustly induced by the ER-stress inducer tunicamycin (Fig. 1B) (37). This could be due to very low levels of CHOP expression that were under the detection limit of our assay or to virus-mediated active inhibition of CHOP expression. Furthermore, like HCMV, MCMV also induced the protein accumulation of Bip, an ATF6-inducible ER chaperone protein that is induced in the ATF6-independent manner during HCMV infection (Fig. 1B) (9, 21). Finally, the activated IRE1 pathway induces Xbp1 transcript splicing to produce the spliced protein product, Xbp1(s). HCMV induces limited Xbp1 transcript splicing, but Xbp1(s)-dependent expression of downstream ERAD enzymes is not detected (21, 42). We found that Xbp1 transcript splicing was also induced by MCMV, but it was very limited in nature compared to that by tunicamycin (data not shown), and that induction of Xbp1(s) protein accumulation was minimal (Fig. 1B). Overall, MCMV modulates the UPR signaling pathways in a way similar to that described during HCMV infection (21, 42).
MCMV is resistant to exogenous ER stress at late times of infection.
To test whether the ability to modulate the UPR allows MCMV to overcome ER stress, we examined how MCMV responded to the treatment of ER-stress inducers during infection. MCMV-infected MEF10.1 cells were treated with tunicamycin, thapsigargin, or solvent only (dimethyl sulfoxide [DMSO]) for 1 h at 1, 24, or 48 hpi. Drug-containing medium was removed, cells were washed, drug-free medium was replenished, and the production of cell-free virus was determined either at 24 h after drug treatment or at 72 hpi (Fig. 1C). Drug treatment at immediate-early times (i.e., 1 hpi) effectively blocked virus production within the first 24 h, a time period where ∼104 infectious progeny viruses per ml were produced in control cells (Fig. 1D, upper panel). This remarkable inhibition persisted throughout the course of the 72-hour experiment, with virus production remaining undetectable under thapsigargin treatment or reduced by almost 1,000-fold under tunicamycin treatment (Fig. 1D, lower panel). Intriguingly, the inhibitory effects of tunicamycin and thapsigargin on virus production were greatly diminished once MCMV established its infection. A 1-h drug treatment at 24 hpi reduced virus produced in next 24 h by <100-fold and, more strikingly, a 1-h drug treatment at 48 hpi could only reduce virus produced between 49 and 72 hpi by <10-fold. We interpret this dramatic inhibitory effect at early times to suggest the importance of a healthy and functional ER for MCMV to establish its infection. However, once the virus starts to replicate, it develops a means to remodel ER function and enhance the capacity of host cells to manage ER stress. It becomes markedly resistant to ER stress inflicted at late times of infection, a finding reminiscent of that described for HCMV infection (20).
To provide insight into how MCMV remodels the host response to an ER stress, we examined the PERK, ATF6, and IRE1 signaling pathways in MCMV-infected cells in response to tunicamycin treatment (Fig. 1B). In this experiment, we treated MCMV-infected cells with tunicamycin for 1 h at 1, 24, or 48 hpi. Tunicamycin-containing medium was then removed, and the cells were washed and cultured in drug-free medium for another 4 h so we could examine the UPR in these cells as early as 6 hpi. As anticipated, tunicamycin induced the accumulation of ATF4, CHOP, Xbp1(s) and, to a lesser degree, ER chaperone Bip in mock-infected cells. When added to MCMV-infected cells at 1 hpi, a time point prior to viral early gene expression, tunicamycin induced these UPR markers to an extent similar to those in mock-infected cells. In drastic contrast, when added at 24 to 48 hpi, tunicamycin induced less ATF4 and, more significantly, CHOP and Xbp1(s) accumulation compared to those in mock-infected cells. These results support the hypothesis that MCMV remodels the UPR, enhances ER function, and desensitizes host cells to deleterious consequence of ER stress at late times of infection. It is worth noting that MCMV infection induced Bip accumulation at levels much greater than tunicamycin did (Fig. 1B), reminiscent of the virus-mediated, ATF6-independent induction of Bip reported for HCMV (9).
MCMV activates the PERK pathway during infection.
In the present study, we primarily focused on the PERK pathway since our initial analysis indicated an MCMV-mediated induction of ATF4 accumulation (Fig. 1B). This induction was evident at 4 hpi and peaked at 24 hpi (Fig. 2A). Given that HCMV activates the PERK pathway and stimulates ATF4 accumulation (21, 42), we speculated that MCMV might similarly modulate this pathway. To test this theory, we examined eIF2α, which is phosphorylated by PERK to induce ATF4 expression during the UPR. The levels of eIF2α phosphorylation increased as early as 4 hpi and remained elevated over the 48-h course of the experiment (Fig. 2A). Increased eIF2α phosphorylation was more pronounced at 24 to 48 hpi, perhaps in part due to the increase in total eIF2α levels at late times of infection (Fig. 2A), an observation reminiscent of our and others' previous reports for HCMV (21, 42). Interestingly, whereas eIF2α phosphorylation appeared to largely correlate with ATF4 accumulation, CHOP induction during MCMV infection consistently remained low compared to that by tunicamycin treatment. To test whether other ATF4 target genes were upregulated during MCMV infection, we examined transcription of four additional cellular genes by reverse transcription-coupled qPCR (RT-qPCR) at 8 and 24 hpi. These were the transcription factor C/EBPβ, the amino acid transporter Slc3a2, oxidoreductase ERO1-like protein (ERO1L), and asparagine synthetase (ASNS). Transcription of these four genes is known to be induced by ATF4 during UPR (15). Consistently, we found that their transcription was markedly upregulated at 8 h after tunicamycin treatment (Fig. 2B). Interestingly, MCMV did not induce C/EBPβ transcription during infection (Fig. 2B). At 8 and 24 hpi, MCMV elevated Slc3a2 transcript levels by 1.5- and 0.5-fold, respectively. Moreover, at these time points, MCMV elevated ASNS transcript levels by 2.5- and 9.5-fold and ERO1L transcript levels by 3.5-fold and 10.5-fold. Therefore, MCMV appears to selectively induce the expression of a subset of ATF4-responsive genes during infection, albeit at levels somewhat less robust than those induced by tunicamycin treatment.
