

Abstract
Epstein-Barr virus (EBV) infection of primary human B cells drives their indefinite proliferation into lymphoblastoid cell lines (LCLs). B cell immortalization depends on expression of viral latency genes, as well as the regulation of host genes. Given the important role of microRNAs (miRNAs) in regulating fundamental cellular processes, in this study, we assayed changes in host miRNA expression during primary B cell infection by EBV. We observed and validated dynamic changes in several miRNAs from early proliferation through immortalization; oncogenic miRNAs were induced, and tumor suppressor miRNAs were largely repressed. However, one miRNA described as a p53-targeted tumor suppressor, miR-34a, was strongly induced by EBV infection and expressed in many EBV and Kaposi's sarcoma-associated herpesvirus (KSHV)-infected lymphoma cell lines. EBV latent membrane protein 1 (LMP1) was sufficient to induce miR-34a requiring downstream NF-κB activation but independent of functional p53. Furthermore, overexpression of miR-34a was not toxic in several B lymphoma cell lines, and inhibition of miR-34a impaired the growth of EBV-transformed cells. This study identifies a progrowth role for a tumor-suppressive miRNA in oncogenic-virus-mediated transformation, highlighting the importance of studying miRNA function in different cellular contexts.
INTRODUCTION
Epstein-Barr virus (EBV) is a DNA tumor virus belonging to the human gammaherpesvirus family capable of establishing a latent infection in nearly 90% of the adult human population worldwide. EBV infection occurs mainly in human B lymphocytes and oropharyngeal epithelial cells and is associated with several human lymphoid- and epithelial-cell malignancies, including endemic African Burkitt's lymphoma (BL), Hodgkin's disease (HD), posttransplant lymphoproliferative diseases (PTLD), diffuse large B cell lymphoma (DLBCL), and nasopharyngeal carcinoma (NPC) (24).
In vitro, EBV infection of B cells results in proliferation and outgrowth of indefinitely proliferating lymphoblastoid cell lines (LCLs) that represent a viable model for the pathogenesis of EBV-associated malignancies. EBV-mediated growth transformation is characterized by the expression of a subset of viral gene products (latency III), including latent membrane protein 1 (LMP1) and LMP2A/B, as well as the nuclear proteins EBNA1, -2, -3A, -3B, and -3C and LP (24). These proteins coordinately regulate host signaling pathways to drive resting B cells to proliferate and ensure cell survival by inducing strong antiapoptotic signals. In addition to these viral proteins, LCLs and naturally occurring EBV+ DLBCLs also express several EBV-encoded viral regulatory RNAs, including the EBER RNAs, which are thought to act as inhibitors of host innate immune responses, as well as several virus-encoded microRNAs (miRNAs) (40).
miRNAs are small noncoding RNAs expressed by all multicellular eukaryotes that negatively regulate gene expression by targeting >30% of human mRNAs. These regulatory RNAs have been demonstrated to play key roles in a variety of processes, including development, the cell cycle, and immunity, and their dysfunction has been associated with several human pathologies, including cancer (12). A number of oncogenes, including c-Myc, directly alter miRNA expression, which can contribute to tumorigenesis (30, 34). EBV infection of B cells also deregulates cellular miRNA expression (1, 14, 23, 27). Among EBV-upregulated miRNAs are miR-155 and miR-21, both of which can function as progrowth onco-miRs, i.e., miRNAs that predispose cells to transformation (21), and miR-146a, which acts as an inhibitor of interferon response gene induction (2). Another miRNA induced by EBV, miR-34a, has been reported to be activated by the host transcription factor p53 and to participate in growth arrest and apoptosis downstream of DNA damage (3, 20, 39, 42, 43, 45).
The changes in miRNA expression observed between EBV-infected and uninfected cell lines (1) prompted us to ask whether and to what extent these changes occurred during primary B cell immortalization. We analyzed miRNA expression by microarray analysis of purified human peripheral blood B cells, EBV-infected proliferating B cells early after infection, and monoclonal LCLs. Our analysis confirmed previously reported changes and identified novel EBV-regulated miRNAs. Finally, given the role of the strongly EBV-induced miR-34a as a tumor suppressor in other cell types, we studied its regulation and function in the context of EBV-transformed B cell growth.
MATERIALS AND METHODS
DNA constructs.
An miR-34a indicator was constructed by inserting 4 perfect target sequences in the lentiviral vector pNL-SIN-CMV-FLuc. Oligonucleotides carrying two perfect miR-34a target sequences, pNL-miR34-sense (5′ CGGGAACAACCAGCTAAGACACTGCCAGTTTTGAACAACCAGCTAAGACACTGCCAAATCGATGATCT 3′) and pNL-miR34-antisense (5′ CTAGAGATCATCGATTTGGCAGTGTCTTAGCTGGTTGTTCAAAACTGGCAGTGTCTTAGCTGGTTGTTCC 3′) (the target sequences are underlined), were annealed and inserted between the ClaI and XbaI sites of the above-described backbone. The ClaI site was destroyed by ligation of the insert, and a second ClaI site upstream of the XbaI site was used to insert two more target sites. The empty vector and pNL-SIN-CMV-RLuc were used as internal controls (15).
The lentiviral miR-34a sponge, containing 6 imperfect matches for miR-34a, was cloned in the vector pLCE as previously described (8, 16) using the following oligonucleotides: Spng miR34 fw primer (5′ CTAGCAACAACCAGGATAGACACTGCCAGTTTTGAACAACCAGGATAGACACTGCCAGTTTTGAACAACCAGGATAGACACTGCCATCTAGATTG 3′ [targets are underlined; mismatch is in boldface]) and Spng miR34 rev primer (5′ AATTCAAATCTAGATGGCAGTGTCTATCCTGGTTGTTCAAAACTGGCAGTGTCTATCCTGGTTGTTCAAAACTGGCAGTGTCTATCCTGGTTGTTG 3′). The murine stem cell virus (MSCV)-based sponge was cloned by removing the DNA fragment containing the miR-34a binding sites from the pLCE-based sponge with NheI (then blunt)/EcoRI restriction enzymes and ligating it into the HpaI (then blunt)/EcoRI-digested pMSCV-Puro. The expression plasmid for miR-34a (pcDNA-pm34-EST) was kindly provided by M. Oren (The Weizmann Institute of Science, Rehovot, Israel) (39).
pSG5-FLMP1 wild type (WT), P204A/Q206A (ΔCTAR1) and YYD384-386ID (ΔCTAR2) mutants, and a double carboxy-terminus-activating region (CTAR) mutant (DM) were previously described (6, 22), and the retroviral constructs MSCV-internal ribosome entry site (IRES)-green fluorescent protein (GFP) and MSCV-LMP1-IRES-GFP were kind gifts of E. Cahir-McFarland, Harvard Medical School, Boston, MA.
Cell lines and culture conditions.