Fig 2.

MCMV activates the PERK signaling pathway. (A) Analyses of activation of key components of the PERK pathway during MCMV infection. MEF10.1 cells were mock infected or infected with MCMV at an MOI of 2. Total cell lysates were collected at the indicated times and analyzed by immunoblotting. Lysate of cells treated with tunicamycin was included as the positive control. Viral protein IE1 and actin were used as the infection control and loading control, respectively. In the ATF4 blot, an arrow indicates the ATF4, whereas other bands represented nonspecific protein species in total cell lysates that cross-reacted with the ATF4 antibody. (B) ATF4-responsive genes are selectively induced during MCMV infection. MEF10.1 cells were mock infected or infected with MCMV at an MOI of 2. The cells were collected at 8 and 24 hpi, and transcription of representative cellular genes downstream of ATF4 was measured by RT-qPCR and normalized to that of actin. The normalized amount of transcript in mock-infected cells at 8 hpi was set at 1. Cells that were treated with tunicamycin (TM) for 8 h were included as the positive control. (C) shRNA knockdown of PERK. MEF10.1 cells were transduced with lentivirus expressing shRNA targeting luciferase (shLuc, negative control) or two independent shRNAs targeting PERK (shPERK-1 and shPERK-2). The efficacy of shRNA knockdown was determined by RT-qPCR analysis of the PERK transcript. The product from actin transcript was included as the internal control. UT (untransduced), normal MEF10.1 cells. (D) eIF2α phosphorylation and ATF4 protein accumulation during MCMV infection were PERK dependent. Normal or shRNA-expressing MEF10.1 cells were mock infected, infected with MCMV, or treated with tunicamycin. Cell lysates were collected at 4 hpi or after drug treatment and then analyzed by immunoblotting.
To confirm that eIF2α phosphorylation and ATF4 accumulation induced by MCMV infection was dependent on PERK, we attempted to analyze PERK and its phosphorylation by immunoblotting. However, endogenous PERK proteins were difficult to detect in MEF10.1 cells, even though overexpressed PERK were readily detectable by antibodies (data not shown). Alternatively, we depleted endogenous PERK by shRNAs targeting two different regions of the PERK coding sequence. Both shRNAs efficiently depleted PERK transcripts (Fig. 2C). In mock cells, the expression of PERK-targeting shRNAs slightly induced eIF2α phosphorylation and ATF4 accumulation, perhaps due to a negative-feedback regulation (Fig. 2D) (33). Importantly, both PERK-targeting shRNAs markedly reduced the levels of eIF2α phosphorylation and ATF4 accumulation induced by tunicamycin or MCMV. Therefore, similar to HCMV, MCMV activates the PERK pathways to induce ATF4 accumulation.
MCMV uses both viral gene expression-dependent and -independent mechanisms to activate the PERK pathway.
To determine what viral factors triggered activation of the PERK pathway, we first determined whether virus entry or viral gene expression was required for this modulation. We infected MEF10.1 cells with UV-inactivated MCMV and determined the induction of eIF2α phosphorylation and ATF4 accumulation in infected cells. UV treatment effectively inactivated the virus, preventing viral immediate-early gene expression (Fig. 3A) and virus production (data not shown). However, in UV-inactivated MCMV-infected cells, eIF2α phosphorylation and ATF4 accumulation was reduced albeit not completely abrogated (Fig. 3A). This suggests that both viral entry and viral gene expression activate the PERK pathway. The viral gene expression-dependent activation was evident as early as 4 hpi, suggesting the involvement of viral immediate-early or early genes. To test this, cells were infected with MCMV in the presence of phosphonoacetic acid (PAA), a specific inhibitor of viral DNA synthesis. PAA blocked the expression of viral late genes (e.g., gB), as expected (Fig. 3C), but had no effects on virus-induced eIF2α phosphorylation and ATF4 accumulation (Fig. 3B). We conclude that MCMV uses two mechanisms to activate the PERK pathway of the UPR. One is mediated by virus entry, and the other is dependent on viral immediate-early or early gene expression.
Fig 3.

MCMV uses both viral gene expression-dependent and -independent mechanisms to activate the PERK pathway. (A) UV inactivation partially reduced the ability of MCMV to activate the PERK pathway. MEF10.1 cells were infected with wild-type or UV-inactivated MCMV. Cell lysates were collected at 4 and 8 hpi, and eIF2α phosphorylation and ATF4 accumulation were determined by immunoblotting. (B) Viral DNA synthesis and viral late gene expression were not required to activate the PERK pathway. MEF10.1 cells were mock infected or infected with MCMV at an MOI of 2 in the presence or absence of PAA (100 μg/ml). Cell lysates were collected and analyzed as described in panel A. (C) PAA treatment completely blocked viral late gene gB expression.
Depletion of ATF4 inhibits MCMV growth at a low MOI.
Since the PERK pathway was activated by MCMV, we wanted to determine whether it was important for virus replication. To address this, we focused on the transcription factor ATF4, the key effector mediating the ability of the PERK pathway to enhance ER function. We depleted functional ATF4 by overexpressing a dominant-negative variant of ATF4, ATF4ΔRK, which lacked the arginine- and lysine-rich DNA-binding motif (18) (Fig. 4D). ATF4 functions as a homodimer, so ATF4ΔRK dimerizes with wild-type ATF4 to disrupt its transcriptional activity. As a control, we also overexpressed wild-type ATF4 (Fig. 4D). We determined how MCMV replicated in these expressing cells under high- or low-MOI conditions by performing infection at an MOI of 2 or 0.001, respectively. At an MOI of 2, MCMV growth was indistinguishable in normal cells or cells overexpressing ATF4ΔRK, wild-type ATF4, or vector control (Fig. 4A). Strikingly, however, at an MOI of 0.001, whereas MCMV grew rapidly in normal cells or cells overexpressing wild-type ATF4 or vector control, it barely replicated in cells overexpressing ATF4ΔRK, resulting in a >3-log growth defect (Fig. 4B). These overexpressing cells showed no appreciable difference in proliferation and viability compared to control cells (Fig. 4C and D), and they supported efficient MCMV replication at a high MOI (Fig. 4A). Therefore, this reduction in viral growth under the low-MOI condition was not due to any potential nonspecific toxicity of ATF4ΔRK overexpression.
Fig 4.

Expression of dominant-negative ATF4 inhibits MCMV growth at a low MOI. (A) Growth analysis of MCMV in MEF10.1 cells expressing dominant-negative ATF4 (ATF4ΔRK) at a high MOI. MEF10.1 cells overexpressing empty vector, wild-type ATF4, or ATF4ΔRK were made by retroviral transduction. Normal cells or expressing cells were infected with MCMV at an MOI of 2. Cell-free virus was collected, and titers were determined at the indicated times postinfection. (B) Growth analysis of MCMV in MEF10.1 cells expressing dominant-negative ATF4 (ATF4ΔRK) at a low MOI. The experiment was performed the same as for panel A except that cells were infected at an MOI of 0.001. (C) ATF4ΔRK overexpression had no deleterious effect on cell proliferation. MEF10.1 cells were seeded onto 12-well dishes at 105 cells/well. Cell numbers at 1, 2, and 3 days after seeding were scored as described in Materials and Methods. (D) ATF4ΔRK overexpression had no deleterious effect on cell viability. MEF10.1 cells were seeded as described in panel C, and cell viability at 3 days after seeding was determined as described in Materials and Methods. Also included were cells treated with PBS for 6 h as the cell death control, as well as immunoblots of overexpressed ATF4 and ATF4ΔRK proteins in these cells.
As an alternative approach to validate the result from dominant-negative ATF4 expression, we knocked down endogenous ATF4 by shRNA. Two independent shRNAs targeting different regions of the ATF4 coding sequence were used to minimize the off-target effect. Both shRNAs efficiently reduced ATF4 protein accumulation that was induced by tunicamycin or MCMV infection (Fig. 5A). Consistent with the result from ATF4ΔRK overexpression, MCMV replicated indistinguishably in normal cells (UT), cells expressing control shRNA (shLuc), or cells expressing ATF4-targeting shRNA (shATF4-1, shATF4-2) at an MOI of 2, but it almost completely failed to replicate in cells expressing ATF4-targeting shRNA at the MOI of 0.001 (Fig. 5B). Taken together, our results indicate that ATF4 is required for efficient MCMV replication in an MOI-dependent manner.
Fig 5.
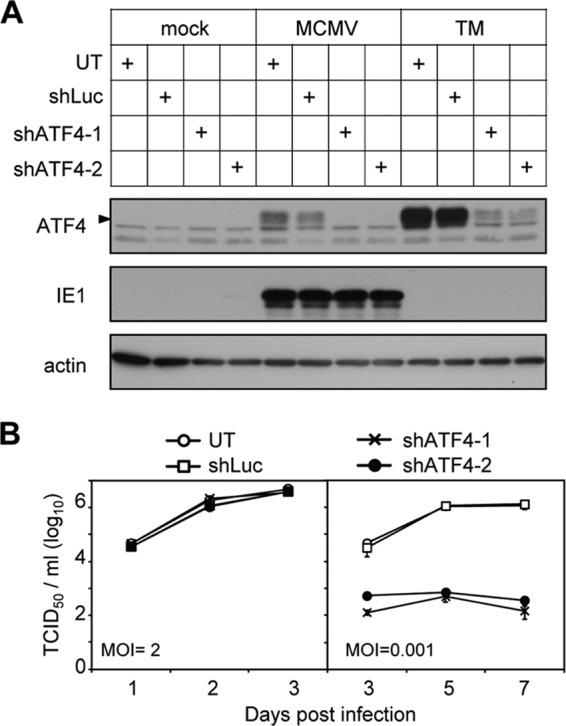
shRNA knockdown of ATF4 inhibits MCMV growth at a low MOI. (A) Expression of two independent shATF4 constructs effectively reduced ATF4 accumulation. MEF10.1 cells were transduced with lentivirus expressing shRNA targeting the luciferase (shLuc) or two independent shRNAs targeting ATF4 (shATF4-1 or shATF4-2). Normal cells (UT) or transduced cells were mock infected, infected with MCMV, or treated with tunicamycin. Cell lysates were collected at 4 hpi or after drug treatment and analyzed by immunoblotting. (B) High- and low-MOI growth curve analyses of MCMV in ATF4-knockdown cells. Cells as indicated were infected with MCMV at an MOI of 2 or 0.001. Cell-free virus was collected, and titers were determined at the indicated times postinfection.