Human peripheral blood mononuclear cells (PBMCs) were obtained by Ficoll purification (Histopaque-1077 column; Sigma) of buffy coats from healthy donors (North Carolina Red Cross) and kept in RPMI 1640 medium (Invitrogen) supplemented with 15% fetal bovine serum (100-500; Gemini Bio Product), 100 U/ml penicillin, and 2 mM l-glutamine (G6784; Invitrogen). B cells were separated using the Human B Lymphocyte Enrichment Set-DM (Imag; BD) as recommended by the manufacturer. PBMCs were infected with limiting amounts of B95-8 virus for 1 h at 37°C (0.4 to 12 μl EBV B95-8 Z-HT supernatant/1 × 106 PBMCs) in the presence of cyclosporine (0.5 μg/ml). The cells were washed once in phosphate-buffered saline (PBS) and then resuspended in fresh medium. For each condition of infection, 105 cells were plated in a 96-well plate (total, 10 wells) to grow out as LCLs. Microarray samples were generated from monoclonal LCLs produced from the smallest amount of virus used. For the purification of EBV-positive and proliferating B cells, 1.5 × 108 PBMCs were stained with 4 μM 6-carboxyfluorescein succinimidyl ester (CFSE) for 3 min and washed three times in washing buffer (PBS plus 5% fetal bovine serum). Stained PBMCs (106/ml) were infected with 25 μl/1 × 106 PBMCs of B95-8 virus as described above. The cells were washed once in PBS and then resuspended in fresh medium. At 6 days postinfection, CFSE-labeled PBMCs were washed in washing buffer and stained with allophycocyanin (APC)-labeled α-CD19 (555415; 2 μl/3 × 105 PBMCs; BD Biosciences) for 30 min on ice. After 3 washes in washing buffer, CD19+/CFSElow cells were sorted on a FACSVantage cell sorter flow cytometer.
36-6, EF3D, and RC-1 LCLs were established in our laboratory, as well as the three LCLs used for the miRNA microarray. GM15807 cells were purchased from the Coriell Cell Repository, while IB-4 and E2-HT (48) cells were provided by Elliot Kieff (Harvard Medical School, Boston, MA). All LCLs were kept in RPMI 1640 medium supplemented with 10% fetal bovine serum, while E2-HT cells were also cultured with 200 nM 4-hydroxytamoxifen (4HT). The same conditions were used for the EBV− germinal-center-derived B cell lymphoma line BJAB and EBV− BL41 (obtained from George Mosialos, Aristotle University, Thessaloniki, Greece), BL2 and BL40 (provided by Geoffrey Wahl), and EBV+ Akata (provided by Elliott Kieff, Harvard Medical School, Boston, MA) Burkitt's lymphoma cell lines. Kaposi's sarcoma-associated herpesvirus-positive (KSHV+)/EBV− primary effusion lymphoma (PEL) cell lines VG-1, BC-3, BCLM, and BCBL1 and the KSHV+/EBV+ PEL cell line BC-1 were provided by Dirk Dittmer (University of North Carolina [UNC], Chapel Hill, NC) or Bryan Cullen (Duke University, Durham, NC) with permission from the original authors and kept in RPMI 1640 medium supplemented with 15% fetal bovine serum, 10 mM HEPES, 1 mM sodium pyruvate, and 0.05 mM β-mercaptoethanol (Sigma). The EBV-positive cell line derived from an AIDS immunoblastic lymphoma, IBL-1, was kindly provided by Ethel Cesarman (Weill Cornell Medical College, New York, NY) and kept in RPMI 1640 supplemented with 20% fetal bovine serum. HCT-116 cells were obtained from ATCC and grown in McCoy's 5A medium supplemented with 10% fetal bovine serum. All cells were maintained in the presence of 100 U/ml penicillin and 2 mM l-glutamine.
Clonogenicity assays were performed in technical triplicate by plating sorted GFPhi HCT-116 cells transduced with control or sponge vectors into 6-well plates. Seven days later, when individual colonies were visible by eye, the cells were washed with 1× PBS and then fixed and stained with 2 ml of 0.1% crystal violet and 20% methanol for 30 min at room temperature. Following a 20% methanol wash, the cell colonies were counted by eye.
Compounds.
IKKβ inhibitors IV and VIII were purchased from Calbiochem as a 10 mM stock solution.
Generation of viral transductants, cell sorting, and DNA transfection.
All retroviral and lentiviral vectors were produced by cotransfection into HEK 293T cells using polyethylenimine (PEI). pLCE-based vectors were produced by cotransfection with pMDLgpRRE, pRSV-Rev, and pVSV-G (7). MSCV-IRES-GFP (38) and MSCV-LMP1-IRES-GFP (a kind gift of E. Cahir-McFarland, Harvard Medical School, Boston, MA) vectors were cotransfected with pMSCV-Gag/Pol and pHIT-G. Forty-eight hours after transfection, the supernatants were collected, filtered (0.45 μm), concentrated using Amicon 100-kDa cutoff spin columns, titrated for GFP levels after BJAB cell infection, and used to infect logarithmically growing cells at a multiplicity of infection (MOI) of 2.
LCLs and HCT-116 cells transduced with lentiviral miR-34 sponge expression or control constructs were sorted for the same mean fluorescence intensities (MFI) of GFP on a FACSVantage cell sorter (top 10 to 25% GFP+, depending on the experiment). After sorting, the cells were maintained in RPMI 1640 or McCoy's 5A medium supplemented with 10% fetal bovine serum in the presence of 100 U/ml penicillin and 2 mM l-glutamine and expanded. BJAB cells were transduced with retroviral LMP-1 expression or control constructs. After 24 h, the cells were treated with IKKβ inhibitors IV and VIII (2 μM) and kept for 4 days in culture in the presence of the inhibitors or dimethyl sulfoxide (DMSO). GFP+ cells were sorted on a FACSVantage cell sorter.
The ectopic expression of miR-34a was obtained by introducing PvuI-linearized pcDNA3-pm34-EST or the empty vector in EF3D LCL, BJAB, or Akata cells by electroporation and in HCT-116 cells by PEI-mediated transfection. After 48 h, cells were selected with G418 (0.3 mg/ml). For the overexpression of LMP-1 WT, the P204A/Q206A (ΔCTAR1) and YYD384–386ID (ΔCTAR2) mutants, and the double CTAR mutant (DM), the pSG5-based vectors were linearized with AgeI and electroporated in BJAB cells. After 48 h, cells were selected with G418 (0.6 mg/ml).
Growth experiments.
LCLs and HCT-116 cells were seeded at 2 × 105 cells/ml, and viable cells were counted by trypan blue exclusion. When the cells reached confluence, they were split into the original 2 × 105 cells/ml.
miRNA activity assays.