ATF4 is not required for early events of the MCMV replication cycle at a low MOI.
We next determined where ATF4 acted during the viral replication cycle to promote MCMV infection. We first examined virus infectivity in control cells or ATF4-depleted cells (i.e., cells overexpressing ATF4ΔRK) during both low- and high-MOI infections. We infected cells with MCMV at an MOI of 0.01, 0.1, or 2. At 4 hpi, the cells were fixed, permeabilized, and stained with antibody against the MCMV IE1 protein. The percentages of IE1-positive cells were determined by flow cytometry (Fig. 6A). We chose the MOI of 0.01 instead of 0.001 as the low-MOI infection condition because this was the limit that allowed us to detect infected cells within a single round of infection. At an MOI of 0.01, the severe defect of MCMV growth persisted in ATF4-depleted cells (Fig. 6C). However, at all of the MOIs tested, we found no appreciable difference in numbers of IE1-positive cells between control cell and ATF4-depleted cells (Fig. 6A and B). Therefore, loss of ATF4 does not compromise the ability of MCMV to enter host cells and initiate the gene expression program.
Fig 6.

Ablation of ATF4 does not alter viral infectivity during a low MOI. (A) Percentage of IE1-positive cells at 4 hpi. MEF10.1 cells expressing ATF4ΔRK or vector control were infected with MCMV at an MOI of 0.01, 0.1, or 2. At 4 hpi, the cells were fixed, permeabilized, stained with antibody against the viral IE1 protein, and analyzed by flow cytometry. Also included in the analysis is the signal of GFP that was expressed from the viral genome, and the strength of GFP fluorescence correlated with the MOI used in the experiment. (B) Quantification of the results from two independent experiments as described in panel A. (C) Virus production in ATF4ΔRK-expressing or control cells infected at an MOI of 0.1 or 0.01 at indicated number of days postinfection (dpi).
To test whether ATF4 is required for MCMV to continue efficient viral gene expression, we analyzed the product accumulation of representative viral immediate-early (e.g., IE1), early (e.g., E1), and late (e.g., gB) genes. At an MOI of 2, the IE1 and E1 proteins accumulated efficiently in ATF4-depleted cells even though gB was slightly reduced at 24 hpi (Fig. 7A, right panel). At an MOI of 0.01, the IE1 protein was detectable at 8 hpi and accumulated to the same levels in control cells and ATF4-depleted cells (Fig. 7A, left panel). This is consistent with the result from virus infectivity analysis (Fig. 6) and indicates that ATF4 is not required for the virus to initiate immediate-early gene expression. Viral E1 and gB proteins were undetectable until 24 and 36 hpi, respectively, during the low-MOI infection (Fig. 7A). At these late time points when the second round of infection could initiate, E1 and gB protein levels were markedly reduced in ATF4-depleted cells, as was the IE1 protein. In addition, viral DNA synthesis was also reduced by 2- to 3-fold in ATF4-depleted cells relative to that in control cells (Fig. 7B and C). These defects could occur within a single round of infection or result from the failure to initiate the second round of infection at these time points.
Fig 7.

ATF4 is not required for MCMV to initiate its immediate-early and early gene expression during a low MOI. (A) Viral protein accumulation in ATF4-depleted cells at a low or high MOI. MEF10.1 cells expressing ATF4ΔRK or control vector were infected with MCMV at an MOI of 0.01 or 2, and the cell lysates were collected at the indicated times and analyzed for viral IE1 (immediate early), E1 (early), and gB (late) gene expression by immunoblotting. (B) Viral DNA synthesis in ATF4-depleted cells at a low or high MOI. Cells were infected with MCMV at an MOI of 0.01 or 2, intracellular DNA was extracted at the indicated times, and the amounts of viral DNA were measured by qPCR and normalized to that of actin. The normalized amount of viral transcript at 4 hpi in the vector cells was set at 1. (C) Reduction in viral early gene expression was secondary to viral DNA synthesis in ATF4-depeleted cells. The cells were infected with MCMV at an MOI of 0.01 in the presence or absence of PAA. Intracellular viral DNA and transcripts of viral early genes (E1) or late genes (gB, M99) at 20 hpi were measured by qPCR and RT-qPCR, respectively, and then normalized to that of actin. The normalized amount of viral DNA or transcript in ATF4ΔRK-expressing cells in the absence of PAA was set as 1. (D) Cells were infected with MCMV at the MOI of 0.01 in the presence or absence of PAA, and the accumulation of the viral IE1 and E1 proteins at 24 hpi was analyzed by immunoblotting.