Sponge-expressing and miR-34a-overexpressing LCLs and HCT-116 cells were assayed for miRNA activity using a dual-luciferase reporter system, as previously described (17), except that the pL-CMV-GL3 (firefly luciferase [Fluc]) vector and pL-CMV-Rluc (Renilla luciferase) vectors were used as described by Linnstaedt et al. (26). pL-CMV-GL3-34a has four perfect target sites for miR-34a in the 3′ untranslated region (UTR) of Fluc, so that Fluc expression will decrease when miR-34a binds. Specifically, viral particles containing Rluc or Fluc and their derivatives were produced in 293T cells (using packaging constructs identical to those for pLCE-s34a) and then used to transduce LCLs or HCT-116 cells. Two days posttransduction, cells were harvested and lysed, and Fluc and Rluc activities were measured according to the manufacturer's protocol (dual-luciferase assay; Promega). Values are expressed as a ratio of Fluc to Rluc activity and normalized to an miR-34a-negative cell line.
Western blotting.
Cells were washed with ice-cold PBS, incubated with lysis buffer (20 mM Tris, pH 7.5, 100 mM NaCl, 1% Triton X-100, 10% glycerol, 1 mM EDTA, 1 mM dithiothreitol [DTT], 0.1 mM sodium orthovanadate, 20 μM NaF, 10 μM pyrophosphate, and Complete EDTA-free protease inhibitors [Roche]) for 30 min on ice, and centrifuged at 21,000 × g for 30 min at 4°C. The protein concentrations in the extracts were determined by Bradford assay (Bio-Rad), and the presence of LMP-1 was confirmed by Western blotting using the S12 antibody (a kind gift from E. Kieff), horseradish peroxidase-conjugated secondary antibodies (Pierce), and the ECL detection system (Amersham). Anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (Santa-Cruz) was used as a control. Expression of miR-34a targets was performed as described above in HCT-116 or EF3D cells stably expressing pcDNA3 or pcDNA3-miR34a. The antibodies used were cyclin D1 and D2 (BD; 554203), cyclin E2 (Cell Signaling; 4132), cdk4 (Santa Cruz; H-22), cdk6 (Santa Cruz; C-21), and Met (Cell Signaling; 4560). Quantitation was performed using Gene Tools software (Syngene) following scanning using the chemiluminescence settings in the G-Box gel documentation system.
RNA extraction and miRNA profiling with the use of microarrays.
Total RNA was prepared using a mir-Vana miRNA isolation kit (Ambion) according to the manufacturer's instructions. miRNA expression profiling of human B cells, EBV-infected proliferating B cells, and monoclonal LCLs from 3 different donors were conducted with the use of up to 2 μg total RNA per sample, as previously described (47). Briefly, RNAs were labeled with Cy3 or Cy5 fluorescent dye, using the miRNA/LNA labeling kit (Exiqon, Vedbæk, Denmark). The fluorescently labeled samples were hybridized to an miRNA microarray (version 10.0; Exiqon) in a nitrogen atmosphere. The microarray slides were scanned with a GenePix 4100 scanner (Axon Instruments, Union City, CA). The quantified signals were normalized using the global Lowess algorithm, using Genespring (Agilent Technologies, Santa Clara, CA) software. The intensity values for multiple spots were averaged, and the normalized values were log2 transformed. Using significance analysis of microarrays (SAM) (44), differentially expressed microRNAs (false discovery rate, <5%) that were differentially expressed among the 3 groups (resting B cells, proliferating B cells, and LCLs) were identified.
Quantitative real-time (qRT) PCR.
miR-34a, miR-15b, and miR-155 were reverse transcribed using Applied Biosystems individual stem-loop primers designed to detect only mature miRNA and measured by TaqMan real-time PCR normalized to the small nucleolar RNA RNU48. Ten nanograms of RNA per reaction was used for all the cell lines except BJAB, for which 50 ng was used. To assess TRAF1 mRNA expression, 500 ng total RNA was reverse transcribed with the ABI High Capacity cDNA Reverse Transcription kit (Applied Biosystems). mRNA expression was measured using an exon-spanning TaqMan probe against TRAF1 (Applied Biosystems; Hs00194639) and normalized to RNU48 expression. Fifty nanograms of RNA was used for each reaction.
For validation of miRNA microarray data, we used a SYBR green-based real-time PCR assay. We utilized Escherichia coli poly(A) polymerase (Epicentre) to add a poly(A) tail at the end of every RNA molecule in the total RNA pool that lacked a poly(A) tail. Before oligo(dT) annealing, we introduced a universal tag at the 3′ ends of cDNAs synthesized by reverse transcriptase Superscript III (Invitrogen). Using this tag, we performed qPCR with an miRNA-specific forward primer and reverse universal primer mixture (Quanta Biosystems).
Microarray data accession number.
miRNA microarray data are available at the Gene Expression Omnibus (GEO) website under accession number GSE36926.
RESULTS
Epstein-Barr virus infection of primary B cells leads to dynamic changes in cellular miRNA expression levels.
To gain insight into the role of miRNAs in EBV transformation, we profiled the expression of ∼800 cellular miRNAs from (i) uninfected, purified CD19+ B cells; (ii) EBV-infected, proliferating B cells sorted at 6 days postinfection; and (iii) monoclonal LCLs derived from three healthy donors (Fig. 1a). Analysis of cellular miRNA expression across these samples revealed a pattern of changes that defined each state (Fig. 1b). EBV infection of resting B cells induced the expression of 42 cellular miRNAs and repressed the expression of 60 miRNAs (see Table S1 in the supplemental material). Of these, 22 miRNAs were induced and 39 were repressed from resting B cells to EBV-infected, early-proliferating cells. Furthermore, a set of 17 cellular miRNAs were specifically regulated from early proliferation through long-term LCL outgrowth.
Fig 1.

EBV regulates cellular miRNA expression. (a) Schematic of samples used for miRNA microarray analysis. (Left) CD19+ B cells were purified by negative selection from peripheral blood mononuclear cells (North Carolina Red Cross). The dot plots show CD19-phycoerythrin (PE) staining on the x axis and nonspecific fluorescein isothiocyanate (FITC) channel on the y axis from PBMCs or negatively isolated cells. Purity was consistently >95% CD19+ cells. (Middle) Dot plots of CD19-PE (y axis) versus CFSE (x axis) indicate B cells proliferating after EBV infection. At 3 days postinfection, few cells are dividing (i.e., CD19+/CFSElow), while by 6 days postinfection, proliferating B cells are readily visible. The red circle indicates the population of cells that was sorted for each donor. (Right) Schematic diagram of a plate in which EBV infection of PBMCs leads to monoclonal LCL outgrowth. The amount of virus plated was diluted from top to bottom through a plate, and outgrowth, indicated by the green wells, was monitored at 3 to 4 weeks. Cells growing out at the lowest virus dilutions were grown until cell lines were established (red box). LCLs were derived in this manner for each of the three healthy donors analyzed for miRNA expression. (b) Expression profiles of resting CD19+ B cells; EBV-infected early-proliferating, i.e., CD19+/CFSElow, cells (Prolif); and monoclonal LCLs. Differentially expressed miRNAs that define each group are depicted over 4-fold levels in a color scale. (c) miRNAs that are upregulated by EBV are shown in a color scale. (d) MiRNAs that are downregulated by EBV infection are shown in a color scale. (e) MiRNAs selectively upregulated or downregulated in early-proliferating EBV-infected cells are shown in a color scale. The asterisks indicate miRNA-star strands (putative unincorporated miRNA strands) detected on the array.