To differentiate these two possibilities, we determined viral early gene expression at a low-MOI infection in the presence of PAA, which prevented viral DNA synthesis and consequently the second round of infection. We first quantified the accumulation of viral early gene transcripts by RT-qPCR analysis at 20 hpi. As anticipated, in the absence of PAA, viral DNA synthesis, early transcript accumulation, and late transcript accumulation were all reduced in ATF4-depleted cells relative to those in control cells (Fig. 7C). However, when viral DNA synthesis (Fig. 7C, left panel) and accumulation of viral late gB and M99 transcripts (Fig. 7C, right panel) were blocked by PAA, the E1 transcript accumulated to similar levels in ATF4-depleted cells and control cells (Fig. 7C, right panel). Consistently, at 24 hpi in the presence of PAA, the IE1 and E1 proteins in ATF4-depleted cells also accumulated to the levels similar to those in control cells (Fig. 7D). Collectively, our results suggest that ATF4 is not required for MCMV to initiate immediate-early and early gene expression prior to viral DNA synthesis and that the reduction in their expression at late times of infection is secondary to viral DNA synthesis or the subsequent round of infection.
ATF4 is required for efficient viral DNA synthesis and late gene expression during a low-multiplicity MCMV infection.
During low-MOI infection, the reduction of viral DNA synthesis and late gene expression in ATF4-depleted cells was evident as early as 20 hpi (Fig. 7C), raising the possibility that ATF4 promotes these late events of the MCMV replication cycle. To directly test this, we first determined whether ATF4 was required for efficient viral late gene expression within a single round of infection. We constructed a mutant MCMV BAC clone, pSMsubM86, which lacked viral gene M86 encoding the major capsid protein. We transfected pSMsubM86 into ATF4-depleted cells or control cells to recapitulate the condition of a low-MOI infection. We anticipated that pSMsubM86 transfection would reconstitute a single round of the viral replication cycle to the stage of viral late gene expression but no capsid assembly. Indeed, no viral plaques were produced even after several weeks upon pSMsubM86 transfection, whereas plaques were readily observed within 4 to 5 days upon a parallel transfection of a wild-type MCMV BAC, confirming that pSMsubM86 did not reconstitute infection beyond the first round (data not shown). At 48 h posttransfection, IE1 transcript was readily detected, accumulating to the same levels in ATF4-depleted cells and control cells (Fig. 8A). However, gB transcript accumulation in ATF4-depleted cells was markedly reduced relative to that in control cells. We interpret this result to indicate that ATF4 is required for efficient expression of viral late genes within a single round of infection.
Fig 8.

ATF4 is required for efficient viral DNA synthesis and late gene expression during a low multiplicity of infection. (A) Viral late gene expression in ATF4-depleted cells during reconstitution of M86-deficient MCMV virus from BAC transfection. MEF10.1 cells expressing ATF4ΔRK or control vector were transfected with the recombinant MCMV BAC clone lacking the gene of viral major capsid protein (M86). BAC-transfected cells were collected at 48 h posttransfection, and viral IE1 and gB transcripts were measured by RT-qPCR and normalized to that of actin. The normalized amount of viral transcript in ATF4ΔRK-expressing cells was set as 1. (B) Viral DNA synthesis in ATF4-depleted cells infected with M79-deficient MCMV virus. MEF10.1 cells expressing ATF4ΔRK or control vector were infected with M79-deficient MCMV virus at an MOI of 0.01. Intracellular viral DNA was measured by qPCR at 2 and 20 hpi and normalized to that of actin. The normalized amount of viral DNA in ATF4ΔRK-expressing cells at 2 hpi was set as 1.
Finally, we determined whether ATF4 also promoted efficient viral DNA synthesis within one round of infection. However, due to the low efficiency of BAC-based virus reconstitution, it was difficult to differentiate newly synthesized viral DNA from the high background of BAC DNA input following transfection (data not shown). Consequently, we utilized a mutant MCMV virus, SMinM79, in which M79 was abrogated. In a separate study, we found that M79 was essential for MCMV replication and that SMinM79 synthesized viral DNA at wild-type levels but failed to produce viral late gene products (Chapa and Yu, unpublished). We infected cells with SMinM79 at an MOI of 0.01 to determine the effect of ATF4 on viral DNA synthesis within a single round of infection. At 2 hpi, an equal amount of intracellular viral DNA was detected in ATF4-depleted cells and control cells, confirming the equal infectivity in both cells (Fig. 8B). At 20 hpi, however, viral DNA levels in ATF4-depleted cells were reduced by 2-fold compared to that in control cells, suggesting that ATF4 is required for efficient viral DNA synthesis.
Taken together, our results indicate that under a high-MOI condition, MCMV can tolerate the loss of ATF4 for its infection. However, under a low-MOI condition, loss of ATF4 has an inhibitory effect on viral DNA synthesis and markedly reduces viral late gene expression. Combined, these ATF4-mediated effects play a critical role to promote MCMV growth in an MOI-dependent manner.
DISCUSSION
This is the first study to examine the function and regulation of the UPR in MCMV infection. Previous works from our lab and others have established that HCMV modulates the UPR for the establishment of a successful infection (21, 42). These studies have also started to unravel the role of individual UPR components during HCMV virus replication (8, 42). However, the UPR is a complex signaling network, and a body of recent literature indicates that it may be involved in innate immunity in addition to its established role in cell metabolic regulation (12). Many questions about whether and how each component or function of the UPR contributes to CMV biology and pathogenesis remain outstanding. In the present study we established MCMV as a valuable model to explore the involvement of the UPR and its regulation in CMV biology. We have shown that MCMV modulates the UPR in a way similar to HCMV. Both viruses activate the PERK signaling pathway and, in particular, induce ATF4 accumulation (Fig. 1 and 2) (21, 42). Moreover, even though HCMV does not induce proteolytic activation of ATF6, it induces the expression of Bip, a target gene of ATF6, via the viral protein IE1 (9). Similarly, we have observed that Bip expression is markedly induced by MCMV to a level even greater than that by tunicamycin, a widely used ER stress inducer (Fig. 1). Furthermore, HCMV induces limited Xbp1 transcript splicing but Xbp1(s)-mediated expression of downstream genes is not observed (21, 42). Consistently, our preliminary study also suggests that induction of Xbp1(s) protein accumulation is minimal during HCMV infection (data not shown), which is likely due to the very limited nature of virus-induced Xbp1 splicing. Similarly, MCMV also induces only very limited Xbp1 splicing (data not shown) and minimal accumulation of the Xbp1(s) protein, if any at all (Fig. 1). We anticipate that Xbp1(s)-mediated downstream events are likely blocked as well during MCMV infection. Overall, the key features of virus-mediated modulation of the UPR are conserved in both MCMV and HCMV infection. Understanding how and why MCMV regulates this key cellular stress response will expedite our effort to determine the contribution of the UPR to CMV biology and pathogenesis.
The present study presents several new findings on the interplay between the UPR and CMV infection. Despite the induction of ATF4, MCMV prevented high-level expression of CHOP, an apoptotic factor downstream of ATF4 (Fig. 1 and 2). Our preliminary data also suggest that HCMV similarly prevents the high-level CHOP induction during its infection (data not shown). This comes as no surprise: it has been well documented that CMVs have evolved many mechanisms to block apoptosis during their infection (7). Intriguingly, overexpression of CHOP has been shown to suppress the lytic replication program of EBV (19). This raises the possibility that active inhibition of CHOP expression or activity may be a conserved means that herpesviruses use to aid their infection. Work is under way to identify the viral mechanism to attenuate CHOP expression and establish the functional consequence of CHOP activity on CMV replication. In addition, we have also found that MCMV actively blocks Xbp1(s) protein accumulation, even in the presence of exogenous ER stress (Fig. 1). It is tempting to speculate this as a possible mechanism underlying the previous observation that HCMV does not induce downstream events of Xbp1(s) during its infection (21). Further studies will be needed to uncover the mechanism by which CMVs inhibit Xbp1(s) production and dissect the biological importance of this regulation in CMV infection.
Importantly, we have shown that ATF4 is required for MCMV infection, particularly a low-MOI infection. The earliest defects within a single viral replication cycle in the absence of ATF4 are viral DNA synthesis and late gene expression. It is possible that a relatively modest, 3-fold decrease in viral DNA synthesis leads to a 5-fold reduction in viral late gene expression (Fig. 8). Alternatively, ATF4 may have a direct role in viral late gene expression in addition to promoting viral DNA synthesis. How do these seemingly nonlethal defects on viral DNA synthesis and late gene expression result in an almost complete loss of viral growth (Fig. 4 and 5)? It is possible that these single-round defects are amplified during multistep infection so that viral growth becomes severely attenuated. However, we cannot rule out the possibility that ATF4 is required for the advance of the MCMV replication cycle at additional steps after viral late gene expression, for example, capsid assembly, virion maturation, or even initiation of the second round of infection. Combined, these effects may markedly inhibit viral growth in a multistep growth analysis.
How does ATF4 promote viral DNA synthesis and late gene expression? ATF4 is a key transcriptional factor induced by the PERK pathway of the UPR. In response to stress, ATF4 upregulates transcription of the genes involved in amino acid transport and biosynthesis, and enhances production of the key intracellular reducing agent glutathione to maintain redox chemistry and proper protein folding. These activities help cells recover from stress, reinitiate protein translation, and maintain a healthy cellular environment and thus may contribute to efficient viral replication. In addition, as a transcription factor, ATF4 (14) may directly promote MCMV late gene expression. Indeed, ATF4 can interact with the human T-cell leukemia virus type 1 protein Tax to transactivate the viral promoter (13, 36), and it can also activate transcription of the viral oncogene LMP-1 during EBV latent infection (26). Alternatively, ATF4 may induce cellular profactors or repress antiviral factors to facilitate viral DNA synthesis or gene expression. Finally, cross talk between the UPR and innate antiviral immunity has been recently described; for example, ATF4 has been reported to inhibit IFN responses by negatively regulating IRF7 (27). IFN responses can inhibit virus replication at multiple steps, including viral DNA synthesis and viral gene expression (5, 38). It is tempting to entertain the possibility that MCMV may efficiently replicate at low MOIs, at least in part, by exerting ATF4-mediated control on host innate antiviral responses. At high MOIs, an increased input abundance of viral tegument proteins, virion proteins, or genome copies may activate redundant signaling pathways to compensate for the loss of ATF4 function, such as its possible antagonizing activity in innate antiviral response. The similar MOI-dependent phenotypes have been documented for recombinant HCMV virus lacking genes such as IE1, pp71, or UL69 (6, 17, 31), since the functions of these proteins are more relevant at a low MOI than at a high MOI. Work is under way to test the potential roles of ATF4 in CMV biology.