EBV infection induced miRNAs involved in B cell activation and oncogenesis, including miR-155, -21, and -146b, supporting previous findings (Fig. 1c; see Table S1 in the supplemental material) (1). However, the most robustly induced miRNAs included several newly identified EBV-regulated genes, e.g., miR-7, -193b, -301a, and -302c (see Table S1 in the supplemental material). These miRNAs are likely associated with proliferation, as they were upregulated early after infection during the transition from resting to proliferating B cells. An additional set of EBV-induced miRNAs, including miR-138, -146a, and -130a, were delayed in their kinetics, and upregulation was observed only during the transition from early proliferation to LCLs (see Table S1 in the supplemental material). Detection of EBV-derived miRNAs served as an internal validation of our method (Fig. 1c; see Table S1 in the supplemental material).
EBV infection also led to the repression of a large number of miRNAs during B cell outgrowth. Early repressed miRNAs included miR-223, -150, and -154 and let-7b (Fig. 1d), while those miRNAs whose expression was not changed in early proliferation but was repressed through LCL outgrowth included miR-147, -151, and -195 (see Table S1 in the supplemental material). One previous study suggested that EBV infection leads to global and potent miRNA downregulation (14). While we observed many similar EBV-repressed miRNAs, including miR-143, -145, and -29c and let-7b, the extent and breadth of downregulation were not as severe in our system (see Table S1 in the supplemental material).
Expression of another group of miRNAs was unique to the period of early proliferation relative to resting B cells and LCLs. This set contains miRNAs regulated by c-Myc, the major oncoprotein driving Burkitt's lymphoma cell proliferation (34, 36). Among them, we found that miR-17, miR-18a, and miR-106b were all transiently induced in early proliferating cells and then attenuated in LCLs (Fig. 1e; see Table S1 in the supplemental material). Similarly, the Myc-repressed miR-23a (11) was repressed in early proliferating cells and then increased during LCL outgrowth (see Table S1 in the supplemental material). These observations are consistent with our recent findings demonstrating that a c-Myc-driven transcriptional program is robustly induced early after infection and is attenuated through long-term LCL outgrowth (32).
Contributions of EBV and c-Myc to miRNA expression in B cells.
We next validated these dynamic miRNA expression changes during EBV transformation of primary B cells using qRT-PCR of mature miRNA species. The expression levels of EBV-induced, EBV-repressed, and proliferation-defining miRNAs in uninfected B cells, EBV-infected early-proliferating B cells, and monoclonal LCLs were analyzed. Furthermore, a previous study compared latently EBV-infected cells to EBV-infected or uninfected Burkitt's lymphoma cells (1). Given the profound effects of c-Myc on BL gene expression, we were interested in comparing the EBV-regulated changes in miRNA expression in primary B cells and latency III-expressing tumors to miRNA expression in BL cell lines. Therefore, we also analyzed miRNA expression in the EBV-infected latency III-expressing AIDS-DLBCL line IBL-1 and two Burkitt's lymphoma cell lines: Akata, an EBV-infected latency I-expressing cell line, and BL41, a non-EBV-infected BL cell line.
We first confirmed that miR-146a, miR-21, and miR-155 were induced by EBV infection of primary B cells (1, 2, 28, 29, 37, 46). In our microarray analysis, miR-146a, miR-21, and miR-155 were induced 5.9-, 3.4-, and 6.0-fold, respectively (see Table S1 in the supplemental material). By qRT-PCR, these miRNAs increased 9.8-, 1.8-, and 7.1-fold (Fig. 2a). Consistently, we found that these miRNAs were also expressed in the latency III DLBCL line IBL-1, while they were poorly or not expressed in the BL cell lines Akata and BL41 (Fig. 2a). In addition, EBV potently downregulated the expression of miR-150 and miR-223, which were also not expressed in IBL-1, Akata, and BL-41 cells (Fig. 2b). miR-15a was repressed upon primary B cell infection, as previously reported (1), and IBL-1 expressed lower levels of this miRNA than Akata and BL41 cells.
Fig 2.

Validation of EBV- and c-Myc-regulated miRNAs. miRNAs were analyzed by qRT-PCR in two healthy donors, EBV-positive IBL-1 AIDS DLBCLs, Burkitt's lymphoma-derived EBV-positive Akata cells, and EBV-negative BL41 cells. The average expression of each miRNA plus the standard error of the mean (SEM) is shown for EBV-induced (a), EBV-repressed (b), and Prolif-defining (c) miRNAs. Each miRNA is indicated above the graph. Expression values were normalized to RNU48 or U6 levels and are shown relative to the level of one of the healthy B cell donors.
Finally, we validated the expression of miRNAs whose levels changed dynamically during LCL outgrowth. Myc-induced miR-18a was upregulated by EBV in early proliferating cells and then attenuated in LCLs (Fig. 2c). The levels of this and other Myc-induced miRNAs were considerably higher in the BL cell lines Akata and BL41 than in LCLs and IBL-1 (Fig. 2c and data not shown). Conversely, miR-23a expression was reduced upon EBV infection and then modestly increased through LCL outgrowth (Fig. 2c). However, levels in Akata and BL41 cells were significantly lower than in LCLs or IBL-1.
EBV infection upregulates miR-34a.
The characterization of miRNA expression in numerous tumor tissues and cancer cell lines has led to the identification of putative oncogenic and tumor suppressor miRNAs, or onco-miRs (12). During the progression of EBV-infected B cells into lymphoblastoid cell lines, several well-characterized progrowth onco-miRs were upregulated, including miR-155 and miR-21 (Fig. 1c and 2a). Conversely, several tumor suppressor miRNAs were downregulated, including miR-29, miR-15, and let-7 family members (Fig. 1d and 2b). However, one prominent tumor suppressor miRNA, miR-34a, was induced upon EBV infection of primary B cells (Fig. 3a) (1).
Fig 3.

miR-34a is upregulated during EBV infection of primary B cells and is expressed at elevated levels in EBV- and KSHV-infected lymphoma cell lines. (a) Quantitative RT-PCR of mature miR-34a expression levels from resting B cells through EBV infection and LCL outgrowth. MiR-34a expression is normalized to RNU48 levels under each condition. The data are the average values of experiments with three independent healthy donors. The error bars represent standard errors of the mean. (b) qRT-PCR expression of mature miR-34a levels in resting B cells and B lymphoma cell lines. The plus signs under each cell line indicate whether they are BL derived, EBV infected, KSHV infected, or HIV/AIDS associated. The +(I) under Akata indicates EBV positive but latency I expressing, while all other EBV-positive cells are latency III expressing. The miR-34a expression values plotted are normalized to RNU48 levels in each cell line and expressed relative to CD19+ B cell levels. The y axis is in logarithmic scale and split between 1 and 5, while the lower portion is more compact than the upper portion to more easily display the differences between miR-34a-expressing cell lines.