ACKNOWLEDGMENTS
We thank Herbert Virgin and the members of his laboratory for helpful discussions and invaluable advice; Martin Messerle (Hanover Medical School, Hanover, Germany), Wolfram Brune (Heinrich Pette Institute, Leibniz Institute for Experimental Virology, Hamburg, Germany), and Ulrich Koszinowski (Max von Pettenkofer Institute, Ludwig Maximilian University, Munich, Germany) for the MCMV BAC clone pSM3fr; Anthony Scalzo (University of Western Australia) for gB antibody; Stipan Jonjic (University of Rijeka, Rijeka, Croatia) for IE1 and E1 antibodies; Jawed Alam (Ochsner Clinic Foundation) for the ATF4ΔRK-expressing plasmid; the Children's Discovery Institute, the RNAi Consortium, and the Genome Institute at Washington University for ATF4- and PERK-targeting shRNAs; the High Speed Cell Sorter Core at the Siteman Cancer Center for excellent technical assistance; and members of the Yu lab for critical readings of the manuscript.
This study was supported by a Public Health Service grant (R01CA120768). D.Y. holds an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund.
Footnotes
Published ahead of print 11 April 2012
REFERENCES
- 1. Ambrose RL, Mackenzie JM. 2011. West Nile virus differentially modulates the unfolded protein response to facilitate replication and immune evasion. J. Virol. 85:2723–2732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Asselah T, et al. 2010. In vivo hepatic endoplasmic reticulum stress in patients with chronic hepatitis C. J. Pathol. 221:264–274 [DOI] [PubMed] [Google Scholar]
- 3. Baird GS, Zacharias DA, Tsien RY. 2000. Biochemistry, mutagenesis, and oligomerization of DsRed, a red fluorescent protein from coral. Proc. Natl. Acad. Sci. U. S. A. 97:11984–11989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bhende PM, Dickerson SJ, Sun X, Feng WH, Kenney SC. 2007. X-box-binding protein 1 activates lytic Epstein-Barr virus gene expression in combination with protein kinase D. J. Virol. 81:7363–7370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bonjardim CA, Ferreira PC, Kroon EG. 2009. Interferons: signaling, antiviral and viral evasion. Immunol. Lett. 122:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bresnahan WA, Shenk TE. 2000. UL82 virion protein activates expression of immediate-early viral genes in human cytomegalovirus-infected cells. Proc. Natl. Acad. Sci. U. S. A. 97:14506–14511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brune W. 2011. Inhibition of programmed cell death by cytomegaloviruses. Virus Res. 157:144–150 [DOI] [PubMed] [Google Scholar]
- 8. Buchkovich NJ, et al. 2008. Human cytomegalovirus specifically controls the levels of the endoplasmic reticulum chaperone BiP/GRP78, which is required for virion assembly. J. Virol. 82:31–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Buchkovich NJ, Yu Y, Pierciey FJ, Jr, Alwine JC. 2010. Human cytomegalovirus induces the endoplasmic reticulum chaperone BiP through increased transcription and activation of translation by using the BiP internal ribosome entry site. J. Virol. 84:11479–11486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carpenter JE, Jackson W, Benetti L, Grose C. 2011. Autophagosome formation during varicella-zoster virus infection following endoplasmic reticulum stress and the unfolded protein response. J. Virol. 85:9414–9424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chrispeels MJ, Higgins TJ, Craig S, Spencer D. 1982. Role of the endoplasmic reticulum in the synthesis of reserve proteins and the kinetics of their transport to protein bodies in developing pea cotyledons. J. Cell Biol. 93:5–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cnop M, Foufelle F, Velloso LA. 2012. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol. Med. 18:59–68 [DOI] [PubMed] [Google Scholar]
- 13. Gachon F, Devaux C, Mesnard JM. 2002. Activation of HTLV-1 transcription in the presence of Tax is independent of the acetylation of CREB-2 (ATF-4). Virology 299:271–278 [DOI] [PubMed] [Google Scholar]
- 14. Hai T, Hartman MG. 2001. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene 273:1–11 [DOI] [PubMed] [Google Scholar]
- 15. Harding HP, et al. 2003. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11:619–633 [DOI] [PubMed] [Google Scholar]
- 16. Harvey DM, Levine AJ. 1991. p53 alteration is a common event in the spontaneous immortalization of primary BALB/c murine embryo fibroblasts. Genes Dev. 5:2375–2385 [DOI] [PubMed] [Google Scholar]
- 17. Hayashi ML, Blankenship C, Shenk T. 2000. Human cytomegalovirus UL69 protein is required for efficient accumulation of infected cells in the G1 phase of the cell cycle. Proc. Natl. Acad. Sci. U. S. A. 97:2692–2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He CH, et al. 2001. Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J. Biol. Chem. 276:20858–20865 [DOI] [PubMed] [Google Scholar]