To ascertain how broadly EBV latent infection was associated with increased miR-34a, we measured miR-34a expression in several EBV-transformed B cell lines, as well as EBV-infected AIDS lymphomas that express the EBV latency III gene expression program. Furthermore, since EBV and the gammaherpesvirus KSHV have commonalities in constitutive activation of signaling pathways in B lymphomas, such as NF-κB (5), we also assayed KSHV-infected lymphoma cell lines. In all EBV- and KSHV-infected AIDS lymphoma and primary infected cell lines tested, miR-34a was highly expressed (Fig. 3b). In comparison, Burkitt's lymphoma cells, which depend on the c-Myc oncoprotein for proliferation and largely repress NF-κB (10), did not express high levels of miR-34a (Fig. 3b). These data highlight the potential importance of miR-34a in the progression of gammaherpesvirus-associated tumors.
EBV LMP1 induces miR-34a expression in an NF-κB-dependent manner.
Since EBV infection increased the steady-state level of mature miR-34a, we were interested in defining the viral oncoprotein responsible for this regulation. We queried the two major EBV transcriptional effectors, EBNA2 and LMP1. EBNA2 failed to induce mature miR-34a in BJAB cells or in LCLs using a regulatable EBNA2-HT allele (data not shown). However, expression of LMP1 was sufficient to increase miR-34a levels ∼10-fold upon transient expression in BJAB cells (Fig. 4a). Two known targets of LMP1, miR-155 and TRAF1, were also induced in these cells, as expected (Fig. 4a) (13).
Fig 4.

Latent membrane protein 1 induces miR-34a through IKKβ-dependent canonical NF-κB activation. (a) BJAB cells transiently transduced with GFP (BJAB) or LMP1-IRES-GFP (BJAB-LMP1) retroviral constructs were sorted 48 h postinfection and subjected to qRT-PCR for miR-34a, miR-155, or TRAF1 as indicated. The data are plotted as expression levels normalized to RNU48 in each cell line and relative to BJAB cells. Relative expression averages from three independent experiments plus SEM are shown. The inset is a representative Western blot of LMP1 (S12) and GAPDH from one of the experiments. (b) qRT-PCR of mature miR-34a levels normalized to RNU48 in BJAB cells (−) or BJAB cells expressing LMP1 in the absence or presence of two distinct IKK-2 (or IKKβ) inhibitors (IV and VIII). Averages and standard deviations from two experiments are shown. (c) qRT-PCR of mature miR-34a normalized to RNU48 and relative to BJAB levels for control (−), wild-type LMP1 (WT), P204A/Q206A (ΔCTAR1), YYD384–386ID (ΔCTAR2), and double CTAR mutant (DM) stable LMP1 transductants. Average expression plus SEM from three independent experiments is shown. (d) Relative levels of mature miR-34a and TRAF1 mRNA for WT and LMP2A knockout (KO) LCLs. The error bars indicate SEM.
LMP1 induction of miR-34a required IKKβ-dependent canonical NF-κB activation, since pharmacological inhibition of IKKβ precluded miR-34a upregulation (Fig. 4b). Furthermore, stable LMP1 expression modestly induced miR-34a, and this activity required carboxy-terminus-activating region 2 (CTAR2), consistent with the importance of canonical NF-κB activation (22) (Fig. 4c). Since p53 is mutated in BJAB cells (9), we concluded that LMP1-mediated NF-κB-dependent miR-34a upregulation did not require p53. This is consistent with recent evidence indicating p53-independent regulation of miR-34a (4, 31).
Further corroborating a role for NF-κB in miR-34a expression, LCLs lacking EBV LMP2A consistently expressed 2-fold less mature miR-34a than WT LCLs, which was accompanied by a 2-fold reduction in the NF-κB target gene TRAF1 (Fig. 4d). These data are consistent with previous studies implying a role for LMP2A in LMP1-mediated NF-κB activation (18) and our findings that EBV-induced miR-34a expression was NF-κB dependent.
Overexpression of miR-34a does not impact LCL or B lymphoma cell growth.
In a number of studies, miR-34a was shown to function as a potent tumor suppressor (3, 20, 39, 42, 43, 45). However, EBV infection of primary B cells increased miR-34a expression, and many AIDS lymphoma cell lines expressed elevated levels of miR-34a (Fig. 3). To determine whether miR-34a plays a growth-suppressive role in EBV-transformed cells, we overexpressed the primary miR-34a transcript in LCLs. As controls, we also overexpressed miR-34a in EBV-infected latency I Akata cells and in the EBV-negative BJAB cell line, both of which express very low levels of miR-34a. Stable clones of these cell lines expressed levels of mature miR-34a comparable to those of LCLs (Fig. 5a). While miR-34a modestly, though not significantly, increased the growth of LCLs (Fig. 5b), no effect on the growth or survival of Akata or BJAB cells was observed (Fig. 5c and d). In contrast and as previously described, miR-34a overexpression in the colon carcinoma cell line HCT-116 led to profound and rapid growth arrest (Fig. 5e). These results highlight an intrinsic difference in the functional consequences of miR-34a expression in different cell types.
Fig 5.

Overexpression of miR-34a does not alter B lymphoma cell growth. (a) qRT-PCR of mature miR-34a expression levels normalized to RNU48 in control plasmid (CTRL) or miR-34a stably transfected LCL EF3D, Akata, BJAB, and HCT-116 cells. Data from one representative transfection are shown. (b) Growth curves of EF3D cells stably transfected with pCDNA3 plasmid (CTRL) or an miR-34a expression plasmid (miR-34a). Data from three independent transfections are plotted as average numbers of viable cells ± SEM/ml as measured by trypan blue exclusion. (c) Growth curves as in panel b for Akata cells. (d) Growth curves as in panel b for BJAB cells. (d) Growth curves as in panel b for HCT-116 cells.
Growth control miR-34a targets are not affected in EBV-transformed cells.
Several growth-promoting miR-34a targets have been identified, including cyclin D1 and cyclin E2, D-type cyclin-dependent kinases cdk4 and cdk6, and the growth factor receptor c-Met (20, 39). Given the lack of phenotype upon miR-34a overexpression in EBV-transformed B cells, we sought to assess the sensitivity of these targets in LCLs relative to HCT-116 cells to decipher the role of miR-34a in the two cell types. It was immediately apparent that a significant difference between the cell lines is the steady-state mRNA levels of miR-34a targets (Fig. 6a). LCLs expressed high levels of cyclin D2 mRNA, but not the miR-34a target cyclin D1, and only minimally expressed cyclin E2. In contrast, HCT-116 cells expressed higher levels of cyclin D1 and E2 than LCLs. Finally, c-Met was expressed in HCT-116 cells, but not in LCLs.
Fig 6.
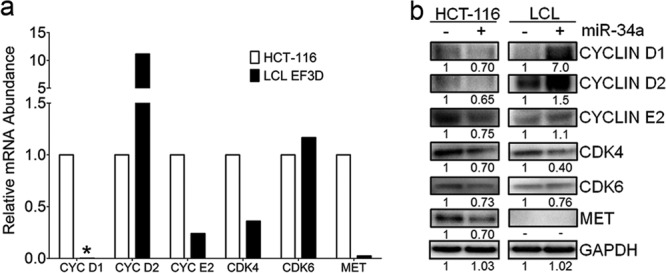
Canonical miR-34a growth control targets are poorly expressed in LCLs relative to HCT-116 cells. (a) Relative mRNA levels as determined by qRT-PCR of cyclin D1, D2, and E2; cdk4; cdk6; and c-Met in LCLs and HCT-116 cells. The asterisk indicates a value below the threshold for detection. (b) Western analysis of miR-34a targets in HCT-116 cells and EF3D LCLs expressing pcDNA3 vector (−) or pCDNA3-miR-34a (+). Quantitation of protein expression is noted below each band.
Overexpression of miR-34a reduced the protein levels of most canonical targets in HCT-116 cells, but not in LCLs. HCT-116 cells overexpressing miR-34a displayed reduced levels of cyclin D1, cyclin E2, cdk4, cdk6, and c-Met proteins relative to vector control-expressing cells (Fig. 6b). In LCLs, cdk4 was minimally reduced by miR-34a overexpression, while cdk6 and cyclin E2 were not affected. Unexpectedly, but consistently, cyclin D1 and D2 were both increased upon miR-34a overexpression in LCLs, possibly due to the modest increase in growth of these cells (Fig. 5b). Finally, c-Met protein was not detected in LCLs. Therefore, canonical miR-34a growth control targets were less affected or poorly expressed in LCLs relative to HCT-116 cells, providing a rationale for the distinct growth phenotypes upon miR-34a overexpression.
miR-34a is important for EBV-transformed cell growth.
Given the increased expression of miR-34a in LCLs and EBV-positive DLBCLs relative to primary B cells, we assessed whether this miRNA was important for maintenance of the EBV-transformed cell state using a “sponge” construct targeting miR-34a. Sponge constructs encode an artificial mRNA target containing multiple imperfect miRNA binding sites in the 3′ UTR of GFP (Fig. 7a) (8). LCLs were transduced with a GFP control or an miR-34a sponge (s34a), and the top 20% of GFP-positive cells were sorted. By luciferase indicator assay and qRT-PCR, we assessed that s34a-expressing cells had ∼75% reduced miR-34a activity and ∼80% reduced steady-state mature miR-34a levels, respectively (Fig. 7b and c) (8). These cells were not compromised for the expression or activity of other cellular miRNAs, indicating specificity of the miR-34a sponge (Fig. 7c and data not shown).
Fig 7.

EBV-transformed cells require miR-34a for efficient growth. (a) Schematic diagram of miR-34a sponge and control (CTRL) construct indicating features of the pL-CMV backbone and sorting plan. CMV, cytomegalovirus; EGFP, enhanced GFP; LTR, long terminal repeat. (b) Dual luciferase miR-34a indicator assay. BJAB cells, EF3D cells not transduced, and sorted EF3D cells transduced with GFP control vector (GFP) or miR-34a sponge (s34a) were each coinfected with a control firefly luciferase lentiviral construct and a Renilla luciferase construct containing 2 perfect miR-34a binding sites in the 3′ UTR. The Renilla/firefly luciferase ratio was determined for each cell line, and the values are plotted relative to BJAB cells as 100. The averages plus SEM of two indicator assays from independent transductions are shown. (c) qRT-PCR expression levels of mature miR-34a, miR-16, miR-15b, and miR-155 normalized to RNU48 for control (CTRL) and miR-34a sponge-transduced and sorted cells are shown. The averages of three independent transductions plus SEM are shown. (d) Growth curves of EF3D cells expressing GFP CTRL, sCXCR4, or s34a. The asterisks signify a P value of <0.05 for CTRL versus s34a. (e) Growth curves similar to those in panel d, except the cells were allowed to grow until saturation without changing the culture medium. The asterisks signify a P value of <0.01 for CTRL versus s34a. (f) EF3D LCLs were transduced with the MSCV-Puro based miR-34a sponge or control vector. Cells were selected in 1.25 μg/ml puromycin for 1 week prior to sorting for GFPhi cells. Growth of the sorted cells was measured as for panel d. The average values from three independent experiments plus SEM are plotted. (g) Growth curve as in panel d following transduction and sorting of HCT-116 cells. Averages and SEM from three independent experiments are shown.
Relative to GFP-expressing LCLs, cells expressing the miR-34a sponge were deficient in growth at normal dilution, as well as in achieving high density at saturation (Fig. 7d and e). Surprised by these observations, given the previously described role of miR-34a as a tumor suppressor, we constructed an additional sponge vector (MSCV based) and confirmed the growth deficiency in two independent LCL lines (Fig. 7f and data not shown). We also compared the growth of s34a cells with that of sCXCR4 cells, which express an independent control sponge containing binding sites for a small interfering RNA (siRNA) against CXCR4 (8). Consistently, s34a-expressing cells were deficient in growth at normal and saturating densities relative to sCXCR4- and GFP-expressing LCLs (Fig. 7d and e). As a final control, we transduced and sorted HCT-116 cells expressing the miR-34a sponge and GFP control vector. As predicted, miR-34a depletion in these cells led to a subtle increase in growth and clonogenicity (Fig. 7g and data not shown). Therefore, miR-34a functions in a cell-type-specific manner and is important for the growth of EBV-transformed cells.
DISCUSSION
In the study, we investigated the dynamic changes in miRNA expression during EBV-mediated primary B cell growth transformation. We identified sets of cellular miRNAs by expression profiling that were induced or repressed in long-term outgrowth, as well as miRNAs uniquely regulated early after infection. Although other groups have observed miRNA expression changes mediated by EBV infection (1, 14, 25, 29), our data define the global miRNA changes during EBV-mediated primary B cell outgrowth. Several miRNA expression changes were confirmed by qRT-PCR, and we included the first expression analysis of miRNAs in the EBV-positive AIDS diffuse large B cell lymphoma cell line IBL-1. The induction of putative progrowth onco-miRs, including miR-155 and -21, and repression of putative tumor suppressor onco-miRs, such as let-7 and miR-29 family members, confirmed and extended previous observations. We also defined a set of previously unrecognized EBV-regulated miRNAs. Furthermore, a set of miRNAs whose expression was regulated specifically during early proliferation was highly enriched for c-Myc-regulated miRNAs (34, 36). This is consistent with recent data from our laboratory indicating a transient period of high-level c-Myc mRNA and activity that is attenuated through LCL outgrowth (32). These data indicate that miRNA expression differences between EBV-transformed cells and BL cells (1) reflect changes due to latent EBV oncoproteins, as well as the potent effects of c-Myc in regulating miRNA expression. For example, miR-18a was induced by EBV during B cell outgrowth, though not to the extent observed in BL cell lines. Thus, EBV dynamically regulates the expression of miRNAs after primary B cell infection, and understanding the physiological relevance of these changes will be important in defining the role of miRNAs in virus-induced oncogenesis.