- 19. Huang J, et al. 2006. Contribution of C/EBP proteins to Epstein-Barr virus lytic gene expression and replication in epithelial cells. J. Virol. 80:1098–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Isler JA, Maguire TG, Alwine JC. 2005. Production of infectious human cytomegalovirus virions is inhibited by drugs that disrupt calcium homeostasis in the endoplasmic reticulum. J. Virol. 79:15388–15397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Isler JA, Skalet AH, Alwine JC. 2005. Human cytomegalovirus infection activates and regulates the unfolded protein response. J. Virol. 79:6890–6899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Joyce MA, et al. 2009. HCV induces oxidative and ER stress, and sensitizes infected cells to apoptosis in SCID/Alb-uPA mice. PLoS Pathog. 5:e1000291 doi:10.1371/journal.ppat.1000291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kinsella TM, Nolan GP. 1996. Episomal vectors rapidly and stably produce high-titer recombinant retrovirus. Hum. Gene Ther. 7:1405–1413 [DOI] [PubMed] [Google Scholar]
- 24. Klomporn P, Panyasrivanit M, Wikan N, Smith DR. 2011. Dengue infection of monocytic cells activates ER stress pathways, but apoptosis is induced through both extrinsic and intrinsic pathways. Virology 409:189–197 [DOI] [PubMed] [Google Scholar]
- 25. Lee DY, Lee J, Sugden B. 2009. The unfolded protein response and autophagy: herpesviruses rule! J. Virol. 83:1168–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee DY, Sugden B. 2008. The LMP1 oncogene of EBV activates PERK and the unfolded protein response to drive its own synthesis. Blood 111:2280–2289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liang Q, Deng H, Sun CW, Townes TM, Zhu F. 2011. Negative regulation of IRF7 activation by activating transcription factor 4 suggests a cross-regulation between the IFN responses and the cellular integrated stress responses. J. Immunol. 186:1001–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lindl KA, Akay C, Wang Y, White MG, Jordan-Sciutto KL. 2007. Expression of the endoplasmic reticulum stress response marker, BiP, in the central nervous system of HIV-positive individuals. Neuropathol. Appl. Neurobiol. 33:658–669 [DOI] [PubMed] [Google Scholar]
- 29. Maingat F, et al. 2011. Inflammation and epithelial cell injury in AIDS enteropathy: involvement of endoplasmic reticulum stress. FASEB J. 25:2211–2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Martinon F, Glimcher LH. 2011. Regulation of innate immunity by signaling pathways emerging from the endoplasmic reticulum. Curr. Opin. Immunol. 23:35–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mocarski ES, Kemble GW, Lyle JM, Greaves RF. 1996. A deletion mutant in the human cytomegalovirus gene encoding IE1(491aa) is replication defective due to a failure in autoregulation. Proc. Natl. Acad. Sci. U. S. A. 93:11321–11326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mulvey M, Arias C, Mohr I. 2007. Maintenance of endoplasmic reticulum (ER) homeostasis in herpes simplex virus type 1-infected cells through the association of a viral glycoprotein with PERK, a cellular ER stress sensor. J. Virol. 81:3377–3390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Novoa I, Zeng H, Harding HP, Ron D. 2001. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J. Cell Biol. 153:1011–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Qian Z, Xuan B, Hong TT, Yu D. 2008. The full-length protein encoded by human cytomegalovirus gene UL117 is required for the proper maturation of viral replication compartments. J. Virol. 82:3452–3465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Reddehase MJ, Simon CO, Seckert CK, Lemmermann N, Grzimek NK. 2008. Murine model of cytomegalovirus latency and reactivation. Curr. Top. Microbiol. Immunol. 325:315–331 [DOI] [PubMed] [Google Scholar]
- 36. Reddy TR, Tang H, Li X, Wong-Staal F. 1997. Functional interaction of the HTLV-1 transactivator Tax with activating transcription factor-4 (ATF4). Oncogene 14:2785–2792 [DOI] [PubMed] [Google Scholar]
- 37. Ron D, Walter P. 2007. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8:519–529 [DOI] [PubMed] [Google Scholar]
- 38. Saikia P, Fensterl V, Sen GC. 2010. The inhibitory action of P56 on select functions of E1 mediates interferon's effect on human papillomavirus DNA replication. J. Virol. 84:13036–13039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Scalzo AA, Corbett AJ, Rawlinson WD, Scott GM, Degli-Esposti MA. 2007. The interplay between host and viral factors in shaping the outcome of cytomegalovirus infection. Immunol. Cell Biol. 85:46–54 [DOI] [PubMed] [Google Scholar]
- 40. Tardif KD, Waris G, Siddiqui A. 2005. Hepatitis C virus, ER stress, and oxidative stress. Trends Microbiol. 13:159–163 [DOI] [PubMed] [Google Scholar]
- 41. Wagner M, Jonjic S, Koszinowski UH, Messerle M. 1999. Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J. Virol. 73:7056–7060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xuan B, Qian Z, Torigoi E, Yu D. 2009. Human cytomegalovirus protein pUL38 induces ATF4 expression, inhibits persistent JNK phosphorylation, and suppresses endoplasmic reticulum stress-induced cell death. J. Virol. 83:3463–3474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang K, Kaufman RJ. 2008. From endoplasmic-reticulum stress to the inflammatory response. Nature 454:455–462 [DOI] [PMC free article] [PubMed] [Google Scholar]