One miRNA whose expression increased through EBV-driven B cell proliferation was miR-34a. We detected expression of miR-34a in several EBV- and KSHV-derived tumor cell lines, indicating a plausible role for this miRNA in tumorigenesis. To discern this role, we investigated the regulation and function of miR-34a in EBV infection. EBV regulation of host gene expression during latency is largely accomplished by the oncoproteins EBNA-2 and LMP-1. While EBNA-2 was not capable of inducing miR-34a expression, LMP-1 was sufficient to induce miR-34a in B cells, similar to what was observed for miR-146a (2). LMP-1-mediated miR-34a induction required CTAR2 and canonical IKKβ-dependent NF-κB activation. Furthermore, LMP2A, which facilitates LMP1-mediated NF-κB activation, was also important to maintain efficient miR-34a expression. Although previous studies indicated that miR-34a expression is primarily regulated by the transcription factor p53, several recent reports suggest that p53-independent induction of miR-34a occurs in various cell types (4, 31). Our findings suggest that p53 is not required for miR-34a expression, since LMP1 induced miR-34a in BJAB cells, which lack functional p53. Furthermore, p53 inducing stimuli were largely ineffective at driving miR-34a expression in primary B cells or LCLs (data not shown).
Increased expression of miR-34a upon EBV infection of primary B cells and elevated expression in EBV- and KSHV-positive B lymphoma cell lines strongly implicate this miRNA in tumorigenesis. While this is contrary to what had been observed in other tumor types, we hypothesized that miR-34a is important for LCL maintenance. Overexpression of miR-34a in LCLs did not alter growth, while depletion of mature miR-34a impaired the growth of these cells. As miRNA function is mediated through targeting specific sets of mRNAs, it will be important to define the miR-34a targets in LCLs that may be sensitive to its loss. Many targets of miR-34a have been identified, including several critical growth control nodes, such as c-Myc, cyclin D1, E2F family members, cdk4, and cdk6 (4, 31, 41). We did not detect changes in c-Myc levels in miR-34a sponge-expressing LCLs (data not shown). Moreover, several of the canonical progrowth targets of miR-34a such as cyclin D1, cyclin E2, and c-Met are poorly or not expressed in LCLs. Rather, since the growth control properties of an LCL are dictated by expression of the latency III gene products, we propose that LCLs are more resistant to the toxic effects of miR-34a target repression than other cell or tumor lines and that a set of miR-34a targets must be repressed in LCLs for normal cell growth.
Two signaling pathways are likely targets for EBV-mediated miR-34a effects. First, others have shown, and we have preliminary evidence, that miR-34a targets multiple components of the Notch signaling pathway (19, 33, 35). Given that EBNA2 depends on the intracellular Notch downstream DNA binding factor RBP-Jκ (or CBF1) for transcriptional activity, derepression and potential activation of endogenous Notch may result in impaired LCL growth (49). Second, miR-34a sponge-expressing cells were less efficient in homotypic aggregation than their control counterparts (data not shown); therefore, adhesion molecules and cytoskeletal signaling proteins regulating homotypic aggregation in LCLs are also plausible miR-34a targets in LCLs. We are intensely investigating whether targets in these pathways are responsible for the miR-34a growth deficiency. In conclusion, the newly discovered progrowth function of miR-34a in B cell transformation contrasts with its canonical role as a tumor suppressor and highlights the importance of studying miRNA functions in different cell types.
Supplementary Material
ACKNOWLEDGMENTS
We thank Moshe Oren for kindly providing the miR-34a expression construct. We thank Dirk Dittmer, Geoffrey Wahl, Elliott Kieff, George Mosialos, Ethel Cesarman, and Rich Longnecker for generously providing cell lines and Kenneth Murphy and Ellen Cahir-McFarland for the pMSCV-IRES-GFP and pMSCV-LMP1-IRES-GFP constructs, respectively. We also thank the Duke Cancer Institute Flow Cytometry core, including Lynn Martinek. Bryan Cullen and his laboratory provided helpful discussions regarding miRNA reagents and analysis.
This work was supported by pilot grants to M.A.L. from the Stewart Trust, the American Cancer Society, and the Duke Cancer Institute, as well as an NCI-supported pilot award for collaborations between CFARs and Cancer Centers (iCHARM) (awarded from Penn CFAR; P30-AI-045008). S.D.L. was supported by NIH training grant T32-AI007392. E.G. was supported by K99-CA137860.
Footnotes
Published ahead of print 11 April 2012
Supplemental material for this article may be found at http://jvi.asm.org.
REFERENCES
- 1. Cameron JE, et al. 2008. Epstein-Barr virus growth/latency III program alters cellular microRNA expression. Virology 382:257–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cameron JE, et al. 2008. Epstein-Barr virus latent membrane protein 1 induces cellular MicroRNA miR-146a, a modulator of lymphocyte signaling pathways. J. Virol. 82:1946–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chang TC, et al. 2007. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell 26:745–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Christoffersen NR, et al. 2010. p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Cell Death Differ. 17:236–245 [DOI] [PubMed] [Google Scholar]
- 5. DE Oliveira DE, Ballon G, Cesarman E. 2010. NF-kappaB signaling modulation by EBV and KSHV. Trends Microbiol. 18:248–257 [DOI] [PubMed] [Google Scholar]
- 6. Devergne O, et al. 1996. Association of TRAF1, TRAF2, and TRAF3 with an Epstein-Barr virus LMP1 domain important for B-lymphocyte transformation: role in NF-kappaB activation. Mol. Cell. Biol. 16:7098–7108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dull T, et al. 1998. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72:8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ebert MS, Neilson JR, Sharp PA. 2007. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 4:721–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Farrell PJ, Allan GJ, Shanahan F, Vousden KH, Crook T. 1991. p53 is frequently mutated in Burkitt's lymphoma cell lines. EMBO J. 10:2879–2887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Faumont N, et al. 2009. c-Myc and Rel/NF-kappaB are the two master transcriptional systems activated in the latency III program of Epstein-Barr virus-immortalized B cells. J. Virol. 83:5014–5027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gao P, et al. 2009. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458:762–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Garzon R, Calin GA, Croce CM. 2009. MicroRNAs in cancer. Annu. Rev. Med. 60:167–179 [DOI] [PubMed] [Google Scholar]
- 13. Gatto G, et al. 2008. Epstein-Barr virus latent membrane protein 1 trans-activates miR-155 transcription through the NF-kappaB pathway. Nucleic Acids Res. 36:6608–6619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Godshalk SE, Bhaduri-McIntosh S, Slack FJ. 2008. Epstein-Barr virus-mediated dysregulation of human microRNA expression. Cell Cycle 7:3595–3600 [DOI] [PubMed] [Google Scholar]
- 15. Gottwein E, Cai X, Cullen BR. 2006. A novel assay for viral microRNA function identifies a single nucleotide polymorphism that affects Drosha processing. J. Virol. 80:5321–5326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gottwein E, Cullen BR. 2010. A human herpesvirus microRNA inhibits p21 expression and attenuates p21-mediated cell cycle arrest. J. Virol. 84:5229–5237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gottwein E, et al. 2007. A viral microRNA functions as an orthologue of cellular miR-155. Nature 450:1096–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guasparri I, Bubman D, Cesarman E. 2008. EBV LMP2A affects LMP1-mediated NF-kappaB signaling and survival of lymphoma cells by regulating TRAF2 expression. Blood 111:3813–3820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hashimi ST, et al. 2009. MicroRNA profiling identifies miR-34a and miR-21 and their target genes JAG1 and WNT1 in the coordinate regulation of dendritic cell differentiation. Blood 114:404–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. He L, et al. 2007. A microRNA component of the p53 tumour suppressor network. Nature 447:1130–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Iorio MV, Croce CM. 2009. MicroRNAs in cancer: small molecules with a huge impact. J. Clin. Oncol. 27:5848–5856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Izumi KM, Kieff ED. 1997. The Epstein-Barr virus oncogene product latent membrane protein 1 engages the tumor necrosis factor receptor-associated death domain protein to mediate B lymphocyte growth transformation and activate NF-kappaB. Proc. Natl. Acad. Sci. U. S. A. 94:12592–12597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jiang J, Lee EJ, Schmittgen TD. 2006. Increased expression of microRNA-155 in Epstein-Barr virus transformed lymphoblastoid cell lines. Genes Chromosomes Cancer 45:103–106 [DOI] [PubMed] [Google Scholar]
- 24. Kieff E, Rickinson A. 2007. Epstein-Barr virus and its replication, p 2603–2654 In Knipe DM, Howley PM. (ed), Fields virology, 5 ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 25. Lawrie CH, et al. 2008. MicroRNA expression in lymphocyte development and malignancy. Leukemia 22:1440–1446 [DOI] [PubMed] [Google Scholar]
- 26. Linnstaedt SD, Gottwein E, Skalsky RL, Luftig MA, Cullen BR. 2010. Virally induced cellular miR-155 plays a key role in B-cell immortalization by EBV. J. Virol. 84:11670–11678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lu F, et al. 2008. Epstein-Barr virus-induced miR-155 attenuates NF-kappaB signaling and stabilizes latent virus persistence. J. Virol. 82:10436–10443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Motsch N, Pfuhl T, Mrazek J, Barth S, Grasser FA. 2007. Epstein-Barr virus-encoded latent membrane protein 1 (LMP1) induces the expression of the cellular microRNA miR-146a. RNA Biol. 4:131–137 [DOI] [PubMed] [Google Scholar]
- 29. Mrazek J, Kreutmayer SB, Grasser FA, Polacek N, Huttenhofer A. 2007. Subtractive hybridization identifies novel differentially expressed ncRNA species in EBV-infected human B cells. Nucleic Acids Res. 35:e73 doi:10.1093/nar/gkm244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mu P, et al. 2009. Genetic dissection of the miR-17∼92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev. 23:2806–2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Navarro F, et al. 2009. miR-34a contributes to megakaryocytic differentiation of K562 cells independently of p53. Blood 114:2181–2192 [DOI] [PubMed] [Google Scholar]
- 32. Nikitin PA, et al. 2010. An ATM/Chk2-mediated DNA damage-responsive signaling pathway suppresses Epstein-Barr virus transformation of primary human B cells. Cell Host Microbe 8:510–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Njie R, et al. 2009. The effects of acute malaria on Epstein-Barr virus (EBV) load and EBV-specific T cell immunity in Gambian children. J. Infect. Dis. 199:31–38 [DOI] [PubMed] [Google Scholar]
- 34. O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. 2005. c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435:839–843 [DOI] [PubMed] [Google Scholar]
- 35. Pang RT, et al. 2010. MicroRNA-34a suppresses invasion through downregulation of Notch1 and Jagged1 in cervical carcinoma and choriocarcinoma cells. Carcinogenesis 31:1037–1044 [DOI] [PubMed] [Google Scholar]
- 36. Petrocca F, Vecchione A, Croce CM. 2008. Emerging role of miR-106b-25/miR-17-92 clusters in the control of transforming growth factor beta signaling. Cancer Res. 68:8191–8194 [DOI] [PubMed] [Google Scholar]
- 37. Rahadiani N, Takakuwa T, Tresnasari K, Morii E, Aozasa K. 2008. Latent membrane protein-1 of Epstein-Barr virus induces the expression of B-cell integration cluster, a precursor form of microRNA-155, in B lymphoma cell lines. Biochem. Biophys. Res. Commun. 377:579–583 [DOI] [PubMed] [Google Scholar]
- 38. Ranganath S, et al. 1998. GATA-3-dependent enhancer activity in IL-4 gene regulation. J. Immunol. 161:3822–3826 [PubMed] [Google Scholar]
- 39. Raver-Shapira N, et al. 2007. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol. Cell 26:731–743 [DOI] [PubMed] [Google Scholar]
- 40. Sullivan CS, Cullen BR. 2009. Non-coding regulatory RNAs of the DNA tumor viruses, p 645–682 In Damania B, Pipas J. (ed), DNA Tumor Viruses. Springer, New York, NY [Google Scholar]
- 41. Sun F, et al. 2008. Downregulation of CCND1 and CDK6 by miR-34a induces cell cycle arrest. FEBS Lett. 582:1564–1568 [DOI] [PubMed] [Google Scholar]
- 42. Tarasov V, et al. 2007. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle 6:1586–1593 [DOI] [PubMed] [Google Scholar]
- 43. Tazawa H, Tsuchiya N, Izumiya M, Nakagama H. 2007. Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc. Natl. Acad. Sci. U. S. A. 104:15472–15477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tusher VG, Tibshirani R, Chu G. 2001. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U. S. A. 98:5116–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Welch C, Chen Y, Stallings RL. 2007. MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene 26:5017–5022 [DOI] [PubMed] [Google Scholar]
- 46. Yin Q, et al. 2008. MicroRNA-155 is an Epstein-Barr virus-induced gene that modulates Epstein-Barr virus-regulated gene expression pathways. J. Virol. 82:5295–5306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang J, et al. 2009. Patterns of microRNA expression characterize stages of human B cell differentiation. Blood 113:4586–4594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhao B, et al. 2006. RNAs induced by Epstein-Barr virus nuclear antigen 2 in lymphoblastoid cell lines. Proc. Natl. Acad. Sci. U. S. A. 103:1900–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zweidler-McKay PA, et al. 2005. Notch signaling is a potent inducer of growth arrest and apoptosis in a wide range of B-cell malignancies. Blood 106:3898–3906 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.